

Abstract
We studied how combination antiviral therapy affects B cell abnormalities associated with HIV-1 infection, namely elevated circulating immunoglobulin (Ig)G antibody-secreting cell (ASC) frequencies and hypergammaglobulinemia. Within a few weeks of starting antiviral therapy, there is a marked decline in IgG-ASC frequency in both acutely and chronically infected people, whereas the hypergammaglobulinemia often present during chronic infection is more gradually resolved. These reductions are sustained while HIV-1 replication is suppressed. HIV-1 antigen–specific B cell responses are also affected by therapy, manifested by a rapid decline in circulating gp120-specific ASCs. Anti-gp120 titers slowly decrease in chronically infected individuals and usually fail to mature in acutely infected individuals who were promptly treated with antiretroviral therapy. Long-term nonprogressors have high titer antibody responses to HIV-1 antigens, but no detectable gp120-specific IgG-ASC, and normal (or subnormal) levels of total circulating IgG-ASC. Overall, we conclude that HIV-1 infection drives B cell hyperactivity, and that this polyclonal activation is rapidly responsive to decreases in viral replication caused by combination antiviral therapy.
Keywords: hypergammaglobulinemia, human immunodeficiency virus 1, B cells, antiretroviral therapy, antibody
Infection with human immunodeficiency virus type 1 (HIV-1) is associated with several abnormalities of B cell function (1–6). In consequence, individuals who have been infected with HIV-1 for several years (chronic infection) are often hypergammaglobulinemic, with elevations in plasma IgG of up to twice the normal level (1, 3, 7, 8). Most of the excess antibody is not HIV-1 antigen–specific, reflecting instead a generalized, polyclonal activation of B cell functions caused by systemic HIV-1 infection (1–5). Consistent with this, B cells from HIV-1–infected people spontaneously secrete IgG in culture to a much greater extent than cells from uninfected individuals, yet respond relatively poorly to B cell mitogens in vitro (1, 3). Once they develop, these abnormalities are sustained throughout the course of infection. However, what drives them is unclear.
Superimposed on these polyclonal responses are strong, antigen-specific B cell responses to multiple HIV-1 antigens, the most dominant being to the viral Env (gp120 and gp41) and Gag (p24 and p17) proteins (9, 10). The antigen-specific responses are initiated within a few weeks of infection (seroconversion) and increase in magnitude for ∼1 yr (11–13). However, the anti-Env and anti-Gag responses differ in the extent to which they are sustained throughout the course of HIV-1 infection. Anti-Env antibodies are usually present at high titers even in individuals who progress to disease, often right until death (11, 14–20). In contrast, anti-Gag responses are lost (or in some cases never fully develop) during disease progression; a low anti-Gag titer is a poor prognostic indicator, associated with low CD4 T cell counts and high viral loads (11, 14–20). This is paradoxical considering the lack of antiviral function associated with anti-Gag antibodies, but it is thought that the decline in the anti-Gag response is a surrogate marker for the loss of T cell help caused by HIV-1 infection of CD4+ T cells (11, 16, 18, 20). Why the anti-Env response is so strongly sustained under these T cell help–depleted conditions is unresolved, but we have suggested that it may reflect the existence of alternative antigen presentation pathways for the highly glycosylated envelope glycoproteins (11).
Although the abnormalities in B cell function and the antigen-specific production of anti–HIV-1 antibodies are both responses to HIV-1 infection, their dependence on sustained viral replication has not been defined. Nor is it clear that they are driven by the same mechanisms. The development of combinations of potent antiretroviral drugs (protease and reverse transcriptase inhibitors) that can suppress HIV-1 replication by several orders of magnitude in vivo has now allowed us to examine these questions. Specifically, we have assessed how the different B cell responses change when viremia is suppressed. We show that sustained reductions in plasma viremia after antiviral therapy cause the numbers of gp120-specific and total IgG antibody-secreting cells (ASC)1 to decline within a few days. Plasma anti-gp120 and anti-p24 antibody titers in chronically infected individuals decline much more gradually, and hypergammaglobulinemia is also eventually resolved. When therapy is initiated during acute HIV-1 infection, the IgG-ASC frequency again decreases rapidly, and in most cases antigen-specific responses are also diminished or fail to increase.
Materials and Methods
Study Subjects.
Cohort 1 comprised 19 drug-naive individuals, 16 of whom were screened for entry into a phase I clinical trial of an antiviral regimen involving three reverse transcriptase inhibitors (zidovudine, lamivudine, and abacavir) and one experimental protease inhibitor (141W94) (Table 1). Of these individuals, 4 had been infected with HIV-1 for <90 d (acute infection) and 12 had been infected for between 4 mo and 12 yr (chronic infection). The four acutely infected cases and six of the chronically infected cases met the criteria for entry into the trial, initiated therapy, and were followed longitudinally for up to 30 wk (21). The remaining six chronically infected individuals did not participate in the trial, so provide only baseline, pretherapy data. Also included in cohort 1 are three long-term nonprogressors (LTNPs), two of whom have been described previously (individuals AD-19 and AD-65; references 11, 22). An additional individual (S006) who had been infected for 1–2 yr had no detectable plasma viremia (<100 RNA copies/ml in the branched DNA [bDNA] assay), with a CD4 count of 413. He was not formally included among the LTNPs in cohort 1 because of the short duration of his infection, but is referred to in the context of studies on LTNPs as a prospective slow progressor with undetectable plasma viremia. All participants gave written informed consent for all the studies described below. Adherence to therapy was confirmed by interview and by the absence of any resurgence in viral loads, measured using the bDNA assay. Nonadherence is noted in the text below.
Table 1.
Baseline CD4 T Cell Counts and Plasma Viral Loads for Cohort 1
| Patient groups | Patient ID | Duration of infection | CD4 count | Plasma viral load | Treatment | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| cells/μl | HIV-1 RNA copies/ml | |||||||||
| Acute infection | No. 902 | <90 d | 999 | 383,000 | Combination | |||||
| No. 909* | <90 d | 734 | 509 | antiretroviral | ||||||
| No. 910 | <90 d | 476 | 9,168 | therapy | ||||||
| No. 911 | <90 d | 685 | 21,800 | |||||||
| Chronic infection | No. 889 | 4 mo | 333 | 9,399 | Combination | |||||
| No. 893 | 12 yr | 181 | 25,700 | antiretroviral | ||||||
| No. 894 | 6 mo | 484‡ | 6,535 | therapy | ||||||
| No. 895 | <1yr | 392 | 16,010 | |||||||
| No. 896§ | >5 yr | 12 | 164,800 | |||||||
| No. 897 | <1yr | 392 | 18,560 | |||||||
| Chronic infection | S001 | 1 yr | 317 | 23,000 | Untreated | |||||
| S002 | 15 mo | 224 | 6,000 | |||||||
| S003 | <1 yr | 417 | 7,800 | |||||||
| No. 891 | 2–3 yr | 288 | 27,360 | |||||||
| S004 | >8 yr | 459 | 4,000 | |||||||
| S005 | 2–3 yr | 513 | 27,570 | |||||||
| LTNP | LTS-6 | >12 yr | 967 | <100 | Untreated | |||||
| AD-65‖ | >15 yr | 825 | <100 | |||||||
| AD-19‖ | >14 yr | 1,053 | <100 |
Of the 19 members of cohort 1, 10 were followed longitudinally after starting therapy with three reverse transcriptase inhibitors and one protease inhibitor. The CD4 counts given for the LTNPs were measured within the 3-mo period before the assays for ASCs.
Subject was first analyzed within 1 wk of starting therapy.
CD4 count was determined at 3 wk.
No baseline PBMC sample was available. The first sample obtained for ELISPOT assay was taken 3 wk after therapy began.
Longitudinal studies that required fresh cells were performed only on 10 members of cohort 1. However, an additional cohort was also formed. Cohort 2 comprises 13 participants in a phase I clinical trial of a different 4-drug regimen (reverse transcriptase inhibitors zidovudine and lamivudine; protease inhibitors saquinavir and ritonavir; Table 2). 3 of the participants were acutely infected with HIV-1, and 10 were chronically infected (23). The median CD4 count for the 10 chronically infected individuals was 434 cells/μl (range 260–910), and for the 3 acutely infected individuals, it was 399 cells/μl (range 136–1,039). Each individual had been treated for between 16 and 36 wk before this immunological study was initiated, so samples were only available for assays that could be performed prospectively. All 13 participants had undetectable levels of plasma viremia (<100 RNA copies/ml in the reverse transcriptase PCR assay) at the time we analyzed their circulating IgG-ASC frequencies.
Table 2.
CD4 Counts, Plasma Viral Loads, Plasma IgG Concentrations, and HIV-1–specific Midpoint Antibody Titers for Cohort 2
| Patient ID | Duration of infection | CD4 count | Plasma viral load | Plasma IgG | Anti-gp120 titer | Anti-p24 titer | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before therapy | After therapy | Before therapy | After therapy | Before therapy | After therapy | Difference | Before therapy | After therapy | Fold reduction | Before therapy | After therapy | Fold reduction | ||||||||||||||||
| cells/μl | RNA copies/ml | (mg/ml) | ||||||||||||||||||||||||||
| Chronic | ||||||||||||||||||||||||||||
| No. 1002 | 4 mo | 537 | 565 | 76,916 | <100 | 15.5 | 12.5 | −3.0 | 38,000 | 11,000 | 3.5 | 30,000 | 2,100 | 14.3 | ||||||||||||||
| No. 1003 | 13 mo | 318 | 517 | 87,000 | <100 | 16.7 | 12.8 | −3.9 | 19,000 | 3,000 | 6.3 | 2,100 | 250 | 8.4 | ||||||||||||||
| No. 1004 | 9 mo | 402 | 462 | 40,468 | <100 | 15.3 | 8.9 | −6.4 | 38,000 | 10,000 | 3.8 | 580 | <100 | 5.8 | ||||||||||||||
| No. 1006 | 3 yr | 376 | 260 | 84,814 | <100 | 13.5 | 10.7 | −2.7 | 28,000 | 11,000 | 2.5 | 1,300 | 360 | 3.6 | ||||||||||||||
| No. 1007 | unknown | 177 | 442 | 133,256 | <100 | 29.5 | 18.5 | −11.0 | 150,000 | 52,000 | 2.9 | 4,286,000 | 710,000 | 6.0 | ||||||||||||||
| No. 1008 | >7 yr | 467 | 426 | 721,855 | <100 | 23.8 | 18.6 | −5.2 | 170,000 | 120,000 | 1.4 | 170,000 | 50,000 | 3.4 | ||||||||||||||
| No. 1009 | >8 yr | 665 | 910 | 22,288 | <100 | 15.1 | 9.8 | −5.4 | 100,000 | 45,000 | 2.2 | <100 | <100 | – | ||||||||||||||
| No. 1010 | 11 yr | 284 | 412 | 248,433 | <100 | 17.6 | 8.7 | −8.9 | 58,000 | 28,000 | 2.1 | 48,000 | 14,000 | 3.4 | ||||||||||||||
| No. 1011 | 4 yr | 354 | 412 | 7,955 | <100 | 15.3 | 12.7 | −2.5 | 110,000 | 60,000 | 1.8 | 16,000 | 5,000 | 3.2 | ||||||||||||||
| No. 1013 | unknown | 124 | 299 | 786,372 | <100 | 19.0 | 10.0 | −9.0 | 14,000 | 3,000 | 4.7 | 140 | <100 | 1.4 | ||||||||||||||
| 365 * | 434 | 85,907 | <100 | 16.1 | 11.6 | −5.3 | 48,000 | 19,500 | 2.7 | 9,050 | 1,230 | 3.6 | ||||||||||||||||
| Acute | ||||||||||||||||||||||||||||
| No. 2001 | <90 d | 290 | 399 | 38,940 | <100 | 8.6 | 7.3 | −1.2 | <100 | <100 | – | 13,000 | 200 | 65.0 | ||||||||||||||
| No. 2002 | <90 d | 580 | 1,039 | 125,283 | <100 | 12.4 | 9.6 | −2.8 | 9,000 | 2,400 | 3.8 | 210 | <100 | 2.1 | ||||||||||||||
| No. 2004 | <90 d | 47 | 136 | 397,700 | <100 | 20.6 | 18.5 | −2.1 | 900 | 250 | 3.6 | <100 | <100 | – | ||||||||||||||
Each member of cohort 2 was treated with two reverse transcriptase and two protease inhibitors. Data are derived from 10 chronically and 3 acutely infected individuals before and after therapy. The post-therapy sample was taken after 36 wk, except for subjects no. 1013 and no. 2002, who had been treated for 32 wk, and subject no. 2004, who had been treated for 16 wk. Antibody titers <1:100 (i.e., barely detectable) are scored as 1:100 for the purpose of calculation.
Median values of chronic patients are shown in bold.
From cohorts 1 and 2, individuals no. 896 (cohort 1) and no. 2004 (cohort 2) can be defined as having progressive HIV-1 infection, based on their CD4 counts and viral loads (Tables 1 and 2). Six uninfected laboratory workers formed a control group relevant to both cohorts 1 and 2.
Clinical Assays.
Plasma HIV-1 RNA levels were determined using the bDNA assay (cohort 1; Chiron Mimotopes, San Diego, CA) or a reverse transcriptase PCR assay from Roche Labs. (cohort 2; Nutley, NJ). CD4+ T cell counts were determined by flow cytometry using directly conjugated mAbs and a FACSCalibur® instrument (Becton Dickinson, La Jolla, CA).
PBMC Isolation.
Venous blood was collected into heparin-coated tubes. PBMCs were isolated within 4 h of collection by Ficoll gradient separation. The cells were washed three times (at 1,000 rpm for 10 min) in cold PBS to remove platelets and resuspended at 106/ml in RPMI 1640 medium containing 10% FCS (R-10 medium).
ELISPOT Assay.
MultiScreen 96-well filtration plates (Millipore, Bedford, MA) were coated overnight at 4°C with either goat anti–human IgM Fc or goat anti–human IgG Fc (Sigma Chemical Co., St. Louis, MO) at 10 μg/ml in PBS (pH 7.4) for detection of total IgM-specific or IgG-specific ASCs, respectively. For detection of anti-gp120 ASCs, the wells were coated with the sheep anti-gp120 antibody D7324 (Aalto BioReagents, Dublin, Ireland) followed by rgp120 JR-FL (a gift from Paul Maddon, Progenics Pharmaceuticals Inc., Tarrytown, NY) at 100 ng/ml in PBS. To detect anti-p24 ASCs, the wells were directly coated with glutathione-S-transferase–p24 fusion protein at 5 μg/ml (the vector construct was a gift from Ian Jones, NERC Institute of Virology, Oxford, UK). All plates were washed three times with 200 μl of PBS containing 0.5% Tween 20 (PBS-T), blocked with 3% BSA in PBS for 30 min at 37°C, and then washed with R-10 medium. Four serial threefold dilutions of PBMCs from a stock at 106/ml were prepared in R-10 medium, and 200 μl aliquots were added to each well. The plates were incubated overnight at 37°C in a humidified incubator, and the cells were removed by washing the wells six times with PBS-T. Antibodies secreted from the cells and bound to the plate were detected by incubation for 2 h at room temperature with alkaline phosphatase–conjugated anti–human IgG Fc (Accurate Chemicals, Westbury, NY) or anti–human IgM μ chain (Sigma Chemical Co.), followed by three washes in PBS-T and one in PBS, and the addition of NBT/BCIP (nitroblue tetrazolium chloride/5-bromo-4 chloro-3-indoyl phosphate p-toluidine salt) substrate for 15 min (GIBCO BRL, Gaithersburg, MD). The plates were rinsed in tap water and allowed to dry before blue spots (positive reactions) were counted using a dissecting microscope. The ELISPOT assay was highly reproducible; duplicate samples provided ASC frequencies within twofold of each other. Control experiments showed that there was negligible binding of cells to wells in the absence of coating antibodies or antigen.
To monitor the efficiency of the ELISPOT assay, B cell lines secreting IgG mAbs to gp120 (A32, an Epstein Barr virus–transformed B cell line from James Robinson, Tulane University, New Orleans, LA) or p24 (1D7, a human heterohybridoma line from Herman Katinger, Institute of Applied Microbiology, Vienna, Austria) were used as positive controls. Approximately 5% of the cells from the A32 line and >50% of the cells from the ID7 line produced ELISPOTS on gp120- or p24-coated plates, respectively; these differing sensitivities are probably attributable to the much larger size and higher levels of antibody production of the 1D7 cells. The numbers of spots were approximately twofold lower when wells were coated with anti-IgG Fc to detect total IgG. Thus the antigen-specific assay was slightly more sensitive than the total IgG assay. Subsequent studies showed that use of an anti-IgG Fab coating antibody (Accurate Chemicals) instead of the anti-IgG Fc allowed more sensitive detection. However, this reagent was not used routinely in the clinical studies, so our total ASC numbers are an underestimate by a factor of approximately two. This does not affect any of the relative values recorded below.
Measurement of Antibodies in Culture Supernatants and Plasma.
PBMCs were cultured at 106/well in 24-well plates in 2 ml of R-10 medium. After 8 d, supernatants were collected and anti-gp120 and anti-p24 antibody titers were measured by ELISA as previously described (11). Plasma anti-gp120 and anti-p24 titers were also determined by this method. Titers are presented as the plasma dilution at which the mid-point of the antibody–antigen titration curve was reached; low titers were calculated by extrapolation, as previously described (11). Total IgG titers in culture supernatants were measured in the same way except that the plates were coated with goat anti–human IgG Fc (5 μg/ml; Sigma Chemical Co.). Total IgG concentrations in plasma were determined by a commercial laboratory (SmithKline Beecham Laboratories, Syosset, NY).
Statistical Analyses.
IgG-ASC and IgM-ASC numbers and levels of secreted IgG in different groups in cohort 1 were compared using a Mann-Whitney nonparametric test, and P values <0.05 are reported. IgG-ASC levels were compared with viral load and CD4 count using Pearson's correlation, and r values and P values (<0.05) are recorded. Statistical estimates for the half-life of IgG-ASC for eight members of cohort 1 were obtained either by logarithmic linear regression analysis or by determining the rate of descent between two consecutive time points. Half-lives for the rate of clearance of excess plasma IgG and the decay in anti-gp120 and anti-p24 titers were calculated by nonlinear fitting of an exponential model with a nonzero asymptote.
Results
Correlation between Circulating IgG-ASC Frequency and Plasma Viremia in HIV-1–infected Individuals.
To measure the number of circulating ASCs, including those specific for HIV-1 antigens, we used an ELISPOT assay since this allows direct quantification of the numbers of B cells activated in vivo without further cultivation and stimulation (24, 25). Preliminary experiments indicated that it was necessary to use freshly isolated PBMCs in the ELISPOT assay; a freeze–thaw cycle or even storing the cells, refrigerated, overnight resulted in a significant reduction in the ASC frequencies recorded. This practical consideration meant that we could not analyze blood samples retrospectively with the ELISPOT assay. We therefore studied individuals who were willing to provide fresh blood on a regular basis.
Baseline ASC frequencies were measured using samples from 18 of the 19 drug-naive HIV-1–infected individuals (cohort 1). Subject no. 896 was not included in this analysis as he had already been receiving therapy for 3 wk before the availability of the first blood sample so no baseline determination was possible. Of the 18 people studied, 4 had been infected with HIV-1 for <90 d (acute infection), 11 for between 4 mo and 12 yr (chronic infection), and 3 were LTNPs (Table 1). ASC frequencies were also determined in 6 uninfected individuals for comparison. The frequencies of IgG-ASC in both acutely (median 1,683/106 PBMCs) and chronically (2,185/106 PBMCs) infected individuals were significantly elevated (P <0.05) compared with uninfected donors (177/106 PBMCs), although the range was very broad (Fig. 1 a). However, the LTNPs had low levels of circulating IgG-ASC (91/106 PBMCs) that were slightly lower than those in uninfected individuals. On average, IgG-ASC constituted around 0.2% of total PBMCs (range 0.03–0.46%) for acutely and chronically infected members of cohort 1.
Figure 1.

Baseline ASC frequencies and spontaneous IgG secretion in cohort 1. ASC frequencies were determined in freshly isolated PBMCs from uninfected individuals (n = 6) and from three groups of individuals who had been infected with HIV-1 for varying lengths of time. All study subjects were drug naive at the time of assay except for one individual whose baseline value could only be determined after 1 wk of therapy (indicated by asterisk). ELISPOT plates were coated with either (a) anti–human IgG Fc to detect IgG-ASCs or (b) anti–human IgM Fc to detect IgM-ASCs. (c) Spontaneous IgG secretion after 8 d of culture was also measured by ELISA for the same individuals. Bars represent the median values for each group.
Unlike IgG-ASC frequencies, IgM-ASC levels were not significantly elevated in HIV-1–infected individuals; only three chronically infected people had IgM-ASC frequencies that were higher than those in normal donors (Fig. 1 b). The LTNPs (and also the aviremic individual no. S006) had no measurable IgM-ASC, whereas all the acutely and most of the chronically infected individuals and the uninfected controls had low, but detectable, levels. Thus, B cell hyperactivity in HIV-1 infection predominantly comprises memory B cells expressing surface IgG, reactivated in a largely non–antigen-specific manner, and is not a more generalized phenomenon.
In addition to measuring IgG-ASC frequencies by ELISPOT, we also quantitated spontaneous IgG secretion from B cells cultured for 8 d. B cells from HIV-1–infected individuals (other than the LTNPs) secreted significantly more IgG than B cells from uninfected people (P <0.05; Fig. 1 c). There was considerable donor-to-donor variation in the extent of IgG secretion, which correlated with the variable IgG-ASC frequencies in the same individuals. Only a small fraction of the secreted IgG was gp120-specific, and even less was p24-specific (data not shown). B cells from the LTNPs did not secrete detectable amounts of IgG in culture, consistent with the almost complete absence of IgG-ASC in these cultures.
Comparing the IgG-ASC frequencies in the blood with plasma HIV-1 RNA levels showed that there was a positive correlation between these parameters (r = 0.52, P <0.05; Fig. 2). The highest IgG-ASC frequency was found in an acutely infected individual (no. 902; 4,640 IgG-ASC/106 PBMCs) who also had the highest viral load (383,000 RNA copies/ml). The individuals with the lowest viral loads (the LTNP and slow progressor no. S006) had almost undetectable levels of IgG-ASC. A positive correlation was still evident when the three LTNPs were excluded, but this no longer reached statistical significance (r = 0.51, P = 0.052). In addition, there was no correlation between CD4 count and IgG-ASC frequency, both for the group as a whole (n = 18) and when the three LTNPs were excluded.
Figure 2.

Correlation between the number of IgG-ASCs and plasma viral load. The 18 drug-naive HIV-1–infected individuals from cohort 1 were studied as in Fig. 1. Viral load was determined using the bDNA assay and IgG-ASC frequencies were determined by ELISPOT.
Antiviral Therapy Rapidly Reduces the Numbers of Circulating IgG-ASCs.
The baseline studies indicated that B cell hyperactivity (elevated IgG-ASC frequency) was associated with the extent of HIV-1 replication (plasma viremia). To determine whether a reduction in viral replication also lowered the IgG-ASC frequency, we studied four acutely and six chronically infected individuals from cohort 1 who participated in a combination antiviral therapy trial involving one protease inhibitor and three reverse transcriptase inhibitors. IgG-ASC frequencies were measured longitudinally in these 10 individuals over a 6–20-wk period.
Plasma viremia was rapidly reduced in each recipient of combination therapy. None of the 10 participants had a detectable viral load after 6 wk of therapy, as determined using a bDNA assay with a sensitivity limit of ∼100 HIV-1 RNA copies/ml. This was associated with a substantial reduction in the number of circulating IgG-ASCs in each individual who had an initially elevated level, irrespective of whether they were acutely or chronically infected (Fig. 3). By 6–10 wk after therapy was initiated, the median IgG-ASC frequency for all 10 participants at baseline (2,185/106 PBMCs) had been reduced by 5.8-fold to 378/106 PBMCs, only slightly elevated compared with the frequency in uninfected controls (177/106 PBMCs). However, the median reduction for the seven individuals who had the highest baseline IgG-ASC frequencies was 8-fold, and the extent of the reduction could be as high as 29-fold (e.g., no. 902) (Fig. 3 a). The extent of spontaneous IgG secretion from B cells in culture was also reduced after antiviral therapy, consistent with the reduction in the circulating IgG-ASC frequency. Thus, in 10 individuals from cohort 1 who were included in the trial, the median pretherapy IgG-secretion level was 215 ng/ml, and the post-therapy level was 13 ng/ml (data not shown).
Figure 3.

Longitudinal analysis of the effects of antiretroviral therapy on IgG-ASC frequency. Four acutely infected and six chronically infected individuals from cohort 1 were studied. Baseline data were not available from subjects no. 909 and no. 896, so the first data points are from wk 1 and 3 after therapy was initiated, respectively.
Subject no. 902's IgG-ASC frequency declined rapidly as his viral load dropped (Fig. 3 a). However, before the visit at week 20 he discontinued the therapeutic regimen for 10 d, resuming treatment 1 wk before returning to the clinic. A transient resurgence in IgG-ASC frequency was then noted at week 20 (Fig. 3 a; asterisk). All other participants reported strict adherence to the regimen and their levels of IgG-ASC remained depressed during the study period. The individual with the highest IgG-ASC frequency (no. 896; 751/106 cells) after 8 wk of therapy was chronically infected, with advanced disease and a CD4 count of 12 cells/ μl (Table 1).
The decline in IgG-ASC frequency was usually rapid, either coincident with the decline in plasma viremia or commencing shortly thereafter, although the kinetics of the decline varied between individuals (Fig. 4). Data derived from 8 of the 10 recipients were used to estimate the half-life of circulating IgG-ASCs (too few data points were available for no. 909, and reliable data could not be obtained from no. 895). This was calculated to be between 7 and 26 d (1–3.5 wk) with a median of 16 d, and was similar in both acutely and chronically infected individuals. These are likely to be minimum estimates, given that viral RNA can still be measured during the first few weeks of treatment. More frequent sampling of blood during the early stages of therapy would have refined our estimates, but this was not always possible.
Figure 4.


Effects of antiretroviral therapy on IgG-ASC frequencies and viral load. Longitudinal IgG-ASC and viral load data for (a) four acutely and (b) six chronically infected individuals from cohort 1. Antiviral therapy was initiated on day 0. Baseline ASC data were not available from subjects no. 909 and no. 896, so the first data points are from wk 1 and 3 after therapy was initiated, respectively. The half-lives of IgG-ASCs are indicated on the figures for nine of the individuals. Note that, unlike in Fig. 3, the IgG-ASC frequencies are plotted on different scales for each individual, in order to depict the rates of change of ASC frequency with maximum clarity.
It should be noted that the above half-lives reflect changes in the IgG-ASC frequency as a dynamic population in the peripheral blood. Thus, we have not determined whether the decline in IgG-ASC frequency in the blood, calculated as a half-life above, is due to the death of these cells, to their reversion to a quiescent state, to their redistribution into other tissue compartments, or to a reduction in the generation of new IgG-ASCs. Any or all of these factors could contribute to the overall reduction in IgG-ASC frequency observed, and to the rate at which it occurs.
Sustained Reduction in IgG-ASC Frequency after Antiviral Therapy.
To determine whether the reduction in circulating IgG-ASCs caused by combination therapy was a prolonged effect, we studied a separate cohort who had been receiving combination therapy for up to 36 wk (cohort 2). The 10 chronically infected individuals had been treated with a 4-drug regimen for between 32 and 36 wk, including two reverse transcriptase inhibitors and two protease inhibitors. Since the ELISPOT assay can only be performed on freshly isolated PBMCs, we were unable to determine baseline IgG-ASC frequencies retrospectively for cohort 2. However, it is reasonable to assume that it would have been broadly comparable to the baseline level in the chronically infected members of cohort 1 (2,185 IgG-ASC/ 106 PBMCs). The median IgG-ASC frequency in cohort 2 after 32–36 wk of therapy was low (339/106 PBMCs), and comparable to that in cohort 1 after 6–8 wk of therapy (378/ 106 PBMCs). This suggests that the rapid reduction in B cell hyperactivity upon therapy is indeed a sustained effect.
The median IgG-ASC frequency in three acutely infected individuals treated for 16–36 wk with this second 4-drug regimen (cohort 2) was also low (292/106 PBMCs), and very similar to the median for individuals treated during acute infection in cohort 1 (291/106 PBMCs). However, one individual (no. 2004) still had a high IgG-ASC frequency after 16 wk of therapy (898 IgG-ASC/106 PBMCs). This individual also had a low CD4 T cell count (136 cells/ μl). This profile is similar to individual no. 896 in cohort 1 who also had progressive infection (Figs. 3 b and 4 b); whether this poor response to antiretroviral therapy will be the norm for individuals with advanced disease will require further studies. The presence of elevated number of IgG-ASCs in these two individuals after therapy does, however, suggest that the reductions in IgG-ASC frequencies seen in other cases are not due to a direct effect of the antiviral drugs on B cells.
HIV-1 Antigen–specific IgG-ASC Frequencies and Specific Antibody Titers: Responses to Antiviral Therapy.
To see whether antiviral therapy also affected HIV-1 antigen–specific B cell responses in our study cohorts, we determined the frequencies of IgG-ASC specific for gp120 and p24, and the plasma antibody titers to the same proteins. Specific IgG-ASC frequencies were measured by ELISPOT using plates coated with recombinant gp120 or p24 proteins. Antibody titers were determined by ELISA, using the same proteins.
At baseline, most members of cohort 1 had low levels of gp120-specific IgG-ASC, which constituted only a minor proportion of the total number of IgG-ASCs (Fig. 5 and data not shown). Only two of the four acutely infected individuals had a detectable level of gp120-specific IgG-ASCs (16 and 5 anti-gp120 ASC/106 PBMCs, respectively; <0.2% of total IgG-ASCs), whereas none of the LTNPs did. Anti-gp120 IgG-ASCs were present at greater frequency in the chronically infected individuals (Fig. 5 b and data not shown); 8 of the 11 had from 15 to 429 anti-gp120 ASCs/106 PBMCs, equivalent to 0.6–14.9% of the total IgG-ASCs.
Figure 5.


Effects of antiretroviral therapy on HIV-1–specific antibody titer and ASC frequency. Depicted are plasma anti-gp120 (•) and anti-p24 (▴) midpoint antibody titers and gp120- (○) and p24-specific (▵) ASC frequencies, in (a) four acutely infected and (b) six chronically infected individuals from cohort 1. Antiviral therapy was initiated on day 0. The first data point for subject no. 909 is from 1 wk after therapy began. Fresh PBMCs were not available from individual no. 896 on day 0 for ASC frequency determination.
The three chronically infected individuals whose circulating gp120-specific IgG-ASC frequency exceeded 5% of the total IgG-ASCs responded to antiviral therapy by losing all detectable gp120-specific IgG-ASCs within 3 wk, and this loss persisted for the duration of the study (Fig. 5 b). Consistent with this, only 3 of the 10 chronically infected individuals who had been treated for 36 wk (cohort 2) had detectable gp120-specific IgG-ASCs, and then only at very low levels (<20/106 PBMCs). The proportion of IgG-ASC specific for p24 was even lower than that for gp120; these cells were detected in only four drug-naive subjects (one acute and three chronic) from cohort 1 (Fig. 5 and data not shown). After treatment, anti-p24 IgG-ASCs were no longer detectable in these individuals, nor could any be detected in either the members of cohort 2 who had been treated for a longer period or the LTNPs.
Despite the reductions in HIV-1 antigen–specific IgG-ASC frequencies after therapy was initiated, the plasma anti-gp120 and anti-p24 antibody titers in most members of cohort 1 did not decline significantly over the 12–30-wk study period (Fig. 5). In one subject (no. 902), the antibody titers actually continued to increase at a time when the numbers of circulating anti-gp120 ASCs were declining. However, this individual was poorly adherent to antiviral therapy and showed a later resurgence in both total IgG-ASCs (Fig. 3 a) and gp120-specific IgG-ASCs, as well as the sustained anti-gp120 titer increase (Fig. 5 a). This may limit any inferences that can be drawn.
The data presented in Fig. 5 suggest that there is little relationship between plasma antibody levels and the frequency of antigen-specific and -nonspecific ASCs in peripheral blood. We confirmed this by directly comparing these parameters. Total plasma IgG concentrations and the frequency of total IgG-ASCs in the 18 examined members of cohort 1 (pretherapy values) are not correlated (Fig. 6 a). This is because infection for <1 yr is often associated with a high circulating IgG-ASC frequency in the absence of hypergammaglobulinemia, which presumably takes some time to develop. For most individuals, there was no correlation between the anti-gp120 IgG-ASC frequency and the anti-gp120 antibody titers (Fig. 6 b). Individuals with the highest anti-gp120 and anti-p24 titers (LTNPs) had no circulating gp120- or p24-specific IgG-ASCs (Fig. 6 b and data not shown). However, one acutely infected individual (no. 911) with no detectable anti-gp120 antibodies also had no detectable gp120-specific IgG-ASCs. In addition, the chronically infected individual with the highest anti-gp120 titer (no. 893) also had the highest gp120-specific IgG-ASC frequency. Both these outliers (no. 911 and no. 893) are marked by asterisks on Fig. 6 b. Their inclusion in the data set does allow a correlation between anti-gp120 IgG-ASC frequency and anti-gp120 titer to be made (r = 0.81; P <0.01). However, we have reservations about the generality of this apparent correlation since omission of the outliers generated an r value of −0.17, which is not significant.
Figure 6.
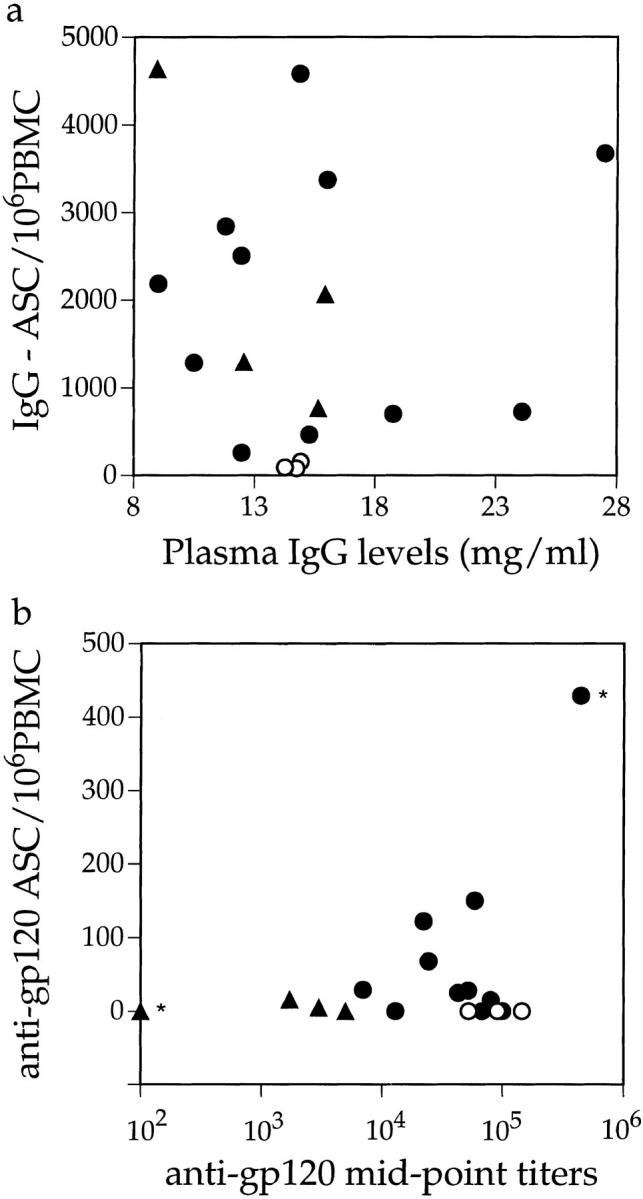
Relationship between circulating IgG-ASC frequencies and plasma antibody titers. (a) Baseline IgG-ASC frequencies are compared with plasma IgG concentrations; (b) circulating anti-gp120 IgG-ASC frequencies are compared with anti-gp120 antibody titers; for 18 members of cohort 1, including 4 acutely (▴) and 11 chronically (•) infected individuals and 3 LTNPs (○). See text for information on asterisked individual.
Antiretroviral Therapy Resolves Hypergammaglobulinemia and Diminishes HIV-1–specific Antibody Responses.
Hypergammaglobulinemia (plasma IgG levels >16.2 mg/ml in Caucasians) is a manifestation of chronic HIV-1 infection but usually takes time to develop. Only the two members of cohort 1 who had been infected for at least 5 yr (nos. 893 and 896) were hypergammaglobulinemic; neither the acutely infected individuals nor those infected for <5 yr had elevated plasma IgG levels despite having high IgG-ASC frequencies (Fig. 1). After antiviral therapy was initiated, both of these individuals remained hypergammaglobulinemic over a 16-wk period, despite the rapid reduction in IgG-ASC frequency (Figs. 3 and 4). No later samples were available from these individuals.
Both baseline and longitudinal studies of plasma IgG levels could be performed on cohort 2, since IgG concentrations can be determined retrospectively from frozen samples. This allowed us to study changes in plasma IgG concentrations over a longer period of time. At baseline, 5 of the 10 chronically infected individuals had IgG levels >16.2 mg/ml (Table 2). After 32–36 wk of therapy, plasma IgG concentrations were reduced in all 10 participants, including those not originally considered to be hypergammaglobulinemic (Table 2). The most pronounced falls in plasma IgG were observed in the two African-American participants (nos. 1007 and 1008), although their levels were still high at the end of the study (18.5 and 18.6 mg/ml, respectively). However, African-Americans have naturally higher plasma IgG concentrations than do Caucasians (7, 8), so the post-therapy level observed may not be abnormal for these two individuals. There were also minor reductions in plasma IgG concentrations in the three acutely infected individuals after therapy (Table 2).
Taken together, the studies on cohorts 1 and 2 indicate that hypergammaglobulinemia is resolved by combination antiviral therapy, but not as rapidly as is the reduction in IgG-ASC frequency. This is not surprising since the half-life of IgG in plasma is ∼3 wk (26), so it would take some time for a reduction in plasma IgG to be manifested, even if all excess IgG production were simultaneously stopped. However, little or no reduction in plasma IgG levels was observed in two members of cohort 1 after 16 wk of therapy (five IgG half-lives), implying that hypergammaglobulinemia is not as quickly resolved as it theoretically could be if all the excess IgG production were due to circulating IgG-ASCs. Since plasma IgG concentrations were reduced in all members of cohort 2 after 32–36 wk (Table 2), we determined more accurately the kinetics of the decline in the four individuals with the greatest initial plasma IgG levels (nos. 1007, 1008, 1010, and 1013; Fig. 7). After therapy, plasma IgG concentrations decayed to a relatively stable level that was within the normal range for Caucasians (or African-Americans, as appropriate). Although there was a degree of scatter in the longitudinal profiles, calculations of the rate of clearance of the excess IgG indicated that the minimal estimate for the half-life was in the range of 4–12 wk for three individuals, and 34 wk for the fourth (no. 1008; Fig. 7; see legend for individual values).
Figure 7.
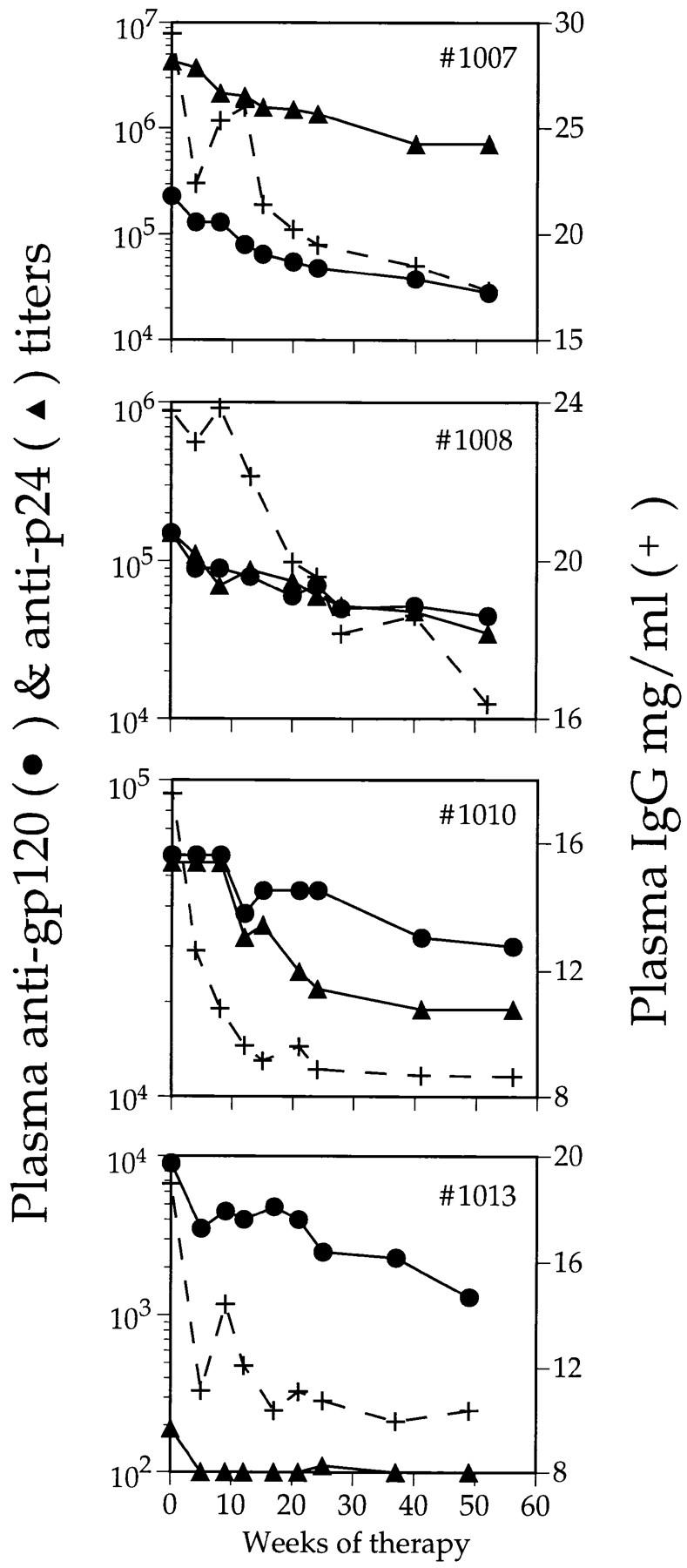
Effect of combination antiviral therapy on plasma IgG concentration and specific antibody titers in cohort 2. Four chronically infected individuals were treated for ∼1 yr. Plasma anti-gp120 titers (•), anti-p24 titers (▴), and total plasma IgG concentrations (+) are shown. Estimated half-lives for the decay in excess total IgG, anti-gp120, and anti-p24 and for each individual were, respectively: no. 1007, 12, 7, and 9 wk; no. 1004, 9 and 15 wk; no. 1010, 4, 21, and 9 wk; no. 1013, 6, 16 and not applicable (no anti-p24 titer).
Longitudinal analyses of the changes in specific antibody titer were also performed on members of cohort 2 (Table 2, Fig. 7). Pre- and post-therapy anti-gp120 and anti-p24 titers are recorded for each member of the cohort, along with the extent of the decreases in each parameter during the period of therapy (Table 2). Anti-gp120 and anti-p24 titers declined in almost all individuals, but generally to only a modest extent (median declines were approximately threefold). The rate of decline in anti-gp120 titer was estimated for the four individuals in whom the kinetics of excess IgG clearance were also studied (Fig. 7). The half-life of the anti-gp120 titer reduction was 7–21 wk, and for those three individuals with measurable titers the half-life of anti-p24 titer reduction was 9–15 wk.
Discussion
The main purpose of this study was to explore how HIV-1–specific and –nonspecific B cell responses were affected by combination antiretroviral therapy. We sampled only the peripheral blood to avoid invasive procedures, so all conclusions we draw refer specifically to this compartment. Nonetheless, the effects of antiviral therapy on the cells of the peripheral blood may mirror, at least qualitatively, effects on cells in other tissues, such as lymph nodes and bone marrow.
B cell abnormalities associated with HIV-1 infection are manifested by a significantly elevated frequency of HIV-1 antigen–nonspecific IgG-ASCs in the peripheral blood and by hypergammaglobulinemia (1–6). These are probably associated phenomena in that overproduction of IgG by circulating ASCs will tend to increase plasma IgG concentrations, but they are not inevitably linked to one another. Thus, IgG-ASC frequencies are significantly elevated in acutely infected individuals, but without hypergammaglobulinemia, whereas both parameters are often increased during chronic infection. Overall, IgG-ASC frequency was not correlated with plasma IgG concentration. Either circulating IgG-ASCs make a negligible contribution to total plasma IgG, so that an increase in their frequency is inconsequential, or else there is an increase in the rate of IgG clearance to compensate; the steady state plasma IgG concentration obviously reflects a balance between IgG production and clearance. The capacity of the IgG-clearance mechanism(s) may, at some stage, be directly or indirectly affected by systemic HIV-1 infection. Defects in the reticuloendothelial system associated with IgG clearance have been reported to be a complication of advanced HIV-1 infection (27, 28); these might contribute to hypergammaglobulinemia.
Antiviral therapy that causes substantial reductions in HIV-1 viremia has a rapid effect on antigen-specific and -nonspecific IgG-ASC frequencies during both acute and chronic infection. Coincident with, or soon after, therapy-induced declines in plasma viremia, IgG-ASCs start to disappear from the blood, with a half-life of only a few weeks. The IgG-ASC frequency then remains at low levels for prolonged periods, provided antiviral therapy continues to suppress HIV-1 replication. In one individual (no. 902) who was poorly compliant with therapy, there was a rapid rebound in the numbers of circulating IgG-ASCs. Hypergammaglobulinemia is also resolved by antiviral therapy, with a half-life of 4–34 wk. This was somewhat longer than the half-life of IgG secretion from the IgG-ASC population isolated from the peripheral blood (1–3.5 wk), but the half-life of plasma IgG molecules (∼3 wk in uninfected people; 26) must also be taken into account. Even if excess IgG production were instantly and completely stopped, which is obviously not the case, it would still take several weeks for a significant reduction in plasma IgG concentration to be manifested. In practice, production of polyclonal IgG molecules in some compartment that is not in equilibrium with the blood is likely to be sustained for some time. Residual viral antigen production due to incompletely effective antiviral therapy could be a contributory factor here, as could the presence of persistent antigen trapped on follicular dendritic cells. In addition, abnormally slow clearance of IgG from plasma could, in principle, contribute to the maintenance of high plasma IgG levels in some chronically infected individuals (see above).
How could a reduction in HIV-1 replication decrease the circulating IgG-ASC frequency? There are several possibilities: (a) these cells die; (b) they are redistributed to another body compartment; (c) cellular activation to secrete IgG is directly or indirectly dependent on continued HIV-1 replication; or (d) new ASCs are not induced. We cannot discriminate between these possibilities, but we note that there is a correlation between the circulating IgG-ASC frequency and the level of plasma viremia before therapy. This tends to argue against the redistribution hypothesis, and in favor of a relationship (direct or otherwise) between the production of HIV-1 antigens and antibody secretion from circulating B cells. The resurgence of the IgG-ASC frequency in no. 902 during a period of poor compliance is also consistent with the latter theory (since no increase in plasma viremia was detected in no. 902 during this period, we presume that HIV-1 replication in lymphoid tissues was responsible).
But how might HIV-1 replication affect B cell function in vivo? HIV-1 antigen load could obviously be an influence on antigen-specific IgG-ASCs, but the majority of circulating IgG-ASCs are not specific for HIV-1 antigens yet are sensitive to a lowering in their concentration. Although HIV-1 has been reported to contain a superantigen(s) (29, 30), this is controversial and the balance of the evidence is not compelling (31, 32). HIV-1 has also been reported to directly activate B cells in vitro (33, 34), and in one study this effect was reported to be mediated by the COOH terminus of gp41 (35). However, nonspecific activation of the immune system appears to be a phenomenon common to many viral infections (36, 37). In particular, simian immunodeficiency virus infection of macaques is associated with a generalized activation of B as well as T cells, leading to a significant increase in the rate of B cell turnover in infected animals (37). Thus, further investigations should not be limited to effects triggered specifically by HIV-1 antigens. Another possibility is that HIV-1 replication in CD4+ T cells or macrophages causes the secretion of a cytokine such as IL-6 that activates IgG secretion from B cells (38–40), and that the reduction in HIV-1 replication interrupts the production of this cytokine(s). In one individual, we have been able to monitor changes in the plasma IL-6 concentrations that mirror changes in plasma viremia (Binley, J.M., and J.P. Moore, unpublished observations), but this requires further investigation.
For most individuals, there was no correlation between circulating anti-gp120 IgG-ASC frequencies and plasma anti-gp120 antibody titers. This is not surprising since most specific antibody production occurs outside the blood, probably by long-lived plasma cells in the bone marrow (41–45), but it does expose the limitations in sampling circulating IgG-ASCs when trying to dissect the specific humoral immune response to HIV-1 infection. Antigen-specific B cell functions were also affected by antiviral therapy. As with the nonspecific IgG-ASCs, gp120-specific IgG-ASC frequencies decreased rapidly when antiviral therapy began, in both acutely and chronically infected individuals, but this was not accompanied by a decrease in plasma anti-gp120 titer over the same time period. Indeed, in one acutely infected individual (no. 902), the anti-gp120 titer continued to increase during a period when there was a marked decline in his circulating gp120-specific IgG-ASC frequency. Since subject no. 902 was not hypergammaglobulinemic, the increase in his anti-gp120 antibody titer has little to do with any defect in IgG clearance; instead, it may reflect the continued production of anti-gp120 antibodies by plasma cells in bone marrow in a manner insensitive to reductions in HIV-1 replication (11). Alternatively, it may be related to this individual's poor adherence to therapy.
The more prolonged therapy provided to the members of cohort 2 was accompanied by a gradual decline in anti-gp120 and anti-p24 titers. But this decrease was usually only modest, approximately threefold on average. Furthermore, it is not yet clear whether the specific titers in chronically infected people are continuing to decline, or have stabilized at a reduced level; the present indications favor the latter interpretation but more protracted studies are required to be sure of this. The half-lives of the anti-gp120 and anti-p24 titer declines were in the range of 7–21 wk. These are somewhat longer than the fall in the circulating gp120- and p24-specific IgG-ASC frequencies, probably for reasons similar to those discussed above in the context of the relationship between total IgG-ASC frequency and hypergammaglobulinemia. Although the relative rates of decline of total IgG and HIV-1 antigen–specific IgG differed between the four individuals, it is likely that the gradual reductions in anti-gp120 and anti-p24 titers are related to the decrease in plasma IgG concentration, rather than to any specific effect on HIV-1 antibody production.
Studies on the three LTNPs were particularly informative. These individuals had essentially undetectable levels of circulating gp120-specific IgG-ASCs, yet very high titers of anti-gp120 and anti-p24 antibodies. This is, in itself, strong evidence that circulating IgG-ASCs make little or no contribution to the overall production of antigen-specific antibodies during HIV-1 infection. Indeed, the appearance of large quantities of antigen-specific ASCs in the blood may be a pathological consequence of HIV-1 replication, and an early one at that, although perhaps of relatively benign consequence. The aviremic individual (no. S006) and the three LTNPs also had normal (indeed slightly subnormal) levels of total IgG-ASCs (and also IgM-ASCs), consistent with their normal plasma IgG concentrations. Thus, although the LTNPs have been HIV-1 infected for up to 15 yr, they show no abnormalities of B cell function. Therefore, the latter is not an inevitable consequence of HIV-1 infection, nor of the antibody response that is generated to HIV-1 antigens. The limited studies on individual no. S006 (infected for only 1–2 yr but also with undetectable plasma viremia) indicated that his B cell–mediated responses were indistinguishable from those in the three LTNPs. Thus, B cell abnormalities are associated with progressive HIV-1 infection and the extensive viral replication that is intimately associated with its pathology. These abnormalities are resolved by effective antiviral therapy, and the effect is sustained while viremia remains suppressed. This seems encouraging for the prospects of repairing at least some of the damage to normal immune functions caused by HIV-1 infection. In addition, the reduction in B cell hyperactivity may also result in a lowering of the incidence of B cell lymphoma during HIV-1 infection.
Acknowledgments
We thank James Robinson and Herman Katinger for providing the A32 and ID7 B cell lines; Paul Maddon for rgp120; and Ian Jones for the gst–p24 fusion vector construct. We appreciate the assistance of Dr. Richard Coico for help in setting up the ELISPOT assay, and we are grateful to Ralph Steinman for his intellectual support. We also thank Simon Monard for help with FACS® analysis and Wen Chen for graphics.
Abbreviations used in this paper
- ASC
antibody-secreting cell
- bDNA
branched DNA
- LTNP
long-term nonprogressor
Footnotes
L. Morris was supported by the Department of Health, the Poliomyelitis Research Foundation, and the Medical Research Council of South Africa. Other support was provided by National Institutes of Health grants R21 AI-42721, U01 AI-41534, and MO1 RR-06102.
Lynn Morris' permanent address is National Institute for Virology, Private Bag X4, Sandringham, Johannesburg 2131, Gauteng, South Africa.
References
- 1.Lane HC, Masur H, Edgar LC, Whalen G, Rook AH, Fauci AS. Abnormalities of B-cell activation and immunoregulation in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1983;309:453–458. doi: 10.1056/NEJM198308253090803. [DOI] [PubMed] [Google Scholar]
- 2.Shirai A, Cosentino M, Leitman-Klinman SF, Klinman DM. Human immunodeficiency virus infection induces both polyclonal and virus-specific B cell activation. J Clin Invest. 1992;89:561–566. doi: 10.1172/JCI115621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yarchoan R, Redfield RR, Broder S. Mechanisms of B cell activation in patients with acquired immunodeficiency syndrome and related disorders. J Clin Invest. 1986;78:439–447. doi: 10.1172/JCI112595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amadori A, DeRossi A, Faulkner-Valle GP, Chieco-Bianchi L. Spontaneous in vitro production of virus-specific antibody by lymphocytes from HIV infected subjects. Clin Immunol Immunopathol. 1988;46:342–351. doi: 10.1016/0090-1229(88)90053-0. [DOI] [PubMed] [Google Scholar]
- 5.Amadori A, Zamarch R, Ciminale V, Del Mistro A, Siervo S, Alberti A, Colombatti M, Chieco-Bianchi L. HIV-1 specific B cell activation. A major constituent of spontaneous B cell activation during HIV-1 infection. J Immunol. 1989;143:2146–2152. [PubMed] [Google Scholar]
- 6.Wolthers KC, Otto SA, Lens SMA, van Lier RAW, Miedema F, Meyaard L. Functional B cell abnormalities in HIV type 1 infection: role of CD40L and CD70. AIDS Res Hum Retrovir. 1997;13:1023–1029. doi: 10.1089/aid.1997.13.1023. [DOI] [PubMed] [Google Scholar]
- 7.Lucey DR, Hendrix CW, Andrzejewski C, Melcher GP, Butzin CA, Henry R, Wians FH, Jr, Boswell RN. Comparison by race of total serum IgG, IgA, and IgM with CD4+ T-cell counts in North American persons infected with the human immunodeficiency virus type 1. J Acquir Immune Defic Syndr. 1992;5:325–332. [PubMed] [Google Scholar]
- 8.Bélec L, Dupré T, Prazuck T, Tévi-Bénissan C, Kanga J-M, Pathey O, Lu X-S, Pillot J. Cervicovaginal overproduction of specific IgG. Human immunodeficiency virus (HIV) contrasts with normal or impaired IgA local response in HIV infection. J Infect Dis. 1995;172:691–697. doi: 10.1093/infdis/172.3.691. [DOI] [PubMed] [Google Scholar]
- 9.Robey WG, Safai B, Oroszlan S, Arthur LO, Gonda MA, Gallo RC, Fischinger PJ. Characterization of envelope and core structural gene products of HTLV-III with sera from AIDS patients. Science. 1985;228:593–595. doi: 10.1126/science.2984774. [DOI] [PubMed] [Google Scholar]
- 10.Allan JS, Coligan JE, Barin F, Sodroski J, Rosen CA, Haseltine WA, Lee T-H, Essex M. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science. 1985;228:1091–1094. doi: 10.1126/science.2986290. [DOI] [PubMed] [Google Scholar]
- 11.Binley JM, Klasse PJ, Cao Y, Jones I, Markowitz M, Ho DD, Moore JP. Differential regulation of the antibody responses to gag and env proteins of human immunodeficiency virus type 1. J Virol. 1997;71:2799–2809. doi: 10.1128/jvi.71.4.2799-2809.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore JP, Cao Y, Ho DD, Koup RA. Development of the anti-gp120 antibody response during seroconversion to human immunodeficiency virus type 1. J Virol. 1994;68:5142–5155. doi: 10.1128/jvi.68.8.5142-5155.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koup RA, Safrit JT, Cao Y, Andrews CA, Wu Y, McLeod G, Borkowsky W, Farthing C, Ho DD. Temporal association of cellular immune response with the initial control of viremia in primary HIV-1 syndrome. J Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown AE, Vahey MT, Zhou SYJ, Chung RC-Y, Ruiz NM, Hofheinz D, Lane JR, Mayers DL the RV43 Study Group. Quantitative relationship of circulating p24 antigen with human immunodeficiency virus (HIV) RNA and specific antibody in HIV-infected subjects receiving antiretroviral therapy. J Infect Dis. 1995;172:1091–1095. doi: 10.1093/infdis/172.4.1091. [DOI] [PubMed] [Google Scholar]
- 15.Cheingsong-Popov R, Panagiotidi C, Bowcock S, Aronstam A, Wadsworth J, Weber J. Relation between humoral responses to HIV gag and env proteins at seroconversion and clinical outcome of HIV infection. Br Med J. 1991;302:23–26. doi: 10.1136/bmj.302.6767.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogervorst E, Jurriaans S, de Wolf F, van Wijk A, Wiersma A, Valk M, Roos M, van Gemen B, Coutinho R, Miedema F, Goudsmit J. Predictors for non- and slow progression in human immunodeficiency virus (HIV) type 1 infection: low viral RNA copy numbers in serum and maintenance of high HIV-1 p24-specific but not V3-specific antibody levels. J Infect Dis. 1995;171:811–821. doi: 10.1093/infdis/171.4.811. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt G, Amiraian K, Frey H, Wethers J, Stevens RW, Berns DS. Monitoring human immunodeficiency virus type 1–infected patients by ratio of antibodies to gp41 and p24. J Clin Microbiol. 1989;27:843–848. doi: 10.1128/jcm.27.5.843-848.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sheppard HW, Ascher MS, McRae B, Anderson RE, Lang W, Allain J-P. The initial response to HIV and immune system activation determine the outcome of HIV disease. J Acquir Immune Defic Syndr. 1991;4:704–712. [PubMed] [Google Scholar]
- 19.Strathdee SA, Frank JW, McLaughlin J, Leblanc M, Major C, O'Shaughnessy MV, Read SE. Quantitative measures of human immunodeficiency virus–specific antibodies predict progression to AIDS. J Infect Dis. 1995;172:1375–1379. doi: 10.1093/infdis/172.5.1375. [DOI] [PubMed] [Google Scholar]
- 20.Zwart G, van der Hoek L, Valk M, Cornelissen MTE, Baan E, Dekker J, Koot M, Kuiken CL, Goudsmit J. Antibody responses to HIV-1 envelope and gag epitopes in HIV-1 seroconvertors with rapid versus slow disease progression. Virology. 1994;201:285–293. doi: 10.1006/viro.1994.1293. [DOI] [PubMed] [Google Scholar]
- 21.Kost, R., Y. Cao, M. Vesanen, A. Talal, A. Hurley, R. Schluger, S. Monard, M. Rogers, J. Johnson, L. Smiley, et al. Combination therapy with Abacavir (1592), 141W94, and AZT/3TC in subjects acutely and chronically infected with HIV. Abstract at 5th Conference on Retroviruses and opportunistic infections, Chicago IL, February 1–5, 1998. Abstract 363:147.
- 22.Cao Y, Qin L, Zhang L, Safrit J, Ho DD. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Engl J Med. 1995;332:201–208. doi: 10.1056/NEJM199501263320401. [DOI] [PubMed] [Google Scholar]
- 23.Talal, A., Y. Cao, A. Hurley, R. Schluger, L. Fischer, M. Salgo, L. Smiley, A. Keller, D.D. Ho, and M. Markowitz. Saquinavir in combination with AZT/3TC and Ritonavir: a convenient BID regimen. Abstract at 37th ICAAC, Toronto, Canada September 28–October 1, 1997. 282.
- 24.Sedgwick JD, Holt PG. A solid-phase immunoenzymatic technique for the enumeration of specific antibody-secreting cells. J Immunol Methods. 1983;57:301–309. doi: 10.1016/0022-1759(83)90091-1. [DOI] [PubMed] [Google Scholar]
- 25.Lee FK, Nahmias AJ, Lowery S, Nesheim S, Reef S, Thompson S, Oleske J, Vahlne A, Czerkinsky C. ELIspot: a new approach to studying the dynamics of virus-immune system interaction for diagnosis and monitoring of HIV infection. AIDS Res Hum Retrovir. 1989;5:517–523. doi: 10.1089/aid.1989.5.517. [DOI] [PubMed] [Google Scholar]
- 26.Zuckier LS, Rodriguez LD, Scharff MD. Immunologic and pharmacologic concepts of monoclonal antibodies. Semin Nucl Med. 1989;19:166–186. doi: 10.1016/s0001-2998(89)80012-1. [DOI] [PubMed] [Google Scholar]
- 27.Bender BS, Frank MM, Lawley TJ, Smith WJ, Brickman CM, Quinn TC. Defective reticuloendothelial system Fc-receptor function in patients with acquired immunodeficiency syndrome. J Infect Dis. 1985;152:409–412. doi: 10.1093/infdis/152.2.409. [DOI] [PubMed] [Google Scholar]
- 28.Bender BS, Bohnsack JF, Sourlis SH, Frank MM, Quinn TC. Demonstration of defective C3-receptor– mediated clearance by the reticuloendothelial system in patients with acquired immunodeficiency syndrome. J Clin Invest. 1987;79:715–720. doi: 10.1172/JCI112876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berberian L, Goodglick L, Kipps TJ, Braun J. Immunoglobulin VH3 gene products: natural ligands for HIV gp120. Science. 1993;261:1588–1591. doi: 10.1126/science.7690497. [DOI] [PubMed] [Google Scholar]
- 30.Karray S, Zouali M. Identification of the B cell superantigen-binding site of HIV-1 gp120. Proc Natl Acad Sci USA. 1997;94:1356–1360. doi: 10.1073/pnas.94.4.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Margolin DH, Reimann KA, Sodroski J, Karlsson GB, Tenner-Racz K, Racz P, Letvin NL. Immunoglobulin VHusage during primary infection of rhesus monkeys with chimeric simian-human immunodeficiency viruses. J Virol. 1997;71:8582–8591. doi: 10.1128/jvi.71.11.8582-8591.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komanduri KV, Salha MD, Sekaly RP, McCune JM. Superantigen-mediated deletion of specific T cell receptor V beta subsets in the SCID-hu Thy/Liv mouse is induced by staphylococcal enterotoxin B, but not HIV-1. J Immunol. 1997;158:544–549. [PubMed] [Google Scholar]
- 33.Pahwa S, Pahwa R, Good RA, Gallo RC, Saxinger C. Stimulatory and inhibitory influences of human immunodeficiency virus on normal B lymphocytes. Proc Natl Acad Sci USA. 1986;83:9124–9128. doi: 10.1073/pnas.83.23.9124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnittman SM, Lane HC, Higgins SE, Folks T, Fauci AS. Direct polyclonal activation of human B lymphocytes by the acquired immune deficiency syndrome virus. Science. 1986;233:1084–1086. doi: 10.1126/science.3016902. [DOI] [PubMed] [Google Scholar]
- 35.Chirmule N, Kalyanaraman VS, Saxinger C, Wong-Staal F, Ghrayeb J, Pahwa S. Localization of B cell stimulatory activity of HIV-1 to the carboxyl terminus of gp41. AIDS Res Hum Retrovir. 1990;6:299–305. doi: 10.1089/aid.1990.6.299. [DOI] [PubMed] [Google Scholar]
- 36.Tough DF, Borrow B, Sprent J. Induction of bystander T cell proliferation by viruses and type 1 interferon in vivo. Science. 1996;272:1947–1950. doi: 10.1126/science.272.5270.1947. [DOI] [PubMed] [Google Scholar]
- 37.Mohri H, Bonhoeffer S, Monard S, Perelson AS, Ho DD. Rapid turnover of T lymphocytes in SIV- infected Rhesus macaques. Science. 1998;279:1223–1227. doi: 10.1126/science.279.5354.1223. [DOI] [PubMed] [Google Scholar]
- 38.Fauci AS. Multifactorial nature of human immunodeficiency virus disease: implications for therapy. Science. 1993;262:1011–1018. doi: 10.1126/science.8235617. [DOI] [PubMed] [Google Scholar]
- 39.Amadori A, Zamarchi R, Veronese ML, Panozzo M, Barelli A, Borri A, Sironi M, Colotta F, Mantovani A, Chieco-Bianchi L. B cell activation during HIV-1 infection. Cell-to-cell interactions and cytokine requirements. J Immunol. 1991;146:57–62. [PubMed] [Google Scholar]
- 40.Reickmann P, Poli G, Kehrl JH, Fauci AS. Activated B lymphocytes from human immunodeficiency virus–infected individuals induce virus expression in infected T cells and a promonocytic cell line, U1. J Exp Med. 1991;173:1–5. doi: 10.1084/jem.173.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slifka MK, Matloubian M, Ahmed R. Bone marrow is a major site of long-term antibody production after acute viral infection. J Virol. 1995;69:1895–1902. doi: 10.1128/jvi.69.3.1895-1902.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. 1996;272:54–60. doi: 10.1126/science.272.5258.54. [DOI] [PubMed] [Google Scholar]
- 43.Bachmann MF, Kundig TM, Odermatt B, Hengarter H, Zinkernagel RM. Free recirculation of memory B cells versus antigen-dependent differentiation to antibody-forming cells. J Immunol. 1994;153:3386–3397. [PubMed] [Google Scholar]
- 44.Paramithiotis E, Cooper MD. Memory B lymphocytes migrate to bone marrow in humans. Proc Natl Acad Sci USA. 1997;94:208–212. doi: 10.1073/pnas.94.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Slifka MK, Antia R, Whitmire JK, Ahmed R. Humoral immunity due to long-lived plasma cells. Immunity. 1998;8:363–372. doi: 10.1016/s1074-7613(00)80541-5. [DOI] [PubMed] [Google Scholar]