

Abstract
A peptide derived from the human papillomavirus L2 protein is recognized by a myelin basic protein (MBP)-specific T cell clone from a multiple sclerosis patient and by MBP-specific autoantibodies purified from multiple sclerosis brain tissue. We now show in mice that low doses of this papillomavirus peptide were optimal in selecting a subpopulation of papillomavirus peptide–specific T cells that cross-reacted with MBP(87–99) and with an unrelated viral peptide derived from the BSLF1 protein of Epstein-Barr virus (EBV). These low dose viral peptide– specific T cell lines were highly encephalitogenic. Splenocytes from mice transferred with viral peptide–specific T cells showed a vigorous response to both the papillomavirus and MBP peptides, indicating that viral antigen–specific T cells survived for a prolonged time in vivo. The EBV peptide, unable to prime and select an autoreactive T cell population, could still activate the low dose papillomavirus peptide–specific cells and induce central nervous system (CNS) autoimmunity. Cytokine profiles of papillomavirus peptide–specific encephalitogenic T cells and histopathology of CNS lesions resembled those induced by MBP. These results demonstrate conserved aspects in the recognition of the self-antigen and a cross-reactive viral peptide by human and murine MBP-specific T cell receptors. We demonstrate that a viral antigen, depending on its nature, dose, and number of exposures, may select autoantigen-specific T cells that survive in vivo and can trigger autoimmune disease after adoptive transfer.
Keywords: autoimmunity, cross-reactivity, experimental autoimmune encephalomyelitis, molecular mimicry, antigenic peptide
utoimmunity has been proposed as the pathogenetic basis for a large number of organ-specific and systemic diseases of unknown etiology. Based on epidemiological studies, it is thought that autoimmunity results from inheritance of susceptibility genes (1–3), as well as environmental factors that play a role in regulating the expression of these genetic traits (for reviews, see references 4–6). For an autoimmune process to occur, self-reactive cells that have escaped thymic deletion have to be activated and expanded. Peptides and superantigens derived from infectious agents are potent activators of T cell–dependent immune responses. For this reason, infectious agents have been suspected for decades to be environmental triggers of autoimmunity. Epidemiological studies, mainly geographic distribution and migration studies (for reviews, see references 5 and 7) and concordance studies between monozygotic and dizygotic twins (8, 9), provide indirect support for this hypothesis. This hypothesis is also supported by several studies in transgenic animal models. Myelin basic protein (MBP)1-specific TCR transgenic mice developed spontaneous central nervous system (CNS) autoimmunity with a significantly higher incidence when the animals were not housed under specific pathogen–free conditions (10). Also, transgenic mice that expressed a viral antigen as a “self”-antigen in islet cells of the pancreas developed diabetes when infected with the respective virus (11, 12). Several mechanisms could account for a role of infections in the etiology of autoimmune diseases. These include autosensitization due to tissue injury in the target organ (13–15), T cell activation by microbial superantigens (16), and T cell activation by cross-reactive microbial peptides (6, 17–19).
Structural similarities between microbial and self-peptides can result in the activation of autoreactive T cells; this concept has been referred to as “molecular mimicry.” In some instances, structural similarities can be identified by sequence alignment between a self-peptide and microbial antigens. This approach has been used to identify microbial peptides that induce histological and/or clinical signs of disease in several animal models, such as experimental autoimmune encephalomyelitis (EAE) in rabbits and autoimmune oophoritis or streptococcal myocarditis in mice (17, 20, 21).
In a recent paper, Zhao et al. (22) demonstrated that an epitope expressed by a coat protein of HSV-1 could be recognized by autoreactive T cells that target corneal antigens in a murine model of autoimmune herpes stromal keratitis. Mutant HSV-1 viruses that lacked this epitope did not induce autoimmune disease. In the herpes stromal keratitis model, the presence of the virus at the site of autoimmune inflammation was required.
Analysis of the structural requirements for MHC class II binding and TCR recognition by MBP(85–99)-specific T cell clones derived from multiple sclerosis (MS) patients revealed that several peptides, including peptides from viral and bacterial origin, that had limited sequence homology with the MBP peptide or to each other could activate the same T cell clone (19, 23–25). For one of these viruses it was possible to demonstrate that a naturally processed viral peptide could also activate MBP-specific T cells (19). In addition, microbial peptides were identified that were bound by affinity-purified MBP-specific autoantibodies from CNS tissue of MS cases. Due to similarities in the T cell and B cell epitopes of MBP, a papillomavirus peptide was identified that was recognized by an MBP-specific T cell clone and by autoantibodies (26, 27). Although studies with individual T cell clones are essential in furthering our understanding of the structural basis of molecular mimicry, it is important to consider the existence in vivo of a multitude of TCRs that can interact with multiple antigens. It is then crucial to investigate the induction of autoimmune disease by viral antigens in an animal model in which the dynamics of polyclonal T cell populations play a major role.
EAE in mice is a well-established model of autoimmune disease that provides an appropriate system to address these questions in vivo (28–31). EAE can be induced either by passive transfer of myelin antigen–specific T cells, or by subcutaneous immunization with a myelin antigen in adjuvant (32–34). It has been shown that even a six-residue peptide, from which five amino acids were derived from the NH2-terminal region of native MBP, could induce EAE in susceptible mice (35). However, the importance of the NH2-terminal acetylation for the encephalitogenicity of MBP(Ac1-11) in the PL mouse has made it difficult to identify cross-reactive microbial peptides in that model. We chose to study the SJL mouse model in which the MBP(87–99) peptide is both immunodominant and encephalitogenic (36); this peptide is also immunodominant for human T cells (37–39), potentially allowing analysis of microbial peptides that activate human T cell clones.
In this study, three viral peptides were identified that activated LN T cells from MBP(87–99)-primed SJL mice. T cells specific for a papillomavirus peptide induced severe EAE after adoptive transfer and could be isolated from mice >11 wk after transfer. In this model, the viral antigen was not present in the target organ, ruling out inflammation mediated by viral peptide–specific T cells activated locally by the same viral peptide. Interestingly, we found that low doses of the papillomavirus peptide (0.004–0.4 μg/ml) were superior to moderate or high doses (10–80 μg/ml) in selecting a subpopulation of viral peptide–specific T cells in vitro that cross-reacted with MBP(87–99) and also with an EBV peptide. In vitro reactivation of papillomavirus-specific T cells by this EBV peptide also resulted in EAE after adoptive transfer. Pathological examination demonstrated that viral peptide–induced disease was indistinguishable from EAE triggered by the self-antigen. The relevance of these results for the induction of autoimmunity by molecular mimicry is discussed.
Materials and Methods
Mice.
Female SJL mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and from the Frederick Cancer Research and Development Center (Frederick, MD). All mice were kept under specific pathogen–free conditions at the animal facilities of the National Institutes of Health and were used between 7 and 12 wk of age according to approved protocols.
Antigens.
MBP was prepared from guinea pig spinal cords (Harlan Sprague Dawley Inc., Indianapolis, IN) according to the protocol from Deibler et al. (40). The peptides used in screening for cross-reactivity to the peptide MBP(87–99) (see Table 1) were synthesized by Quality Controlled Biochemicals, Inc. (Hopkinton, MA) and by Chiron Mimotopes (San Diego, CA). MBP(87–99), MBP(83–100), papillomavirus peptide (pHPV-7), and EBV peptide (pEBV) were prepared by continuous flow solid phase synthesis by the Protein and Nucleic Acid Facility, Beckman Center, Stanford University (Stanford, CA). The peptide NP(260–283) was a gift from Drs. Benjamin Segal and Ethan Shevach (National Institute of Allergy and Infectious Diseases, Bethesda, MD).
Table 1.
Screening of a Panel of Viral and Bacterial Potential Mimicry Peptides for Their Ability to Stimulate LN Cells from SJL Mice Immunized with Peptide MBP(87–99)
| Peptide | Pathogen | Sequence | SImax* | Cmax‡ | ||||
|---|---|---|---|---|---|---|---|---|
| MBP(83–100) | ENPVVHFFKNIVTPRTPP | 35.6 | 50 | |||||
| MBP(87–99) | VHFFKNIVTPRTP | 43.8 | 100 | |||||
| Group 1. Peptides that matched the recognition motif for human MBP(85–99) specific T cell clones. | ||||||||
| pHPV-7 | Human papillomavirus 7 | IGGRVHFFKDISPIASSE | 15.2 | 100 | ||||
| 8.5 | 50 | |||||||
| 6.6 | 10 | |||||||
| 3.6 | 1 | |||||||
| 2.4 | 0.1 | |||||||
| 19 | Human CMV | VVALVNFLRHLTQKP | <1.5 | − | ||||
| 29 | EBV | TGGVYHFVKKEAFNT | 1.8 | 100 | ||||
| 34 | Lymphocytic choriomeningitis virus | ISIFLHFVRIPTHRH | <1.5 | − | ||||
| 41 | Adenovirus 12 | DFEVVTFLKDVLPEF | 2.1 | 60 | ||||
| 43 | Coxsackie virus A9 | TLMVIPFVKLDYADT | <1.5 | − | ||||
| 52 | Influenza A | YRNLVWFIKKNTRYP | <1.5 | − | ||||
| 70 | Pseudomonas | DRLLMLFAKDVVSRN | <1.5 | − | ||||
| 140 | HIV | RQTALFLLKLAARWP | <1.5 | − | ||||
| 149 | Herpes simplex | GGRRLFFVKAHVRES | <1.5 | − | ||||
| 180 | Reovirus | MARAAFLFKTVGFGG | <1.5 | − | ||||
| 186 | Human respiratory syncytium virus | HSFKLWFLKRLNVAE | <1.5 | − | ||||
| 193 | Human astrovirus | QITSIFLIKPLADFK | <1.5 | − | ||||
| Group 2. Peptides that matched the recognition motif for MBP(85–99) specific autoantibodies from MS patients. | ||||||||
| B2 | Human papillomavirus 13 | IGGRVHFFKDISPIS | 2.8 | 35 | ||||
| B3 | Human papillomavirus 40 | IGGRVHFFRDISPIG | <1.5 | − | ||||
| B4 | Human papillomavirus 32 | IGSRVHFFHDISPIT | <1.5 | − | ||||
| pEBV | EBV | RAHPVYFFKSACPPA | 16.9 | 50 | ||||
| 20.8 | 35 | |||||||
| 15.6 | 3.5 | |||||||
| 7.4 | 0.35 | |||||||
| pCMV | Human CMV | DRHPVYFFKSACPPN | 17.5 | 100 | ||||
| 26.6 | 50 | |||||||
| 18.5 | 35 | |||||||
| 9.3 | 3.5 | |||||||
| 1.5 | 0.35 | |||||||
| B7 | Dhori virus | SDDFIHFFKAKSYDD | <1.5 | − | ||||
| B8 | HSV-1 | GGRRLFFVKAHVRES | <1.5 | − | ||||
| B9 | EBV | TGGVYHFVKKHVHES | <1.5 | − | ||||
| B10 | Influenza A virus | KDMTKEFFKNKSETW | <1.5 | − | ||||
| B11 | Human herpes virus 4 | VSGFISFFKNPFGGM | 1.5 | − | ||||
| B12 | Hepatitis A virus | EVKPASFFKNPHNDM | <1.5 | − | ||||
| B13 | Human adenovirus | LATYHIFFKNQRIPL | 4.4 | 50 | ||||
| B14 | Streptococcus agalactiae | RNIGYIFFKNSTIDI | 2.5 | 100 | ||||
| B15 | Bacillus subtilis | RKVVTDFFKNIPQRI | <1.5 | − | ||||
| B16 | Bacillus subtilis | KGTIYTFFKNKEELF | <1.5 | − | ||||
| B17 | Bacillus thuringiensis | PAPADLFFKNADINV | <1.5 | − | ||||
| B18 | Clostridium cellulare | CDAISNFFKNIGYAN | 7.8 | 100 | ||||
| B19 | Vibrio anguillarum | ELLHQRFFKNVESTP | 4.5 | 100 | ||||
| B20 | Pseudomonas aeruginosa | LEIIEEFFKNKSGLK | <1.5 | − | ||||
| B21 | Escherichia coli | LLSAISFFKNTHDFI | <1.5 | − | ||||
| B22 | Staphylococcus | GIDFDKFFKNRIDTF | <1.5 | − | ||||
| B23 | Streptococcus equisimilis | RNKIRQFFKNQDKEL | <1.5 | − | ||||
(continued)
Maximal stimulation index in a proliferation assay using LN cells from mice that had been immunized with 200 μg/mouse of peptide MBP(87–99). Each peptide was tested in triplicate, at five different concentrations ranging from 0.1 to 100 μg/ml, in AIM-V medium.
Concentration of peptide (μg/ml) at which the maximal stimulation occurred. For the viral peptides pHPV-7, pEBV, and pCMV responses to several doses of the antigens are shown.
Cloning.
T cell clones were generated by limiting dilution. LN cells from mice that had been immunized with 200 μg of the papillomavirus peptide pHPV-7 were stimulated twice in vitro with low doses (0.01–0.05 μg/ml) of the priming antigen. At the third in vitro stimulation, T cells were seeded at 0.5 cells/well together with 3 × 105 irradiated splenocytes/well and antigen (papillomavirus peptide) in 96-well round-bottomed plates. On days 4, 7, and 10 after stimulation, the medium was changed for EAE medium containing 10% of medium from Con A–stimulated splenocytes. At day 14, the cells were restimulated using the same dose of antigen and number of APCs as before. The cycle of restimulation was repeated until cells were expanded (the typical cloning efficiency resulted in clonal growth in <1/60 wells seeded).
Induction of EAE by Passive Transfer.
An emulsion, containing equal volumes of CFA (Difco Laboratories, Inc., Detroit, MI) and a solution of antigen in PBS, was used to immunize mice subcutaneously. 200 μl of the emulsion was distributed over four sites at the flanks of the animals. A total of 400 μg of MBP, 200 μg of peptide (MBP(87–99), pHPV-7, pEBV), or just PBS in CFA (referred to as CFA immunization) was injected per mouse. For the influenza nucleoprotein peptide NP(260–283), only 5 μg/mouse was used (optimal dose [41]). The draining LNs were removed 10 d after the immunization, and the cells were stimulated in vitro using the immunizing antigen, a cross-reactive antigen, or an irrelevant antigen. For the stimulation, 2 ml of a suspension of 4 × 106 cells/ml in EAE medium (RPMI 1640 [GIBCO BRL, Gaithersburg, MD] supplemented with 2 mM glutamine, 10 mM Hepes, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate [all from BioWhittaker, Inc., Walkersville, MD], and 10% fetal bovine serum [GIBCO BRL]) was placed in each well of 24-well plates (Costar Corp., Cambridge, MA) with the appropriate concentration of antigen. Supernatants from representative wells were collected for cytokine analysis at 48, 72, and 96 h. 4 d after stimulation, the cells were collected and were either transferred into naive recipients or kept for further experiments. When transferred at this stage, the cells were washed once in EAE medium and once in PBS, and 0.6–1 × 108 cells were injected intraperitoneally into each recipient mouse. Alternatively, the cells were cultured in 80-cm2 flasks. 13–14 d after the first in vitro activation, they were washed and restimulated. For the stimulation of T cell lines, a suspension containing 106 T cells/ml, 3 × 106 irradiated (3,000 rads) splenocytes per ml, and the appropriate concentration of antigen was distributed into 24-well plates (2 ml/well). Supernatants were collected at different time points to measure the concentrations of cytokines. When transferred after two or more rounds of stimulation, the cells were collected and washed at 72 h and injected intraperitoneally into naive recipients.
Clinical Scores.
The clinical signs of EAE were scored using the following scale: 0, no apparent abnormalities; 0.5, weak tail with some muscle tone remaining; 1, completely limp tail; 1.5, hind leg weakness as demonstrated by impaired righting ability and partially limp tail; 2, impaired righting reflex with completely limp tail; 2.5, hind leg weakness to the degree of short lapses of dragging of a leg; 3, complete hind leg paralysis; 4, complete hind leg paralysis and weakness of front legs; 5, moribund (dead mice were scored as 5 if they had previously shown signs of progressive disease).
Purification of T Cells.
In some cases (see Table 2), LN cells were enriched for T cells to levels of >97% purity by passing them through mouse T cell enrichment columns (R&D Systems, Minneapolis, MN) following the manufacturer's instructions. The recovered cells were washed twice and resuspended (106 cells/ml) in EAE medium together with irradiated splenocytes (3 × 106 cells/ml) and the appropriate concentration of antigen. The suspension was plated in 24-well plates (2 ml/well).
Table 2.
Passive Transfer of EAE with Cells that had been Primed with Either the Papillomavirus Peptide (pHPV-7), MBP(87–99), or MBP and Activated with the Specified Dose of the Priming Antigen
| Immunization | In vitro stimulation | Dose | Incidence of EAE | No. of stimulations | Cells (/107) transferred | Maximal EAE score* | Day of onset* | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| μg/ml | ||||||||||||||
| High antigen dose experiments | ||||||||||||||
| MBP(87–99) | MBP(87–99) | 10 | 3/4 | 1 | 6,6,6,6 | 3,3,2,0 | <9,<9,<9,− | |||||||
| pHPV-7 | pHPV-7 | 10 | 0/4 | 1 | 6,6,6,6 | 0,0,0,0 | − | |||||||
| MBP | MBP | 25 | 2/2 | 1 | 6,6 | 2,3 | 8,8 | |||||||
| PHPV-7 | pHPV-7 | 10 | 0/5 | 1 | 6,6,6,6,6 | 0,0,0,0,0 | − | |||||||
| MBP(87–99) | MBP(87–99) | 10 | 4/4 | 1 | 10,10,10,10 | 3,3,3,3 | 7,7,8,8 | |||||||
| pHPV-7 | pHPV-7 | 50 | 1/6 | 1 | 10,10,10 | 1,0,0 | 9,−,−, | |||||||
| 10,10,10 | 0,0,0 | −,−,− | ||||||||||||
| MBP(87–99) | MBP(87–99) | 10 | 2/3 | 3 | 2 | 4,4,0 | 7,7,0 | |||||||
| pHPV-7 | pHPV-7 | 10 | 0/3 | 3 | 2 | 0,0,0 | − | |||||||
| Low antigen dose experiments | ||||||||||||||
| pHPV-7 | pHPV-7 | 0.05 | 5/6 | 2 | 3 (1),‡ 0.6§ | 2,3 | 7,6 | |||||||
| 2 | 3,3 (4)§ | 2,1.5 | 6,7 | |||||||||||
| 5 | 3,3 | 2,0 | 8,− | |||||||||||
| pHPV-7 | pHPV-7 | 0.05 | 4/5 | 2 | 3,3,0.8 | 2.5,2.5,0 | 7,7,− | |||||||
| 2 | 3 (10),§ 1§ | 3,2 | 6,7 | |||||||||||
| pHPV-7 | pHPV-7 | 0.05 | 4/4 | 2 | 3 (12),3,3,3 | 2.5,2.5,2,2 | 7,7,7,7 | |||||||
| pHPV-7 | pHPV-7 | 0.05 | 2/3 | 2 | 3 (16),3,3 | 2,1,0 | 8,8,− | |||||||
| pHPV-7 | pHPV-7 | 0.05 | 4/4 | 2 | 4,4,4,4 | 5,3,3,2 | 6,6,6,8 | |||||||
| MBP(87–99) | MBP(87–99) | 0.05 | 4/4 | 2 | 3,3,3,3 | 3,2.5,2.5,2.5 | 5,5,6,6 | |||||||
| MBP(87–99) | MBP(87–99) | 10 | 4/4 | 2 | 3,3,3,3 | 5,5,5,4 | 5,5,5,5 | |||||||
Data for individual mice are separated by commas.
Numbers in parenthesis relate to histopathological samples (see Fig. 5).
Enriched for T cells at the LN cell stage as described in Materials and Methods.
In Vitro Proliferation Assays.
When LN cells were tested, 4 × 105 cells/well (0.2 ml) were cultured for 4 d in triplicates in 96-well flat-bottomed plates (Costar Corp.) in the presence of various concentrations of antigen or medium alone. In the case of T cell lines, 2 × 104 T cells and 2 × 105 irradiated (3,000 rads) splenocytes were cultured per well in round-bottomed plates (Nunc, Inc., Roskilde, Denmark) for 3 d. In both cases, [3H]thymidine (1 μCi/well) was added for the last 6–12 h of culture. The incorporation of [3H]thymidine was determined in a scintillation counter (1205 Betaplate; Wallac, Gaithersburg, MD) as described previously (34). Assays were performed in EAE medium, except for those represented in Table 1, Fig. 1, and Fig. 7, A2, B2, and C2, for which serum-free lymphocyte culture medium (AIM-V [GIBCO BRL] supplemented with 2 mM glutamine, 10 mM Hepes, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.1 mM nonessential amino acids, and 1 mM sodium pyruvate) was used in order to reduce background responses. The results were always confirmed using EAE medium.
Figure 1.

Stimulation with MBP, but not with moderate doses of papillomavirus peptide, selects a highly cross-reactive subpopulation of viral peptide–specific T cells. Proliferative responses of (A) LN cells or (B and C) T cell lines from SJL mice immunized with 200 μg/mouse of papillomavirus peptide in CFA. (A) Papillomavirus peptide– specific LN cells are able to respond, but only poorly to the self-antigen MBP(87– 99). In vitro proliferative responses are shown for (B) a short-term T cell line established by stimulating viral peptide–primed LN cells once with 10 μg/ml pHPV-7, and (C) a short-term T cell line established by stimulating viral peptide–primed LN cells once with 50 μg/ml MBP. Values represent mean stimulation indices (± SD) for triplicate wells.
Figure 7.
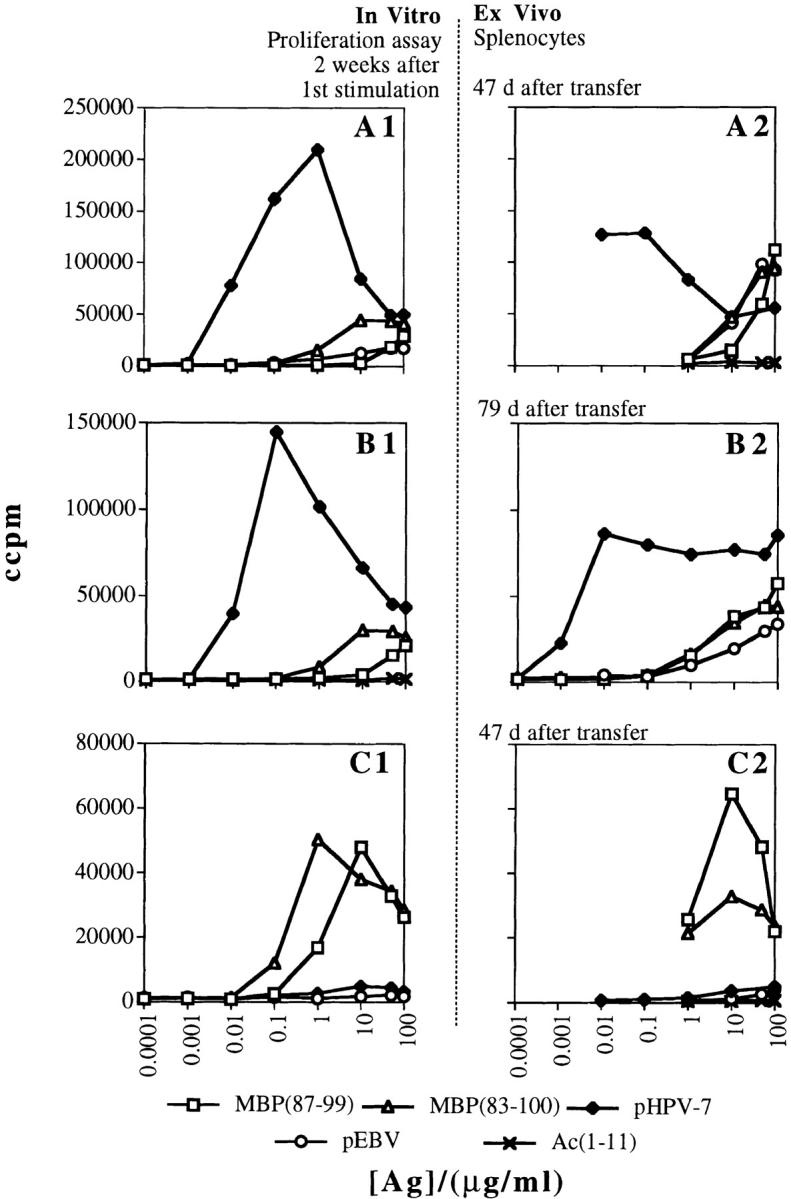
Survival of viral peptide–specific encephalitogenic T cells in vivo. Proliferative responses of (A1, B1, and C1) T cell lines at the time of the second stimulation, and of splenocytes from mice transferred (A2 and C2) 47 d or (B2) 79 d earlier with the cells resulting from the second activation of those T cell lines. Panels represent the in vitro responses of (A1 and B1) two different papillomavirus-specific T cell lines; (A2 and B2) the splenocytes from individual mice transferred with the same T cell lines after reactivation with (A2) 0.05 μg/ml of pHPV-7 (representative of three mice) or with (B2) 0.07 μg/ml of pHPV-7 (representative of three mice); (C1) the MBP(87–99)-specific T cell line; and (C2) the splenocytes from a mouse (representative of two mice) transferred with the same line after stimulation with 10 μg/ml of MBP(87–99).
ELISA.
TNF-α, IFN-γ, IL-4, and IL-10 were measured using a sandwich ELISA technique. DuoSet ELISA Development System kits (Genzyme Corp., Cambridge, MA) were used for the quantification of TNF-α, IFN-γ, and IL-4, following the manufacturer's recommendations. The antibodies for the detection of IL-10 and the recombinant cytokine were obtained from PharMingen (San Diego, CA). The plates were developed with the peroxidase substrate tetramethylbenzidine (TMB), stopped with 1 M sulfuric acid, and read at 450 nm using a microplate reader (model MRX; Dynatech Laboratories, Inc., Chantilly, VA).
Histopathology.
Animals were killed by intracardiac perfusion with PBS under narcosis at various stages of disease (10–96 d after transfer). The brains and spinal cords were removed, fixed by immersion in 10% phosphate-buffered formalin, and paraffin embedded. Three longitudinal sections spanning the entire length of the spinal cord, three cross-sections at different levels in the spinal cord, and four to five sections of brain and brain stem were stained with hematoxylin and eosin (H&E) for each animal. The same number of sections (from adjacent 7-μm sections) were stained with luxol fast blue. Sections were analyzed by a neuropathologist who was not informed of the identity of the specimens. Animals 1, 4, 10, 12, and 16 from Table 2 and three animals transferred with MBP(87–99)-specific short-term T cell lines were examined.
Results
Identification of Pathogen-derived Peptides that Activate MBP(87–99)-specific T Cells In Vitro.
The MBP(87–99) peptide that is presented by the MHC class II molecule I-As is an immunodominant MBP epitope in SJL mice. A panel of microbial peptides with sequence similarity to this MBP peptide was tested for the ability to activate LN T cells from SJL mice primed with MBP(87–99). The following peptides were used (Table 1): group 1, peptides that matched the recognition motif for human MBP(85–99)- specific T cell clones (peptides pHPV-7, 19, 29, 34, 41, 43, 52, 70, 140, 149, 180, 186, and 193; reference 19); and group 2, peptides that matched the recognition motif for MBP(85–99)-specific autoantibodies from MS patients (peptides B2–B23; reference 27); and group 3, peptides that matched the T cell recognition motif for MBP(88–102)- specific T cells from Lewis rats (none of the 42 peptides in this group showed cross-reactivity; data not included).
These peptides were tested in a proliferation assay using LN cells derived from SJL mice immunized with MBP(87– 99). Three peptides were identified which activated MBP(87–99)-specific T cells with a stimulation index >10 (Table 1). These peptides were derived from the L2 minor capsid protein of human papillomavirus type 7 (peptide pHPV-7), the BSLF1 primase of EBV (peptide pEBV), and the UL70 protein of human cytomegalovirus (peptide pCMV). Three other peptides gave a stimulation index >4 and were derived from human adenovirus (peptide B13), Clostridium cellulare (peptide B18), and Vibrio anguillarum (peptide B19). The papillomavirus peptide shares 6 of 17 amino acids with MBP(83–100), whereas the EBV peptide shares 5 residues with the MBP peptide. Several peptides (B2, B3, and B4) that also share five to six amino acids with the MBP peptide induced only marginal or no proliferation of MBP(87–99)-specific LN T cells.
Reactivation of Viral Peptide–specific T Cells with the MBP Peptide Results in Increased Cross-reactivity in the T Cell Population and Clinical Disease.
After testing the ability of viral peptides to activate MBP(87–99)-specific T cells, we performed the reverse experiment. SJL mice were immunized with the papillomavirus peptide and tested for proliferation to the MBP peptide in vitro (Fig. 1 A). LN cells from mice primed with the papillomavirus peptide showed strong responses towards the viral peptide, but only a minimal response to MBP(87–99). Stimulating the papillomavirus peptide–specific LN cells once in vitro with moderate to high doses of the same viral peptide (10–50 μg/ml) increased the proliferative responses (upon restimulation 2 wk later) to the papillomavirus peptide, but it diminished the response to MBP (Fig. 1 B). When we transferred such a population of cells into naive SJL mice, there was almost no clinical effect (Table 2, top). Table 2 summarizes results from independent adoptive transfer experiments using different short-term T cell lines.
Based on the hypothesis that only a subpopulation of the T cells specific for the papillomavirus peptide could recognize MBP(87–99) and vice versa, we decided to expand the cross-reactive population by stimulating with the cross- reactive instead of the priming antigen. LN cells from mice primed with the viral peptide were stimulated in vitro with 50 μg/ml of the autoantigen MBP. 2 wk after in vitro stimulation, a proliferation assay was performed which demonstrated a marked increase in cross-reactivity (Fig. 1 C). Interestingly, stimulation with MBP apparently selected a subpopulation of viral peptide–specific cells that not only showed an increased response to the self-antigen (MBP), but also responded optimally to very low concentrations of the cognate viral peptide (Fig. 1 C).
Since it has been shown that the ability of T cells to respond in vitro does not always correlate with an in vivo effect (42, 43), it was important to test the encephalitogenic potential of the cross-stimulated cells. SJL mice were immunized with either MBP in CFA, papillomavirus peptide in CFA, or CFA alone. 10 d later, the draining LNs were collected. The cells were stimulated with 50 μg/ml of whole MBP for 4 d and transferred into naive animals. The recipients of cells that were primed with the papillomavirus peptide and stimulated in vitro with MBP developed severe relapsing–remitting EAE with an incidence of 100% (Fig. 2 A). The severity was comparable to that of mice receiving T cells which were both primed and stimulated with MBP. Importantly, none of the animals that received cells primed with CFA and cultured with 50 μg/ml of MBP developed any sign of EAE, ruling out the possibility that disease was due to in vitro priming by the autoantigen. Papillomavirus peptide–primed T cells also induced severe autoimmune disease when activated by the MBP(87–99) peptide instead of the whole protein (not shown). Cells primed with the immunogenic influenza virus peptide NP(260–283), which is not cross-reactive with MBP(87– 99), did not induce disease when stimulated with the self-peptide (not shown), demonstrating the specificity of the phenomenon.
Figure 2.

Cross-stimulation of viral peptide–primed LN cells with the self-antigen (and vice versa) can select and activate potently encephalitogenic cells. Mice were immunized with an emulsion in CFA containing either no additional antigen, 400 μg of MBP, 200 μg of pHPV-7 or MBP(87–99), or 5 μg of NP(260–83) (not shown). 10 d later, LN cells were collected and stimulated with either (A) 50 μg/ml of MBP or (B) 10 μg/ml of the specified antigens. 4 d after stimulation, 108 cells were injected intraperitoneally into each naive recipient.
We also tested whether the induction of disease through cross-stimulation was a bidirectional phenomenon, using T cells that were primed with the MBP(87–99) peptide and stimulated in vitro with the viral peptide (Fig. 2 B). These cells also induced EAE. To exclude the possibility that disease was due to nonspecific bystander activation of T cells (i.e., secretion of Th1 cytokines by surrounding cells), we transferred T cells that had been primed with MBP(87–99) and activated with the same strain of Mycobacterium tuberculosis (MT) as that contained in CFA (H37RA). This activation led to a strong proliferative response accompanied by the secretion of high levels of TNF-α and IFN-γ (data not shown), but did not induce disease (Fig. 2 B).
Expansion of Virus-specific T Cells with Low Doses of Viral Peptide Selects a Cross-reactive T Cell Population with a Th1 Phenotype.
Since a subpopulation of papillomavirus-specific T cells that showed a high level of cross-reactivity to MBP responded optimally to very low concentrations of the cognate viral antigen (Fig. 1 C), we tested the hypothesis that different doses of the papillomavirus peptide could select subpopulations of viral peptide–specific T cells with distinct response profiles to both the cognate viral peptide and MBP(87–99). We collected LN cells from mice that had been immunized with the viral peptide, split them into five groups, and stimulated each group with a different dose of viral peptide (0.004, 0.05, 0.4, 10, or 80 μg/ml). 2 wk later, the proliferative responses of the resulting populations of cells to the papillomavirus peptide and MBP(87– 99) were analyzed (Fig. 3 A). It was evident that the lower doses of the viral peptide (0.004–0.4 μg/ml) were selecting a population of cells that responded better to the autoantigen MBP(87–99) than higher doses (10 and 80 μg/ml). The same result was obtained in five different experiments in which low (0.05 μg/ml; Fig. 3 B) versus moderate (10 μg/ml, which is the standard dose used for stimulation of lines with encephalitogenic peptides; Fig. 3 C) doses of viral peptide were compared. Interestingly, the T cell populations expanded by low doses of the papillomavirus peptide also showed a higher degree of cross-reactivity to the EBV peptide (Fig. 3, B and C) and to a peptide from adenovirus (peptide 41, data not shown).
Figure 3.

Very low doses of human papilloma virus peptide (0.004–0.4 μg/ml) select cross-reactive subpopulations of cells compared with moderate and high doses (10–100 μg/ml). (A) LN cells from mice that had been immunized with 200 μg/mouse of papillomavirus peptide were split into five groups and stimulated with various concentrations of the same viral peptide. Each curve represents the response to peptide MBP(87–99) by a particular T cell line. The maximal stimulation index in response to the papilloma virus peptide by each T cell line (SImax) and the concentration at which it occurred (Cmax) are shown. A separate experiment in which papilloma virus peptide–primed LN cells were stimulated with either (B) 0.05 μg/ml or (C) 10 μg/ml of the same viral peptide is also shown. In a similar approach, EBV peptide–primed LN cells were stimulated with either (D) 0.1 μg/ml or (E) 10 μg/ml of EBV peptide. Contrary to the papillomavirus peptide, the EBV peptide was not able to expand an autoreactive population of EBV peptide–specific cells. The short-term T cell lines generated by these means were tested (A–E) in standard proliferation assays, performed 14 d after the first activation. The viral peptide was not toxic at 10 μg/ml; when papillomavirus-specific T cells selected by one in vitro stimulation with 10 μg/ml of the viral peptide were stimulated for a second time in vitro using the same dose of viral peptide, the cell number increased to 145–180% of the original value in 3 d. If papillomavirus-specific T cells selected with 0.05 μg/ml were stimulated a second time with 0.05 μg/ml of viral peptide, the cell yield 3 d later was 160–170% of the initial value.
T cell lines (Table 3) specific for the papillomavirus peptide or peptide MBP(87–99) were tested for cytokine production upon stimulation with different doses of the priming antigen. The viral peptide–specific T cells had a similar pattern of cytokine secretion to that of MBP(87–99)-specific cells (Th1 skewed). However, papillomavirus peptide– specific T cells secreted higher levels of TNF-α and IFN-γ than the MBP(87–99)-specific T cells when stimulated with similar doses of the priming antigen (Table 3). Even low doses of the viral peptide led to this Th1 phenotype (including low production of IL-10). Table 3 shows cytokine levels produced by short-term T cell lines from one representative experiment out of three independent experiments performed using different T cell lines in each experiment.
Table 3.
Cytokine Production at 48 h by Short-term T Cell Lines Stimulated with Different Doses of the Priming Antigen
| Sample | Cytokine concentration | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Immunization | First stimulation | Second stimulation | TNF-α | IFN-γ | IL-4 | IL-10 | ||||||
| μg/ml | μg/ml | pg/ml | ||||||||||
| MBP(87–99) | 10 | No Ag | 0 | 1,605 ± 26 | 0 | 0 | ||||||
| 10 | 25,368 ± 434 | 9,508 ± 116 | 77 ± 5 | 3,693 ± 222 | ||||||||
| pHPV-7 | 0.05 | No Ag | 0 | 97 | 0 | 0 | ||||||
| 0.05 | 6,509 ± 251 | 15,768 ± 84 | 0 | 468 ± 70 | ||||||||
| 10 | No Ag | 0 | 0 | 0 | 0 | |||||||
| 10 | 45,079 ± 405 | 18,595 ± 3,570 | 0 | 1,464 ± 104 | ||||||||
Representative data (± SD) from three independent experiments with different T cell lines in each experimental series.
Specificity and Degeneracy of Antigen Recognition by T Cell Clones from SJL Mice.
To demonstrate recognition of different peptides by the same T cell, T cell clones were raised. Fig. 4 shows representative examples of responses by a panel of three clones specific for human papillomavirus peptide (pHPV-7) that were tested for recognition of two MBP peptides (p87–99 and p83–100) and two virus- derived peptides (pHPV-7 and pEBV). The clones (limiting dilution approach, 0.5 cells/well) were generated from papillomavirus peptide–primed LN cells that were only stimulated with the papillomavirus peptide at low doses (0.01–0.05 μg/ml) during in vitro expansion. Clones RU-1.01 and RU-4.01 proliferated to both the papillomavirus peptide and MBP(87–99), whereas clone RU-1.05 responded only to the papillomavirus peptide (in total, 3 out of 11 clones raised under similar conditions showed cross-reactivity to the MBP peptides).
Figure 4.

T cell clones specific for the human papilloma virus peptide recognize the self-antigens MBP(87–99) and MBP(83–100). T cell clones were generated from HPV-specific T cell lines which had been expanded with low doses of the same viral peptide. These clones were tested for their ability to proliferate in vitro to the self-peptides MBP(87–99) and MBP(83–100) and the viral peptides from HPV and EBV. T cell clones were incubated with irradiated splenocytes and various concentrations of the peptides in EAE medium. The values correspond to mean stimulation indices from triplicate wells.
Low Dose Viral Peptide–specific Cells Induce EAE.
Based on these data, we reasoned that low concentrations of the viral peptide may select a subpopulation of T cells with a high encephalitogenic potential. After stimulation of LN cells specific for the papillomavirus peptide with 0.05 μg/ ml of the same peptide, the cells were cultured for 2 wk, then reactivated with the same antigen/dose (Table 2, bottom). 3 d later, naive animals were injected intraperitoneally with 3 × 107 cells/mouse and observed for the development of clinical signs of EAE (Table 2, summary of results from independent adoptive transfer experiments using different short-term T cell lines). The disease induced by these papillomavirus-specific cells which had been stimulated with low doses of their cognate antigen (and never exposed to the self-antigen in vitro) was consistently early in onset, high in incidence, and moderate to severe in quality (Table 2, bottom). EAE could be transferred by papillomavirus-specific T cell lines that were established after T cell enrichment of LN cells to a level of >97% purity. FACS® analysis of the T cell lines generated after two in vitro stimulations with low doses of the papillomavirus peptide revealed that >97% of the T cells were CD4+ (data not shown). Disease could be transferred, although with decreasing incidence, even after five in vitro stimulations with 0.05 μg/ml of papillomavirus peptide.
The papillomavirus peptide was also tested for active induction of EAE (as described in reference 44) over a dose range of 200 μg to 2 mg of peptide per mouse. Two out of six mice immunized with 2 mg of the papillomavirus peptide developed mild to moderate disease. Clinical onset (day 12) and maximal severity (grade 2 in both mice) of disease were comparable to MBP-induced EAE, whereas disease duration was only 2–3 d. No signs of clinical disease were observed in nine mice immunized with 200 μg of the papillomavirus peptide. Results obtained in vitro and in adoptive transfer experiments suggest that multiple exposures to low doses of the viral peptide are more effective in selecting an autoreactive T cell population.
Histological Features of Disease Induced by T Cell Lines Specific for the Papillomavirus Peptide.
Histological examination demonstrated that disease induced by T cell lines specific for the papillomavirus peptide and the MBP(87–99) peptide was indistinguishable when animals with similar clinical severity were compared. Several lesion types could be identified within individual mice in both groups. Some lesions consisted mainly of lymphocytic infiltrates (Fig. 5 A). These infiltrates typically occurred either in the meninges or in very close proximity to a blood vessel, and were usually not accompanied by significant demyelination. Lesions containing mixed cellular infiltrates in which either macrophages (Fig. 5 B, and central lesion in Fig. 5 D) or polymorphonuclear cells (Fig. 5 C, and subpial lesion in Fig. 5 D) predominated commonly showed inflammatory cells infiltrating deep into the parenchyma and were accompanied by myelin destruction as demonstrated by luxol fast blue stains (Fig. 5 E). In these lesions, demyelination was either limited to the area of infiltration or extended beyond the infiltrate. These histopathological findings are akin to those described by Fallis et al. (45) and are considered characteristic of EAE.
Figure 5.





Representative photomicrographs of EAE histopathology in SJL mice transferred with papillomavirus peptide–specific T cells. (A) A large chronic inflammatory (lymphocytic) infiltrate is identified in the cortical meninges (original magnification: ×20; H&E). (B) A chronic inflammatory infiltrate of the spinal cord deep white matter consisting primarily of macrophages (arrows; original magnification: ×40; H&E). (C) An acute inflammatory infiltrate of the spinal cord subpial white matter consisting of a mixture of polymorphonuclear neutrophils, lymphocytes, and macrophages (original magnification: ×40; H&E). (D) A low power view of a longitudinal section of spinal cord demonstrating both acute (as in C) and chronic (as in B) infiltrates (original magnification: ×5; H&E). (E) Luxol fast blue histochemical stain reveals the loss of myelin in an adjacent section of spinal cord (original magnification: ×5).
Reactivation of Virus-specific T Cells with an Unrelated Viral Peptide Renders them Encephalitogenic.
The observation that the EBV peptide could reactivate both MBP peptide or papillomavirus-specific T cells (Table 1, and Fig. 3 B) was further analyzed. In contrast to the papillomavirus peptide, cross-reactivity with the EBV peptide was largely unidirectional. When SJL mice were immunized with the EBV peptide, LN cells showed very low responses to MBP(87– 99), much lower than those from papillomavirus peptide– primed LN cells. Furthermore, neither low (0.1 μg/ml) nor moderate (10 μg/ml) doses of pEBV could expand a subpopulation of T cells with significant cross-reactivity to MBP(87–99) or MBP(83–100) (Fig. 3, D and E). Transfer of EBV peptide–specific T cells that had been stimulated twice in vitro with either 0.1 or 10 μg/ml of EBV peptide did not induce disease (data not shown).
Since we observed that papillomavirus-specific cells that had been stimulated with low doses of papillomavirus peptide showed an increased response to the EBV peptide, we designed the following experiment (Fig. 6). 2 wk after an in vitro stimulation of papillomavirus-specific LN cells with 0.05 μg/ml of papillomavirus peptide (first in vitro activation), the resulting T cell line was divided into four different groups and reactivated with either 0.002 μg/ml of staphylococcal enterotoxin A (SEA), 0.05 μg/ml of papillomavirus peptide, 50 μg/ml of MBP(87–99), or 50 μg/ml of the EBV peptide. Since activation of T cells is required for their extravasation into the CNS (46, 47) and induction of EAE (48, 49), we tested whether nonantigen-specific stimulation of T cells by the superantigen (SEA) was sufficient to reactivate encephalitogenic T cells through bystander activation. Although the given dose of SEA induced a powerful activation of at least a subpopulation of T cells, as measured by proliferation and the secretion of the Th1 cytokines IFN-γ and TNF-α, the resulting population of cells did not induce disease when transferred 72 h after activation (SEA has been reported to activate T cells expressing a Vβ3 or Vβ17 TCR rearrangement in the SJL mouse [50]). In contrast, T cells activated with either the papillomavirus peptide or MBP(87–99) induced disease. Interestingly, the cells reactivated with the EBV peptide induced EAE with a similar incidence (2/2) and severity (mean maximal clinical severity of 2.5 ± 0.7) as the other two groups. This was the case even when a low dose of the EBV was used (Fig. 6 B).
Figure 6.

An unrelated viral peptide from EBV activates papillomavirus peptide–specific cells resulting in EAE. A short-term T cell line was generated by stimulating papillomavirus–primed LN cells with 0.05 μg/ml of the same viral peptide. 2 wk later, the resulting cells were split into four groups and stimulated with 0.05 μg/ml of papillomavirus peptide, 50 μg/ ml MBP(87–99), 50 μg/ml of the EBV peptide, or 0.002 μg/ml of SEA. Naive mice were transferred intraperitoneally with either 3 × 107 cells (two animals per group for the papillomavirus–, MBP(87–99)-, and EBV peptide–activated cells) or 6 × 107 cells (two animals for the SEA-activated cells).
In Vivo Survival of Viral Peptide–specific Cells that Recognize the Self-antigen.
For T cells selected and expanded by a viral antigen to induce autoimmune disease after a second exposure to the same or a different viral antigen, they need to survive in vivo for a prolonged time after the first activation. To examine this possibility, we transferred naive mice with encephalitogenic T cell lines that had been generated either by stimulation of papillomavirus peptide (pHPV-7)- specific LN cells with 0.05 μg/ml (Fig. 7 A) or 0.07 μg/ml (Fig. 7 B) of pHPV-7 (cells used in B were coming from different LN cells than those in A), or by stimulation of MBP(87–99)-specific LN cells with 10 μg/ml of the same MBP peptide (Fig. 7 C). The proliferative responses to various self- and viral peptides of the resulting T cell lines were assayed 2 wk after the first stimulation (Fig. 7, A1, B1, and C1). At the same time, the T cell lines were reactivated for transfer into naive animals using the same antigen and dose as for the first stimulation. 3 d after activation, 4 × 107 cells were transferred intraperitoneally into each naive recipient. 47 d (Fig. 7, A2 and C2) or 79 d (Fig. 7 B2) after transfer, the animals were killed and the splenocytes tested in a proliferation assay. Surprisingly, not only was it clear that the cells survived in vivo even after >11 wk, but it also seemed that the interactions within the animal had selected a highly self-reactive T cell population from the papillomavirus-specific cells (Fig. 7, A2 and B2). These cells responded vigorously to both the papillomavirus peptide and the EBV peptide.
Discussion
To advance our understanding of the potential role of microbial antigens in the induction of autoimmunity, it is important to consider the complex interactions between pathogen-derived antigens and cross-reactive T cells in polyclonal T cell populations with multiple specificities. In this study, we focused on LN cell populations, short-term T cell lines, and T cell clones. We began by screening pathogen-derived peptides, testing them for their ability to activate populations of MBP(87–99)-primed LN cells in vitro. Despite the lack of a well-defined MHC/TCR recognition motif for SJL mice, we identified three viral peptides that showed significant cross-reactivity (19) with MBP(87–99). Immunization with one of these peptides, from the human papillomavirus L2 capsid protein, resulted in a T cell population that showed a vigorous response to the viral peptide and significant cross-reactivity to MBP(87–99). Stimulation of LN cells from mice immunized with the papillomavirus peptide with different doses of the same viral peptide appeared to select different T cell subpopulations. The cells resulting from stimulation with moderate to high doses of the papillomavirus peptide (10– 80 μg/ml) responded optimally to high doses of papillomavirus peptide and showed very low responses to MBP(87– 99) and the EBV peptide. In contrast, stimulation with low doses of papillomavirus peptide (0.004–0.4 μg/ml) selected cells that responded optimally to low doses of the viral peptide and in addition showed an increased response to MBP(87–99) and to other cross-reactive viral peptides (EBV peptide and peptide 41). This “low dose” population of cells induced severe EAE at a high incidence. Moreover, the highly promiscuous population of cells selected by low doses of the papillomavirus peptide, different from that selected by high doses, could also be reactivated and rendered encephalitogenic by an unrelated EBV peptide. Of note, only four amino acids (V-FFK) were conserved among the papillomavirus peptide, the EBV peptide, and the MBP peptide. The low doses of viral peptide apparently selected a population of T cells with higher affinity for the I-As/papillomavirus peptide complex. High affinity for the low dose selecting viral antigen may explain the increased cross-reactivity of this T cell population with MBP(87–99) and the unrelated EBV peptide, since T cells with a higher affinity for their MHC/peptide ligands may tolerate nonconservative substitutions of the peptide. Interestingly, similar observations have been made for human MBP-specific T cell clones (19). In addition, it was shown that antibodies with higher affinity for their cognate ligand display a broader cross-reactivity to other antigens (51). We conclude that cross-reactivity and pathogenicity of selected T cell populations are dependent on the dose of the selecting antigen.
The papillomavirus peptide was previously found to activate an MBP-specific HLA-DQ1–restricted T cell clone from an MS patient; this T cell clone recognized four microbial peptides (derived from HSV, adenovirus type 12, human papillomavirus type 7, and Pseudomonas aeruginosa). The papillomavirus peptide was an effective stimulator of this T cell clone (background proliferation 1,059 ± 48 cpm, MBP(85-99) 11,661 ± 468 cpm, papillomavirus peptide 7,675 ± 10 cpm; our unpublished observations); among the other peptides, the adenovirus peptide induced the strongest stimulation of the T cell clone. Since murine I-A molecules are the homologues of HLA-DQ, these peptides were candidates for the induction of EAE in SJL mice. Of the four peptides tested, the papillomavirus peptide induced strong stimulation of MBP(87–99)-specific T cells, whereas the adenovirus peptide showed only a low level of cross-reactivity. These results indicate that there are similarities, as well as differences, in the specificity of human and murine TCRs specific for this immunodominant MBP peptide.
The concept that T cell population dynamics are important in the development of an autoimmune process is supported by another set of results. Different pathogen-derived antigens, and even different doses of the same antigen, might contribute in diverse ways to the development of autoimmunity by molecular mimicry, depending on their effect on self-reactive populations of T cells: (a) some viral peptides may prime and, on their own, activate T cells that will induce autoimmunity; (b) particular combinations of antigen and dose may predominantly expand and/or maintain a population of self-reactive memory T cells. Continuous or recurrent exposure to antigenic stimulation either by the cognate peptide or a cross-reactive peptide may be necessary to maintain T cell memory (52–54); and (c) some viral peptides (e.g., EBV peptide) which are unable to select and expand a self-reactive population of T cells on their own (because they preferentially expand a noncross-reactive population of viral peptide–specific cells) may still be capable of activating a self-reactive population that had already been selected and expanded by another viral peptide (Fig. 6) or the self-antigen. Certain combinations of antigen and dose (low doses of papillomavirus peptide) might preferentially expand a subpopulation of virus-specific T cells that show cross-reactivity to both the self-antigen and other viral antigens. The result would be an increase in the population of self-reactive cells that is available for activation either by the original viral antigen, or by a different antigen (e.g., EBV peptide).
In that view, the observation that viral peptide–specific cells survived in vivo for a long time and were enriched for recognition of the self-antigen and the unrelated EBV peptide (Fig. 7) was particularly interesting. In our model, in vivo survival of an autoreactive population of viral peptide– specific cells was observed in the absence of the cognate viral antigen in the animal. These results led us to speculate that exposure to the self-antigen in vivo (compare to exposure to self-antigen in vitro, Fig. 1 C) might again reshape the viral peptide–specific T cell population in a manner that would increase its potential for both (a) reactivation by a second exposure to the same or a different viral peptide and (b) induction of autoimmune disease.
Although it is not possible to draw conclusions about the role of a specific pathogen in triggering a particular human autoimmune disease based on studies in this animal model, our data suggest a role of molecular mimicry in the induction of T cell–mediated autoimmunity. Activation of self-reactive T cells by viral antigens could be relevant in two important steps in the development of inflammatory CNS diseases: (a) enhanced interaction with and transmigration through the blood–brain barrier (46, 47), and (b) facilitated reactivation of pathogenic cells by the self-antigen upon encounter in the CNS due to reduced requirements for costimulation of activated T cells (55–61). The fact that it has not been possible to identify a single pathogen as a culprit of MS may indicate that (sub)clinical infections may be involved. Furthermore, given the ability of viral antigen–primed autoreactive T cells to survive in vivo, it is possible to envision that a series of infections with different pathogens may be responsible for selection, maintenance, and/or expansion of a cross-reactive and pathogenic subpopulation of T cells.
Acknowledgments
The authors thank Drs. William Biddison, Henry McFarland, and Jack L. Strominger for support and discussions, and Drs. Thomas P. Leist, Michael Lenardo, Ethan Shevach, Ronald Schwartz, Andreas Steinbrecher, and Lawrence Steinman for critical comments.
This work was supported by a grant from the National Institutes of Health (AI-41641) and by the National Multiple Sclerosis Society (to K.W. Wucherpfennig). K.W. Wucherpfennig is a Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society. S. Hausmann is the recipient of a postdoctoral fellowship from the Deutscher Akademischer Austauschdienst.
Abbreviations used in this paper
- CNS
central nervous system
- EAE
experimental autoimmune encephalomyelitis
- H&E
hematoxylin and eosin
- MBP
myelin basic protein
- MS
multiple sclerosis
- SEA
Staphylococcal enterotoxin A
References
- 1.Shoenfeld Y, Schwartz RS. Immunologic and genetic factors in autoimmune diseases. N Engl J Med. 1984;311:1019–1029. doi: 10.1056/NEJM198410183111605. [DOI] [PubMed] [Google Scholar]
- 2.Ebers GC, Kukay K, Bulman DE, Sadovnick AD, Rice G, Anderson C, Armstrong H, Cousin K, Bell RB, Hader W, et al. A full genome search in multiple sclerosis. Nat Genet. 1996;13:472–476. doi: 10.1038/ng0896-472. [DOI] [PubMed] [Google Scholar]
- 3.Dyment DA, Sadovnick AD, Ebers GC. Genetics of multiple sclerosis. Hum Mol Genet. 1997;6:1693–1698. doi: 10.1093/hmg/6.10.1693. [DOI] [PubMed] [Google Scholar]
- 4.Ross RT, Cheang M. Geographic similarities between varicella and multiple sclerosis: an hypothesis on the environmental factor of multiple sclerosis. J Clin Epidemiol. 1995;48:731–737. doi: 10.1016/0895-4356(94)00184-r. [DOI] [PubMed] [Google Scholar]
- 5.Kurtzke JF. Epidemiologic evidence for multiple sclerosis as an infection. Clin Microbiol Rev. 1993;6:382–427. doi: 10.1128/cmr.6.4.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brocke S, Hausmann S, Steinman L, Wucherpfennig KW. Microbial peptides and superantigens in the pathogenesis of autoimmune diseases of the central nervous system. Semin Immunol. 1998;10:57–67. doi: 10.1006/smim.1997.0105. [DOI] [PubMed] [Google Scholar]
- 7.Ebers GC, Sadovnick AD. The geographic distribution of multiple sclerosis: a review. Neuroepidemiology. 1993;12:1–5. doi: 10.1159/000110293. [DOI] [PubMed] [Google Scholar]
- 8.James WH. Review of the contribution of twin studies in the search for non-genetic causes of multiple sclerosis. Neuroepidemiology. 1996;15:132–141. doi: 10.1159/000109900. [DOI] [PubMed] [Google Scholar]
- 9.Leslie RD, Hawa M. Twin studies in auto- immune disease. Acta Genet Med Gemellol. 1994;43:71–81. doi: 10.1017/s000156600000297x. [DOI] [PubMed] [Google Scholar]
- 10.Goverman J, Woods A, Larson L, Weiner LP, Hood L, Zaller DM. Transgenic mice that express a myelin basic protein-specific T cell receptor develop spontaneous autoimmunity. Cell. 1993;72:551–560. doi: 10.1016/0092-8674(93)90074-z. [DOI] [PubMed] [Google Scholar]
- 11.Oldstone MB, Nerenberg M, Southern P, Price J, Lewicki H. Virus infection triggers insulin-dependent diabetes mellitus in a transgenic model: role of anti-self (virus) immune response. Cell. 1991;65:319–331. doi: 10.1016/0092-8674(91)90165-u. [DOI] [PubMed] [Google Scholar]
- 12.Ohashi PS, Oehen S, Buerki K, Pircher H, Ohashi CT, Odermatt B, Malissen B, Zinkernagel RM, Hengartner H. Ablation of “tolerance” and induction of diabetes by virus infection in viral antigen transgenic mice. Cell. 1991;65:305–317. doi: 10.1016/0092-8674(91)90164-t. [DOI] [PubMed] [Google Scholar]
- 13.Miller SD, Vanderlugt CL, Begolka WS, Pao W, Yauch RL, Neville KL, Katz LY, Carrizosa A, Kim BS. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat Med. 1997;3:1133–1136. doi: 10.1038/nm1097-1133. [DOI] [PubMed] [Google Scholar]
- 14.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature. 1992;358:155–157. doi: 10.1038/358155a0. [DOI] [PubMed] [Google Scholar]
- 15.Yu M, Johnson JM, Tuohy VK. A predictable sequential determinant spreading cascade invariably accompanies progression of experimental autoimmune encephalomyelitis: a basis for peptide-specific therapy after onset of clinical disease. J Exp Med. 1996;183:1777–1788. doi: 10.1084/jem.183.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brocke S, Gaur A, Piercy C, Gautam A, Gijbels K, Fathman CG, Steinman L. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigen. Nature. 1993;365:642–644. doi: 10.1038/365642a0. [DOI] [PubMed] [Google Scholar]
- 17.Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–1045. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- 18.Davies JM. Molecular mimicry: can epitope mimicry induce autoimmune disease? . Immunol Cell Biol. 1997;75:113–126. doi: 10.1038/icb.1997.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garza KM, Tung KS. Frequency of molecular mimicry among T cell peptides as the basis for autoimmune disease and autoantibody induction. J Immunol. 1995;155:5444–5448. [PubMed] [Google Scholar]
- 21.Huber SA, Cunningham MW. Streptococcal M protein peptide with similarity to myosin induces CD4+ T cell-dependent myocarditis in MRL/++ mice and induces partial tolerance against coxsackieviral myocarditis. J Immunol. 1996;156:3528–3534. [PubMed] [Google Scholar]
- 22.Zhao Z, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279:1344–1347. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 23.Ausubel LJ, Kwan CK, Sette A, Kuchroo V, Hafler DA. Complementary mutations in an antigenic peptide allow for crossreactivity of autoreactive T-cell clones. Proc Natl Acad Sci USA. 1996;93:15317–15322. doi: 10.1073/pnas.93.26.15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vergelli M, Hemmer B, Kalbus M, Vogt AB, Ling N, Conlon P, Coligan JE, McFarland H, Martin R. Modifications of peptide ligands enhancing T cell responsiveness imply large numbers of stimulatory ligands for autoreactive T cells. J Immunol. 1997;158:3746–3752. [PubMed] [Google Scholar]
- 25.Hemmer B, Fleckenstein BT, Vergelli M, Jung G, McFarland H, Martin R, Wiesmuller KH. Identification of high potency microbial and self-ligands for a human autoreactive class II–restricted T cell clone. J Exp Med. 1997;185:1651–1659. doi: 10.1084/jem.185.9.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warren KG, Catz I, Steinman L. Fine specificity of the antibody response to myelin basic protein in the central nervous system in multiple sclerosis: the minimal B-cell epitope and a model of its features. Proc Natl Acad Sci USA. 1995;92:11061–11065. doi: 10.1073/pnas.92.24.11061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wucherpfennig KW, Catz I, Hausmann S, Strominger JL, Steinman L, Warren KG. Recognition of the immunodominant myelin basic protein peptide by autoantibodies and HLA-DR2–restricted T cell clones from multiple sclerosis patients. Identity of key contact residues in the B-cell and T-cell epitopes. J Clin Invest. 1997;100:1114–1122. doi: 10.1172/JCI119622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zamvil SS, Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol. 1990;8:579–621. doi: 10.1146/annurev.iy.08.040190.003051. [DOI] [PubMed] [Google Scholar]
- 29.Dal Canto, M.C., R.W. Melvold, B.S. Kim, and S.D. Miller. Two models of multiple sclerosis: experimental allergic encephalomyelitis (EAE) and Theiler's murine encephalomyelitis virus (TMEV) infection. A pathological and immunological comparison. Microsc Res Tech. 1995;32:215–229. doi: 10.1002/jemt.1070320305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf SD, Dittel BN, Hardardottir F, Janeway CA., Jr Experimental autoimmune encephalomyelitis induction in genetically B cell–deficient mice. J Exp Med. 1996;184:2271–2278. doi: 10.1084/jem.184.6.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hafler DA, Weiner HL. Immunologic mechanisms and therapy in multiple sclerosis. Immunol Rev. 1995;144:75–107. doi: 10.1111/j.1600-065x.1995.tb00066.x. [DOI] [PubMed] [Google Scholar]
- 32.Fritz RB, Chou CH, McFarlin DE. Relapsing murine experimental allergic encephalomyelitis induced by myelin basic protein. J Immunol. 1983;130:1024–1026. [PubMed] [Google Scholar]
- 33.Mokhtarian F, McFarlin DE, Raine CS. Adoptive transfer of myelin basic protein-sensitized T cells produces chronic relapsing demyelinating disease in mice. Nature. 1984;309:356–358. doi: 10.1038/309356a0. [DOI] [PubMed] [Google Scholar]
- 34.Brocke S, Quigley L, McFarland HF, Steinman L. Isolation and characterization of autoreactive T cells in experimental autoimmune encephalomyelitis of the mouse. Methods. 1996;9:458–462. doi: 10.1006/meth.1996.0053. [DOI] [PubMed] [Google Scholar]
- 35.Gautam AM, Lock CB, Smilek DE, Pearson CI, Steinman L, McDevitt HO. Minimum structural requirements for peptide presentation by major histocompatibility complex class II molecules: implications in induction of autoimmunity. Proc Natl Acad Sci USA. 1994;91:767–771. doi: 10.1073/pnas.91.2.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakai K, Zamvil SS, Mitchell DJ, Lim M, Rothbard JB, Steinman L. Characterization of a major encephalitogenic T cell epitope in SJL/J mice with synthetic oligopeptides of myelin basic protein. J Neuroimmunol. 1988;19:21–32. doi: 10.1016/0165-5728(88)90032-x. [DOI] [PubMed] [Google Scholar]
- 37.Ota K, Matsui M, Milford EL, Mackin GA, Weiner HL, Hafler DA. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature. 1990;346:183–187. doi: 10.1038/346183a0. [DOI] [PubMed] [Google Scholar]
- 38.Pette M, Fujita K, Kitze B, Whitaker JN, Albert E, Kappos L, Wekerle H. Myelin basic protein-specific T lymphocyte lines from MS patients and healthy individuals. Neurology. 1990;40:1770–1776. doi: 10.1212/wnl.40.11.1770. [DOI] [PubMed] [Google Scholar]
- 39.Martin R, Jaraquemada D, Flerlage M, Richert J, Whitaker J, Long EO, McFarlin DE, McFarland HF. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J Immunol. 1990;145:540–548. [PubMed] [Google Scholar]
- 40.Deibler GE, Martenson RE, Kies MW. Large scale preparation of myelin basic protein from central nervous tissue of several mammalian species. Prep Biochem. 1972;2:139–165. doi: 10.1080/00327487208061467. [DOI] [PubMed] [Google Scholar]
- 41.Segal BM, Shevach EM. IL-12 unmasks latent autoimmune disease in resistant mice. J Exp Med. 1996;184:771–775. doi: 10.1084/jem.184.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Speiser DE, Kyburz D, Stubi U, Hengartner H, Zinkernagel RM. Discrepancy between in vitro measurable and in vivo virus neutralizing cytotoxic T cell reactivities. Low T cell receptor specificity and avidity sufficient for in vitro proliferation or cytotoxicity to peptide-coated target cells but not for in vivo protection. J Immunol. 1992;149:972–980. [PubMed] [Google Scholar]
- 43.Aichele P, Bachmann MF, Hengartner H, Zinkernagel RM. Immunopathology or organ-specific autoimmunity as a consequence of virus infection. Immunol Rev. 1996;152:21–45. doi: 10.1111/j.1600-065x.1996.tb00909.x. [DOI] [PubMed] [Google Scholar]
- 44.Brocke S, Veromaa T, Weissman IL, Gijbels K, Steinman L. Infection and multiple sclerosis: a possible role for superantigens? . Trends Microbiol. 1994;2:250–254. doi: 10.1016/0966-842x(94)90630-0. [DOI] [PubMed] [Google Scholar]
- 45.Fallis RJ, Raine CS, McFarlin DE. Chronic relapsing experimental allergic encephalomyelitis in SJL mice following the adoptive transfer of an epitope-specific T cell line. J Neuroimmunol. 1989;22:93–105. doi: 10.1016/0165-5728(89)90039-8. [DOI] [PubMed] [Google Scholar]
- 46.Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci. 1986;6:271–277. [Google Scholar]
- 47.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 48.Panitch HS. Adoptive transfer of experimental allergic encephalomyelitis with activated spleen cells: comparison of in vitro activation by concanavalin A and myelin basic protein. Cell Immunol. 1980;56:163–171. doi: 10.1016/0008-8749(80)90091-x. [DOI] [PubMed] [Google Scholar]
- 49.Ben-Nun A, Wekerle H, Cohen IR. The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol. 1981;11:195–199. doi: 10.1002/eji.1830110307. [DOI] [PubMed] [Google Scholar]
- 50.Hamad AR, Marrack P, Kappler JW. Transcytosis of staphylococcal superantigen toxins. J Exp Med. 1997;185:1447–1454. doi: 10.1084/jem.185.8.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis, B.D., R. Dulbecco, H.N. Eisen, H.S. Ginsberg, and W.B. Wood. 1970. Microbiology. 6th ed. Hoeber Medical Division, New York.
- 52.Gray D, Matzinger P. T cell memory is short-lived in the absence of antigen. J Exp Med. 1991;174:969–974. doi: 10.1084/jem.174.5.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beverley PC. Is T-cell memory maintained by crossreactive stimulation? . Immunol Today. 1990;11:203–205. doi: 10.1016/0167-5699(90)90083-l. [DOI] [PubMed] [Google Scholar]
- 54.Matzinger P. Immunology. Memories are made of this? . Nature. 1994;369:605–606. doi: 10.1038/369605a0. [DOI] [PubMed] [Google Scholar]
- 55.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 56.Miller SD, Vanderlugt CL, Lenschow DJ, Pope JG, Karandikar NJ, Dal CM, Bluestone JA. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity. 1995;3:739–745. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- 57.Vanderlugt CL, Karandikar NJ, Lenschow DJ, Dal CM, Bluestone JA, Miller SD. Treatment with intact anti-B7-1 mAb during disease remission enhances epitope spreading and exacerbates relapses in R-EAE. J Neuroimmunol. 1997;79:113–118. doi: 10.1016/s0165-5728(97)00108-2. [DOI] [PubMed] [Google Scholar]
- 58.Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157:1333–1336. [PubMed] [Google Scholar]
- 59.Perrin PJ, Scott D, June CH, Racke MK. B7-mediated costimulation can either provoke or prevent clinical manifestations of experimental allergic encephalomyelitis. Immunol Res. 1995;14:189–199. doi: 10.1007/BF02918216. [DOI] [PubMed] [Google Scholar]
- 60.Grewal IS, Foellmer HG, Grewal KD, Xu J, Hardardottir F, Baron JL, Janeway CA, Jr, Flavell RA. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science. 1996;273:1864–1867. doi: 10.1126/science.273.5283.1864. [DOI] [PubMed] [Google Scholar]
- 61.McRae BL, Nikcevich KM, Karpus WJ, Hurst SD, Miller SD. Differential recognition of peptide analogs by naive versus activated PLP 139-151-specific CD4+ T cells. J Neuroimmunol. 1995;60:17–28. doi: 10.1016/0165-5728(95)00048-7. [DOI] [PubMed] [Google Scholar]