

Abstract
Chronic obstructive pulmonary disease (COPD) is a common chronic inflammatory disease of the lungs with little or no response to glucocorticoids and a high level of oxidative stress. Histone deacetylase (HDAC) activity is reduced in cells of cigarette smokers, and low concentrations of theophylline can increase HDAC activity. We measured the effect of theophylline on HDAC activity and inflammatory gene expression in alveolar macrophages (AM) from patients with COPD. AM from normal smokers showed a decrease in HDAC activity compared with normal control subjects, and this was further reduced in COPD patients (51% decrease, P < 0.01). COPD AMs also showed increased basal release of IL-8 and TNF-α, which was poorly suppressed by dexamethasone. Theophylline induced a sixfold increase in HDAC activity in COPD AM lysates and significantly enhanced dexamethasone suppression of induced IL-8 release, an effect that was blocked by the HDAC inhibitor trichostatin A. Therefore, theophylline might restore steroid responsiveness in COPD patients.
Keywords: histone deacetylase, theophylline, steroid resistance, COPD, interleukin-8
Introduction
Chronic obstructive pulmonary disease (COPD) is a common and debilitating chronic inflammatory disease characterized by progressive airflow limitation that is poorly reversible (1). Cigarette smoking is the major causal factor for the ongoing inflammation in the airways and lung parenchyma, and the severity of airflow limitation is correlated with degree of pulmonary inflammation (2). Inflammation is amplified in the lungs of COPD patients with a striking increase in macrophage numbers as the disease becomes more severe (3). In addition, neutrophil chemotactic mediators, such as IL-8 and proinflammatory cytokines, such as TNF-α, are increased in the sputum of patients with COPD (4). Importantly, the inflammation in COPD is unresponsive to corticosteroids. Even high doses of inhaled and oral glucocorticoids have no effect on the inflammatory cell and cytokine profile and fail to reverse the protease-antiprotease imbalance (5). Furthermore, the antiinflammatory response to glucocorticoids is reduced in alveolar macrophages (AM) from normal smokers compared with nonsmokers and is absent in cells from patients with COPD (6).
Histone acetytransferases (HAT) and histone deacetylase (HDAC) are families of enzymes that regulate chromatin structure and thereby affect inflammatory gene expression (7). Acetylation of core histones by coactivator proteins that possess intrinsic HAT activity leads to unwinding of chromatin, which subsequently allows transcription factors and RNA polymerase II to switch on gene transcription. Conversely, deacetylation of core histones is generally associated with transcriptional repression. We have shown previously that glucocorticoid suppression of inflammatory genes requires recruitment of HDAC2 to the transcriptional activation complex by the glucocorticoid receptor (GR) (8).
The lack of response to glucocorticoids may be secondary to the increased oxidative stress as a result of cigarette smoking (9) leading to reduced HDAC activity (10). Theophylline is a bronchodilator at high doses; however, there is increasing evidence that at lower concentrations it has antiinflammatory effects in asthma and COPD (11), possibly due to an effect on HDAC activity, resulting in suppression of inflammatory genes and enhancement of the antiinflammatory effects of glucocorticoids (12). We investigated the effect of theophylline on HDAC activity and glucocorticoid antiinflammatory action in alveolar macrophages from COPD patients.
Materials and Methods
Patients.
19 patients with COPD diagnosed according to GOLD guidelines (13), 15 current smokers without airway obstruction (FEV1 of >70% predicted), and 13 nonsmokers with normal lung function were recruited. All patients studied underwent elective bronchoscopy for diagnostic purposes, and normal subjects were volunteers recruited through advertisement. Subjects with COPD and smokers both had a smoking history of >20 packs per year. COPD subjects receiving inhaled corticosteroids or oral theophylline and those with an acute exacerbation in the previous 6 wk were excluded. Exclusion criteria for smokers and nonsmokers were suspicion of infective or interstitial disease, age <35 yr. The study was approved by the Ethics Committees of the Harefield and Royal Brompton, St Mary's and Riverside Hospital NHS Trusts, and all the subjects gave their signed consent.
After initial recruitment, six patients in the COPD group, three patients in the smoker group, and one patient in the nonsmoking group were excluded. The reasons for exclusion were: infection of the cultured macrophages (4) and insufficient macrophage yield (6). Data from all the remaining subjects are included (Table I). There were significant differences in the number of macrophages and neutrophils in smokers and COPD patients compared with nonsmokers (Table I).
Table I.
Patient Characteristics and BAL Fluid Measurements
|
|
COPD (n = 13) |
Smokers (n = 12) |
Nonsmokers (n = 12) |
|---|---|---|---|
| Age (yr) | 69.4 ± 2.6 | 64.2 ± 3.5 | 62.2 ± 4.4 |
| Sex (male:female) | 7:7 | 9:3 | 6:6 |
| FEV1 (L) | 1 .15 ± 0.13a | 2.67 ± 0.1 | 3.27 ± 0.3 |
| FEV1 (% predicted) | 49.8 ± 3.3a | 83 ± 1.6 | 91.8 ± 7.5 |
| FEV1:FVC ratio | 0.59 ± 0.06c | 0.70 ± 0.02 | 0.74 ± 0.03 |
| Pack-years | 39 ± 4 | 48 ± 9 | 0 |
| Diagnosis: cancer | 7 | 7 | 2 |
| Volume of BAL recovered (ml) | 42.5 ± 5 | 46 ± 8.8 | 57 ± 9.3 |
| Macrophages (×103/ml) BAL fluid recovered | 204.9 ± 35c | 220.4 ± 39c | 101.1 ± 25 |
| Macrophages (%) | 88.9 ± 0.9 | 91.2 ± 0.8 | 93 ± 0.4 |
| Neutrophils (%) | 4.1 ± 0.3c | 3.9 ± 0.6 | 2.8 ± 0.5 |
| IL-8 (pg/μg protein) | 3.4 ± 0.7b | 1.2 ± 0.5c | 0.40 ± 0.3 |
| Glutathione (μM/μg protein) | 0.36 ± 0.03b | 0.15 ± 0.02 | 0.12 ± 0.03 |
FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; BAL, bronchoalveolar lavage.
Significance of difference compared to nonsmokers:
P < 0.001,
P < 0.01,
P < 0.05.
Bronchoscopy and Bronchoalveolar Lavage.
Bronchoscopy and bronchoalveolar lavage (BAL) was collected according to a standard protocol from the right middle lobe or the contralateral lobe to pathology as previously described (10).
Isolation and Culture of AM.
BAL cells were centrifuged (500 g for 10 min) and washed twice with Hanks buffered salt solution. Cell viability was assessed by trypan blue exclusion, and cytospins were prepared as described previously (10). BAL macrophages were either lysed or isolated by plastic adhesion as previously described (10), and cells (3 × 105) were incubated in 24-well plates in the presence of theophylline (10 μM), dexamethasone (1 μM), and/or Salmonella enteritidis LPS (10 μg/ml; Sigma-Aldrich).
U937 Cell Culture.
Cells were cultured exactly as described previously (10).
Cytokine ELISAs.
IL-8 and TNF-α concentrations were determined by sandwich ELISA according to the manufacturer's instructions (R&D Systems Europe).
Glutathione Assay.
Total glutathione was determined in BAL fluid using a colorimetric assay kit (Oxford Biomedical Research) according to the manufacturer's instructions.
Western Blotting.
Nuclear extracts were measured by Western blotting using specific antibodies obtained from Santa Cruz Biotechnology as previously described (8).
HDAC Activity.
HDAC activity of nuclear extracts was measured with a nonisotopic assay (BIOMOL) as recommended by the manufacturer. Specific HDAC isoforms were immunoprecipitated before analysis of HDAC activity as described previously (8).
Immunocytochemistry.
Cytospins were stained using antibodies against HDAC2 and p65 subunit of NF-κB (Santa Cruz Biotechnology). All antibodies were used at dilutions of 1:50–1:200 of a 200 mg/ml solution. Bronchial biopsies were used as control. For the negative control slides, normal rabbit nonspecific immunoglobulins (Dako) were used.
Statistics.
Results are expressed as means ± SEM. Changes in AM secretory products were compared with control subjects using analysis of variance (ANOVA). Comparison between experimental groups was performed using the Mann-Whitney U test. All statistical testing was performed at a two-sided 5% level of significance using GraphPad Prism software (GraphPad Software Inc.).
Results and Discussion
Patients with COPD showed significantly higher levels of IL-8 and glutathione in BAL fluid than smokers and nonsmokers (Table I). Baseline secretion of IL-8 was increased in AM from smokers and COPD patients compared with normal (P < 0.05 versus normal) and increased significantly more in AM from smokers and COPD patients after LPS stimulation (P < 0.05 versus normal) (Fig. 1 A). The NF-κB inhibitor AS602868 inhibited LPS-induced IL-8 production by 76% (14.0 ± 1.2 versus 58.7 ± 0.9 ng/ml, P < 0.01). Dexamethasone was less effective in reducing IL-8 and TNF-α release in COPD patients and smokers compared with controls (Fig. 1, A and B). Using immunocytochemistry (not depicted) and Western blotting of nuclear extracts from AM, we confirmed that there was enhanced nuclear translocation of the p65 subunit of NF-κB in COPD patients compared with normal subjects (0.38 ± 0.06 versus 0.11 ± 0.01 arbitrary units, P < 0.05) and to a lesser extent also in smokers (0.25 ± 0.05, P < 0.05).
Figure 1.

Effect of dexamethasone on cytokine release from AM. AM from normal subjects, healthy smokers, and patients with COPD were incubated with dexamethasone (Dex; 10−6 M) for 30 min before being stimulated with LPS (10 μg/ml). LPS-induced IL-8 release (A) and TNFα release (B) after 18 h were measured. *P < 0.05 versus control subjects; #P < 0.05 versus unstimulated cells; ¶P < 0.05 versus LPS-stimulated cells; n ≥ 6 in each group.
HDAC activity was significantly reduced in AM from COPD patients (2788 ± 339 AFU/10 μg) compared with smokers (3562 ± 392 AFU/10 μg, P < 0.05) and normal subjects (5664 ± 521 AFU/10 μg, P < 0.01) (Fig. 2 A). This correlated with a reduction in HDAC2 protein expression observed by immunocytochemistry (Fig. 2 B) and Western blotting (2.1 ± 0.1 versus 1.4 ± 0.2 versus 1.0 ± 0.1 ratio to β-actin expression) (Fig. 2 C).
Figure 2.
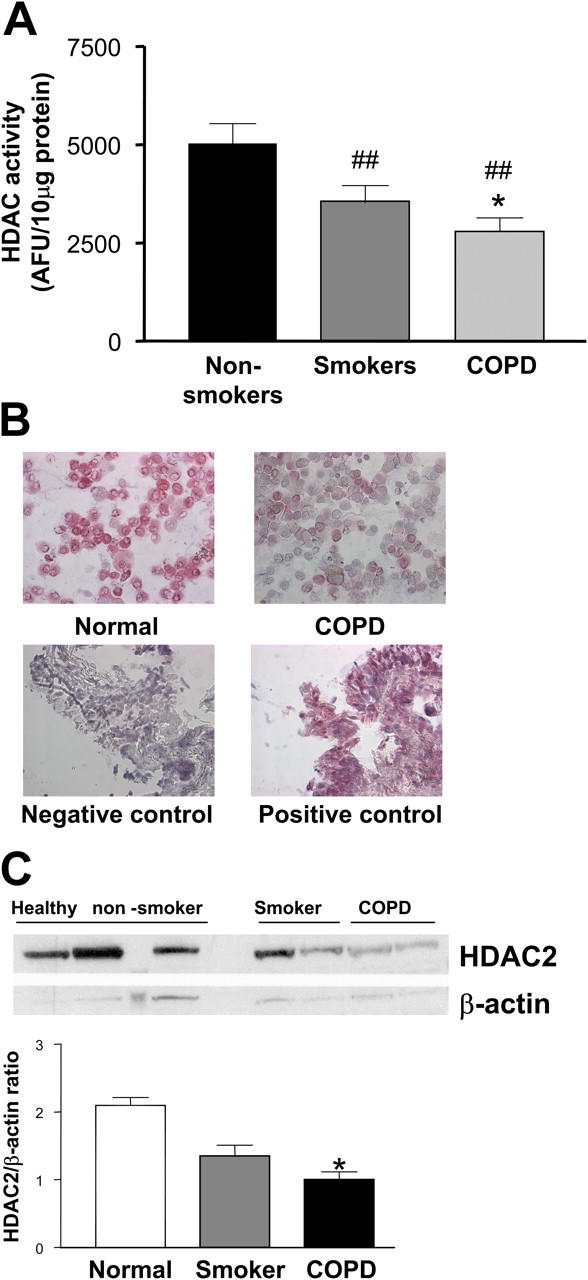
HDAC activity and expression in AM. (A) HDAC activity was measured in protein extracts from AM obtained from normal subjects, healthy smokers, and COPD patients and expressed according to protein content. ##P < 0.01 versus nonsmoker; *P < 0.05 versus smoker; n ≥ 6 in each group. (B) Immunocytochemical analysis of HDAC2 expression in AM from normal subjects and patients with COPD. Results are representative of at least six subjects in each group. (C) Western blot analysis of HDAC2 expression in AM from normal subjects, healthy smokers, and COPD patients and expressed according to protein content. *P < 0.05 versus nonsmoker, n = 3 in each group.
We have reported previously in cell lines that theophylline enhances HDAC activity (12). Therefore, we examined whether we could detect the same phenomena in AM from smokers and patients with COPD. Nuclear extracts from COPD and smokers directly exposed to theophylline (10−5 and 10−6 M) showed an approximate sixfold increase in HDAC activity (16,015 ± 509 versus 2,788 ± 339 AFU/10 μg, P < 0.001) (Fig. 3 A). Functionally, in cultured AM obtained from COPD patients, theophylline (10−6 M) alone had no effect on LPS-stimulated IL-8 release. In contrast, high concentrations of dexamethasone (10−6 M) partially suppressed LPS-stimulated IL-8 release by 36% (117 ± 11 ng/ml versus 184 ± 19 ng/ml, P < 0.05). Co-incubation of theophylline (10−6 M) and dexamethasone (10−6 M) enhanced the suppressive effect of dexamethasone on LPS-stimulated IL-8 release (64 ± 10 versus 184 ± 19 ng/ml, P < 0.01) (Fig. 3 B). This effect was significantly greater than that seen with dexamethasone (1 μM) alone.
Figure 3.

Effect of theophylline effect on HDAC activity and dexamethasone sensitivity in AM. (A) Nuclear extracts obtained from AM of heavy smokers and patients with COPD were incubated with theophylline (10−5 and 10−6 M) for 20 min, and HDAC activity was measured. **P < 0.01, n = 3. (B) Effect of theophylline (Theo; 10−6 M) and dexamethasone (Dex; 10−6 M) alone and in combination on LPS-stimulated IL-8 release from AM obtained from COPD patients cultured overnight. #P < 0.05 versus LPS; ##P < 0.01 versus LPS; *P < 0.05 versus LPS plus dexamethasone (n = 6).
Due to the lack of AM available from patients, we used PMA-treated U937 cells as a macrophage cell model to further study the interactions between glucocorticoids and theophylline since we have shown similar effects of theophylline in this cell line to those now reported in primary cells. After preincubation of these cells with hydrogen peroxide (H2O2, 100 μM) for 4 h, there was a marked increase in IL-1β–stimulated IL-8 release (2.41 ± 0.18 versus 1.88 ± 0.38 ng/ml, P < 0.05) and a reduced ability of dexamethasone (10−8 M) to suppress IL-8 release, thus mimicking the situation in COPD macrophages (Fig. 4 A). This was also reflected in the dexamethasone IC50 (IL-1β 6.6 ± 0.4 nM versus IL-1β + H2O2 > 1 μM, P < 0.01) and was accompanied by a marked decrease in HDAC activity in these cells after H2O2 (100 μM) treatment (46 ± 3 versus 130 ± 8 AFU/μg protein, P < 0.01). This effect of H2O2 was completely blocked by 10 mM N-acetyl cysteine (basal; 182 ± 24 versus H2O2; 52 ± 13 versus H2O2 + NAC; 163 ± 45 versus AFU/μg protein, P < 0.01). Similar effects on HDAC activity were also seen with cigarette smoke extract (Fig. 4 B) and resulted in enhanced IL-8 release (1.36 ± 0.24 versus 0.28 ± 0.04 ng/ml IL-8). This was completely blocked by addition of 10 mM NAC (0.34 ± 0.08 versus 1.36 ± 0.24 ng/ml). Addition of theophylline (10−6 M) alone had a small effect on IL-1β–stimulated IL-8 release (1.24 ± 0.07 versus 1.88 ± 0.38, P < 0.05) and on the ability of dexamethasone to suppress IL-8 release (Fig. 4 A). There was also a small effect on the dexamethasone IC50 (6.6 ± 0.4 versus 1.0 ± 0.2 nM, P < 0.05). However, theophylline restored dexamethasone sensitivity in IL-1β + H2O2–stimulated cells (Fig. 4 A) and reduced IL-8 expression down to levels seen with IL-1β + Dex–stimulated cells. There was also a marked shift in the dexamethasone IC50 (4.9 ± 0.44 nM versus > 1 μM, P < 0.01). In AM from smokers, the enhancing effect of theophylline was completely blocked by the addition of the HDAC inhibitor trichostatin A (Fig. 4 C). Immunoprecipitation of HDACs 1 and 2 indicated that the activity of both of these enzymes was increased by theophylline in U937 cells (Fig. 4 D) and also in BAL macrophages from smokers (Fig. 4 E).
Figure 4.

Theophylline effect on HDAC activity and steroid sensitivity in oxidant-stressed U937 cells and AM. (A) Effect of theophylline (Theo, 10−6 M) and dexamethasone (Dex; 10−8 M) alone and in combination on IL-1β (1 ng/ml) and IL-1β + hydrogen peroxide (H2O2; 100 μM)–stimulated IL-8 release from U937 cells. Theophylline (10−6 M) enhanced sensitivity compared with cells treated with IL-1β and H2O2. *P < 0.05 versus IL-1β; #P < 0.01 versus IL-1β + H2O2· (B) Effect of theophylline (Theo; 10−6 M) on cigarette smoke extract (CSE; 0.15 dilution)–induced reduction in HDAC activity. ***P < 0.001 versus control; ##P < 0.01 versus CSE in U937 cells. (C) Effect of theophylline (Theo; 10−5 M) and dexamethasone (Dex; 10−10 M) alone and in combination on LPS (10 ng/ml)-induced IL-8 release from AM obtained from smokers. Theophylline enhanced Dex sensitivity, an effect that was blocked by addition of the HDAC inhibitor trichostatin A (TSA; 10 ng/ml). *P < 0.05 versus LPS, n = 3. (D) Specific HDAC isoforms were immunoprecipitated from U937 cells and incubated with theophylline (10−6 M) for 20 min before HDAC activity was measured. ***P < 0.001 (n = 4). (E) Specific HDAC isoforms were immunoprecipitated from AM from smokers and incubated with theophylline (10−6 M) for 20 min before HDAC activity was measured. n = 3.
We have shown that AM from smokers and patients with COPD show increased release of the neutrophil-selective chemokine IL-8 and the proinflammatory cytokine TNF-α, which are associated with enhanced activation of the transcription factor NF-κB. We have also shown that the expression and activity of the corepressor protein HDAC, which inhibits NF-κB, is reduced in AM from smokers and that this is significantly further reduced in COPD patients. This reduction in HDAC activity was reversed by theophylline in AM from both smokers and patients with COPD. Furthermore, theophylline enhanced the ability of dexamethasone to suppress LPS-induced IL-8 release in macrophages and restored dexamethasone sensitivity. In U937 cells, oxidative stress reduced HDAC activity, and we hypothesize that oxidative stress is the causative agent for the reduced HDAC activity seen primary macrophages from smokers and that this leads to enhanced inflammatory gene transcription and a reduced response to glucocorticoids. Under these conditions, where we see a 70% reduction in HDAC activity, the sensitivity to glucocorticoids is reduced as well as the maximal response resulting in a marked 2–3-log shift in the concentration response curve. Achieving a 100–1,000-fold difference in glucocorticoid dosage in patients is difficult due to side effects and the reduced maximal response would still result in less suppression of inflammatory indices. Theophylline restored the antiinflammatory effects of glucocorticoids by increasing HDAC activity an effect that was completely blocked by the HDAC inhibitor trichostatin A in AM from smokers.
Macrophages are critical cells in COPD and contribute to the airway inflammation in smokers and COPD patients by secreting neutrophil and lymphocyte chemotactic factors, proteases, and reactive oxygen species (5). In this study, we found significantly higher numbers of AM and neutrophils in BAL fluid of smokers and COPD patients, in agreement with previous studies (3). We found increased nuclear localization of p65, indicating NF-κB activation in AM of smokers and to an even greater extent in COPD patients, confirming previous reports (14). The presence of nuclear p65 and enhanced basal and enhanced IL-8 release suggests that these cells were in a more active state than those obtained from normal subjects. In addition, an indirect sign of oxidative stress was the increased concentration of glutathione (GSH) in BAL fluid. Oxidative stress has been correlated previously with a defective thiol status in alveolar macrophages, with an increase of GSH in epithelial lining fluid (15), and is an important regulator of IL-8 gene expression (16).
IL-8 and TNFα are important inflammatory mediators in COPD, and both mediators are increased in sputum from patients with COPD (4). Neither inhaled nor oral glucocorticoids had any suppressive effect on their regulation in COPD patients (5). In our study, AM from smokers and COPD patients were activated and relatively resistant to the suppressive actions of glucocorticoids confirming previous data previously. We have reported previously that cytokine expression is enhanced and glucocorticoid effectiveness is reduced in alveolar macrophages from normal smokers and that this is correlated with a decreased HDAC activity and, in particular, reduced expression of HDAC2 (17). We have also shown previously that maximal repressive actions of glucocorticoids require recruitment of HDAC2 to a p65-HAT complex (11). In the present study, with a different population of smokers, we confirm that HDAC activity is reduced and for the first time show an even greater reduction in HDAC activity and HDAC2 expression in AM from COPD patients. In this study, 6 of 19 COPD subjects were excluded due to a failure to obtain sufficient viable AM. Although we obtained a large number of AM from almost of all patients, in some of the more severe patients all, or most, of the AM died. This would suggest that there may be a bias toward a less severe group of patients. In preliminary studies we have found that the reduction in HDAC activity in the peripheral lung of COPD patients correlated with increasing stage of disease (unpublished data). This suggests that if we were able to obtain AM consistently from these more severe patients that there may be a greater effect of theophylline on the parameters measured here.
In the macrophage-like U937 cells, we were able to demonstrate that oxidative stress (H2O2) markedly enhanced the release of IL-8 and reduced the antiinflammatory action of glucocorticoids, thus mimicking closely the behavior of COPD macrophages. Moreover, these abnormalities were completely reversed by low concentrations of theophylline. Trichostatin A inhibited the action of theophylline, confirming that it was mediated via HDAC activation rather than some other mechanism. It is of interest that in patients with mild asthma who are normally steroid sensitive, a complete resistance the antiinflammatory action of inhaled and oral glucocorticoids is observed in smokers (18, 19), and this may be explained by a similar mechanism. We have shown recently (20) that oxidative stress can induce HDAC nitration and that this correlates with reduced HDAC activity possibly as a result of targeting for proteosomal degradation. Furthermore, in preliminary data we have shown that HDAC2 isolated from macrophages from COPD patients has a greater level of tyrosine nitration of HDAC2 than normal subjects (21). We speculate, therefore, that one possible mechanism that accounts for the inhibitory effect of oxidative stress on HDAC2 activity is nitration and inactivation/degradation of the enzyme.
In an attempt to reverse the reduced HDAC activity, we explored the activity of theophylline as a HDAC stimulator in COPD (12). Theophylline activates HDAC activity and therefore suppresses the expression of inflammatory genes through a mechanism independent of cAMP modulation (12). This would explain the antiinflammatory effects of theophylline observed previously in asthma (11) and more recently in COPD (11). The effect is distinct from that of glucocorticoids, as there appears to be a relatively direct activation of HDAC, whereas glucocorticoid effects are due to recruitment of HDAC2 to the active site of transcription (8). In the present study, we saw a marked (over sixfold) increase in the depressed HDAC activity in COPD macrophages. Enhanced HDAC activity alone is not effective in suppressing inflammatory gene transcription unless it is recruited to the active proinflammatory transcriptional complex by the glucocorticoid receptor. This would explain the relative lack of effect of theophylline alone in suppressing IL-8 release and the potentiation of this effect when the cells are pretreated with dexamethasone. This effect is seen at therapeutic concentrations of theophylline (10−6–10−5 M) and is not mediated by either phosphodiesterase inhibition or by adenosine receptor antagonism (12). Theophylline enhances HDAC1 and HDAC2 activity in U937 cells and in AM from smokers, and this may play a role in the regulation of NF-κB–induced inflammatory gene expression (8, 22, 23). However, HDAC2 is the relevant isoform for the enhancement of steroid responsiveness.
In conclusion, we have shown an important explanation to the glucocorticoid-resistant inflammatory process observed in COPD (9). Reduction of HDAC activity may be responsible for the enhanced inflammatory gene transcription, and this is not directly related to an increase in NF-κB activation, but as a direct effect of oxidative stress. Low dose theophylline may “unlock” the glucocorticoid resistance of COPD and potentiate its suppressive effects. This suggests that theophylline may restore the antiinflammatory effect of glucocorticoids and thus control the underlying disease process in COPD. This needs to be confirmed in clinical studies in COPD patients, but theophylline might provide a relatively inexpensive way of managing this common and globally increasing disease.
Acknowledgments
The authors are indebted to Dr. Pallav Shah and Dr. Onn Min Kon for their contribution as bronchoscopists in this study.
The study was supported by grants from National Institutes of Health and the British Lung Foundation. B. Cosio was the recipient of a European Respiratory Society and a Separ Fellowship.
P.J. Barnes, I.M. Adcock, and K. Ito have received nonrestricted funding from GlaxoSmithKline and Mitsubishi Pharma to fund part of this work.
References
- 1.Barnes, P.J. 2000. Chronic obstructive pulmonary disease. N. Engl. J. Med. 343:269–280. [DOI] [PubMed] [Google Scholar]
- 2.Saetta, M., G. Turato, P. Maestrelli, C.E. Mapp, and L.M. Fabbri. 2001. Cellular and structural bases of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 163:1304–1309. [DOI] [PubMed] [Google Scholar]
- 3.Retamales, I., W.M. Elliott, B. Meshi, H.O. Coxson, P.D. Pare, F.C. Sciurba, R.M. Rogers, S. Hayashi, and J.C. Hogg. 2001. Amplification of inflammation in emphysema and its association with latent adenoviral infection. Am. J. Respir. Crit. Care Med. 164:469–473. [DOI] [PubMed] [Google Scholar]
- 4.Keatings, V.M., P.D. Collins, D.M. Scott, and P.J. Barnes. 1996. Differences in interleukin-8 and tumor necrosis factor-α in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am. J. Respir. Crit. Care Med. 153:530–534. [DOI] [PubMed] [Google Scholar]
- 5.Barnes, P.J. 2003. New concepts in COPD. Annu. Rev. Med. 54:113–129. [DOI] [PubMed] [Google Scholar]
- 6.Culpitt, S.V., D.F. Rogers, P. Shah, C. de Matos, R.E. Russell, L.E. Donnelly, and P.J. Barnes. 2003. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 167:24–31. [DOI] [PubMed] [Google Scholar]
- 7.Urnov, F.D., and A.P. Wolffe. 2001. Chromatin remodeling and transcriptional activation: the cast (in order of appearance). Oncogene. 20:2991–3006. [DOI] [PubMed] [Google Scholar]
- 8.Ito, K., P.J. Barnes, and I.M. Adcock. 2000. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits IL-1 β-induced histone H4 acetylation on lysines 8 and 12. Mol. Cell. Biol. 20:6891–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barnes, P.J., K. Ito, and I.M. Adcock. 2004. A mechanism of corticosteroid resistance in COPD: inactivation of histone deacetylase. Lancet. 363:731–733. [DOI] [PubMed] [Google Scholar]
- 10.Ito, K., S. Lim, G. Caramori, K.F. Chung, P.J. Barnes, and I.M. Adcock. 2001. Cigarette smoking reduces histone deacetylase 2 expression, enhances cytokine expression and inhibits glucocorticoid actions in alveolar macrophages. FASEB J. 15:1100–1102. [PubMed] [Google Scholar]
- 11.Barnes, P.J. 2003. Theophylline: new perspectives on an old drug. Am. J. Respir. Crit. Care Med. 167:813–818. [DOI] [PubMed] [Google Scholar]
- 12.Ito, K., S. Lim, G. Caramori, B. Cosio, K.F. Chung, I.M. Adcock, and P.J. Barnes. 2002. A molecular mechanism of action of theophylline: induction of histone deacetylase activity to decrease inflammatory gene expression. Proc. Natl. Acad. Sci. USA. 99:8921–8926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pauwels, R.A., A.S. Buist, P.M. Calverley, C.R. Jenkins, and S.S. Hurd. 2001. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am. J. Respir. Crit. Care Med. 163:1256–1276. [DOI] [PubMed] [Google Scholar]
- 14.Di Stefano, A., G. Caramori, A. Capelli, M. Lusuardi, I. Gnemmi, F. Ioli, K.F. Chung, C.F. Donner, P.J. Barnes, and I.M. Adcock. 2002. Increased expression of NF-κB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 20:556–563. [DOI] [PubMed] [Google Scholar]
- 15.Tager, M., A. Piecyk, T. Kohnlein, U. Thiel, S. Ansorge, and T. Welte. 2000. Evidence of a defective thiol status of alveolar macrophages from COPD patients and smokers. Chronic obstructive pulmonary disease. Free Radic. Biol. Med. 29:1160–1165. [DOI] [PubMed] [Google Scholar]
- 16.Rahman, I., and W. Macnee. 1999. Lung glutathione and oxidative stress: implications in cigarette smoke-induced airway disease. Am. J. Physiol. 277:L1067–L1088. [DOI] [PubMed] [Google Scholar]
- 17.Ito, K., S. Watanabe, S. Kharitonov, T. Hanazawa, I.M. Adcock, and P.J. Barnes. 2001. Histone deacetylase activity and gene expression in COPD patients. Eur. Respir. J. 18:316S. [Google Scholar]
- 18.Chalmers, G.W., K.J. Macleod, S.A. Little, L.J. Thomson, C.P. McSharry, and N.C. Thomson. 2002. Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax. 57:226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaudhuri, R., E. Livingston, A.D. McMahon, L. Thomson, W. Borland, and N.C. Thomson. 2003. Cigarette smoking impairs the therapeutic response to oral corticosteroids in chronic asthma. Am. J. Respir. Crit. Care Med. 168:1265–1266. [DOI] [PubMed] [Google Scholar]
- 20.Ito, K., T. Hanazawa, K. Tomita, P.J. Barnes, and I.M. Adcock. 2004. Oxidative stress reduces histone deacetylase (HDAC)2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem. Biophys. Res. Commun. 315:240–245. [DOI] [PubMed] [Google Scholar]
- 21.Ito, K., G. Caramori, A. Papi, P. Casolari, A. Ciaccia, L.M. Fabbri, P.J. Barnes, and I.M. Adcock. 2002. Histone deacetylase expression and activity in COPD. Am. J. Respir. Crit. Care Med. 168:B5 (Abstr). [Google Scholar]
- 22.Viatour, P., S. Legrand-Poels, C. Van Lint, M. Warnier, M.P. Merville, J. Gielen, J. Piette, V. Bours, and A. Chariot. 2003. Cytoplasmic IkappaBalpha increases NF-kappaB-independent transcription through binding to histone deacetylase (HDAC) 1 and HDAC3. J. Biol. Chem. 278:46541–46548. [DOI] [PubMed] [Google Scholar]
- 23.Zhong, H., M.J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell. 9:625–636. [DOI] [PubMed] [Google Scholar]