

Abstract
An in vitro fusion assay uses fluorescence microscopy of labeled lipids to monitor single v-SNARE vesicle docking and fusion events on a planar lipid bilayer containing t-SNAREs. For vesicles and bilayer comprising phosphatidylcholine (POPC, 84–85% by mol) and phosphatidylserine (DOPS, 15% by mol), previous work demonstrated prompt, full fusion (τfus = 25 ms). Substitution of 20–60% phosphatidylethanolamine (DOPE) for phosphatidylcholine in the v-SNARE vesicle with either 0 or 20% DOPE included in the t-SNARE bilayer gives rise to hemifusion events. Labeled lipids diffuse into the planar bilayer as two temporally distinct waves, presumably hemifusion of the outer leaflet followed by inner leaflet (core) fusion. The fusion kinetics with DOPE is markedly heterogeneous. Some vesicle/docking site pairs exhibit prompt, full fusion while others exhibit hemifusion. Hemifusion events are roughly half productive (leading to subsequent core fusion within 20 s) and half dead-end. In qualitative accord with expectations from studies of protein-free vesicle-vesicle fusion, the hemifusion rate khemi is 15–20 times faster than the core fusion rate kcore, and the fraction of hemifusion events increases with increasing percentage of DOPE. This suggests similar underlying molecular pathways for protein-free and neuronal SNARE-driven fusion. Removal of phosphatidylserine from the v-SNARE vesicle has no effect on docking or fusion.
INTRODUCTION
Fusion of two biological membranes is a fundamentally important process in the trafficking of lipids, proteins, and vesicle content as well as in the infection of host cells by viruses. The detailed mechanism remains unclear. Protein-free lipid bilayer fusion is a high-barrier process that does not occur at 37°C unless stimulated by osmotic forces or membrane tension (1–5). The reaction pathway to protein-free fusion involves hemifusion (Fig. 1), the mixing of lipids between the two proximal (outer) vesicle leaflets but not the distal (inner) leaflets (6,7). Accordingly, physical models of the protein-free fusion process that treat the lipid bilayer as a double sheet of continuous elastic material (4,6,7) predict a stable hemifusion intermediate state along the lowest energy pathway to formation of the fusion pore. In such models, the rate-limiting barrier for protein-free fusion lies between the stalk intermediate (in which hemifusion has occurred) and full fusion products. Recent x-ray diffraction studies of the structure of lipid multilayers provide direct experimental confirmation of a stable, stalklike structure (9).
FIGURE 1.

Schematic of hemifusion intermediate (the stalk) and nascent fusion pore. Negative curvature of phosphatidylethanolamine lipids (DOPE, red) might be expected to enhance hemifusion (fusion of proximal leaflets) as suggested at a and b, but to inhibit core fusion (fusion of the distal leaflets) as suggested at d.
It is an open question to what extent mechanistic ideas gleaned from studies of protein-free fusion are relevant to membrane fusion in vivo (5). Both intracellular and intercellular membrane fusion require catalytic protein machinery, as evidenced by knockout and mutation studies of neuronal exocytosis, endocrine cell secretion, and yeast and viral fusion with host cells (10). In neuronal exocytosis, various protein components of the fusion machinery evidently target synaptic vesicles to specific membrane sites, confer Ca2+ triggering, and lower the barrier to membrane fusion (12). A seemingly universal component of membrane fusion machines is the SNARE complex (soluble n-ethyl-maleimide-sensitive fusion protein attachment protein receptor) or a close analog (13,14). In Ca2+-triggered neuronal exocytosis, formation of ternary SNARE complexes is thought to be directly coupled to vesicle fusion (15–19). Such SNARE complexes comprise a highly stable four-helix bundle (20) anchored in the synaptic vesicle by the v-SNARE synaptobrevin (syb or vesicle-associated membrane protein, i.e., VAMP) and in the presynaptic plasma membrane by the binary t-SNARE comprising syntaxin (syx) plus SNAP-25 (25 kDa synaptosome-associated protein). Recent in vitro assays have measured enhanced rates of fusion between v-SNARE vesicles and t-SNARE vesicles, further implicating SNAREs as the minimal fusion machinery (13,21,22).
Direct observation of hemifusion intermediate states that are productive in SNARE-driven membrane fusion (i.e., that lead to full fusion) would argue in favor of a close mechanistic connection between protein-free and protein-catalyzed membrane fusion. There is evidence of hemifusion intermediates in the intercellular viral fusion process, which involves a single type of fusion protein anchored in the virus particle (e.g., influenza hemagglutinin (5,23,24)). The yeast intercellular fusion machinery is closely analogous to the complementary v-SNARE/t-SNARE motif used by neurons and endocrine cells. A recent study of fusion between yeast vacuoles was interpreted to indicate that a chemical inhibitor of full fusion kinetically traps hemifusion intermediates (25). In vitro studies of vesicle-vesicle fusion induced in bulk solution by yeast SNAREs show hemifusion at low protein copy number but full fusion at higher copy number (26). However, it is difficult in bulk fusion assays to determine the extent to which hemifusion intermediates lie on the pathway to full fusion. Most recently, a new single-event fusion assay between yeast t-SNARE vesicles and tethered yeast v-SNARE vesicles enabled direct observation of productive hemifusion intermediate states (27).
For neuronal SNARE-driven fusion, several previous studies suggest that hemifusion intermediates are physiologically relevant. Electron tomography recently found evidence of stable hemifusion intermediates for small synaptic vesicles docked at rat cortical synapses, albeit in chemically fixed samples (28). A cell-cell fusion assay with v-SNAREs and t-SNAREs expressed facing outward from the plasma membrane exhibited both full fusion and hemifusion events (29). A vesicle-vesicle bulk in vitro assay using neuronal SNAREs found that hemifusion states (proximal leaflet mixing) formed on a timescale of ∼1000 s, while full fusion products (distal leaflet mixing) formed on an ∼3000 s timescale (30). It remains difficult to prove to what extent these hemifusion intermediates lead to full fusion.
We and two other groups (31–33) recently introduced a single-vesicle fusion assay in which fluorescently labeled vesicles containing neuronal v-SNAREs interact with a lipid bilayer containing neuronal t-SNAREs and supported on glass. Such an assay enables direct observation of individual v-SNARE vesicle docking and fusion as separate kinetic steps, providing a great simplification compared with bulk vesicle-vesicle assays. With 1% of the vesicle's lipids fluorescently labeled, fusion is detected as a prompt dequenching of fluorescence intensity and subsequent radially symmetric diffusion of the labeled lipids into the planar bilayer (33). The hallmark of our assay compared with the others is the fast kinetics of SNARE-driven fusion. Vesicles dock efficiently on sparse t-SNARE bilayers and fuse with rate constant kfus = 40 s−1 (τfus = 25 ms), 103–104 times faster than any other in vitro fusion assay. Our initial study used a standard lipid mixture of 84% phosphatidylcholine (POPC), 15% phosphatidylserine (DOPS), and 1% fluorescently labeled phosphatidylethanolamine (DOPE) in the v-SNARE vesicles. The planar bilayer was 85:15 POPC:DOPS. We found no clear evidence of hemifusion intermediates either in high signal/noise movies using 40-ms camera frames or in lower signal/noise movies using 5-ms frames.
In this work, we begin to explore the effects of lipid composition on the fast fusion assay. Replacement of DOPS by POPC in the v-SNARE vesicle has no effect on docking and fusion kinetics. This argues against an inhibitory role of basic residues in the linker region between the v-SNARE membrane anchor and four-helix SNARE bundle (34–38). In contrast, substitution of DOPE for POPC in the v-SNARE vesicles gives rise to hemifusion events for a significant fraction of the vesicle-docking site pairs. Hemifusion is directly observed as the sequential release of two distinct waves of labeled lipids separated by a measurable delay time. The fraction of hemifusion events increases to 0.22 for 60% PE in the v-SNARE vesicle without PE in the t-SNARE bilayer and to 0.45 for 60% PE in the v-SNARE vesicle with 20% DOPE in the t-SNARE bilayer. The hemifusion timescale is ∼80 ms, approximately two times slower than that of prompt, full fusion events occurring in the same sample. The single-vesicle assay shows directly that some two-thirds of the hemifusion events are productive. Subsequent fusion of the vesicle core is much slower (1–5 s), indicating a significantly larger barrier to core fusion in the presence of PE. This is in accord with expectations from protein-free fusion studies (5).
Our new results further demonstrate the power of single-vesicle methodology to observe fast events and directly dissect kinetic pathways. The effects of PE in our fast, neuronal SNARE-driven fusion assay are qualitatively similar to its effects on protein-free fusion, suggesting the importance of a stalk intermediate in both processes. These new results do not, however, directly address the important question of whether the nascent fusion pore is lipid- or protein-lined (4,10).
MATERIALS AND METHODS
Protein expression and proteoliposome reconstitution
SNARE proteins were expressed, purified, and reconstituted into vesicles as described previously (33). The specific proteins are mouse synaptobrevin-2 (syb, the v-SNARE) and the full-length binary complex of rat syntaxin 1A (syx) and mouse SNAP25B (the t-SNARE). The synthetic phospholipids are 1-palmitoyl,2-oleoyl phosphatidylcholine (POPC), 1,2-dioleoyl phosphatidylserine (DOPS), 1,2-dioleoyl phosphatidylethanolamine (DOPE), and 1,2-dipalmitoyl phosphatidylcholine. All phospholipids were purchased from Avanti Polar Lipids (Alabaster, AL) except the fluorescent probe, n-(tetramethylrhodamine)-1,2-diheptadecanoyl phosphatidylethanolamine (TRITC-DHPE, Molecular Probes, Eugene, OR) used to label v-SNARE vesicles.
To study the effect on fusion of the percentage of PS within v-SNARE vesicles, two types of v-SNARE vesicles were reconstituted: v0PS vesicles containing 99% POPC and 1% TRITC-DHPE, and v15PS vesicles containing 84% POPC, 15% DOPS, and 1% TRITC-DHPE. All percentages are in mol/mol ratio. To study the effect of the percentage of PS within the t-SNARE bilayer, six types of t-SNARE vesicles were reconstituted: t0PS, t5PS, t10PS, t15PS, t20PS, and t25PS, where txxPS denotes xx% DOPS. In each case, the remainder of the lipids (100 − xx)% was POPC. Similarly, to study the effect of DOPE within v-SNARE vesicles, five types of v-SNARE vesicles were reconstituted: v0PE, v20PE, v40PE, and v60PE, where vxxPE denotes xx% DOPE. Each type also contained 15% DOPS, 1% TRITC-DHPE, and the remainder POPC. To study the effect of DOPE within the t-SNARE bilayer, two kinds of t-SNARE vesicles were reconstituted: t0PE and t20PE, where txxPE denotes xx% DOPE. Each of these types contained 15% DOPS and the remainder POPC. Note that v15PS and v0PE are the same vesicle type but with different names, as are t15PS and t0PE.
The v-SNARE and t-SNARE vesicles were reconstituted by rapid dilution and dialysis and subsequently purified by flotation in an Accudenz (Accurate Chemical, Westbury, NY) step gradient as described previously (22). The difference in lipid components has little effect on syb incorporation into vesicles, as measured by SDS-PAGE of liposomes detergent-solubilized after all purification steps. The mean copy number of syb for all types of v-SNARE vesicles is ∼100 copies/vesicle. In contrast, increasing DOPE decreases the recovery of t-SNARE binary complexes. Therefore, 10.2 μg binary t-SNARE in elution buffer was added to a lipid mixture containing 20% DOPE to make t20PE vesicles. This is three times what was added to a lipid mixture containing 0% DOPE for t0PE. This yielded the same mean t-SNARE complex copy number of ∼0.8 copy/vesicle for both t0PE and t20PE. The final buffer was 25 mM HEPES-KOH (pH 7.4) in 100 mM KCl in all cases.
Formation of t-SNARE bilayers
Supported lipid bilayers are formed by vesicle fusion on a clean, hydrophilic glass coverslip, a well-established technique pioneered by Tamm and McConnell (39). Coverslips were cleaned by sonication in detergent for 1 h (CONTRAD 70; Decon Laboratories, King of Prussia, PA), thoroughly rinsed in Millipore water (Simplicity 185; Millipore, Billerica, MA), sonicated in Millipore water for 30 min, and exhaustively rinsed in Millipore water. Then they were stored overnight at 90°C in Nano-Strip (Cyantek, Fremont, CA), a commercial mixture of H2O2 and concentrated H2SO4, and again rinsed thoroughly immediately before use. The total lipid concentration during t-SNARE bilayer deposition was 25 μM. After deposition at 4°C for 2.5 h, the bilayers were warmed to 37°C for 1 h and gently washed three times with buffer (60 cell volumes total) just before docking and fusion studies. We have explored the integrity of lipid bilayers deposited using the standard t0PE vesicles by both wide-field fluorescence microscopy and tapping mode atomic force microscopy (33). Lipid bilayers exhibit no defects on a 10 nm–20 μm scale. Analogous fluorescence and atomic force microscopy studies of bilayers from t20PE vesicles show similar integrity. As before, protein free vesicles do not fuse with t-SNARE bilayers, and v-SNARE vesicles do not fuse with protein free bilayers, regardless of lipid composition.
Syx and SNAP-25 are coexpressed in the same bacteria. Only SNAP-25 has a His tag but both proteins are pulled down in the purification step, indicating that all syx is bound to SNAP-25. Any isolated SNAP-25 would not be reconstituted into the t-SNARE vesicles because it is not palmitoylated and does not bind lipid bilayers by itself. Analysis by SDS-PAGE shows a roughly 1:1 mol ratio of syx:SNAP-25 within the t-SNARE vesicles. The t-SNARE vesicles presumably contain mostly 1:1 complexes, but we cannot rule out the possible presence of complexes of different stoichiometry.
Docking and fusion assay by fluorescence microscopy
A modified commercial wide-field microscope (Eclipse TE2000-U; Nikon, Melville, NY) enables excitation of fluorophores at the glass/water interface by “through the objective” total internal reflection (40). A 0.1 mW cw Ar+ laser at 514 nm illuminates a 200-nm thick, 50-μm diameter layer including the lipid bilayer. We use a 1.45 NA, 60× oil-immersion objective (Olympus, Melville, NY). When used in a Nikon microscope, the magnification of the Olympus objective becomes 100×. Fluorescence from TMR-DHPE passes a 565–595 nm bandpass filter (D580/30; Chroma, Rockingham, VT) and is imaged onto a fast CCD camera (I-Pentamax; Roper Scientific, Trenton, NJ), yielding digital movies of fluorescence intensity versus time. The camera pixels are square, 15 μm × 15 μm, corresponding to 150 nm × 150 nm in real space at the sample.
Docking movies enable measurement of the absolute number of v-SNARE vesicles per μm2 that have docked on the surface versus time. Movies up to 16 min long were acquired in time-lapse mode, for which the laser illuminates the sample for 40 ms during each 2-s frame. The v-SNARE vesicle solution is ∼0.9 μM in total lipid concentration. This corresponds to a vesicle concentration of ∼5 × 10−11 M, assuming 18,000 lipids/vesicle (50-nm outer diameter, 40-nm inner diameter, ∼0.7 nm2/lipid head). Using a pipette, we flow four times the cell volume of v-SNARE vesicle solution through the cell, fully exchanging solution in ∼2 s. Docking/fusion movies begin 10–20 s later, after manually focusing the microscope. As before, we measure the total fluorescence intensity versus time, Itot(t), calibrate the mean intensity per fused vesicle, and apply a correction for slow photobleaching. The resulting absolute number density of fused vesicles is accurate to ±10%.
The fusion movies used a 250 × 250 pixel region of interest and 40-ms frames for a period of 20 s, with no dead time between frames. For each vesicle, we measure integrated fluorescence intensity above background within a circular region of radius of 0.74 μm centered at the point where each vesicle will eventually fuse. We denote this intensity I0.7 μm (t). Fusion causes a sudden increase in I0.7 μm by a factor of 1.5–3 due to dequenching of TMR fluorescence and to excitation and emission polarization effects, as detailed earlier. Both the intensity versus time trace and visual inspection of the sequence of images were used to determine the onset of docking and of fusion.
Quantitative modeling of histograms of the time between docking and fusion is carried out as follows. For homogeneous fusion kinetics, the probability of a vesicle fusing with the t-SNARE bilayer at time tfus after firm docking would follow a single exponential decay (33):
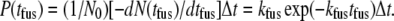 |
(1) |
Here N0 is the total number of vesicles, kfus is the unimolecular decay rate in s−1, and Δt is the width of the time bins into which events are distributed (here the camera frame duration, 40 ms). Equation 1 holds in the limit 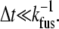 However, this inequality typically does not hold for 40-ms frames.
However, this inequality typically does not hold for 40-ms frames.
We improved the model first by recognizing that docking events may begin at any time within the first 40-ms camera frame. This attenuates the probability that fusion is observed in that same camera frame, which slows the apparent fusion rate. This effect can be simulated by numerically averaging Eq. 1 over a uniform distribution of docking times within the first camera frame, as described earlier for the 5-ms data (33). Inclusion of this effect gave best-fit decay rates of kfus = 84 ± 15 s−1 for the v15PS vesicles and 74 ± 15 s−1 for the v0PS vesicles, almost a factor-of-two faster than the earlier result from 5-ms camera frames. In the final model, we recognize that when a vesicle docks late in frame n, we will often assign its docking time to frame (n + 1). This may occur because the intensity in that frame is too low to be considered firm docking or because the image in frame n is blurred due to searching behavior before firm docking. The fusion time tfus is systematically underestimated in such cases. We model this bias by distributing true docking events uniformly in time, but assigning docking events that occur in the last half of frame n to frame (n + 1).
RESULTS
No effect of DOPS in v-SNARE vesicles on docking or fast fusion
The docking and fusion of v-SNARE vesicles with 15% DOPS (v15PS) and without DOPS (v0PS) on standard t-SNARE bilayers (t15PS) was investigated. Typical examples of docking kinetics data are shown in Supplementary Material Fig. S1. The docking kinetics are very similar with and without 15% DOPS in the v-SNARE vesicles; differences between the two curves lie well within the range of variation of docking curves for different runs under the same conditions. We showed earlier (33) that the docking rate constant on the t-SNARE bilayer containing 15% DOPS was within a factor of two of the diffusion-limited rate constant. Evidently SNARE formation is highly efficient with or without PS in the v-SNARE vesicle.
The fusion kinetics of v0PS and v15PS v-SNARE vesicles on standard t-SNARE bilayers containing 85% POPC and 15% DOPS were also studied. To obtain good signal/noise ratio we acquired 20 s movies of v-SNARE vesicles docking and fusing into t-SNARE bilayers with 40 ms exposure time for each frame. Histograms of tfus for v0PS and v15PS vesicles are plotted in Fig. 2, a and b. The histograms decay very similarly in each case, which implies that v15PS and v0PS fuse at similar rates. In both cases, some 80% of the docked vesicles fused on the 0–0.6 s timescale shown. There is a strong hint of a low-amplitude tail extending from 0.1 to 0.5 s in the v15PS data that is absent in the v0PS data.
FIGURE 2.

Histogram of fusion times tfus on standard t-SNARE bilayers containing 15% DOPS for v-SNARE vesicles containing (a) no DOPS and (b) 15% DOPS. The numerically simulated best fit to the first three data bins (open circles) uses a single exponential kinetics model corrected to include effects of the finite camera frame time, as discussed in the text.
We carried out a numerical simulation including the timing effects described in Materials and Methods, with kfus an adjustable parameter varied to best fit the first three time bins. As shown in Fig. 2 a, the data are well modeled using kfus = 37 ± 15 s−1 (τfus = 27 ± 15 ms) for the v15PS vesicles, in good agreement with the earlier result of 40 ± 15 s−1 from the data with 5-ms camera frames (33). This corroborates the model. Using the same model, we obtain kfus = 30 ± 15 s−1 (τfus = 33 ± 15 ms) for the fusion rate of v0PS vesicles. The errors are estimated from the range of results among different trials/membranes. The difference between the unimolecular fusion rates for v15PS and v0PS lies within the experimental error, i.e., there is no discernible effect of PS within the v-SNARE vesicle on the fusion rate with the standard 15% PS t-SNARE bilayer. A single exponential, least-squares fit to the data of Fig. 2, a and b, yields kfus = 29 ± 10 s−1 (τfus = 35 ± 15 ms) for v15PS vesicles and kfus = 26 ± 10 s−1 (τfus = 38 ± 15 ms) for v0PS. In the case of the v15PS vesicles studied earlier with shorter camera frames (Δt = 5 ms) but lower signal/noise ratio, we obtained kfus = 40 s−1, some 1.5 times faster and surely a better estimate of the true decay rate.
Hemifusion induced by DOPE in v-SNARE vesicles or t-SNARE bilayer
Observation of hemifusion events
Our assay detects the motion of lipids but not content release. Thus hemifusion (mixing of the proximal monolayers, i.e., the outer leaflet of the vesicle and the upper leaflet of the planar bilayer) followed by core fusion (mixing of the distal monolayers, the inner leaflet of the vesicle and the lower leaflet of the planar bilayer) would manifest itself as two temporally distinct waves of labeled lipids diffusing radially outward into the planar bilayer.
In the earlier work without PE (33), full fusion occurred promptly, i.e., all labeled lipids were apparently released from the docked vesicle in a single burst. For such full fusion events we measure the time delay tfus between firm docking and fusion. Supplementary Material Fig. S2 and Supplementary Material Movie 1 show an example of such a prompt, fast fusion event for the lipid combination t0PE + v60PE. As shown earlier, for such events the integrated fluorescence intensity versus time, I0.7 μm(t), increases by a factor of 1.5–3 upon fusion due to fluorescence dequenching of the lipid probes and polarization effects (Materials and Methods). The nonexponential decay of I0.7 μm(t) is well modeled by the diffusion equation. For these prompt events we cannot rule out the possibility of short-lived hemifusion intermediates that survive less than a few camera frames.
When DOPE is included in the v-SNARE vesicle, some of the vesicles exhibit full fusion, but others exhibit hemifusion. For the hemifusion events, the single-vesicle method enables measurement of both the time between firm docking and hemifusion (which we call themi) and the time between hemifusion and subsequent fusion of the core (tcore). An example is shown as a sequence of still images in Fig. 3, and also in Supplementary Material Movie 2. In Fig. 3, the vesicle approaches the bilayer at the second frame and docks firmly in the fifth frame for a total of five frames, or 200 ms. Hemifusion occurs at frame 10. After part of the fluorescence has spread into the planar bilayer, the core becomes apparent at approximately frame 14. It survives some 1.4 s and then fuses abruptly at the 45th frame. After fusion of the core, no punctal image is left behind and the intensity drops to a level close to the baseline.
FIGURE 3.

Sequence of 60 consecutive 40-ms movie frames for a productive hemifusion event using the lipid combination t0PE + v60PE.
Fig. 4 shows I0.7 μm(t) for a different hemifusion event that occurred on a very dark background. In this case, there is an obvious second intensity rise signaling core fusion, but in other cases there is no obvious second intensity rise due to large local background fluorescence and poor signal/noise ratio. In the latter cases we use the images rather than the intensity trace to measure tcore. In still other cases, the core survives for the remainder of the 20-s movie. We call these dead-end hemifusion events; such cores might or might not fuse in a longer movie. We can only set a lower bound on tcore for these events.
FIGURE 4.

An example of the integral fluorescence intensity time trace I0.7μm(t) for a hemifusion event. Dequenching of lipid labels occurs in two successive bursts. The solid line is a least-squares fit of the data by Eq. S4 with D1 = 0.85 μm2 s−1, D2 = 0.45 μm2 s−1, I2/(I1 + I2) = 0.31, and tcore = 2.08 s.
In addition to measuring themi and tcore, we can fit I0.7 μm(t) by a solution of the time-dependent diffusion equation for a double-burst of lipid release and thus estimate the intensity of the core relative to that of the full vesicle. Details of the procedure are given in the Supplementary Material. The fits are qualitatively good; an example is shown in Fig. 4. This procedure yields two diffusion coefficients (which are not constrained to match) plus the intensity ratio I2/(I1 + I2), which is the fraction of labeled lipids released in the second step. Results are described below. In the Supplementary Material, we also show that the two lipid release events arise from centers that are colocalized in space within 15 nm, which tends to rule out the possibility that two 50-nm vesicles were stuck together, docked side by side, and fused fully but sequentially.
Dependence of hemifusion frequency on DOPE
As described in Materials and Methods, DOPE was included in the v-SNARE vesicles at 0–60 mol % and in the t-SNARE bilayer at 0 and 20 mol %. We tested eight combinations of v-SNARE vesicles and t-SNARE bilayers: t0PE + v0PE, t0PE + v20PE, t0PE + v40PE, t0PE + v60PE, t20PE + v0PE, t20PE + v20PE, t20PE + v40PE, and t20PE + v60PE. The statistics of 1318 full fusion or hemifusion events are provided in Table 1. For each lipid combination, some 100–250 individual events were analyzed quantitatively for the time to prompt full fusion (tfus) or for the time to hemifusion (themi) and the subsequent time to core fusion (tcore). The table includes the number of events analyzed (Ntotal) and the percentage of those events characterized as full fusion (ffus) versus hemifusion (fhemi). The hemifusion events are further classified according to whether the core fuses during the 20-s movie (fcore) or does not (fdead).
TABLE 1.
Effects of DOPE on fusion kinetics
| Full fusion
|
Hemifusion
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Lipid composition | Ntotal* | ffus* (%) | kfus†(s−1) | fhemi*(%) | khemi† (s−1) | fcore*(%) | kcore† (s−1) | fdead*(%) | |
| t0PE | v0PE | 138 | 96 | 37 ± 15 | 4 | — | — | — | — |
| v20PE | 97 | 92 | 32 ± 15 | 8 | — | — | — | — | |
| v40PE | 121 | 79 | 30 ± 15 | 21 | 3.2 ± 1.0† | 14 | — | 7 | |
| v60PE | 195 | 79 | 34 ± 15 | 21 | 12 ± 5† | 13 | 0.6 ± 0.2 | 8 | |
| t20PE | v0PE | 257 | 96 | 28 ± 15 | 4 | — | — | — | — |
| v20PE | 225 | 81 | 44 ± 15 | 19 | 15 ± 3† | 14 | 1.1 ± 0.3 | 5 | |
| v40PE | 129 | 62 | 50 ± 15 | 38 | 14 ± 4† | 22 | 0.8 ± 0.2 | 16 | |
| v60PE | 156 | 55 | 31 ± 15 | 45 | 17 ± 3† | 12 | 0.6 ± 0.2 | 33 | |
Total number of fusion events observed from three to five different t-SNARE bilayers at each lipid composition. These are classified as full (prompt) fusion or hemifusion events, occurring with fractions ffus and fhemi respectively, according to whether or not a bright core remains behind after the first wave of lipids is released. The fraction fhemi is further partitioned into productive or dead-end hemifusion events for which, respectively, the core does (fcore) or does not (fdead) decay as a second wave during the 20-s movie. Normalization is chosen so that fhemi = fcore + fdead.
Best fits of a single exponential decay model to the first three bins of the histograms shown in Supplementary Material Figs. S4–S6. See text for model details.
The hemifusion percentage fhemi depends on the concentration of DOPE in both v-SNARE vesicles and t-SNARE bilayers (Fig. 5). In the baseline experiments with DOPE absent from both v-SNARE vesicles and t-SNARE bilayers (t0PE + v0PE), only 4% of the events exhibit the characteristics of hemifusion. Addition of 20% DOPE to the t-SNARE vesicles that form the planar bilayer (v0PE + t20PE) has little or no effect. However, v-SNARE vesicles containing 20%, 40%, and 60% DOPE exhibit larger fhemi, up to 22% for the t0PE + v60PE combination. Addition of 20% DOPE to the t-SNARE bilayer further enhances fhemi. The most extreme t20PE + v60PE combination exhibits 46% hemifusion events. Qualitatively, the docking efficiencies are similar for all lipid combinations studied.
FIGURE 5.

Bar graph of the fraction of hemifusion events fhemi versus lipid composition of the v-SNARE vesicles and the t-SNARE bilayer. Shaded bars at left have no DOPE in the t-SNARE bilayer, while solid bars at the right have 20% DOPE in the t-SNARE bilayer.
Full fusion and hemifusion kinetics
We binned the times to full fusion (tfus) or hemifusion (themi) for all events for each different lipid combination. The first three 40-ms time bins of the histograms of tfus and of themi were fit by the model incorporating homogeneous kinetics (single-exponential decay) and taking account of the uniform distribution of docking times within the initial camera frame and docking time measurement bias, as described in Materials and Methods. For productive hemifusion events, we also obtain a histogram of tcore, the time between hemifusion and core fusion. These times are so long that the histogram is fit by a simple exponential decay (Eq. 1). The histograms for all lipid combinations are collected in Supplementary Material Figs. S4–S6. Some lipid combinations yielded fewer than 20 hemifusion or core fusion events, which was deemed too few to form a useful histogram. The best-fit values of kfus, khemi, and kcore are collected in Table 1.
Fig. 6 compares representative prompt fusion histograms for the lipid combinations t0PE + v40PE and t20PE + v40PE. The histogram for t0PE + v0PE is repeated from Fig. 2 a (where it is labeled t15PS + v15PS). The kinetics is reasonably homogeneous in both cases. The long-time tail on the t0PE + v40PE distribution indicates kinetic heterogeneity due to variations among v-SNARE vesicles or t-SNARE docking sites. The best-fit unimolecular rate constants all lie in the range 30–50 s−1 and overlap within the estimated error limits. The fastest fitted decay rate occurs for the distribution with the smallest-amplitude long-time tail (t20PE + v40PE). The more heterogeneous distribution of t0PE + v40PE (Fig. 6 a) biases the fit to the first three time bins toward somewhat smaller kfus.
FIGURE 6.

Two examples of histograms of tfus for the lipid compositions shown. The open circles show the best fit to the first three bins as in Fig. 2. See Supplementary Material Fig. S4 for histograms of tfus for all lipid combinations.
A histogram of hemifusion times themi for the case t0PE + v40PE is displayed in Fig. 7 a. The first three bins were simulated numerically to yield the best-fit rate constant khemi listed in Table 1. Supplementary Material Fig. S5 compares histograms of themi for all lipid combinations studied. The number of events is sometimes quite small. Nevertheless, the histograms of themi are often less smooth than expected for Poisson binning statistics. Using Ni and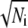 to estimate the mean and standard deviation of the number of counts in a channel, roughly 68% of the data should lie within
to estimate the mean and standard deviation of the number of counts in a channel, roughly 68% of the data should lie within 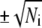 of the smooth curve and 96% should lie within
of the smooth curve and 96% should lie within 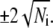 By this criterion, the kinetics appear reasonably homogeneous for the cases t0PE + v40PE (khemi = 3.2 s−1), t20PE + v20PE (15 s−1), and t20PE + v60PE (17 s−1). The kinetics appear more heterogeneous for the cases t0PE + v60PE (khemi = 12 s−1) and t20PE + v40PE (khemi = 14 s−1). There is no clear-cut trend of khemi with DOPE content (Table 1), but we can safely conclude that for each lipid combination, khemi is at least 2–3 times slower than kfus for the population of vesicle/docking sites that lead to prompt, full fusion events. We emphasize that both types of events are occurring in the same sample.
By this criterion, the kinetics appear reasonably homogeneous for the cases t0PE + v40PE (khemi = 3.2 s−1), t20PE + v20PE (15 s−1), and t20PE + v60PE (17 s−1). The kinetics appear more heterogeneous for the cases t0PE + v60PE (khemi = 12 s−1) and t20PE + v40PE (khemi = 14 s−1). There is no clear-cut trend of khemi with DOPE content (Table 1), but we can safely conclude that for each lipid combination, khemi is at least 2–3 times slower than kfus for the population of vesicle/docking sites that lead to prompt, full fusion events. We emphasize that both types of events are occurring in the same sample.
FIGURE 7.

(a) Histogram of themi for the t0PE + v40PE lipid composition. (b) Histogram of tcore for the t20PE + v40PE lipid composition. In these slowly decaying cases, open circles show the best fit by a single exponential decay. See Supplementary Material Figs. S5 and S6 for a histograms of themi and tcore for all lipid combinations.
Supplementary Material Fig. S6 displays histograms of the time to core fusion tcore for those lipid combinations that yielded 20 or more such events. The distribution for the case t20PE + v40PE is shown in Fig. 7 b. For all lipid combinations, the timescale of core fusion is ∼1–5 s, much slower than that of either tfus or themi. In all cases, the model of homogeneous, single-exponential kinetics fits the data very well by the Poisson criterion, lending confidence to comparisons of the resulting values of kcore across lipid compositions (Table 1). The rates kcore are 15–30 times slower than the corresponding values of khemi, and 40–60 times slower than kfus. Comparing the series of v-SNARE vesicles v20PE, v40PE, and v60PE docking on t20PE bilayers, kcore decreases monotonically from 1.1 ± 0.3 s−1 to 0.8 ± 0.2 s−1 to 0.6 ± 0.2 s−1 as the % DOPE in the vesicle increases. The combination t0PE + v60PE exhibits kcore = 0.6 ± 0.2 s−1, the same as for t20PE + v60PE. The data provide substantial evidence that increasing the DOPE percentage in the vesicle inner leaflet from 0% to 60% substantially increases the fraction of vesicle/docking site pairs that undergo hemifusion. For hemifusion events, increasing %DOPE from 20% to 60% seems to gradually increase the barrier to fusion between the distal monolayers of the v-SNARE vesicle and the t-SNARE bilayer.
Finally, we discuss the quantitative fits of I0.7 μm(t) by Eq. S4 for five cases of spatially well isolated hemifusion events for which core fusion occurred before the end of the movie. One example is shown in Fig. 4; details of the best-fit parameters are included in the caption. In all cases the intensity traces are well fit by the model function. For the five events, D1 varies from 0.6–1.2 μm2/s with a mean of 0.90 ± 0.26 μm2 s−1, and D2 varies from 0.4 to 1.0 μm2/s with a mean of 0.61 ± 0.27 μm2 s−1 (±1 SD). In every case, D1 > D2. These diffusion coefficients are approximately two times smaller than typical lipid diffusion coefficients in protein-free planar bilayers at 37°C (41). The data mildly suggest that the second wave of lipids diffuses more slowly than the first, perhaps because a substantial amount of protein from the vesicle is deposited locally in the bilayer as the core lipids emerge. In addition, core fusion should preferentially deposit head-labeled TMR-DHPE with its dye molecule contiguous to the glass substrate, which also may diminish its diffusion constant.
The best-fit fractional intensity released by the core fusion event, I2/(I1 + I2), varies from 0.19 to 0.42, with a mean of 0.28 ± 0.09 (±1σ). The first event is always more intense than the second; this is further evidence against the possibility of sequential fusion of two side-by-side vesicles. In a simple geometric estimate for a 50-nm vesicle with bilayer thickness of 5 nm, the fraction of total lipid residing in the inner leaflet should be ∼202/(202 + 252) = 0.39, somewhat larger than the mean fractional intensity of the core fusion events. This may indicate a propensity for the TMR-DHPE labeled lipids to preferentially partition to the outer leaflet of the v-SNARE vesicles. The bulky TMR label on the head may in effect confer positive curvature to the labeled lipids, matching the curvature of the outer leaflet. We do not expect crowding of the labels inside the vesicle to be a consideration, based on the rough estimate of 70 nm2 of available interior surface area per labeled lipid head. Alternatively, there is a chance that some labels from the inner leaflet leak out between the two noticeable bursts of lipid release. Such behavior would be reminiscent of the flickering events inferred in the recent tethered vesicle study using yeast SNAREs (27). This bears further study using both lipid and content labels.
In summary, the colocalization of the original docked vesicle and the residual core within 15 nm, the good fits of the intensity traces by the double-burst model of Eq. S4, and the semiquantitative agreement between the ratio of best-fit burst intensities and expectations based on geometric calculations all support our interpretation of the double-burst events as arising from hemifusion followed by core fusion. Evidently DOPE within the v-SNARE vesicle and the t-SNARE bilayer enhances the probability of hemifusion relative to prompt, full fusion primarily by inhibiting the rate of core fusion. The rate constant for formation of hemifusion intermediates is some 2–3 times slower than the rate constant for full fusion events, and core fusion is another factor of 15–30 slower. DOPE introduces measurable heterogeneity into the population of docked v-SNARE vesicle and their corresponding t-SNARE binding sites, as discussed below.
Controls
For the single lipid combination of 20% PE in the t-SNARE bilayer with 40% PE in the v-SNARE vesicles, we tested the effect of omitting SNAP-25 (syx-only bilayer). There was no clearly discernible effect on either the branching between full fusion and hemifusion or the timescale of full fusion and hemifusion events. As in the original work without DOPE (33), it seems that formation of “promiscuous” SNARE complexes leads to fast fusion or hemifusion in this low-barrier assay.
As before (33), docking and fusion of v-SNARE vesicles are blocked by incubating the t-SNARE bilayer with the soluble part of syb (cd-VAMP) before addition of the v-SNARE vesicles. This holds true both in the presence and in the absence of SNAP-25.
DISCUSSION
No effect of DOPS in v-SNARE vesicles on docking or fusion
In both syntaxin and synaptobrevin, the trans-membrane domain (C-terminus) is separated from the SNARE motif by a linker that contains a cluster of basic amino acids (34). The linker-region sequences of syx 1A and syb 2 are 261-ARRKK-265 and 85-KRKYWWKNLK-94, respectively; both are highly positively charged. An EPR relaxation study of syb reconstituted into liposomes containing 15% PS showed that the two Trp residues insert into the membrane (37). The authors propose that insertion allows the basic residues to interact with negatively charged lipids and that this binding enhances the SNARE complex-membrane mechanical coupling. Such insertion also inhibited SNARE complex formation (42). Similarly, a fusion assay between reconstituted t-SNARE vesicles and either synaptic vesicles or reconstituted v-SNARE vesicles containing PS suggested that accessibility of syb was inhibited by interaction with vesicular lipids (35). A surface plasmon resonance study revealed that syb 277–94 binds liposomes containing phosphatidylserine (43); the authors proposed that interaction between PS at the vesicle surface and the membrane proximal region of syb inhibits SNARE complex assembly (36).
In contrast, a recent bulk vesicle-vesicle fusion assay (44) using the same materials as this work found that the presence or absence of PS within the v-SNARE vesicles had no effect on membrane fusion driven by SNAREs alone. However, PS was an essential factor for Ca2+-regulated membrane fusion in the presence of both SNAREs and synaptotagmin I.
In our fast fusion assay, within experimental error we observe the same docking rate and the same unimolecular fusion rate kfus for v-SNARE vesicles in which DOPS is present at 15% or absent (Figs. 2 and S1). There is no evidence that an interaction between PS and the basic linker region of syb evidently regulates either docking or fusion. The magnitude of kdock lies within a factor-of-two of the estimated diffusion-limited docking rate, indicating that the efficiency of SNARE complex formation is high with or without PS in the v-SNARE vesicle. Insertion of the syb linker region into a PS-containing v-SNARE vesicle bilayer apparently does not inhibit SNARE complex formation on the sub-ms timescale of a vesicle/docking site encounter. Nor does insertion of the syb linker into a PS-containing vesicle bilayer enhance the fusion rate, within the 25-ms time resolution of this study.
Effects of DOPE on fusion
Lipid spontaneous curvature
The mechanics of protein-free membrane fusion have been investigated experimentally and theoretically for approximately two decades (6,7,45). The key reaction intermediate is evidently the fusion stalk or hemifusion intermediate (Fig. 1), and the rate-limiting step is the transition from the stalk to the opening of the fusion pore. Experimentally, the rate of formation of the fusion pore has been found to depend on the composition of the distal monolayers (5,46). Conical lipids such as lyso-PC having positive spontaneous curvature (head region wider than tail region) promote fusion pore opening. Conical lipids such as PE and arachidonic acid having negative curvature (head region narrower than tail region) inhibit fusion pore opening (46).
This is consistent with a simple geometric argument based on the structure of the stalk intermediate, shown schematically in Fig. 1. The seam between proximal leaflets on the pathway toward the stalk intermediate (hemifusion) has strong negative curvature that matches that of DOPE (as shown at point a). However, the inner leaflet of the incipient fusion pore has strong positive curvature (point d), opposite to that of DOPE. To best avoid unfavorable interactions between water and hydrophobic tails, the tails must be able to form a quasi-continuous sheet without gaps between lipid heads. Within the proximal leaflets, the negative curvature of DOPE lipids should help accomplish this en route to the stalk intermediate. However, the same negative curvature of DOPE within the distal leaflets should be unfavorable for overcoming the barrier between the stalk and the fusion pore.
The data clearly support the idea that 20% DOPE in the vesicle slows core fusion (distal leaflet mixing) and that additional DOPE in the planar bilayer further slows core fusion, but only in a subset of vesicle/docking site pairs. The subpopulation that undergoes hemifusion as an isolable step is itself heterogeneous. In productive hemifusion events, the core fuses with rate constant kcore in the range 0.6–1.1 s−1, while for dead-end hemifusion events we can set an approximate upper limit kcore < 0.1 s−1. In all cases, kcore is much slower than kfus ∼ 40 s−1.
These considerations are relevant to fusion in vivo. PE accounts for ∼35% of total phospholipids in both adult rat brain synaptic vesicles and presynaptosomal plasma membranes (47,48,50), compared with ∼41% phosphatidylcholine (PC). In cholinergic synaptic vesicle membranes, ∼77% of total PE resides on the outer (cytoplasmic) surface (51). Within the plasma membrane, PE is concentrated on the cytoplasmic side (52,53). It may be inappropriate to combine facts derived from different vesicle types, but they suggest that as much as 50% of both proximal leaflets of the vesicle/plasma membrane could be PE. A much lower percentage of the distal leaflets is PE. Moreover, the PE lipid tails are highly unsaturated (highly curved). In both synaptic vesicles and plasma membranes ∼65% of the tails on PE contain at least one double bond, and ∼86% of those are highly unsaturated with at least four double bonds. In contrast, for PC and sphingomyelin the fatty acids 16:0, 18:0, and 18:1 account for ∼90% of the lipid tails (52).
Heterogeneous kinetics
Our data suggest that the concepts of the stalk intermediate and lipid curvature derived from protein-free vesicle-vesicle fusion provide a useful starting point for building molecular-level models of SNARE-driven fusion as well. In this study of PE effects, we have divided observed events into two categories: prompt, full fusion characterized by kfus and sequential hemifusion/core fusion characterized by khemi and kcore. The latter events are somewhat arbitrarily subdivided into productive hemifusion events (for which core fusion occurs during the 20-s movie duration, typically within ∼1–2 s of hemifusion) and dead-end hemifusion events (for which core fusion never occurs during the movie and kcore is nominally zero). Inclusion of 20%, 40%, and 60% DOPE in the v-SNARE vesicles causes the percentage of observable hemifusion events to increase from the 0%PE value of 5% to 8%, 21%, and 22%, respectively (Fig. 5). When, in addition, 20% DOPE is included in the t-SNARE bilayer, the percentage of hemifusion events is further enhanced to 19%, 38%, and 46%, respectively. These values of fhemi should be viewed as lower limits; we may be missing additional hemifusion events due to limits on the speed and signal/noise ratio of the experiment. More generally, we cannot rule out the possibility that many or all of the full fusion events observed in this work and in the previous study would be revealed as sequential hemifusion/core fusion events when observed with faster camera frames at higher signal/noise ratio.
Among the majority of vesicle/docking site pairs that undergo prompt full fusion, DOPE has little or no observable effect at this signal/noise ratio using 40-ms frames. However, for each lipid composition that includes DOPE, we observe not only full fusion events with a fast rate constant kfus in the range 30–50 s−1, but also the hemifusion events with a slower rate constant khemi in the range 3–17 s−1. That is, DOPE introduces substantial kinetic heterogeneity within the population of v-SNARE vesicle/t-SNARE docking site pairs. Different vesicle/docking site pairs have intrinsically different kinetics. Naively, one might consider full fusion and stepwise hemifusion/core fusion as parallel kinetic pathways available to all vesicle/docking site combinations. However, such parallel, homogeneous kinetics would give rise to a single population decay rate ktotal = kfus + khemi, the sum of the two rates, which is not observed.
We have no clear insight at present into what variable factor(s) among vesicle/docking site pairs causes fast, full fusion versus slower, productive hemifusion versus dead-end hemifusion. The v-SNARE vesicles vary substantially in size. However, we have observed no clear correlation of fusion or hemifusion timescales with vesicle brightness (as measured by TMR-DOPE intensity). Electron microscope images indicate a mean vesicle diameter of ∼50 nm. To estimate the volume distribution, in the case of v-SNARE vesicles containing 30% DOPE we prepared vesicles containing calcein dye at 50 mM and judged the distribution of vesicle volumes from the distribution of docked vesicle fluorescence intensities on excitation at 488 nm. The distribution is broad, peaking at ∼25 arbitrary units (au) with a mean of 31 au and a standard deviation of 21 au. This is roughly consistent with earlier estimates of the surface area distribution in v-SNARE vesicles from TMR-DOPE intensities (33). There are enough lipids per vesicle (∼18,000, assuming 0.7 nm2/lipid head) that the statistics of vesicle formation would yield only minor variation in lipid composition across vesicles, assuming the lipids are well mixed.
Instead, we suspect that the primary source of kinetic heterogeneity lies in the t-SNARE docking sites. By its very nature the deposition of t-SNARE vesicles (having a mean copy number of ∼0.8 t-SNARE/vesicle, but an unknown distribution of copy numbers of the sticky t-SNAREs across vesicles) on a heterogeneous glass surface must lead to heterogeneity among the docking sites. They may differ in the number of t-SNAREs, how many t-SNAREs are facing up or down, the degree of t-SNARE entanglement and mobility, and the local morphology of the glass substrate. Better characterization of the t-SNARE docking sites remains an important goal for future work.
Finally, it is important to appreciate that the kinetic differences observed here could arise from rather small energetic differences along the reaction pathway to vesicle fusion products. By expressing each rate constant in the Arrhenius form 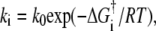 with k0 assumed constant and
with k0 assumed constant and 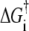 the free energy of activation for a particular kinetic step, we can calculate rough estimates of the differences in activation energies among different vesicle/docking site pairs. The kinetic data show that khemi measured for those vesicle/docking site pairs that undergo hemifusion is typically approximately three times smaller than kfus measured for fast-fusing pairs. Taking kfus as a lower bound on the rate of proximal leaflet fusion during full fusion events, we estimate that the activation energy for proximal leaflet fusion is ∼1.1 RT = 1.4 kJ/mol larger for the hemifusion events than for the full fusion events at T = 37°C. Similarly, for those vesicle/docking site pairs that undergo productive hemifusion, kcore is typically ∼40 times smaller than kfus for fast fusion events, suggesting that the barrier to distal leaflet fusion is ∼3.7 RT = 5.5 kJ/mol larger at 37°C. For vesicle/docking site pairs that undergo dead-end hemifusion, the ratio kfus/kcore is at least 400, suggesting a barrier to distal leaflet fusion that is at least 9 kJ/mol larger at 37°C.
the free energy of activation for a particular kinetic step, we can calculate rough estimates of the differences in activation energies among different vesicle/docking site pairs. The kinetic data show that khemi measured for those vesicle/docking site pairs that undergo hemifusion is typically approximately three times smaller than kfus measured for fast-fusing pairs. Taking kfus as a lower bound on the rate of proximal leaflet fusion during full fusion events, we estimate that the activation energy for proximal leaflet fusion is ∼1.1 RT = 1.4 kJ/mol larger for the hemifusion events than for the full fusion events at T = 37°C. Similarly, for those vesicle/docking site pairs that undergo productive hemifusion, kcore is typically ∼40 times smaller than kfus for fast fusion events, suggesting that the barrier to distal leaflet fusion is ∼3.7 RT = 5.5 kJ/mol larger at 37°C. For vesicle/docking site pairs that undergo dead-end hemifusion, the ratio kfus/kcore is at least 400, suggesting a barrier to distal leaflet fusion that is at least 9 kJ/mol larger at 37°C.
Comparison with other studies
Recently four groups have demonstrated that SNARE-mediated bilayer fusion can involve hemifusion structures. Rothman and co-workers (29) used fluorescence microscopy with both lipid and content labels to directly observe individual fusion events between cells having neuronal SNAREs expressed on the outer surface of the plasma membrane, a construct not found in nature. Mayer and co-workers (25) mixed rhodamine-PE labeled yeast vacuoles containing yeast SNAREs with unlabeled vacuoles containing yeast SNAREs. Dequenching of rhodamine-PE fluorescence signaled membrane fusion. A post-docking fusion inhibitor GTP-ϒS was found to arrest fusion at an intermediate state that permitted lipid flow between the fusion partners but no content mixing, suggesting it had trapped a hemifusion intermediate. In a bulk, vesicle-vesicle fusion study using yeast SNAREs, Shin and co-workers found that a v-SNARE mutant Snc2p lacking half of its trans-membrane domain caused hemifusion between t-SNARE vesicles and v-SNARE vesicles (26,54). At low t-SNARE copy number, both hemifusion and full fusion were observed between v-SNARE and t-SNARE vesicles using wild-type yeast SNAREs. For neuronal SNAREs, the analogous vesicle-vesicle bulk in vitro assay found hemifusion states (proximal leaflet mixing) on a timescale of ∼1000 s, while full fusion products (distal leaflet mixing) rose on a ∼3000 s timescale (54). While a sequential model was shown capable of fitting the data, it is very difficult in bulk studies to prove that hemifusion is productive, i.e., that it leads to subsequent full fusion.
The results most directly comparable to our work come from a recent single-vesicle fusion assay combining solution-phase yeast t-SNARE vesicles containing donor-labeled lipids with yeast v-SNARE vesicles containing acceptor-labeled lipids (27). The v-SNARE vesicles are tethered to an inert surface. A time-resolved FRET assay enabled dissection of individual vesicle-vesicle encounters into partial and full fusion events with 100-ms time resolution. Productive hemifusion events were observed directly. In addition, some v-SNARE/t-SNARE vesicle pairs exhibited pulses of lipid mixing, suggesting transient fusion pore openings, although contents were not imaged. In our assay, analogous events might account for the low apparent fraction of fluorescent labels released by core fusion compared with a geometric estimate.
This work is significant for its direct observation of a short-lived hemifusion intermediate state for neuronal SNARE-mediated fusion that occurs on a fast timescale and leads directly to core fusion (i.e., is productive). For productive hemifusion events, the movies directly show that hemifusion is an intermediate on the path to complete fusion. This is an important advantage of the single-vesicle method over bulk methods. The fact that the probability of hemifusion events increases as the percentage of PE increases underscores the important role that specific lipids play in SNARE-mediated membrane fusion. The curvature of the two fusing bilayers is physiologically appropriate. The v-SNARE vesicle mean diameter of 50 nm is comparable to that of small synaptic vesicles, and the planar bilayer approximates the essentially flat plasma membrane.
The fact that these observations are consistent with the qualitative effects of PE on protein-free vesicle fusion suggests that SNARE-driven and protein-free fusion proceed along roughly analogous reaction paths but with very different energetics. Indeed, the hemifusion state may be an intermediate shared by protein-free membrane fusion, viral entry, and also SNARE-mediated intracellular membrane fusion. However, we do not believe our results provide clear guidance on the question of whether the nascent fusion pore is lined with lipids or SNARE protein anchors (the lipidic versus the proteinaceous fusion pore) (4,10,55). In one view, the fusion pore is purely lipidic, and the pathway to fusion passes through the stalk intermediate. The SNAREs, perhaps in concert with additional proteins, serve as catalysts that lower the intrinsic barrier to bilayer fusion. In the other view, the fusion pore is formed by the cylindrical stacking of a ring of 5–8 v-SNARE anchors with a ring of 5–8 t-SNARE anchors. Our data are consistent with the lipidic viewpoint, but do not rule out a primarily protein-lined fusion pore or a mixed lipid-protein pore. Hemifusion can presumably occur around a protein-lined pore, with proximal-leaflet lipid tails diffusing around or between the hydrophobic residues of the protein pore (10). The new data do point the way to potentially more incisive experiments that will simultaneously measure lipid mixing and content release on single vesicles with ∼5-ms time resolution, our next goal.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Acknowledgments
We thank members of the Weisshaar and Chapman labs for helpful discussions and comments.
E.R.C. is an Investigator of the Howard Hughes Medical Institute. This study was supported by grants from the National Institutes of Health (NINDS No. NS051518 to J.C.W., and NIGMS No. GM56827 and NIMH No. MH61876 to E.R.C.) and from the American Heart Association (grant No. 0440168N to E.R.C.). The UW-Madison Department of Chemistry and Graduate School supported the initial stages of this work.
Tingting Liu's present address is Dept. of Molecular & Cell Biology, University of California, Berkeley, CA 94720-3206.
Editor: Helmut Grubmuller.
References
- 1.Malinin, V. S., and B. R. Lentz. 2004. Energetics of vesicle fusion intermediates: comparison of calculations with observed effects of osmotic and curvature stresses. Biophys. J. 86:2951–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finkelstein, A., J. Zimmerberg, and F. S. Cohen. 1986. Osmotic swelling of vesicles: its role in the fusion of vesicles with planar phospholipid bilayer membranes and its possible role in exocytosis. Annu. Rev. Physiol. 48:163–174. [DOI] [PubMed] [Google Scholar]
- 3.Lentz, B. R., and J. K. Lee. 1999. Poly(ethylene glycol) (PEG)-mediated fusion between pure lipid bilayers: a mechanism in common with viral fusion and secretory vesicle release? Mol. Membr. Biol. 16:279–296. [DOI] [PubMed] [Google Scholar]
- 4.Chernomordik, L. V., and M. M. Kozlov. 2005. Membrane hemifusion: crossing a chasm in two leaps. Cell. 123:375–382. [DOI] [PubMed] [Google Scholar]
- 5.Chernomordik, L. V., and M. M. Kozlov. 2003. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72:175–207. [DOI] [PubMed] [Google Scholar]
- 6.Kozlovsky, Y., and M. M. Kozlov. 2002. Stalk model of membrane fusion: solution of energy crisis. Biophys. J. 82:882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozlovsky, Y., L. V. Chernomordik, and M. M. Kozlov. 2002. Lipid intermediates in membrane fusion: formation, structure, and decay of hemifusion diaphragm. Biophys. J. 83:2634–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reference deleted in proof.
- 9.Yang, L., and H. W. Huang. 2003. A rhombohedral phase of lipid containing a membrane fusion intermediate structure. Biophys. J. 84:1808–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson, M. B., and E. R. Chapman. 2006. Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu. Rev. Biophys. Biomol. Struct. 35:135–160. [DOI] [PubMed] [Google Scholar]
- 11.Reference deleted in proof.
- 12.Jahn, R. 2004. Principles of exocytosis and membrane fusion. Ann. N. Y. Acad. Sci. 1014:170–178. [DOI] [PubMed] [Google Scholar]
- 13.Weber, T., B. V. Zemelman, J. A. McNew, B. Westermann, M. Gmachl, F. Parlati, T. H. Sollner, and J. E. Rothman. 1998. SNAREpins: minimal machinery for membrane fusion. Cell. 92:759–772. [DOI] [PubMed] [Google Scholar]
- 14.Jahn, R., and T. C. Sudhof. 1999. Membrane fusion and exocytosis. Annu. Rev. Biochem. 68:863–911. [DOI] [PubMed] [Google Scholar]
- 15.Chen, Y. A., and R. H. Scheller. 2001. Snare-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2:98–106. [DOI] [PubMed] [Google Scholar]
- 16.Mayer, A. 2001. What drives membrane fusion in eukaryotes? Trends Biochem. Sci. 26:717–723. [DOI] [PubMed] [Google Scholar]
- 17.Brunger, A. T. 2001. Structural insights into the molecular mechanism of calcium-dependent vesicle-membrane fusion. Curr. Opin. Struct. Biol. 11:163–173. [DOI] [PubMed] [Google Scholar]
- 18.Sollner, T. H. 2003. Regulated exocytosis and SNARE function (Review). Mol. Membr. Biol. 20:209–220. [DOI] [PubMed] [Google Scholar]
- 19.Lin, R. C., and R. H. Scheller. 2000. Mechanisms of synaptic vesicle exocytosis. Annu. Rev. Cell Dev. Biol. 16:19–49. [DOI] [PubMed] [Google Scholar]
- 20.Sutton, R. B., D. Fasshauer, R. Jahn, and A. T. Brunger. 1998. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Ångström resolution. Nature. 395:347–353. [DOI] [PubMed] [Google Scholar]
- 21.Schuette, C. G., K. Hatsuzawa, M. Margittai, A. Stein, D. Riedel, P. Kuster, M. Konig, C. Seidel, and R. Jahn. 2004. Determinants of liposome fusion mediated by synaptic SNARE proteins. Proc. Natl. Acad. Sci. USA. 101:2858–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tucker, W. C., T. Weber, and E. R. Chapman. 2004. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 304:435–438. [DOI] [PubMed] [Google Scholar]
- 23.Kemble, G. W., T. Danieli, and J. M. White. 1994. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell. 76:383–391. [DOI] [PubMed] [Google Scholar]
- 24.Zavorotinskaya, T., Z. H. Qian, J. Franks, and L. M. Albritton. 2004. A point mutation in the binding subunit of a retroviral envelope protein arrests virus entry at hemifusion. J. Virol. 78:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reese, C., F. Heise, and A. Mayer. 2005. Trans-SNARE pairing can precede a hemifusion intermediate in intracellular membrane fusion. Nature. 436:410–414. [DOI] [PubMed] [Google Scholar]
- 26.Xu, Y. B., F. Zhang, Z. L. Su, J. A. McNew, and Y. K. Shin. 2005. Hemifusion in SNARE-mediated membrane fusion. Nat. Struct. Mol. Biol. 12:417–422. [DOI] [PubMed] [Google Scholar]
- 27.Yoon, T. Y., B. Okumus, F. Zhang, Y. K. Shin, and T. Ha. 2006. Multiple intermediates in SNARE-induced membrane fusion. Proc. Natl. Acad. Sci. USA. 103:19731–19736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zampighi, G. A., L. M. Zampighi, N. Fain, S. Lanzavecchia, S. A. Simon, and E. M. Wright. 2006. Conical electron tomography of a chemical synapse: vesicles docked to the active zone are hemi-fused. Biophys. J. 91:2910–2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giraudo, C. G., C. Hu, D. Q. You, A. M. Slovic, E. V. Mosharov, D. Sulzer, T. J. Melia, and J. E. Rothman. 2005. SNAREs can promote complete fusion and hemifusion as alternative outcomes. J. Cell Biol. 170:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu, X., F. Zhang, J. A. McNew, and Y. K. Shin. 2005. Membrane fusion induced by neuronal SNAREs transits through hemifusion. J. Biol. Chem. 280:30538–30541. [DOI] [PubMed] [Google Scholar]
- 31.Fix, M., T. J. Melia, J. K. Jaiswal, J. Z. Rappoport, D. Q. You, T. H. Sollner, J. E. Rothman, and S. M. Simon. 2004. Imaging single membrane fusion events mediated by SNARE proteins. Proc. Natl. Acad. Sci. USA. 101:7311–7316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bowen, M. E., K. Weninger, A. T. Brunger, and S. Chu. 2004. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys. J. 87:3569–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, T. T., W. C. Tucker, A. Bhalla, E. R. Chapman, and J. C. Weisshaar. 2005. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys. J. 89:2458–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weimbs, T., K. Mostov, S. H. Low, and R. Hofmann. 1998. A model for structural similarity between different SNARE complexes based on sequence relationships. Trends Cell Biol. 8:260–262. [DOI] [PubMed] [Google Scholar]
- 35.Hu, K., J. Carroll, S. Fedorovich, C. Rickman, A. Sukhodub, and B. Davletov. 2002. Vesicular restriction of synaptobrevin suggests a role for calcium in membrane fusion. Nature. 415:646–650. [DOI] [PubMed] [Google Scholar]
- 36.De Haro, L., S. Quetglas, C. Iborra, C. Leveque, and M. Seagar. 2003. Calmodulin-dependent regulation of a lipid binding domain in the v-SNARE synaptobrevin and its role in vesicular fusion. Biol. Cell. 95:459–464. [DOI] [PubMed] [Google Scholar]
- 37.Kweon, D. H., C. S. Kim, and Y. K. Shin. 2003. Insertion of the membrane-proximal region of the neuronal SNARE coiled coil into the membrane. J. Biol. Chem. 278:12367–12373. [DOI] [PubMed] [Google Scholar]
- 38.Kweon, D. H., C. S. Kim, and Y. K. Shin. 2002. The membrane-dipped neuronal SNARE complex: a site-directed spin labeling electron paramagnetic resonance study. Biochemistry. 41:9264–9268. [DOI] [PubMed] [Google Scholar]
- 39.Tamm, L. K., and H. M. McConnell. 1985. Supported phospholipid bilayers. Biophys. J. 47:105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Axelrod, D. 2001. Selective imaging of surface fluorescence with very high aperture microscope objectives. J. Biomed. Opt. 6:6–13. [DOI] [PubMed] [Google Scholar]
- 41.Naumann, C. A., O. Prucker, T. Lehmann, J. Ruhe, W. Knoll, and C. W. Frank. 2002. The polymer-supported phospholipid bilayer: tethering as a new approach to substrate-membrane stabilization, Biomacromolecules. 3:27–35. [DOI] [PubMed] [Google Scholar]
- 42.Kweon, D. H., C. S. Kim, and Y. K. Shin. 2003. Regulation of neuronal SNARE assembly by the membrane. Nat. Struct. Biol. 10:440–447. [DOI] [PubMed] [Google Scholar]
- 43.Quetglas, S., C. Leveque, R. Miquelis, K. Sato, and M. Seagar. 2000. Ca2+-dependent regulation of synaptic SNARE complex assembly via a calmodulin- and phospholipid-binding domain of synaptobrevin. Proc. Natl. Acad. Sci. USA. 97:9695–9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhalla, A., W. C. Tucker, and E. R. Chapman. 2005. Synaptotagmin isoforms couple distinct ranges of Ca2+, Ba2+, and Sr2+ concentration to SNARE-mediated membrane fusion. Mol. Biol. Cell. 16:4755–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chernomordik, L., M. M. Kozlov, and J. Zimmerberg. 1995. Lipids in biological membrane-fusion. J. Membr. Biol. 146:1–14. [DOI] [PubMed] [Google Scholar]
- 46.Chernomordik, L., A. Chanturiya, J. Green, and J. Zimmerberg. 1995. The hemifusion intermediate and its conversion to complete fusion—regulation by membrane-composition. Biophys. J. 69:922–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Breckenridge, W. C., I. G. Morgan, J. P. Zanetta, and G. Vincendon. 1973. Adult rat brain synaptic vesicles. II. Lipid composition. Biochim. Biophys. Acta. 320:681–686. [DOI] [PubMed] [Google Scholar]
- 48.Breckenridge, W. C., G. Gombos, and I. G. Morgan. 1972. The lipid composition of adult rat brain synaptosomal plasma membranes. Biochim. Biophys. Acta. 266:695–707. [DOI] [PubMed] [Google Scholar]
- 49.Reference deleted in proof.
- 50.Takamori, S., M. Holt, K. Stenius, E. A. Lemke, M. Gronborg, D. Riedel, H. Urlaub, S. Schenck, B. Brugger, P. Ringler, S. A. Muller, B. Rammner, F. Grater, J. S. Hub, B. L. De Groot, G. Mieskes, Y. Moriyama, J. Klingauf, H. Grubmuller, J. Heuser, F. Wieland, and R. Jahn. 2006. Molecular anatomy of a trafficking organelle. Cell. 127:831–846. [DOI] [PubMed] [Google Scholar]
- 51.Michaelson, D. M., G. Barkai, and Y. Barenholz. 1983. Asymmetry of lipid organization in cholinergic synaptic vesicle membranes. Biochem. J. 211:155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westhead, E. W. 1987. Lipid-composition and orientation in secretory vesicles. Ann. N. Y. Acad. Sci. 493:92–100. [DOI] [PubMed] [Google Scholar]
- 53.Devaux, P. F. 1991. Static and dynamic lipid asymmetry in cell membranes. Biochemistry. 30:1163–1173. [DOI] [PubMed] [Google Scholar]
- 54.Lu, X. B., F. Zhang, J. A. McNew, and Y. K. Shin. 2005. Membrane fusion induced by neuronal SNAREs transits through hemifusion. J. Biol. Chem. 280:30538–30541. [DOI] [PubMed] [Google Scholar]
- 55.Han, X., C. T. Wang, J. H. Bai, E. R. Chapman, and M. B. Jackson. 2004. Transmembrane segments of syntaxin line the fusion pore of Ca2+-triggered exocytosis. Science. 304:289–292. [DOI] [PubMed] [Google Scholar]