

Abstract
In tumor transplantation models in mice, cytotoxic T lymphocytes (CTLs) are typically the primary effector cells. CTLs recognize major histocompatibility complex (MHC) class I–associated peptides expressed by tumors, leading to tumor rejection. Peptides presented by cancer cells can originate from viral proteins, normal self-proteins regulated during differentiation, or altered proteins derived from genetic alterations. However, many tumor peptides recognized by CTLs are poor immunogens, unable to induce activation and differentiation of effector CTLs. We used MHC binding motifs and the knowledge of class I:peptide:TCR structure to design heteroclitic CTL vaccines that exploit the expression of poorly immunogenic tumor peptides. The in vivo potency of this approach was demonstrated using viral and self-(differentiation) antigens as models. First, a synthetic variant of a viral antigen was expressed as a tumor antigen, and heteroclitic immunization with peptides and DNA was used to protect against tumor challenge and elicit regression of 3-d tumors. Second, a peptide from a relevant self-antigen of the tyrosinase family expressed by melanoma cells was used to design a heteroclitic peptide vaccine that successfully induced tumor protection. These results establish the in vivo applicability of heteroclitic immunization against tumors, including immunity to poorly immunogenic self-proteins.
Keywords: cytotoxic T lymphocyte, tumors, peptide vaccines, heteroclitic peptides, major histocompatibility complex class I
Cytotoxic T lymphocytes (CTLs) can play a central role in rejecting tumors (1). Tumor antigens recognized by CTL generally originate from three sources: (a) viruses; (b) self-proteins expressed during development or differentiation; and (c) mutant or aberrantly expressed proteins (1). Because many, if not most, tumor antigens are products of normal or altered cellular genes, they are typically not efficient at initiating immune responses. Thus, a central problem in cancer immunotherapy is how to efficiently prime CTLs against poorly immunogenic tumor antigens.
CTLs recognize target antigens in the form of short intracellularly processed peptides, presented by self-MHC– encoded class I molecules (pep:class I). Upon binding of the antigen-specific TCRs on a CTL to its cognate peptide– MHC complex on the tumor cell, the target cell is lysed and the tumor eliminated. To develop into effector CTLs capable of tumor lysis, naive precursor CTLs (pCTLs)1 have to be activated. This pCTL activation requires two signals: the first, or stimulatory signal (signal 1), transmitted via the TCR–CD3 complex, and the second, or costimulatory signal (signal 2), delivered by professional APCs (2–4). In the thymus, a strong signal 1 will induce negative selection of immature thymocytes, regardless of signal 2 (5, 6). In the periphery, the same strong signal 1 will induce immunity (including pCTL → CTL differentiation) or anergy, depending on the presence or absence of signal 2 (2–4). By contrast, even a weak signal 1 without signal 2 can be sufficient for target cell lysis by differentiated CTLs (7). This means that a whole class of antigenic peptides exists that, although poorly immunogenic (i.e., unable to induce CTL immunity), can readily serve as molecular targets for lysis by differentiated effector CTLs. Such antigenic, but poorly immunogenic, peptides remain invisible to the naive pCTL.
One strategy to exploit the presence of such poorly immunogenic or nonimmunogenic peptides at the surface of tumor cells is to design immunogenic variants of these peptides that would prime CTL response that cross-reacts to the original targeting peptide. If successful, this strategy could be attractive, since the relative invisibility of poorly immunogenic self-peptides to the immune system could be advantageous. Namely, unlike self-peptides that provide strong signal 1 (8), poorly immunogenic peptides would most likely fail to induce tolerance, and the T cell repertoire reactive against them should be intact and available for activation with immunogenic peptide variants. According to the nomenclature used for variants of a pigeon cytochrome C peptide (9), peptides of higher biological potency than the original peptide were called heteroclitic. Crystal structure analysis revealed that, of the 8–10 amino acid residues of a class I-bound peptide, roughly half point into the solvent and can interact directly with the TCR via their side chains (10–12). The other half are buried by class I and are not directly accessible to the TCR (10–12). Heterocliticity has been achieved by substituting amino acids that contact class I, the TCR, or both (13, 14).
The aim of this study was to produce heteroclitic immunogens that elicit antitumor CTL responses that cross-react to the original poorly immunogenic antigens. Therefore, to design peptides that are heteroclitic for polyclonal CTL responses, one would like to optimize pep:class I binding, since this property correlates with immunogenicity (7, 15). At the same time, the peptide:TCR contact should not be disturbed, to maximize the potential crossreactivity between the heteroclitic and the original, nonimmunogenic targeting peptide. We sought to test whether this strategy could induce antitumor CTLs reactive to nonimmunogenic targeting peptides. We demonstrate successful in vivo induction of cross-reactive CTLs in two tumor models, using an engineered viral peptide variant expressed as a tumor antigen (a model of a tumor antigen of viral origin) and a self-antigen expressed in melanomas (a model of nonmutated, differentiation antigen). Induced CTLs were biologically active in vivo, and were able to effect rejection of both newly implanted and established day 3 tumors.
Materials and Methods
Mice.
Female C57BL/6 (B6) mice were purchased from the National Cancer Institute breeding program (Frederick, MD). B6.C-H-2bm8 (bm8) mice were bred in the MSKCC vivarium from a breeding stock obtained from The Jackson Laboratory (Bar Harbor, ME) via Dr. J. Sprent (The Scripps Research Institute, La Jolla, CA). All mice entered the study between 7 and 10 wk of age.
Antibodies, In Vivo CD8 Depletion, Flow Cytometry and Class I Stabilization Assays.
The anti-CD8 mAb, 53.6.7 (rat IgG) and the anti-Kb mAb, Y3 (mouse IgG2b), both obtained from the American Type Culture Collection (Rockville, MD) were produced as ascitic fluid in our lab. For in vivo CD8 depletion, 100 μl of ascitic fluid was injected intraperitoneally on days −7 and −3 relative to tumor challenge, which was denoted as day 0. PE-conjugated anti–mouse IgG2b was purchased from Fisher Biotech (Malvern, PA). Flow cytometry and the class I stabilization assays were performed exactly as previously described (16) using a FACScan® instrument equipped with Lysys II software (Becton Dickinson, Mountain View, CA).
Construction of Minigenes.
Inserts coding for the endoplasmic reticulum (ER) insertion sequence (17, 18) (amino acid sequence: MRYMILGLLALAAVCSA) followed by the peptides SEI (SEIEFARL) and SSI (SSIEFARL), based on the immunodominant sequence 498–505 of the Herpes simplex virus glycoprotein B, or peptides TWH (TWHRYHLL) or TAY (TAYRYHLL), based upon the sequence 222–229 of the melanoma gp75 protein, were produced by multistep PCR. All oligonucleotides were purchased from Retrogen (San Diego, CA). For the construction of pERIS-SSI and pERIS-SEI minigenes, the PCR reactions were done with two common oligomers, C1 (GGG AAG CTT ACC ATG AGA TAC ATG ATC CTG GGC CTG CTG), C2 (GGC CTG CTG GCC CTG GCC GCC GTG TGC AGC GCT GCC AGC), and the specific oligomers SSI (TTT CTC GAG TCA CAG CCT GGC GAA CTC GAT GCT GCT GGC AGC) or SEI (TTT CTC GAG TCA CAG CCT GGC GAA CTC GAT CGA GCT GGC AGC). C2 and SSI or SEI were first joined in 50-μl reactions consisting of 300 μM dNTPs and 20 μg/ml of primers for 30 cycles at 95°C for 30 s, 30°C for 60 s, and 72°C for 30 s. 5 μl of the product was added to 45 μl of C1 and either SSI or SEI at the same primer and dNTP concentrations, and another PCR was performed for 10 cycles as above, followed by 30 cycles at 95°C for 1 min and 72°C for 2 min. For the construction of pERIS-TAY and pERIS-TWH, the common oligomers C1.1 (GGG AAG CTT ACC ATG AGA TAC ATG ATC CTG GGC CTG CTG GCC CTG GCC GC) and C2.1 (GGC CTG CTG GCC CTG GCC GCC GTG TGC AGC GCT GCT) were used with the specific oligomers TAY (TTT CTC GAG TCA CAG CAG GTG GTA TCT GTA GGC GGT GGC AGC GCT) or TWH (TTT CTC GAG TCA CAG CAG GTG GTA TCT GTG CCA GGT GGT AGC GCT). The first step was exactly as described above. However, the second step was done for 40 cycles at 94°C for 20 s, followed by 60°C for 20 s and 72°C for 45 s. Products thus engineered contain the HindIII and XhoI sites, which were used for cloning into a LacZ-containing pCR2 cloning vector (Invitrogen, San Diego, CA). Clones that scored positive by blue/white screening were digested by HindIII and XhoI, and the inserts were recloned into pCDNA3 to obtain the appropriate expression constructs, pcERIS-SSI, pcERIS-SEI, pcERIS-TWH, and pcERIS-TAY. The transfer was confirmed by sequencing.
Cell Transfection.
10 μg of linearized plasmid DNA was electroporated into 107 RMA-S cells in 500 μl of Optimem (GIBCO BRL, Gaithersburg, MD) containing 5% FCS, using a Gene Pulser (Bio-Rad, Hercules, CA) set at 220 V and 960 μF. 48 h later, the cells were incubated in the presence of 600 μg/ml of G418 (GIBCO BRL). G418-resistant clones were selected from the 96-well plates with <30% positive wells.
Peptide and Gene Gun Immunization, In Vitro CTL Restimulation, and CTL Assays.
The five peptides used in this study, HIV-10 (RGPGRAFVTI), SSI (SSIEFARL), SEI (SEIEFARL), TWH (TWHRYHLL), and TAY (TAYRYHLL), were synthesized by the Memorial Sloan Kettering Cancer Center Microchemistry Core Facility, and were HPLC purified to >98% purity. Peptide immunization using the synthetic immune adjuvant TiterMax (CytRx Inc., Norcross, GA), referred to as pep/TM in the text, CTL restimulation, and 51Cr-release assays were performed as previously described (19). In brief, mice were immunized in the footpad with 10 μl of the pep/TM emulsion (mixed according to the manufacturer's instructions) containing 5 μg of the indicated peptide. 7 d later, spleen cells from the immunized mice were restimulated in vitro with syngeneic, irradiated (30 Gy), peptide-coated (1 μg/ml, 2 ml/spleen, 1 h at 37°C followed by three washes) cells. 5 d later, cytolytic activity was assessed in a standard 51Cr-release assay. Genetic immunization using DNA-coated gold particles was performed exactly as previously described using a gene gun provided by Powderject, Inc. (Middleton, WI) (20). 100 μg of DNA from the plasmids described in the previous section was mixed with 0.95–2.6-μm diameter gold particles, in the presence of 0.05–0.1 μM spermidine. CaCl2 (1.5 mM) was added in a dropwise fashion to this mixture during vortexing. After precipitation, the gold plasmid DNA complex was washed three times in 100% ethanol and 7 ml of ethanol was added to achieve a bead-loading rate of 0.5 mg of gold to 1.0 μg plasmid DNA per injection. This solution was instilled into plastic Tefzel tubing, the ethanol was gently drawn off, and the tube was purged under nitrogen gas at 400 ml/min for drying. The tube was then cut into 0.5-inch bullets. The gold particles in the “bullets” were injected into the skin of anesthetized mice using a helium-driven gene gun (Powderject, Inc.). The skin was shaved and depilated before injection (20). Four injections at 400 pounds/square inch were delivered to each mouse, one to each of the abdominal quadrants, for a total of 4 μg of plasmid DNA per mouse. 7 d later, spleen cells were restimulated and CTL activity was determined as described for pep/TM.
Tumor Challenge and Follow Up.
5 × 105 RS-H2E or RS-Null cells or 105 B16F10LM3 (designated B16 in the text) melanoma cells (derived from B16F10 melanoma cells, a gift of Isaiah Fidler, MD Anderson Cancer Center, Houston, TX) were injected into the shaved left flank of the mice. Mice were then monitored three times/wk for tumor growth, initially by palpation and subsequently, when tumor growth was manifest, using Vernier calipers. Measurements were achieved by obtaining the maximum diameter of the tumor and the diameter perpendicular to the maximum, which were then multiplied, and the product of these two values was reported as tumor size. Tumor growth curves are shown for individual mice per experiment. Mice surviving tumor challenge were followed for a minimum of 60–90 d. The mice were killed if the maximum tumor diameter exceeded 10 mm, or if the tumor became ulcerated.
Results
Heteroclitic Vaccination in an Engineered Lymphoma Model.
To establish the principle of heteroclitic immunization against tumors, we used an engineered peptide based on the sequence of the Herpes simplex virus glycoprotein B498–505 peptide, which also served as a model of a tumor antigen of viral origin. This peptide, called SEI (sequence SEIEFARL), binds poorly to the murine MHC I molecule H-2Kb (Kb) due to the electrostatic repulsion between the negatively charged glutamic acid residues at the buried position 2 of the peptide (P2E) and the adjacent position 24 (MHC24E) of Kb (16). Due to poor binding, this peptide cannot elicit a CTL response in Kb-bearing C57BL/6 (B6) mice after immunization with peptide/adjuvant (Fig. 1 A, circles, and Table 1) or with genetic immunization using DNA delivered by particle bombardment (Table 1). However, SEI was a good immunogen in B6.C-H-2.bm8 (bm8) mice (Fig. 1 B, circles), which express a natural Kb variant, Kbm8. This class I molecule has an E24→ S mutation that enables strong SEI binding (16). These results demonstrated that the immunogenicity of the above peptide correlated directly to peptide binding, and that the absence of a response in B6 was not a result of deficient vaccine formulation.
Figure 1.


Efficacy of CTL priming by SSI and SEI peptides. (A) CTL responses to peptide priming of B6 mice by SEI (circles) and SSI (squares). Three mice per group were vaccinated with indicated peptides in adjuvant (pep/TM). 7 d later, spleen cells were restimulated in vitro and CTL responses of individual mice tested in a 51Cr-release assay using Kb- expressing EL-4 target cells pulsed with 10 μM of the immunizing peptide, as previously described (19). The lysis of unpulsed EL-4 cells (always <10%) was subtracted, and results are shown for individual mice at indicated effector/target ratios. Results are representative of >45 mice tested in at least 10 independent experiments. Indistinguishable results were obtained using DNA immunization by particle bombardment (Table 1). (B) Peptide immunogenicity correlates to peptide binding. bm8 mice respond to peptide priming by SSI (squares) and SEI (circles), both of which bind well to Kbm8 (16). Results are representative of at least 25 mice/strain tested in at least six independent experiments. Methods and data representation were as described in A.
Table 1.
Summary of In Vitro CTL Responses in C57BL/6
| Immunogen | Exp. No. | Responding mice/ Immunized mice | ||||
|---|---|---|---|---|---|---|
| DNA | Peptide* | |||||
| SSI | 1 | 5/5 | 5/5 | |||
| 2 | 5/5 | 5/5 | ||||
| 3 | 10/10 | 5/5 | ||||
| SEI | 1 | 0/5 | 0/3 | |||
| 2 | 0/5 | 0/5 | ||||
| 3 | ND | 0/5 | ||||
| TAY | 1 | 5/5 | 3/5 | |||
| 2 | 3/3 | 3/3 | ||||
| 3 | ND | 5/5 | ||||
| TWH | 1 | 0/5 | 0/5 | |||
| 2 | 0/3 | 0/3 | ||||
| 3 | ND | 0/5 | ||||
The responses were assayed 5 d after in vitro stimulation, in a Cr- release assay as previously described (19). Only mice with specific lysis of >35% were scored as responders. Groups responding to the immunogen are shown in bold. ND, Not done.
Refers to pep/TM immunization.
The natural viral peptide from which H2E was derived, SSI (SSIEFARL, also referred to as HSV-8), differs from SEI by having a serine (P2S) instead of the glutamic acid (P2E) in position 2. SSI would be predicted to remove the electrostatic repulsion between the peptide and Kb. Indeed, SSI bound 100-fold better than SEI to Kb (16) and was strongly immunogenic for B6 CTLs (Fig. 1 A, squares, and Table 1). We next asked whether SSI-induced CTLs could lyse cells bearing the SEI peptide in a cross-reactive fashion, and found that this was the case when target cells were coated with high concentrations of SEI (>10 μM) in vitro (Dyall, R., unpublished data).
Although these in vitro results were encouraging, their in vivo relevance for tumor immunity was obscure. In particular, it was unclear whether intracellularly expressed weak MHC binders, such as SEI, would be processed and presented efficiently enough for target cell lysis. To address that issue, we expressed SEI in a B cell lymphoma, RMA-S (21). RMA-S has a chemically induced deletion of one of its transporter associated with peptide processing (TAP) genes, Tap-2. This deletion prevents the vast majority of cytosolically processed peptides from entering the ER and binding to empty class I molecules, which leads to decreased expression of stable class I molecules at the surface of RMA-S cells. The TAP defect was circumvented using a minigene encoding an ER insertion sequence (ERIS) (17) followed by the SEI peptide. Fusion proteins encoded by such ERIS-containing minigenes have been shown previously to insert the attached class I binding peptides into the ER, thereby bypassing the TAP defect and partially restoring the surface expression of pep:class I (17, 18). Indeed, the experimental tumor line, RS-SEI (RMA-S cells transfected with the pERIS-SEI plasmid), had a higher surface level of Kb than RS-Null cells (transfected with the “empty” control plasmid, pcDNA3), as measured by flow cytometry. The mean relative Kb fluorescence intensity for RS-SEI was 123 as compared to only 79 for RS-null (Dyall, R., unpublished data). These observations were consistent with the efficient ERIS-mediated import of SEI into the ER. The fact that RS-SEI was lysed by anti-SSI CTL line, whereas RS-Null was not (Fig. 2), not only confirmed this conclusion, but also demonstrated that CTLs induced by the heteroclitic vaccine cross-reacted on the SEI:Kb expressed by the minigene-transfected cells.
Figure 2.

SSI is a heteroclitic immunogen for the antigenic, but not immunogenic, SEI peptide. Three anti-SSI CTL lines, derived from individual B6 mice by peptide immunization, were tested for the ability to lyse the Kb-expressing target cell lines RS-SEI (closed squares) and RS-Null (open squares), transfected with SEI-encoding and control plasmids, respectively, in a standard 51Cr-release assay. Six more lines were tested and gave identical results.
To test the potency of SSI as a heteroclitic vaccine in vivo, mice were immunized with either the heteroclitic or the parental peptide as previously described (19) and challenged with RS-SEI or RS-Null tumor lines. Tumor growth was then assessed for 90 d. Tumor growth curves among the different groups of challenged mice for a typical experiment are shown in Fig. 2 A and results from all experiments are shown in Table 2. The only group protected was the one vaccinated with the heteroclitic vaccine (SSI) and challenged with the tumor line expressing SEI. Numbers shown above each figure show cumulative tumor survival for all mice within the indicated experiment. These results clearly demonstrate the antigenic specificity of the response, and confirm the in vitro findings that SEI cannot induce a protective immune response in B6 mice (Fig. 3). Heteroclitic protection was dependent on CD8+ cells, because mice depleted of CD8+ cells by antibody treatment were not protected (Fig. 3, Table 2). Identical results were obtained with genetic immunization (Table 2). The ability of the heteroclitic immunogen to induce rejection of 3-d tumors was next investigated, using both peptide (19) and genetic (20) vaccination. Successful rejection was achieved by genetic immunization only (Fig. 4 and Table 2), consistent with our previous findings that genetic vaccination may be more potent than peptide vaccination (20). These results establish the in vivo relevance of heteroclitic immunization.
Table 2.
Summary of In Vivo Responses
| Immunogen | Tumor | Day of immunization | Tumor-free mice/ challenged mice | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DNA | Peptide | DNA | Peptide | |||||||
| SSI | RS-SEI | −14, −7 | −7 | 10/10 | 9/10 | |||||
| 3, 6 | 3, 6 | 18/19 | 0/19 | |||||||
| SSI | RS-Null | −14, −7 | −7 | 0/10 | 1/10 | |||||
| SEI | RS-SEI | −14, −7 | −7 | 0/10 | 0/10 | |||||
| 3, 6 | 3, 6 | 0/17 | 0/19 | |||||||
| SEI | RS-Null | −14, −7 | −7 | 0/10 | 0/10 | |||||
| TAY | B16 | −14, −7 | −7 | 10/10 | 18/20 | |||||
| 3, 6 | 3, 6 | 0/10 | 0/10 | |||||||
| TWH | B16 | −14, −7 | −7 | 0/10 | 2/20 | |||||
| 3, 6 | 3, 6 | 0/10 | 0/10 | |||||||
The day of tumor challenge was day 0. The two last columns show the cumulative number of mice that did not grow the tumor relative to the total number of mice challenged with the tumor. All tumor-free mice were followed for a minimum of 90 d. Groups with significant inhibition of tumor growth are shown in bold. Results of two separate experiments were combined and are shown in italics. Each experiment included 8–10 mice per group.
Figure 3.

In vivo ability of the heteroclitic vaccine SSI to protect mice against a transplantable tumor expressing SEI. 10 B6 mice per group were vaccinated with peptides SSI, SEI, or control PBS (19) emulsified in TM. 7 d later, animals were challenged with 5 × 105 RS-SEI or RS-Null cells subcutaneously. Nodules were palpable 3 d after challenge. Numbers on figures show numbers of tumor-free mice at 90 d. A seventh group also received SSI and was challenged with RS-SEI, but the animals were depleted of CD8+ cells by administration of an anti-CD8 mAb before the challenge. Tumors were measured as described in Materials and Methods, and results are shown as tumor growth curves. All tumor-free mice remained free of tumors for >90 d. DNA vaccination yielded identical results (Table 2).
Figure 4.
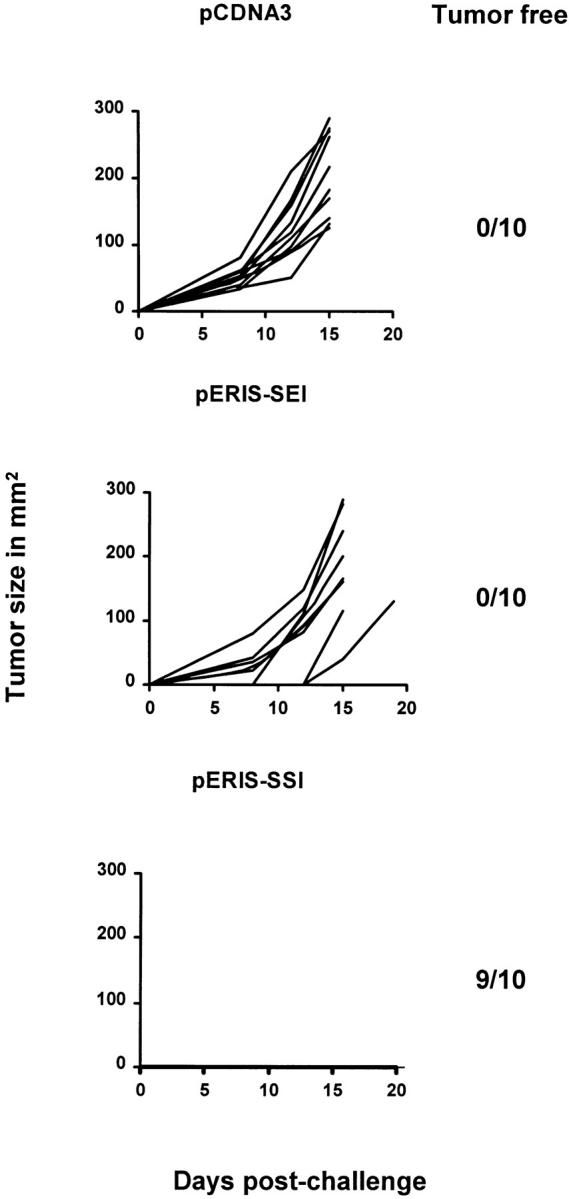
DNA vaccination with a heteroclitic immunogen eradicates 3-d tumors in mice. B6 mice were injected with the RS-SEI tumor, as described in the legend of Fig. 2 A. 3 d later, when palpable tumors appeared (2–3 mm in diameter), the mice were injected with DNA constructs (indicated in the figure) and tumor growth was scored. Tumors were followed and data shown exactly as in Fig. 3, with the figure showing one experiment and the numbers depicting tumor-free animals after 90 d of observation. Together with another similar experiment, this data is also shown in Table 2.
Heteroclitic Vaccination against a Tyrosinase Family Epitope of a Melanoma.
We next sought to test the applicability of this approach using naturally occurring tumor antigens. As a model, we selected the brown locus product, also known as the tyrosinase-related protein 1, or gp75. This glycoprotein is a lineage-specific self-antigen, present in melanocytes and expressed in melanomas (22, 23). The product of the brown locus is a relevant cancer antigen, recognized by both antibodies and T cells in patients with melanoma (22– 24). In mice, passive and active immunization against gp75 results in both melanoma rejection and manifestations of autoimmunity (25, 26). However, in the mouse model, CTL immunity against gp75-expressing melanoma cells was not induced by immunization with tumor cells plus adjuvant, nor tumor cells engineered to express cytokines, or with purified gp75 protein (26).
Using the canonical Kb-binding motif (27) to scan the amino acid sequence of gp 75 (28), five potential epitopes were identified. Synthetic peptides corresponding to these epitopes were used in both CTL inhibition and class I stabilization assays to determine their ability to bind to Kb (Moroi, Y., and R. Dyall, unpublished data). Peptide TWH, corresponding to gp75 residues 222–229 (TWHRYHLL), exhibited similar binding characteristics to SEI and was selected for further experiments. Since this peptide has two bulky side chains not commonly found at the buried, MHC-contacting positions 2 and 3 (P2W and P3H), we suspected that they might sterically impair pep–Kb binding. Based on the available sequence/motif information for Kb-binding peptides, and on the crystallographic data on the pep:Kb structures (10–12, 27), we changed these two residues to A and Y, respectively. The variant designed in this manner, named TAY (TAYRYHLL), turned out to be an excellent Kb binder, comparable to SSI (Fig. 5).
Figure 5.

The TAY peptide is an excellent binder to Kb. An RMA-S stabilization assay was performed and results are shown as previously described (16), using peptides SSI (open diamonds, positive control), HIV-10, a Dd-binding peptide, (RGPGRAFVTI, filled squares, negative control), TWH (filled circles), and its heteroclitic variant, TAY (filled triangles) at indicated concentrations. In this type of assay, the percentage of maximal stabilization provides a direct correlate of peptide binding (16). Experiment is representative of three such assays.
We next immunized B6 mice with the TAY and TWH peptides using both pep/TM and genetic methods. For both immunization protocols, successful CTL priming was obtained only with the engineered, and not the native, peptide (Table 1). Importantly, the anti-TAY CTLs lysed target cells pulsed with TWH in vitro, revealing that TAY exhibits the heteroclitic properties for TWH (Fig. 6 A). The well-characterized melanoma, B16 (29), and its radiation-induced gp75 loss mutant, B78H.1 (29), were next used as in vitro targets for the anti-TAY CTL lines (Fig. 6 B). Despite comparable surface expression of Kb after induction with IFN-γ (data not shown), B16 (Fig. 6 B, closed squares), but not B78H.1 (Fig. 6 B, open circles), was efficiently lysed. Importantly, TWH-peptide–sensitized B78H.1 cells were also efficiently lysed, showing that such cells expressed sufficient levels of MHC class I molecules for CTL lysis. These results show that: (a) peptide priming was antigen specific; (b) gp75 TWH peptide was naturally processed in the class I pathway; and (c) the TWH–Kb complexes were expressed at high enough levels on the melanoma cell surface to serve as targets for CTL attack.
Figure 6.


The TAY peptide is a heteroclitic immunogen for the native gp75 melanoma peptide, TWH. (A) TAY-induced CTLs lyse Kb- expressing target cells pulsed with TWH. CTL activity of anti-TAY CTLs against Kb-expressing target cells pulsed with 1 μM TAY (closed squares) or TWH (open squares). Lysis of control target cells (<10% at any point) was subtracted from the shown values. Target cells were pulsed with peptides and a Cr-release assay was performed as described (19). Cumulative results from several experiments of this type are shown in Table 1. (B) TWH is naturally processed in vivo, and can serve as a target for anti-TAY CTLs. The gp75-positive B16 melanoma line, but not its gp75 negative variant (B78.H1), is efficiently lysed by anti-TAY CTLs. B16 (filled squares), B78.H1 (open circles), or B78H.1 pulsed with 10 μM of the TWH peptide (open squares) were used as targets in a standard Cr-release assay after a 24-h incubation with 10 U/ml of IFN-γ to induce MHC class I expression. The extent of class I induction was confirmed by flow cytometry, and was similar for both tumor lines (data not shown). Similar results were obtained with the ex vivo explanted B16 melanoma (these cells express high levels of MHC class I molecules owing to class I upregulation in vivo). Results are shown for three independent CTL lines, each one depicted by a different type of line, and are representative of six lines tested thus far.
We next investigated the potential of TAY as a heteroclitic gp75 vaccine to protect against tumor challenge. Fig. 7 and Table 2 show tumor growth and incidence in mice vaccinated with TWH or TAY and challenged with B16. Of the mice that were vaccinated with the heteroclitic vaccine, TAY, 100% were protected in that experiment (Fig. 7), and 90% were protected in two experiments (Table 2). By contrast, minimal (if any) protection was conferred upon the mice vaccinated with the wild-type peptide TWH (Fig. 7 and Table 2). In day 3 tumors, the same priming regimen did not result in tumor eradication (Table 2), possibly owing to differences in tumor biology between the melanoma and a lymphoma, differences in inherent immunogenicity of the tumors, and other factors. Together with previous studies in vitro (30–32), the above results confirm the principles and establish the applicability of rationally designed heteroclitic vaccination to tumor immunity in vivo.
Figure 7.

In vivo efficacy of a heteroclitic antimelanoma vaccine. 10 mice per group were vaccinated with TAY/TM or TWH/TM, as previously described (19). 7 d later, they were challenged with 105 B16 melanoma cells per mouse, subcutaneously in the flank. Tumor measurements, number of experiments and result presentation was as in Figs. 3 and 4. The tumors typically became palpable after 9–15 d. Numbers represent the ratio of tumor-free mice to total mice challenged in each group over a period of >90 d. Another experiment yielded comparable results (Table 2).
Discussion
These results demonstrate the applicability and the in vivo efficacy of heteroclitic CTL vaccines. The approach to substitute the buried, MHC-contacting residues of a poorly binding peptide, but leave the solvent-exposed TCR-contacting residues intact, was previously used to generate CTLs that killed virally infected (30) or tumor (31, 32) targets expressing poorly immunogenic peptides in vitro. Furthermore, heteroclitic peptides of the human tyrosinase were shown to be more effective in stimulating CD4+ cells in vitro (31). Most recently, in a heterologous vaccination model, a fortuitous presence of a peptide with heteroclitic properties primed T cells that exhibited antitumor activity upon in vivo adoptive transfer (33). Using rational epitope identification and engineering and in vivo tumor challenge, we now demonstrate the in vivo potency of this strategy. The amino acid sequences of tumor antigens can be easily examined to select candidate MHC-binding peptides that can target the tumor for CTL attack. The increasing wealth of the available structural data about MHC–peptide and MHC–peptide–TCR interactions (10–12, 27) can then be used to rationally design candidates for heteroclitic vaccines. Once the best candidates are identified, they can be used individually or in a cocktail vaccine (reference 19 and Dyall, R., and L. Weber, unpublished data). The latter would maximize the odds of success and minimize the risk of tumor escape by epitope mutation. Of course, similar principles can be applied equally well against intracellular pathogens.
Our data also touch upon the issue of self-tolerance and tumor immunotherapy. As recently discussed (8), it is now clear that many self-differentiation antigens do not induce complete tolerance through deletion of self-reactive lymphocytes from the immune repertoire. In that regard, an important advantage of poor MHC binders derived from self-proteins may be that they are unlikely to tolerize T cells (as the signal 1 they provide is not strong enough), sparing a precursor CTL population that can be activated by an appropriate heteroclitic vaccine. Our results with the gp75 peptide TWH lend experimental support to this view, suggesting that this class of weak differentiation antigens could become a potential target for tumor therapy. Of interest, in the experiments described here, we did not notice any overt signs of autoimmunity (including depigmentation) that one might expect if a melanocyte antigen, such as gp75, is used to target the tumor for lysis by heteroclitic CTLs. Indeed, such autoimmune manifestations frequently accompany productive antimelanoma immunity in mice and have been suggested in humans (26 and the references contained therein). At present, it is unclear whether CTLs induced by heteroclitic vaccination are less prone to induce autoimmunity or whether the results observed were due to the particular peptide chosen. This issue is currently being addressed experimentally.
Acknowledgments
The authors wish to thank Dr. Y. Takechi for help with peptide binding assays; Dr. S. Vukmanovic for perusing the manuscript; Ms. D. Nikolić-Žugić for flow cytometry; and CytRx Inc. and Powderject, Inc. for generously providing the immune adjuvant Titermax and the gene gun, respectively.
This work was supported in part by the Byrne Fund and the DeWitt Wallace Fund awards (to J. Nikolić-Žugić), Swim Across America, the Louis and Anne Abrons Foundation, the Milstein Family Fund, the Lymphoma Foundation, and US Public Health Service grants CA56821, CA59350 (to A.N. Houghton), and CA08253 (to the Memorial Sloan-Kettering Cancer Center) from the National Institutes of Health.
Abbreviations used in this paper
- ER
endoplasmic reticulum
- ERIS
ER insertion sequence
- pCTL
precursor CTL
- SEI
peptide SEIEFARL, a variant of SSI
- SSI
Herpes simplex virus glycoprotein B498–505 peptide SSIEFARL
- TAY
peptide TAYRYHLL, an engineered variant of TWH
- TM
synthetic adjuvant TiterMax
- TWH
melanoma gp75222–229 peptide, TWHRYHLL
References
- 1.Houghton AN. Cancer antigens: immune recognition of self and altered self. J Exp Med. 1994;180:1–4. doi: 10.1084/jem.180.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bretscher P, Cohn M. A theory of self-nonself discrimination. Science. 1970;169:1042–1049. doi: 10.1126/science.169.3950.1042. [DOI] [PubMed] [Google Scholar]
- 3.Jenkins MK, Ashwell JD, Schwartz RH. Allogeneic non-T spleen cells restore the responsiveness of normal T cell clones stimulated with antigen and chemically modified antigen-presenting cells. J Immunol. 1988;140:3324–3330. [PubMed] [Google Scholar]
- 4.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 5.Nossal GVJ. Negative selection of lymphocytes. Cell. 1994;76:229–240. doi: 10.1016/0092-8674(94)90331-x. [DOI] [PubMed] [Google Scholar]
- 6.Vukmanović S, Bevan MJ, Hogquist KA. The specificity of positive selection: MHC and peptides. Immunol Rev. 1993;135:51–66. doi: 10.1111/j.1600-065x.1993.tb00643.x. [DOI] [PubMed] [Google Scholar]
- 7.Sette A, Vitiello A, Reherman B, Fowler P, Nayersina R, Kast WM, Melief CJM, Oseroff C, Yuan L, Ruppert J, et al. The relationship between class I binding affinity and immunogenicity of potential cytotoxic T cell epitopes. J Immunol. 1994;153:5586–5597. [PubMed] [Google Scholar]
- 8.Robbins PF, Kawakami Y. Human tumor antigens recognized by T cells. Curr Opin Immunol. 1996;8:628–636. doi: 10.1016/s0952-7915(96)80078-1. [DOI] [PubMed] [Google Scholar]
- 9.Solinger AM, Ultee ME, Margoliash E, Schwartz RH. T-lymphocyte response to cytochrome c. Demonstration of a T-cell heteroclitic proliferative response and identification of a topographic antigenic determinant on pigeon cytochrome c whose immune recognition requires two complementing major histocompatibility complex-linked immune response genes. J Exp Med. 1979;150:830–848. doi: 10.1084/jem.150.4.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madden DR. The three dimensional structure of peptide-MHC complexes. Annu Rev Immunol. 1995;13:587–622. doi: 10.1146/annurev.iy.13.040195.003103. [DOI] [PubMed] [Google Scholar]
- 11.Garcia CG, Degano M, Stanfield RL, Brunmark A, Jackson MR, Peterson PA, Teyton L, Wilson IA. An αβ T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science. 1996;274:209–216. doi: 10.1126/science.274.5285.209. [DOI] [PubMed] [Google Scholar]
- 12.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 13.Boehncke WH, Takeshita T, Pendleton CD, Houghten RC, Sadegh-Nasseri S, Racioppi L, Berzofsky JA, Germain RN. The importance of dominant negative effects of amino acid side chain substitution in peptide-MHC molecule interactions and T cell recognition. J Immunol. 1993;150:331–339. [PubMed] [Google Scholar]
- 14.England RD, Killberg MC, Conette JL, Berzofsky JA. Molecular analysis of a heteroclitic T cell response to the immunodominant epitope of sperm whale myoglobin. Implications for peptide partial agonists. J Immunol. 1995;155:4295–4306. [PubMed] [Google Scholar]
- 15.van der Burg SH, Visseren MJ, Brandt RM, Kast WM, Melief CJ. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. J Immunol. 1996;156:3308–3314. [PubMed] [Google Scholar]
- 16.Dyall R, Fremont DH, Jameson SC, Nikolić J - Žugić. T cell receptor (TCR) recognition of MHC Class I variants: intermolecular second-site reversion provides evidence for peptide/MHC conformational variation. J Exp Med. 1996;184:253–258. doi: 10.1084/jem.184.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yewdell J, Lapham C, Bacik I, Spies T, Bennink J. MHC-encoded proteasome subunits LMP2 and LMP7 are not required for efficient antigen presentation. J Immunol. 1994;152:1163–1170. [PubMed] [Google Scholar]
- 18.Anderson K, Cresswell P, Gammon M, Hermes J, Williamson A, Zweerink H. Endogenously synthesized peptide with an endoplasmic reticulum signal sequence sensitizes antigen processing mutant cells to class I–restricted cell-mediated lysis. J Exp Med. 1991;174:489–492. doi: 10.1084/jem.174.2.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dyall R, Vasović LV, Molano A, Nikolić-Žugić J. CD4-independent priming of murine CTLs by optimal MHC class I-restricted peptides in vivo. Int Immunol. 1995;7:1205–1212. doi: 10.1093/intimm/7.8.1205. [DOI] [PubMed] [Google Scholar]
- 20.Ross H, Weber LW, Wang S, Takechi Y, Piskun G, Dyall R, Nikolić-Žugić J, Houghton AN, Lewis JJ. Priming for T cell mediated rejection of tumors by cutaneous DNA immunization. Clin Cancer Res. 1997;3:2191–2196. [PubMed] [Google Scholar]
- 21.Ljunggren HG, Karre K. Host resistance directed selectively against H-2 deficient lymphoma variants. Analysis of the mechanism. J Exp Med. 1985;162:1745–1759. doi: 10.1084/jem.162.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vijayasaradhi S, Bouchard B, Houghton AN. The melanoma antigen gp75 is the human homologue of the mouse b (Brown)locus gene product. J Exp Med. 1990;171:1375–1380. doi: 10.1084/jem.171.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houghton AN, Vijayasaradhi S, Bouchard B, Naftzger C, Hara I, Chapman PB. Recognition of autoantigens by patients with melanoma. Ann NY Acad Sci. 1993;690:59–68. doi: 10.1111/j.1749-6632.1993.tb43996.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang RF, Appella E, Kawakami Y, Kang X, Rosenberg SA. Identification of TRP2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med. 1996;184:2207–2216. doi: 10.1084/jem.184.6.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara I, Takechi Y, Houghton AN. Implicating a role for immune recognition of self in tumor rejection: passive immunization against the BROWN locus protein. J Exp Med. 1995;182:1609–1614. doi: 10.1084/jem.182.5.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naftzger C, Takechi Y, Kohda H, Hara I, Vijayasaradhi S, Houghton AN. Immune response to a differentiation antigen induced by altered antigen: a study of tumor rejection and autoimmunity. Proc Natl Acad Sci USA. 1996;93:14809–14814. doi: 10.1073/pnas.93.25.14809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rammensee H-G, Falk K, Rotzschke O. Peptides naturally presented by MHC class I molecules. Annu Rev Immunol. 1993;11:213–244. doi: 10.1146/annurev.iy.11.040193.001241. [DOI] [PubMed] [Google Scholar]
- 28.Shibahara S, Tomita Y, Sakakura T, Nager C, Chaudhuri B, Muller R. Cloning and expression of cDNA encoding mouse tyrosinase. Nucleic Acids Res. 1986;14:2413–2427. doi: 10.1093/nar/14.6.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takechi Y, Hara I, Naftzger C, Xu Y, Houghton AN. A melanosomal protein is a cell surface target for melanoma therapy. Clin Cancer Res. 1996;2:1837–1842. [PubMed] [Google Scholar]
- 30.Lipford GB, Bauer S, Wagner H, Heeg K. Peptide engineering allows cytotoxic T-cell vaccination against human papilloma virus tumor antigen, E6. Immunology. 1995;84:298–303. [PMC free article] [PubMed] [Google Scholar]
- 31.Topalian SL, Gonzales MI, Parkhurst M, Li YF, Southwood S, Sette A, Rosenberg SA, Robbins PF. Melanoma-specific CD4+T cells recognize nonmutated HLA-DR–restricted tyrosinase epitopes. J Exp Med. 1996;183:1965–1971. doi: 10.1084/jem.183.5.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakker ABH, van der Burg SH, Huijbens RJ, Drijfhout J-W, Melief CJM, Adema GJ, Figdor CG. Analogues of CTL epitopes with improved MHC class-I binding capacity elicit anti-melanoma CTL recognizing the wild-type tumor epitope. Int J Cancer. 1997;70:302–309. doi: 10.1002/(sici)1097-0215(19970127)70:3<302::aid-ijc10>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 33.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp 100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive tumoricidal T cells using high-affinity, altered peptide ligand. J Exp Med. 1998;188:277–286. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]