

Abstract
We report the generation and analysis of functional data from multiple, diverse experiments performed on a targeted 1% of the human genome as part of the pilot phase of the ENCODE Project. These data have been further integrated and augmented by a number of evolutionary and computational analyses. Together, our results advance the collective knowledge about human genome function in several major areas. First, our studies provide convincing evidence that the genome is pervasively transcribed, such that the majority of its bases can be found in primary transcripts, including non-protein-coding transcripts, and those that extensively overlap one another. Second, systematic examination of transcriptional regulation has yielded new understanding about transcription start sites, including their relationship to specific regulatory sequences and features of chromatin accessibility and histone modification. Third, a more sophisticated view about chromatin structure has emerged, including its interrelationship with DNA replication and transcriptional regulation. Finally, integration of these new sources of information, in particular with respect to mammalian evolution based on inter- and intra-species sequence comparisons, has yielded novel mechanistic and evolutionary insights about the functional landscape of the human genome. Together, these studies are defining a path forward to pursue a more-comprehensive characterisation of human genome function.
Introduction
The human genome is an elegant but cryptic store of information. Its roughly three billion bases encode, either directly or indirectly, the instructions for synthesizing nearly all the molecules that form each human cell, tissue, and organ. Sequencing the human genome1–3 provided highly accurate DNA sequences for each of the 24 chromosomes. At present, however, we have an incomplete understanding of the protein-coding portions of the genome, and markedly less understanding of both non-protein-coding transcripts and genomic elements that temporally and spatially regulate gene expression. To understand the human genome, and by extension the biological processes it orchestrates and the ways in which its defects can give rise to disease, we need a more transparent view of the information it encodes.
The molecular mechanisms by which genomic information directs the synthesis of different biomolecules has been the focus of much of molecular biology over the last three decades. Previous studies have typically concentrated on individual genes, with the resulting general principles then providing insights into, for example, transcription, chromatin remodeling, mRNA splicing, DNA replication and numerous other genomic processes. Although many such principles appear valid as additional genes are investigated, they typically have not provided genome-wide insights about biological function.
The first genome-wide analyses that shed light on human genome function made use of observing the actions of evolution. The ever-growing set of vertebrate genome sequences4–8 is providing increasing power to reveal the genomic regions that have been most and least acted upon by the forces of evolution. However, while these studies convincingly indicate the presence of numerous genomic regions under strong evolutionary constraint, they have less power in identifying the precise bases that are constrained and provide little, if any, insight into why those bases are biologically important. Further, although we have good models for how protein-coding regions evolve, our present understanding about the evolution of other functional genomic regions is poorly developed. Experimental studies that augment what we learn from evolutionary analyses are key for solidifying our insights about genome function.
The Encyclopedia of DNA Elements (ENCODE) Project9 aims to provide a more biologically informative representation of the human genome by using high-throughput methods to identify and catalogue the functional elements it encodes. In its pilot phase, 35 groups provided more than 200 experimental and computational datasets that examined in unprecedented detail a targeted 29.998 Mb of the human genome. This roughly 30 Mb— ~1% of the human genome— is sufficiently large and diverse to allow for rigorous pilot testing of multiple experimental and computational methods. These 30 Mb are divided among 44 genomic regions; roughly 15 Mb reside in 14 regions for which there is already substantial biological knowledge, while the other roughly 15 Mb reside in 30 regions chosen by a stratified random-sampling method (see http://www.genome.gov/10506161).
The highlights of our findings to date include:
The human genome is pervasively transcribed, such that the majority of its bases are associated with at least one primary transcript and many transcripts link distal regions to established protein-coding loci.
Many novel non-protein-coding transcripts have been identified, with many of these overlapping protein-coding loci and others located in regions of the genome previously thought to be transcriptionally silent.
Numerous previously unrecognised transcription start sites have been identified, many of which show chromatin structure and sequence-specific protein-binding properties similar to well-understood promoters.
Regulatory sequences that surround transcription start sites are symmetrically distributed, with no bias towards upstream regions.
Chromatin accessibility and histone-modification patterns are highly predictive of both the presence and activity of transcription start sites.
Distal DNaseI hypersensitive sites have characteristic histone modification patterns that reliably distinguish them from promoters; some of these distal sites show marks consistent with insulator function.
DNA-replication timing is correlated with chromatin structure.
A total of 5% of the bases in the genome can be confidently identified as being under evolutionary constraint in mammals; for approximately 60% of these constrained bases, there is evidence of function based on the results of the experimental assays performed to date.
While there is general overlap between genomic regions identified as functional by experimental assays and those under evolutionary constraint, not all bases within these experimentally-defined regions show evidence of constraint.
Different functional elements vary greatly in their sequence variability across the human population and in their likelihood of residing within a structurally variable region of the genome.
To our surprise, many functional elements are seemingly unconstrained across mammalian evolution. This suggests the possibility of a large pool of neutral elements that are biologically active but provide no specific benefit to the organism. This pool may serve as a ‘warehouse’ for natural selection, potentially acting as the source of lineage-specific elements and functionally conserved but non-orthologous elements between species.
Below, we first provide an overview of the experimental techniques used for our studies, after which we describe the insights gained from analyzing and integrating the generated datasets. We conclude with a perspective of what we have learned to date about this 1% of the human genome and what we believe the prospects are for a broader and deeper investigation of the functional elements in the human genome. To aid the reader, Box 1 provides a glossary for many of the abbreviations used throughout this paper.
Experimental techniques
Table 1 (expanded in Supplementary Information section S1.1) lists the major experimental techniques used for the studies reported here, relevant acronyms, and references reporting the generated datasets. These datasets reflect over 400 million experimental data points (603 million data points if one includes comparative sequencing bases). In describing the major results and initial conclusions, we seek to distinguish biochemical function from biological role. Biochemical function reflects the direct behaviour of a molecule(s), while biological role is used to describe the consequence(s) of this function for the organism. Genome-analysis techniques nearly always focus on biochemical function but not necessarily on biological role. This is because the former is more amenable to large-scale data-generation methods, while the latter is more difficult to assay on a large scale.
Table 1.
Summary of types of experimental techniques used in ENCODE
| Feature Class | Experimental Technique(s) | Abbreviations | References | Number of Experimental Data Points |
|---|---|---|---|---|
| Transcription | Tiling array, Integrated annotation | TxFrag, RxFrag, GENCODE | Harrow et al 117
Emanuelsson et al118 Rozowsky et al 19 Kapranov et al 119 |
63,348,656 |
| 5′ Ends of transcripts * | Tag sequencing | GIS-PET, CAGE | Ng et al 121
Carninci et al 13 |
864,964 |
| Histone modifications | Tiling array | Histone nomenclature†, RFBR | Koch et al 46 | 4,401,291 |
| Chromatin structure + | QT-PCR, Tiling array | DHS, FAIRE | Dorschner et al42
Sabo et al43 Crawford et al 44 Giresi et al122 |
15,318,324 |
| Sequence- specific factors | Tiling array, tag sequencing, Promoter assays | STAGE, ChIP- Chip, ChIP- PET, RFBR | Bieda et al 52
Bhinge et al120 Euskirchen et al11 Rada-Iglesias et al123 Thurman et al81 Cawley et al 34 Kim et al 41 Kim et al 51 Kim et al 124 Heintzman et al49 Cooper et al 33 Wei et al 40 |
324,846,018 |
| Replication | Tiling array | TR50 | Jeon et al 59
Karnani el al75 |
14,735,740 |
| Computational analysis | Computational methods | CCI, RFBR Cluster | Greenbaum et al80
Halees & Weng 125 Zhang et al 10 Guigo et al 126 Bajic et al 127 Zheng & Gerstein 128 |
NA |
| Comparative sequence analysis * | Genomic sequencing, multi- sequence alignments, computational analyses | CS | Cooper et al 87
Margulies et al 86 Washietl et al26 |
NA |
| Polymorphisms * | Resequencing, copy number variation | CNV | The International HapMap Consortium 103 Stranger et al 129 | NA |
= Not all data generated by ENCODE Project.
= Also contains histone modification.
= Histone code nomenclature follows the Brno nomenclature as described by Turner130
ENCODE aimed to establish redundancy with respect to the findings represented by different datasets. In some instances, this involved the intentional use of different assays based on a similar technique, whereas in other situations, different techniques assayed the same biochemical function. Such redundancy has allowed methods to be compared and the generation of consensus datasets, much of which is discussed in companion papers, such as the ChIP/chip platform comparison10, 11. All ENCODE data have been released after verification but prior to this publication, as befits a ‘community resource’ project (http://www.wellcome.ac.uk/doc_wtd003208.html). Verification is defined as when the experiment is reproducibly confirmed (see Supplementary Information section S1.2). The main portal for ENCODE data is provided on the UCSC Genome Browser (http://genome.ucsc.edu/ENCODE/); this is augmented by multiple other web sites (see Supplementary Information section S1.1).
A common feature of genomic analyses is the need to assess the significance of the co-occurrence of features or other statistical tests. One confounding factor is the heterogeneity of the genome, which can produce uninteresting correlations of variables distributed across the genome. We have developed and used a statistical framework that mitigates many of these hidden correlations by adjusting the appropriate null distribution of the test statistics. We term this correction procedure “Genome Structure Correction” (GSC) (see Supplementary Information section S1.3).
In the next five sections, we detail the various biological insights of the pilot phase of the ENCODE Project.
Transcription
Overview
RNA transcripts are involved in many cellular functions, either directly as biologically active molecules or indirectly by encoding other active molecules. In the conventional view of genome organisation, sets of RNA transcripts (for example, mRNAs) are encoded by distinct loci, with each usually dedicated to a single biological role (for example, encoding a specific protein). However, this picture has substantially grown in complexity in recent years12. Other forms of RNA molecules (such as snoRNAs and microRNAs) are known to exist, and often these are encoded by regions that intercalate with protein-coding genes. These observations are consistent with the well-known discrepancy between the amount of observable mRNAs and large structural RNAs compared to the total RNA in a cell, suggesting that there are numerous RNA species yet to be classified13–15. In addition, studies of specific loci have indicated the presence of RNA transcripts that play a role in chromatin maintenance and other regulatory control. We sought to assay and analyse transcription comprehensively across the 44 ENCODE regions in an effort to understand the repertoire of encoded RNA molecules.
Transcript maps of the ENCODE regions
We used three methods to identify transcripts emanating from the ENCODE regions: hybridisation of RNA (either total or polyA-selected) to unbiased tiling arrays (see Supplementary Information section S2.1), tag sequencing of cap-selected RNA at the 5′ or joint 5′/3′ ends (see Supplementary Information sections S2.2 and S2.3), and integrated annotation of available cDNA and EST sequences involving computational, manual, and experimental approaches16 (see Supplementary Information section S2.4). We abbreviate the regions identified by unbiased tiling arrays as TxFrags (Transcribed Fragments), the cap-selected RNAs as CAGE/Ditags, and the integrated annotation as GENCODE transcripts. When a TxFrag does not overlap a GENCODE annotation, we call it an unannotated TxFrag (Un.TxFrag). Validation of these various studies is described in papers reporting these datasets17 (see Supplementary Information sections S2.1.4 and S2.1.5).
These methods recapitulate previous findings, but provide enhanced resolution due to the larger number of tissues sampled and the integration of results across the three approaches. To begin with, our studies show that 14.7% of the bases represented in the unbiased tiling arrays are transcribed in at least one tissue sample. Consistent with previous work14, 15, many (63%) TxFrags reside outside of GENCODE annotations, both in intronic (40.9%) and intergenic (22.6%) regions. GENCODE annotations are richer than the more-conservative RefSeq or Ensembl annotations, with 2,608 transcripts clustered into 487 loci, leading to an average of 5.4 transcripts per locus. Finally, extensive testing of predicted protein-coding sequences outside of GENCODE annotations was positive in only 2% of cases16, suggesting that GENCODE annotations cover nearly all protein-coding sequences. The GENCODE annotations are categorised both by likely function (mainly, the presence of an open reading frame) and by classification evidence (for example, transcripts based solely on ESTs are distinguished from other scenarios); This classification is not strongly correlated with expression levels (see Supplementary Information sections S2.4.2 and S2.4.3).
Analyses of more biological samples have allowed a richer description of the transcription specificity (see Figure 1 and Supplementary Information section S2.5). We found that 40% of TxFrags are present in only one sample, whereas only 2% are present in all samples. Although exon-containing TxFrags are more likely (74%) to be expressed in more than one sample, 45% of unannotated TxFrags are also expressed in multiple samples. GENCODE annotations of separate loci often (42%) overlap with respect to their genomic coordinates, in particular on opposite strands (33% of loci). Further analysis of GENCODE-annotated sequences with respect to the positions of open read frames revealed that some component exons do not have the expected synonymous vs non synonymous substitution patterns of protein-coding sequence (see Supplementary Information section S2.6) and some have deletions incompatible with protein structure18. Such exons are on average less expressed (25% vs 87% by RT-PCR, see Supplementary Information section S2.7) than exons involved in more than one transcript (see Supplementary Information section S2.4.3), but when expressed have a tissue distribution comparable to well-established genes.
Figure 1.
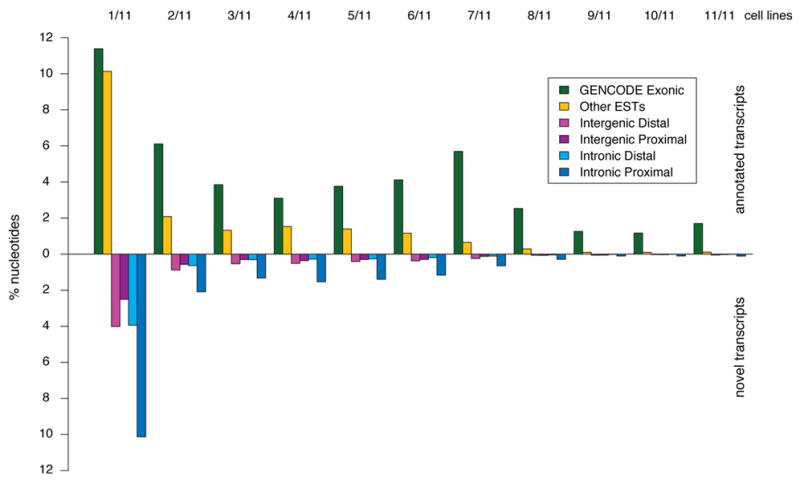
Annotated and unannotated TxFrags detected in different cell lines. The proportion of different types of transcripts detected in the indicated number of cell lines (from 1/11 at the far left to 11/11 at the far right) is shown. The data for annotated and unannotated TxFrags are indicated separately, and also split into different categories based on GENCODE classification: Exonic, Intergenic (Proximal being within 5 kb of a gene and Distal being otherwise), Intronic (Proximal being within 5 kb of an intron and Distal being otherwise), and matching other ESTs not used in the GENCODE annotation (principally because they were unspliced). The y-axis indicates the percent of tiling array nucleotides present in that class for that number of tissues.
Critical questions are raised by the presence of a large amount of unannotated transcription with respect to how the corresponding sequences are organised in the genome – do these reflect longer transcripts that include known loci, do they link known loci, or are they completely separate from known loci? We further investigated these issues using both computational and new experimental techniques.
Computational Analysis of Unannotated Trancription
Consistent with previous findings, the Un.TxFrags did not have evidence of encoding proteins (see Supplementary Information section S2.8). One might expect Un.TxFrags to be linked within transcripts that exhibit coordinated expression and have similar conservation profiles across species. To test this, we clustered Un.TxFrags using two methods. The first method19 used expression levels in 11 cell lines or conditions, dinucleotide composition, location relative to annotated genes, and evolutionary conservation profiles to cluster TxFrags (both unannotated and annotated). By this method, 14% of Un.TxFrags could be assigned to annotated loci, and 21% could be clustered into 200 novel loci (with an average of ~7 TxFrags per locus). We experimentally examined these novel loci to study the connectivity of transcripts amongst Un.TxFrags and between Un.TxFrags and known exons. Overall, about 40% of the connections (18 out of 46) were validated by RT-PCR. The second clustering method involved analysing a time course (0, 2, 8, and 32 hours) of expression changes in HL60 cells following retinonic-acid stimulation. There is a coordinated program of expression changes from annotated loci, which can be shown by plotting Pearson correlation values of the expression levels of exons inside annotated loci versus unrelated exons (see Supplementary Information section S2.8.2). Similarly, there is coordinated expression of nearby Un.TxFrags, albeit lower, though still significantly different from randomised sets. Both clustering methods indicate that there is coordinated behaviour of many Un.TxFrags, consistent with them residing in connected transcripts.
Investigation of transcript connectivity using RACE and tiling arrays
We used a combination of RACE and tiling arrays20 to investigate the diversity of transcripts emanating from protein-coding loci. Analogous to TxFrags, we refer to transcripts detected using RACE followed by hybridization to tiling arrays as “RxFrags.” We performed RACE to examine 399 protein-coding loci (those loci found entirely in ENCODE regions) using RNA derived from 12 tissues, and were able to unambiguously detect 4,573 RxFrags for 359 loci (see Supplementary Information section S2.9). Almost half of these RxFrags (2,324) do not overlap a GENCODE exon, and most (90%) loci have at least one novel RxFrag, which often extends a considerable distance beyond the 5′ end of the locus. Figure 2 shows the distribution of distances between these new RACE-detected ends and the previously-annotated transcription start site (TSS) of each locus. The average distance of the extensions is between 50 kb and 100 kb, with many extensions (>20%) being more than 200 kb. Consistent with the known presence of overlapping genes in the human genome, our findings reveal evidence for an overlapping gene for 224 loci, with transcripts from 180 of these loci (~50% of the RACE-positive loci) appearing to have incorporated at least one exon from an upstream gene.
Figure 2.
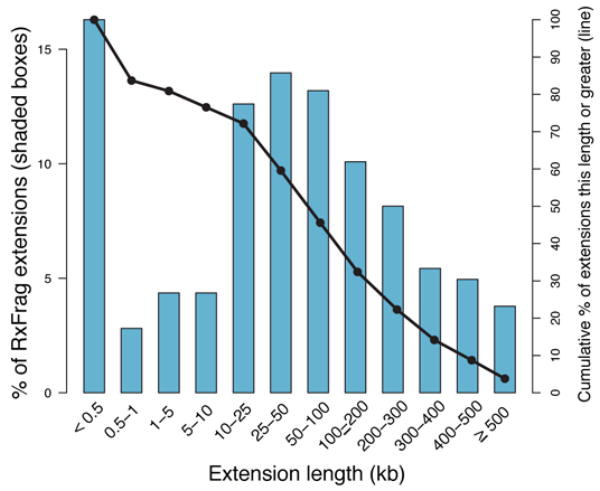
Length of genomic extensions to GENCODE-annotated genes based on RACE experiments followed by array hybridisations (RxFrags). The indicated bars reflect the frequency of extension lengths among different length classes. The solid line shows the cumulative frequency of extensions of that length or greater. Most of the extensions are greater than 50 kb from the annotated gene (see text for details).
To further characterise the 5′ RxFrag extensions, we performed RT-PCR followed by cloning and sequencing for 550 of the 5′ RxFrags (including the 261 longest extension identified for each locus). The approach of mapping RACE products using microarrays is a combination method previously described and validated in several studies14, 17, 20. Hybridization of the RT-PCR products to tiling arrays confirmed connectivity in almost 60% of the cases. Sequenced clones confirmed transcript extensions. Longer extensions were harder to clone and sequence, but 5 of 18 RT-PCR-positive extensions over 100-kb were verified by sequencing (see Supplementary Information section S2.9.7 and Denoeud et al17). The detection of numerous RxFrag extensions coupled with evidence of considerable intronic transcription indicates that protein-coding loci are more transcriptionally complex than previously thought. Instead of the traditional view that many genes have one or more alternative transcripts that code for alternative proteins, our data suggest that a given gene may both encode multiple protein products as well as produce other transcripts that include sequences from both strands and from neighbouring loci (often without encoding a different protein). Figure 3 illustrates such as case, where a new fusion transcript is expressed in the small intestine, and consists of at least three coding exons from the ATP50 gene and at least two coding exons from the DONSON gene, with no evidence of sequences from two intervening protein-coding genes (ITSN1 and CRYZL1).
Figure 3.
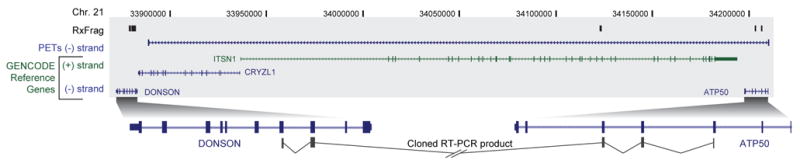
Overview of RACE experiments showing a gene fusion. Transcripts emanating from the region between the DONSON and ATP50 genes. A 330-kb interval of human chromosome 21 (within ENm005) is shown, which contains four annotated genes: DONSON, CRYZL1, ITSN1, and ATP50. The 5′ RACE products generated from small intestine RNA and detected by tiling-array analyses (RxFrags) are shown along the top. Along the bottom is shown the placement of a cloned and sequenced RT-PCR product that has two exons from the DONSON gene followed by three exons from the ATP50 gene; these sequences are separated by a 300-kb intron in the genome. A PET tag shows the termini of a transcript consistent with this RT-PCR product.
Pseudogenes
Pseudogenes, reviewed in Balakirev et al21 and Mighell et al22, are generally considered non-functional copies of genes that are sometimes transcribed and often complicate analysis of transcription due to close sequence similarity to functional genes. We utilised various computational methods to identify 201 pseudogenes (124 processed and 77 non processed) in the ENCODE regions (see Supplementary Information section S2.10 and Zheng et al23). Tiling-array analysis of 189 of these revealed that 56% overlapped at least one TxFrag. However, possible cross-hybridisation between the pseudogenes and their corresponding parent genes may have confounded such analyses. To better assess the extent of pseudogene transcription, 160 pseudogenes (111 processed and 49 non-processed) were examined for expression using RACE/tiling-array analysis (see Supplementary Information section S2.9.2). Transcripts were detected for 14 pseudogenes (8 processed and 6 non-processed) in at least one of the 12 tested RNA sources, the majority (9) being in testis (see Zheng et al23). Additionally, there was evidence for the transcription of 25 pseudogenes based on their proximity (within 100bp of a pseudogene end) to CAGE tags (8), PETs (2), or cDNAs/ESTs (21). Overall, we estimate that at least 19% of the pseudogenes in the ENCODE regions are transcribed, which is consistent with previous estimates24, 25.
Non-protein-coding RNA
Non-protein-coding RNAs (ncRNAs) include structural RNAs (for example, tRNAs, rRNAs, and snRNAs) and more recently-discovered regulatory RNAs (for example, microRNAs). There are only 8 well-characterised ncRNA genes within the ENCODE regions (U70, ACA36, ACA56, mir-192, mir-194-2, mir-196, mir-483 and H19), while representatives of other classes, (for example, box C/D snoRNAs, tRNAs, and functional snRNAs) appear to be completely absent in the ENCODE regions. Tiling-array data provided evidence for transcription in at least one of the assayed RNA samples for all of them, with the exception of mir-483 (expression of mir-483 might be specific to fetal liver, which was not tested). There is also evidence for the transcription of 6 of 8 pseudogenes of non-protein-coding RNAs (mainly snoRNA-derived). Similar to the analysis of protein-pseudogenes, the hybridisation results could also originate from the known snoRNA gene elsewhere in the genome.
Many known ncRNAs are characterised by a well-defined RNA-secondary structure. We applied two de-novo ncRNA-prediction algorithms – EvoFold and RNAz – to predict structured ncRNAs (as well as functional structures in mRNAs) using the multi-species sequence alignments (see below, Supplementary Information section S2.11, and Washietl et al26). Using a sensitivity threshold capable of detecting all known miRNAs and snoRNAs, we identified 4986 and 3707 candidate ncRNA loci with EvoFold and RNAz, respectively. Only 268 loci (5% and 7%, respectively) were found with both programs, representing a 1.6-fold enrichment over that expected by chance; the lack of more extensive overlap is due to the two programs having optimal sensitivity at different levels of GC content and conservation. We experimentally examined 50 of these targets using RACE/tiling-array analysis and brain and testis tissues (see Supplementary Information sections S2.11 and S2.9.3); the predictions were validated at a 56%, 65%, and 63% rate for Evofold, RNAz, and dual predictions, respectively.
Primary transcripts
The detection of numerous unannotated transcripts coupled with increasing knowledge of the general complexity of transcription prompted us to examine the collective span of primary (i.e., unspliced) transcripts across the ENCODE regions. Three data sources provide insight about these primary transcripts: the GENCODE annotation, PET ditags, and RxFrag extensions. Figure 4 summarizes the fraction of bases in the ENCODE regions that overlap transcripts identified by these technologies. Remarkably, 93% of bases are represented in a primary transcript identified by at least two independent observations (but potentially using the same technology); this figure is reduced to 74% in the case of primary transcripts detected by at least two different technologies. These increased spans are not mainly due to cell line rearrangements since they were present in of multiple tissue experiments confirming the spans (see Supplementary Information section S2.12). These estimates assume that the presence of PET ditags or RxFrags defining the terminal ends of a transcript imply that the entire intervening DNA is transcribed and then processed. Other mechanisms, thought to be unlikely in the human genome, such as trans-splicing or polymerase jumping would also produce these long termini and potentially should be reconsidered in more detail.
Figure 4.
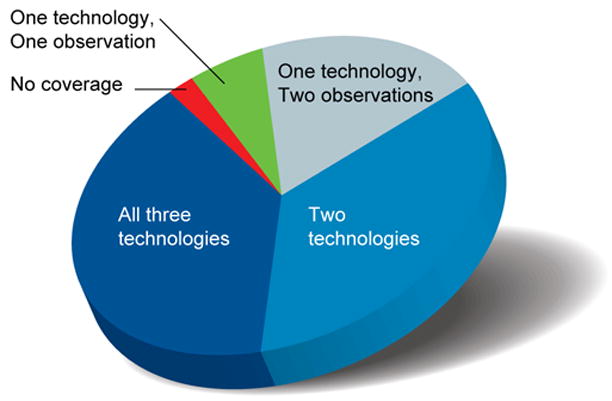
Coverage of primary transcripts across ENCODE regions. Three different technologies [integrated annotation from GENCODE, RACE-array experiments (RxFrags), and PET tags] were used to assess the presence of a nucleotide in a primary transcript. Use of these technologies provided the opportunity to have multiple observations of each finding. The proportion of genomic bases detected in the ENCODE regions associated with each of the following scenarios is depicted: detected by all three technologies, by two of the three technologies, by one technology but with multiple observations, and by one technology with only one observation. Also indicated are genomic bases without any detectable coverage of primary transcripts.
Previous studies have suggested a similar broad amount of transcription across the human14, 15 and mouse27 genomes. Our studies confirm these results, and have investigated the genesis of these transcripts in greater detail, confirming the presence of substantial intragenic and intergenic transcription. At the same time, many of the resulting transcripts are neither traditional protein-coding transcripts nor easily explained as structural non-coding RNAs. Other studies have noted complex transcription around specific loci or chimeric-gene structures (for example refs28–30), but these have often been considered exceptions; our data show that complex intercalated transcription is common at many loci. The results presented in the next section show extensive amounts of regulatory factors around novel TSSs, which is consistent with this extensive transcription. The biological relevance of these unannotated transcripts remains unanswered by these studies. Evolutionary information (detailed below) is mixed in this regard, for example, it indicates that unannotated transcripts show weaker evolutionary conservation than many other annotated features. As with other ENCODE-detected elements, it is difficult to identify clear biological roles for the majority of these transcripts; such experiments are challenging to perform on a large scale, and furthermore, it seems likely that many of the corresponding biochemical events may be evolutionarily neutral (see below).
Regulation of transcription
Overview
A significant challenge in biology is to identify the transcriptional regulatory elements that control the expression of each transcript and to understand how the function of these elements is coordinated to execute complex cellular processes. A simple, commonplace view of transcriptional regulation involves five types of cis-acting regulatory sequences— promoters, enhancers, silencers, insulators, and locus control regions (LCRs)31. Overall, transcriptional regulation involves the interplay of multiple components, whereby the availability of specific transcription factors and the accessibility of specific genomic regions determine whether a transcript is generated31. However, the current view of transcriptional regulation is known to be overly simplified, with many details remaining to be established. For example, the consensus sequences of transcription factor-binding sites (typically 6 to 10 bases) have relatively little information content and are present numerous times in the genome, with the great majority of these not participating in transcriptional regulation. Does chromatin structure then determine whether such a sequence has a regulatory role? Are there complex inter-factor interactions that integrate the signals from multiple sites? How are signals from different distal regulatory elements coupled without affecting all neighbouring genes? Meanwhile, our understanding of the repertoire of transcriptional events is becoming more complex, with an increasing appreciation of alternative TSSs32, 33 and the presence of non-coding27, 34 and anti-sense transcripts35, 36.
To better understand transcriptional regulation, we sought to begin cataloguing the regulatory elements residing within the 44 ENCODE regions. For this pilot project, we mainly focused on the binding of regulatory proteins and chromatin structure involved in transcriptional regulation. We analysed over 150 datasets, mainly from ChIP-chip37–39, Chip-PET and STAGE40, 41 studies (see Supplementary Information sections S3.1 and S3.2). These methods use chromatin immunoprecipitation (ChIP) with specific antibodies to enrich for DNA in physical contact with the targeted epitope. This enriched DNA can then be analysed using either microarrays (ChIP-chip) or high-throughput sequencing (ChIP-PET and STAGE). The assays included 18 sequence-specific transcription factors and components of the general transcription machinery [for example, RNA polymerase II (PolII), TAF1, and TFIIB]. In addition, we tested more than 600 potential promoter fragments for transcriptional activity by transient-transfection reporter assays that utilized 16 human cell lines33. We also examined chromatin structure by studying the ENCODE regions for DNaseI sensitivity (via quantitative PCR42 and tiling arrays43, 44, see Supplementary Information section S3.3), histone composition45, histone modifications (via ChIP-chip assays)37, 46, and histone displacement (using FAIRE, see Supplementary Information section 3.4). Below, we detail these analyses, starting with the efforts to define and classify the 5′ ends of transcripts with respect to their associated regulatory signals. Following that are summaries of generated data about sequence-specific transcription-factor binding and clusters of regulatory elements. Finally, we describe how this information can be integrated to make predictions about transcriptional regulation.
Cataloguing TSSs
We analysed two datasets to catalogue TSSs in the ENCODE regions: the 5′ ends of GENCODE-annotated transcripts and the combined results of two 5′-end-capture technologies— CAGE and PET-tagging. The initial results suggested the potential presence of 16,051 unique TSSs. However, in many cases, multiple TSSs resided within a single small segment (up to ~200 bases); this was due to some promoters containing TSSs with many very close precise initiation sites 47. To normalise for this effect, we grouped TSSs that were 60 or fewer bases apart into a single cluster, and in each case considered the most frequent CAGE or PET tag (or the 5′-most TSS in the case of TSSs identified only from GENCODE data) as that cluster’s representative for downstream analyses.
The above effort yielded 7,157 TSSs clusters in the ENCODE regions. We classified these TSSs into three categories: Known (present at the end of GENCODE-defined transcripts), Novel (supported by other evidence), and Unsupported. The Novel TSSs were further subdivided based on the nature of the supporting evidence (see Table 3 and Supplementary Information section S3.5), with all four of the resulting subtypes showing significant overlap with experimental evidence using the GSC statistic. Although there is a larger relative proportion of singleton tags in the Novel category, when analysis is restricted to only singleton tags, the Novel TSSs continue to have highly significant overlap to supporting evidence (see Supplementary Information section S3.5.1).
Table 3.
Different categories of TSSs defined on the basis of support from different transcript-survey methods.
| Category | Transcript survey method | No. TSS clusters (non-redundant)1 | P-value2 | % singleton clusters3 |
|---|---|---|---|---|
| Known | GENCODE 5′ ends | 1,730 | 2e-70 | 25% (74% overall) |
| GENCODE sense exons | 1,437 | 6e-39 | 64% | |
| Novel | GENCODE antisense exons | 521 | 3e-8 | 65% |
| Unbiased transcription survey | 639 | 7e-63 | 71% | |
| CpG island | 164 | 4e-90 | 60% | |
| Unsupported | None | 2,666 | - | 83.4% |
Number of TSS clusters with this support, excluding TSSs from higher categories.
Probability of overlap between the transcript support and the Pet/CAGE tags, as calculated by the Genome Structure Correction statistic (see Supplementary Information section S1.3).
Percent of clusters with only one tag. For the Known category this was calculated as the percent of GENCODE 5′ ends with Tag support (25%) or overall (74%).
Correlating genomic features with chromatin structure and transcription factor binding
By measuring relative sensitivity to DNaseI digestion (see Supplementary Information section S3.3), we identified DNaseI Hypersensitive Sites (DHSs) throughout the ENCODE regions. DHSs and TSSs both reflect genomic regions thought to be enriched for regulatory information and many DHSs reside at or near TSSs. We partitioned DHSs into those within 2.5kb of a TSS (958; 46.5%) and remaining classified as distal (1,102; 53.5%). We then cross-analysed the TSSs and DHSs with datasets relating to histone modifications, chromatin accessibility, and sequence-specific transcription-factor binding by summarising these signals in aggregate relative to the distance from TSSs or DHSs. Figure 5 shows representative profiles of specific histone modifications, PolII, and selected transcription factor binding for the different categories of TSSs. Further profiles and statistical analysis of these studies can be found in Supplementary Information section S3.6.
Figure 5.
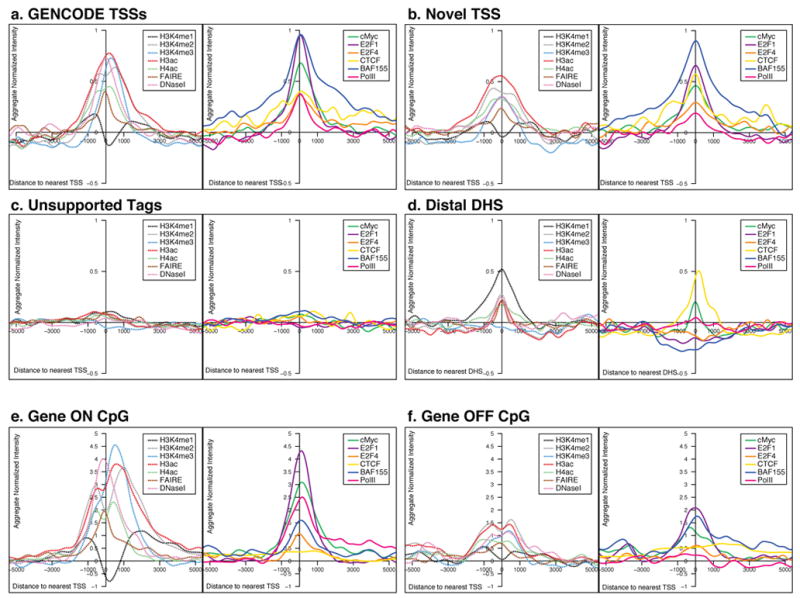
Aggregate signals of tiling-array experiments from either ChIP-chip or chromatin structure assays, represented for different classes of TSS and DHS. For each plot, the signal was first normalised with a mean of 0 and standard deviation of 1, and then the normalised scores were summed at each position for that class of TSS or DHS and smoothed using a kernel density method (see Supplementary Information section S3.6). For each class of sites there are two adjacent plots. The left hand plot depicts the data for general factors: FAIRE and DNaseI sensitivity as assays of chromatin accessibility and H3K4me1, H3K4me2, H3K4me3, H3ac, and H4ac histone modifications (as indicated); the right hand plot shows the data for additional factors, namely cMyc, E2F1, E2F4, CTCF, BAF155, and PolII. The columns provide data for the different classes of TSS class or DHS (unsmoothed data and statistical analysis shown in Supplementary Information section S3.6).
In the case of the three TSS categories (Known, Novel, and Unsupported), Known and Novel TSSs are both associated with similar signals for multiple factors (ranging from histone modifications through DNaseI accessibility), whereas Unsupported TSSs are not. The enrichments seen with chromatin modifications and sequence-specific factors, along with the significant clustering of this evidence, indicate that the Novel TSSs do not reflect false positives and likely utilise the same biological machinery as other promoters. Sequence-specific transcription factors show a marked increase in binding across the broad region that encompasses each TSS. This increase is notably symmetric, with binding equally likely upstream or downstream of a TSS (see Supplementary Information section S3.7 for an explanation of why this symmetrical signal is not an artefact due to the analysis of the signals). Further, there is enrichment of BAF155 binding (a member of the swi/snf chromatin-modifying complex), which persists across a broader extent than other factors. The broad signals with this factor indicate that the ChIP-chip results reflect both specific enrichment at the TSS and broader enrichments across ~5kb regions; (this is not due to technical issues, see Supplementary Information section S3.8).
We selected 577 GENCODE-defined TSSs at the 5′ ends of a protein-coding transcript with over 3 exons to assess expression status. Each transcript was classified as: (1) ’active’ (gene on) or ‘inactive’ (gene off) based on the unbiased transcript surveys, and (2) residing near a ‘CpG island’ or not near a CpG island (‘non-CpG island’) (see Supplementary Information section S3.17). As expected, the aggregate signal of histone modifications is mainly attributable to active TSSs (Figure 5), in particular those near CpG islands. Pronounced doublet peaks at the TSS can be seen with these large signals, similar to previous work in yeast48, due to the chromatin accessibility at the TSS. Many of the histone marks and PolII signals are now clearly asymmetrical, with a persistent level of PolII into the genic region, as expected. However, the sequence specific factors remain largely symmetrically distributed. TSSs near CpG islands show a broader distribution of histone marks than those not near CpG islands (see Supplementary Information section S3.6). The binding of some transcription factors (E2F1, E2F4, and cMyc) is extensive in the case of active genes, and is lower (or absent) in the case of inactive genes.
Chromatin signature of distal elements
Distal DHSs show characteristic patterns of histone modification that are the inverse of TSSs, with high H3K4me1 accompanied by lower levels of H3K4Me3 and H3Ac (Figure 5). Many factors with high occupancy at TSSs (for example, E2F4) show little enrichment at distal DHSs, whereas other factors (for example, cMyc) are enriched at both TSSs and distal DHSs49. A particularly interesting observation is the relative enrichment of the insulator-associated factor CTCF50 at both distal DHSs and TSSs; this contrasts with swi/snf components BAF170 and BAF155, which are TSS-centric. Such differential behaviour of sequence-specific factors points to distinct biological differences, mediated by transcription factors, between distal regulatory sites and TSSs.
Unbiased maps of sequence-specific regulatory factor binding
The previous section focused on specific positions defined by TSSs or DHSs. We then analysed sequence-specific transcription factor-binding data in an unbiased fashion. We refer to regions with enriched binding of regulatory factors as “Regulatory Factor-Binding Regions” (RFBRs). RFBRs were identified based on ChIP-chip data in two ways: first, each investigator developed and used their own analysis method(s) to define high-enrichment regions, and second (and independently), a stringent False Discovery Rate (FDR) method was applied to analyse all data using three cut-offs (1%, 5%, and 10%). The laboratory-specific and FDR-based methods were highly correlated, particularly for regions with strong signals10, 11. For consistency, we used the results obtained using the FDR-based method (see Supplementary Information section S3.10). These RFBRs can be used to find sequence motifs (see Supplementary Information section S3.11).
Many RFBRs are associated with the 5′ ends of transcripts
The distribution of RFBRs is non-random (see Zhang et al10) and correlates with the positions of TSSs. We examined the distribution of specific RFBRs relative to the Known TSSs. Different transcription factors and histone modifications vary with respect to their association with TSSs (Figure 6; see Supplementary Information section S3.12 for modelling random expectation). Factors whose binding sites are most enriched at the 5′ ends of genes include histone modifications, TAF1, and RNA PolII with hypo-phosphorylated C terminal domain51— confirming previous expectations. Surprisingly, we found that E2F1, a sequence-specific factor that regulates the expression of many genes at the G1 to S transition52, is also tightly associated with TSSs52; this association is as strong as that of TAF1, the well-known TATA box-binding protein associated factor 153. These results suggest that E2F1 plays a more general role in transcription than previously suspected, similar to that for cMyc54–56. for which the large-scale assays did not support the promoter binding that was found in smaller-scale studies (for example, on SIRT1 and SPI1 (PU1)).
Figure 6.
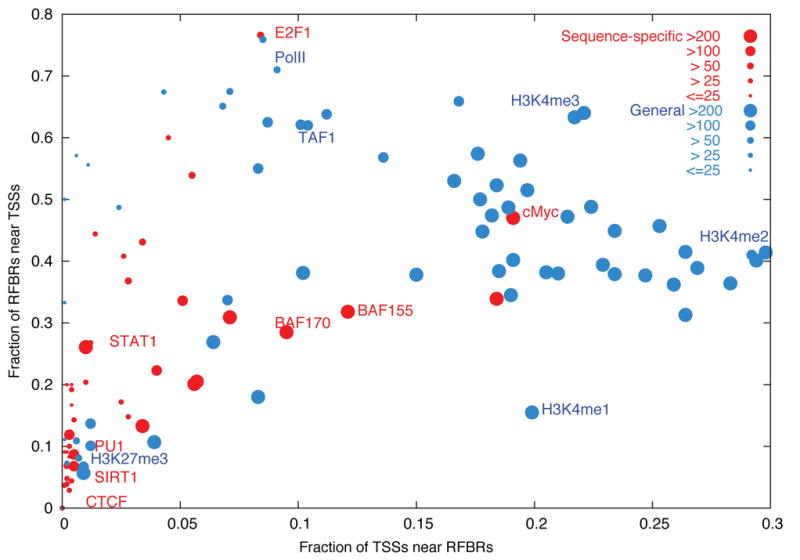
Distribution of RFBRs relative to GENCODE TSSs. Different RFBRs from Sequence Specific factors (Red) or general factors (Blue) are plotted showing their relative distribution near TSSs. The x-axis indicates the proportion of TSSs close (within 2.5KB) to the specified factor. The y-axis indicates the proportion of RFBRs close to TSSs. The size of the circle provides an indication of the number of RFBRs for each factor. A handful of representative factors are labelled.
Integration of data on sequence-specific factors
We expect that regulatory information is not dispersed independently across the genome, but rather is clustered into distinct regions57. We refer to regions that contain multiple regulatory elements as “Regulatory Clusters.” We sought to predict the location of regulatory clusters by cross-integrating data generated using all transcription-factor and histone-modification assays, including results falling below an arbitrary threshold in individual experiments. Specifically, we used four complementary methods to integrate the data from 129 ChIP-chip datasets (see Supplementary Information section S3.13 and Trinklein et al58). These four methods detect different classes of regulatory clusters and as a whole identified 1,393 clusters. Of these, 344 were identified by all four methods, with another 500 found by three methods (see Supplementary Information section S3.13.5). 67% of the 344 regulatory clusters identified by all four methods (or 65% of the full set of 1,393) reside within 2.5 kb of a Known or Novel TSS (as defined above; see Table 3 and Supplementary Information section S3.14 for a breakdown by category). Restricting this analysis to previously-annotated TSSs (for example, RefSeq or Ensembl) reveals that roughly 25% of the regulatory clusters are close to a previously identified TSS. These results suggest that many of the regulatory clusters identified by integrating the ChIP-chip datasets are undiscovered promoters or are somehow associated with transcription in another fashion. To test these possibilities, sets of 126 and 28 non-GENCODE-based regulatory clusters were tested for promoter activity (see Supplementary Information section S3.15) and by RACE, respectively. These studies revealed that 24.6% of the 126 tested regulatory clusters had promoter activity and that 78.6% of the 28 regulatory clusters analyzed by RACE yielded products consistent with a TSS58. The ChIP-chip datasets were generated on a mixture of cell lines, predominantly HeLa and GM06990, and different from the CAGE/diTag data, meaning that tissue specificity contribute to the presence unique TSSs and regulatory clusters. The large increase in promoter-proximal regulatory clusters identified by including the additional Novel TSSs coupled with the positive promoter and RACE assays suggests that most of the regulatory regions identifiable by these clustering methods represent bona fide promoters (see Supplementary Information section S3.16). Although the regulatory-factor assays were more biased towards regions associated with promoters without the more extensive TSS dataset, many of the sites from these experiments would have previously been described as distal to promoters. This suggests that commonplace use of RefSeq or Ensembl based gene definition to define promoter proximity and distal will dramatically over estimate the number of distal sites.
Predicting TSSs and transcriptional activity based on chromatin structure
The strong association between TSSs and both histone modifications and DHSs prompted us to investigate whether the location and activity of TSSs could be predicted based solely on chromatin-structure information. We trained a Support Vector Machine (SVM) by using histone-modification data anchored around DHSs to discriminate between DHSs near TSSs and those distant from TSSs. We used a selected 2,573 DHSs, split roughly between TSS-proximal DHSs and TSS-distal DHSs, as a training set. The SVM performed well, with an accuracy of 83% (see Supplementary Information section S3.17). Using this SVM, we then predicted new TSSs using information about DHSs and histone modifications— of 110 high-scoring predicted TSSs, 81 resided within 2.5 kb of a Novel TSS. As expected, these show a significant overlap to the novel TSS groups (defined above) but without a strong bias towards any particular category (see Supplementary Information section 3.17.1.5).
To investigate the relationship between chromatin structure and gene expression, we examined transcript levels in two cell lines using a transcript-tiling array. We compared this transcript data with the results of ChIP-chip experiments that measured histone modifications across the ENCODE regions. From this, we developed a variety of predictors of expression status using chromatin modifications as variables; these were derived using both Decision Trees and SVMs (see Supplementary Information section S3.17). The best of these correctly predicts expression status (transcribed vs. non-transcribed) in 91% of cases. This success rate did not decrease dramatically when the predicting algorithm incorporated the results from one cell line to predict the expression status of another cell line. Interestingly, despite the striking difference in histone-modification enrichments in TSSs residing near versus those more distal to CpG islands (see Figure 5 and Supplementary Information section S3.6), including information about the proximity to CpG islands did not improve the predictors. This suggests that despite the marked differences in histone modifications among these TSS classes, a single predictor can be made, using the interactions between the different histone modification levels.
In summary, we have integrated many datasets to provide a more complete view of regulatory information, both around specific sites (TSSs and DHSs) and in an unbiased manner. Based on analysing multiple datasets, we find 4,491 Known and Novel TSSs in the ENCODE regions, almost ten-fold more than the number of established genes. This large number of TSSs might explain the extensive transcription described above; it also begins to change our perspective about regulatory information – without such a large TSS catalogue, many of the regulatory clusters would have been classified as residing distal to promoters. In addition to this revelation about the abundance of promoter-proximal regulatory elements, we also identified a considerable number of putative distal regulatory elements, particularly based on the presence of DHSs. Our study of distal regulatory elements was probably most hindered by the paucity of data generated using distal element-associated transcription factors; nevertheless, we clearly detected a set of distal DHS-associated segments bound by CTCF or cMyc. Finally, we showed that information about chromatin structure alone could be used to make effective predictions about both the location and activity of TSSs.
Replication
Overview
DNA replication must be carefully coordinated, both across the genome and with respect to development. On a larger scale, early replication in S phase is broadly correlated with gene density and transcriptional activity59–66; however, this relationship is not universal, as some actively transcribed genes replicate late and vice versa61, 64–68. Importantly, the relationship between transcription and DNA replication emerges only when the signal of transcription is averaged over a large window (>100 kb)63, suggesting that larger-scale chromosomal architecture may be more important than the activity of specific genes69.
The ENCODE Project provided an unique opportunity to examine whether individual histone modifications on human chromatin can be the correlated with the time of replication and whether such correlations support the general relationship of active, open chromatin with early replication. Our studies also tested whether segments showing interallelic variation in time of replication have two different types of histone modifications consistent with an interallelic variation in chromatin state.
Experimental DNA-replication dataset
We mapped replication timing across the ENCODE regions by analysing Brd-U-labelled fractions from synchronised HeLa cells (collected at 2-hour intervals throughout S-phase) on tiling arrays (see Supplementary Information section 4.1). Although the HeLa cell line has a considerably altered karytope, correlation of this data with other cell line data (see below) suggests the results are relevant to other cell types. The results are expressed as the time at which 50% of any given genomic position is replicated (TR50), with higher values signifying later replication times. In addition to the five ‘activating’ histone marks, we also correlated the TR50 with H3K27me3, a modification associated with polycomb-mediated transcriptional repression70–74. To provide a consistent comparison framework, the histone data was smoothed to 100 kb, and then correlated with the TR50 data by a sliding window correlation analysis (see Supplementary Information section S4.2). The continuous profiles of the activating marks, histone H3K4 mono-, di-, and tri-methylation and histone H3 and H4 acetylation, are generally anti-correlated with the TR50 signal (Figure 7a and Supplementary Information section S4.3). In contrast, H3K27me3 marks show a predominantly positive correlation with late-replicating segments (Figure 7a; see Supplementary Information section 4.3 for additional analysis).
Figure 7.
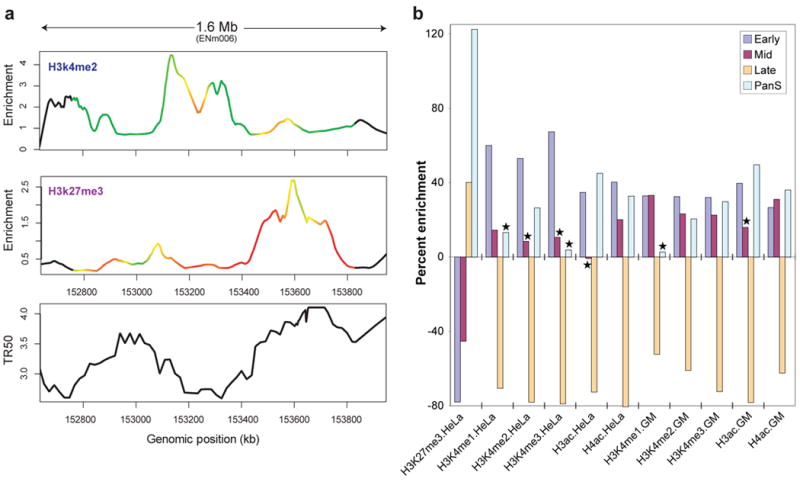
Correlation between replication timing and histone modifications. (a) Comparison of two histone modifications (H3K4me2 and H3K27me3), plotted as enrichment ratio from the Chip-chip experiments and the time for 50% of the DNA to replicate (TR50), indicated for ENCODE region ENm006. The colours on the curves reflect the correlation strength in a sliding 250 kb window. (b) Differing levels of histone modification for different TR50 partitions. The amounts of enrichment or depletion of different histone modifications in various cell lines are depicted (indicated along the bottom as ‘Histone mark.Cell line’; GM= GM06990). Asterisks indicate enrichments/depletions that are not significant based on multiple tests. Each set has four partitions based on replication timing: Early, Mid, Late, and PanS.
While most genomic regions replicate in a temporally specific window in S phase other regions demonstrate an atypical pattern of replication (Pan-S) where replication signals are seen in multiple parts of S phase. We have suggested that such a pattern of replication stems from interallelic variation in the chromatin structure59, 75. If one allele is in active chromatin and the other in repressed chromatin, both types of modified histones are expected to be enriched in the Pan-S segments. An ENCODE region was classified as non-specific (or Pan-S) regions when >60% of the probes in a 10 kb window replicated in multiple intervals in S phase. The remaining, regions were sub-classified into early, mid, or late replicating based on the average TR50 of the temporally-specific probes within a 10kb window75. For regions of each class of replication timing, we determined the relative enrichment of various histone-modification peaks in HeLa cells (Figure 7b; Supplemental material S4.4). The correlations of activating and repressing histone-modification peaks with TR50) are confirmed by this analysis (Figure 7b). Intriguingly, the Pan-S segments are unique in being enriched for both activating (H3K4me2, H3ac, and H4ac) and repressing (H3K27me3) histones, consistent with the suggestion that the Pan-S replication pattern arises from interallelic variation in chromatin structure and time of replication75. This observation is also consistent with the Pan-S replication pattern seen for the H19/IGF2 locus, a known imprinted region with differential epigenetic modifications across the two alleles76.
The extensive rearrangements in the genome of HeLa cells led us to ask whether the detected correlations between TR50 and chromatin state are seen with other cell lines. The histone-modification data with GM06990 cells allowed us to test whether the time of replication of genomic segments in HeLa cells correlated with the chromatin state in GM06990 cells. Early- and late-replicating segments in HeLa cells are enriched and depleted, respectively, for activating marks in GM06990 cells (Figure 7b). Thus, despite the presence of genomic rearrangements (see Supplementary Information section S2.12), the TR50 and chromatin state in HeLa cells are not far from a constitutive baseline also seen with a cell line from a different lineage. The enrichment of multiple activating histone modifications and the depletion of a repressive modification from segments that replicate early in S phase extends previous work in the field at a level of detail and scale not attempted before in mammalian cells. The duality of histone modification patterns in Pan-S areas of the HeLa genome, and the concordance of chromatin marks and replication time across two disparate cell lines (HeLa and GM06990) show the coordination of histone modifications with replication in the human genome.
Chromatin architecture and genomic domains
Overview
The packaging of genomic DNA into chromatin is intimately connected with the control of gene expression and other chromosomal processes. We next examined chromatin structure over a larger scale to ascertain its relation to transcription and other processes. Large domains (50 to >200 kb) of generalised DNaseI sensitivity have been detected around developmentally-regulated gene clusters77, prompting speculation that the genome is organised into ‘open’ and ‘closed’ chromatin territories that represent higher-order functional domains. We explored how different chromatin features, particularly histone modifications, correlate with chromatin structure, both over short and long distances.
Chromatin accessibility and histone modification
We used histone modification studies and DNaseI sensitivity datasets introduced above to examine general chromatin accessibility without focusing on the specific DHS sites (see Supplementary Information sections S3.1, S3.3, and S3.4). A fundamental difficulty in analysing continuous data across large genomic regions is determining the appropriate scale for analysis (e.g, 2 kb, 5 kb, 20 kb, etc.). To address this problem, we developed an approach based on wavelet analysis, a mathematical tool pioneered in the field of signal processing that has recently been applied to continuous-value genomic analyses. Wavelet analysis provides a means for consistently transforming continuous signals into different scales, enabling the correlation of different phenomena independently at differing scales in a consistent manner.
Global correlations of chromatin accessibility and histone modifications
We computed the local correlation between DNaseI sensitivity and each histone modification at multiple scales using a wavelet approach (Figure 8 and Supplementary Information section S4.2). To make quantitative comparisons between different histone modifications, we computed histograms of correlation values between DNaseI sensitivity and each histone modification at several scales and then tested these for significance at specific scales. Figure 8c shows the distribution of correlation values at a 16-kb scale, which is considerably larger than individual cis-acting regulatory elements. At this scale, H3K4me2, H3K4me3, and H3ac show similarly high correlation. However, they are significantly distinguished from H3K4me1 and H4ac modifications (P<1.5 ×10-33; see Supplementary Information section S4.5), which show lower correlation with DNaseI sensitivity. These results suggest that larger-scale relationships between chromatin accessibility and histone modifications are dominated by sub-regions in which higher average DNaseI sensitivity is accompanied by high levels of H3K4me2, H3K4me3, and H3ac modifications.
Figure 8.
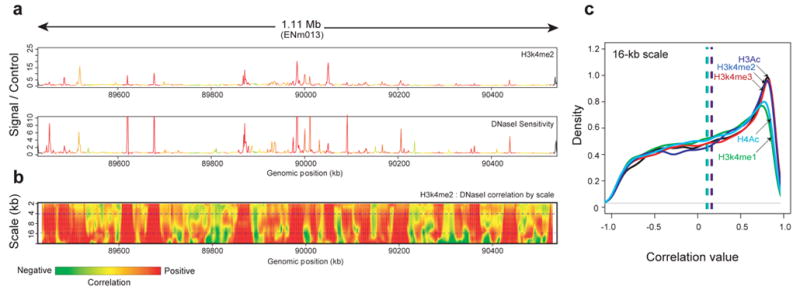
Wavelet correlations of histone marks and DNaseI sensitivity. As an example, correlations between DNaseI sensitivity and H3K4me2 (both in the GM06990 cell line) over a 1.1-Mb region on chromosome 7 (ENCODE region ENm013) are shown. (a) The relationship between histone modification H3K4me2 (upper plot) and DNaseI sensitivity (lower plot) is shown for ENCODE region ENm013. The curves are coloured with the strength of the local correlation at the 4-kb scale (top dashed line in panel b). (b) The same data as in a are represented as a wavelet correlation. The y-axis shows the differing scales decomposed by the wavelet analysis from large to small scale (in kb); the colour at each point in the heatmap represents the level of correlation at the given scale, measured in a 20-kb window centered at the given position. (c) Distribution of correlation values at the 16-kb scale between the indicated histone marks and. The x-axis shows different correlation values. The Y-axis is the density of these correlation values across ENCODE; all modifications show a peak at a positive-correlation value.
Local correlations of chromatin accessibility and histone modifications
Narrowing to a scale of ~2 kb revealed a more complex situation, in which H3K4me2 is the histone modification that is best correlated with DNaseI sensitivity. However, there is no clear combination of marks that correlate with DNaseI sensitivity in a way that is analogous to that seen at a larger scale (Supplementary Information section S4.3). One explanation for the increased complexity at smaller scales is that there is a mixture of different classes of accessible chromatin regions, each having a different pattern of histone modifications. To examine this, we computed the degree to which local peaks in histone methylation or acetylation occur at DHSs (see Supplementary Information section S4.5.1). We found that 84%, 91%, and 93% of significant peaks in H3K4 mono-, di-, and tri-methylation, respectively, and 93% and 81% of significant peaks in H3ac and H4ac acetylation, respectively, coincided with DHSs (see Supplementary Information section S4.5). Conversely, a proportion of DHSs seemed not to be associated with significant peaks in H3K4 mono-, di-, or tri-methylation (37%, 29%, and 47%, respectively), nor with peaks in H3 or H4 acetylation (both 57%). Because only a limited number of histone modification marks were assayed, the possibility remains that some DHSs harbour other histone modifications. The absence of a more-complete concordance between DHSs and peaks in histone acetylation is surprising given the widely accepted notion that histone acetylation plays a central role in mediating chromatin accessibility by disrupting higher-order chromatin folding.
DNA structure at DHSs
The observation that distinctive hydroxyl radical-cleavage patterns are associated with specific DNA structures78 prompted us to investigate whether DHS subclasses differed with respect to their local DNA structure. Conversely, because different DNA sequences can give rise to similar hydroxyl radical-cleavage patterns79, genomic regions that adopt a particular local structure do not necessarily have the same nucleotide sequence. Using a Gibbs-sampling algorithm on hydroxyl radical cleavage patterns of 3,150 DHSs80, we discovered an 8-base segment with a conserved cleavage signature (CORCS; see Supplementary Information section S4.6). The underlying DNA sequences that give rise to this pattern have little primary sequence similarity despite this similar structural pattern. Further, this structural element is strongly enriched in promoter-proximal DHSs (11.3-fold enrichment compared to the rest of the ENCODE regions) relative to promoter-distal DHSs (1.5-fold enrichment); this element is enriched 10.9 fold in CpG islands, but is higher still (26.4 fold) in CpG islands that overlap a DHS.
Large-scale domains in the ENCODE regions
The presence of extensive correlations seen between histone modifications, DNaseI sensitivity, replication, transcript density, and protein factor-binding led us to investigate whether all these features are organised systematically across the genome. To test this, we performed an unsupervised training of a two-state hidden Markov model (HMM) with inputs from these different features (see Supplementary Information section S4.7 and Thurman et al81). No other information except for the experimental variables was used for the HMM-training routines. We consistently found that one state (‘active’) generally corresponded to domains with high levels of H3ac and RNA transcription, low levels of H3K27me3 marks, and early replication timing, whereas the other state (‘repressed’) reflected domains with low H3ac and RNA, high H3K27me3, and late replication. (See Figure 9.) In total, we identified 70 active regions spanning 11.4 Mb and 82 inactive regions spanning 17.8 Mb (median size 136 kb vs 104 kb respectively). The active domains are markedly enriched for GENCODE TSSs, CpG islands and Alu repetitive elements (P<0.0001 for each), while repressed regions are significantly enriched for LINE1 and LTR transposons (P<0.001). Taken together, these results demonstrate remarkable concordance between ENCODE functional data types and provide a view of higher-order functional domains defined by a broader range of factors at markedly higher resolution than previously available82.
Figure 9.
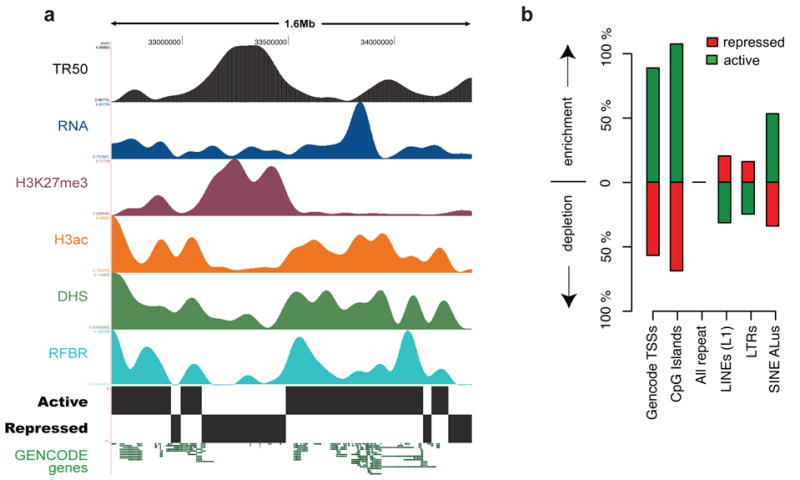
Higher-order functional domains in the genome. The general concordance of multiple data types is illustrated for an illustrative ENCODE region (ENm005). (a) Domains were determined by simultaneous HMM segmentation of replication time (TR50; black), bulk RNA transcription (blue), H3K27me3 (purple), H3ac (orange), DHS density (green), and RFBR density (light blue) measured continuously across the 1.6-Mb ENm005. All data were generated using HeLa cells. The histone, RNA, DHS, and RFBR signals are wavelet-smoothed to an approximately 60 kb scale (see Supplementary Information section S4.7). The HMM segmentation is shown as the blocks labeled “active” and “repressed” and the structure of GENCODE genes (not used in the training) is shown at the end. (b) Enrichment or depletion of annotated sequence features (GENCODE TSSs, CpG islands, different types of repetitive elements, and non-exonic CSs) in active versus repressed domains. Note the marked enrichment of TSSs, CpG islands, and Alus in active domains, and the enrichment of LINE and LTRs in repressed domains.
Evolutionary and population-genetic insights into genome function
Overview
Functional genomic sequences can also be identified by examining evolutionary changes across multiple extant species and within the human population. Indeed, such studies complement experimental assays that identify specific functional elements83–85. Evolutionary constraint (i.e., the rejection of mutations at a particular location) can be measured by either (i) comparing observed substitutions to neutral rates calculated from multi-sequence alignments 86–88, or (ii) determining the presence and frequency of intra-species polymorphisms. Importantly, both approaches are indifferent to any specific function that the constrained sequence might confer.
Previous studies comparing the human, mouse, rat, and dog genomes examined bulk evolutionary properties of all nucleotides in the genome, and provided little insight about the precise positions of constrained bases. Interestingly, these studies indicated that the majority of constrained bases reside within the non-coding portion of the human genome. Meanwhile, increasingly rich datasets of polymorphisms across the human genome have been used extensively to establish connections between genetic variants and disease, but far fewer analyses have sought to use such data for assessing functional constraint85.
The ENCODE Project provides an excellent opportunity for more fully exploiting inter- and intra-species sequence comparisons to examine genome function in the context of extensive experimental studies on the same regions of the genome. We consolidated the experimentally-derived information about the ENCODE regions and focused our analyses on 11 major classes of genomic elements. These classes are listed in Table 4 and include two non-experimentally-derived datasets: Ancient Repeats (ARs; mobile elements that inserted early in the mammalian lineage, have subsequently become dormant, and are assumed to be neutrally evolving) and Constrained Sequences (CSs; regions that evolve detectably more slowly than neutral sequences).
Table 4.
Eleven classes of genomic elements subjected to evolutionary and population-genetics analyses.
| Abbreviation | Description |
|---|---|
| CDS | Coding exons, as annotated by GENCODE |
| 5′UTR | 5′ Untranslated region, as annotated by GENCODE |
| 3′ UTR | 3′ Untranslated region, as annotated by GENCODE |
| Un.TxFrag | Unannotated region detected by RNA hybridisation to tiling array (i.e., unannotated TxFrag) |
| RxFrag | Region detected by RACE and analysis on tiling array |
| Pseudogene | Pseudogene identified by consensus pseudogene analysis |
| RFBR | Regulatory Factor Binding Region identified by ChIP-chip assay |
| RFBR-SeqSp | Regulatory Factor Binding Region identified only by ChIP-chip assays for factors with known sequence-specificity |
| DHS | DNaseI hypersensitive sites found in multiple tissues |
| FAIRE | Region of open chromatin identified by the FAIRE assay |
| TSS | Transcription start site |
| AR | Ancient repeat inserted early in the mammalian lineage and presumed to be neutrally evolving |
| CS | Constrained sequence identified by analysing multi-sequence alignments |
Comparative sequence analysis
We generated 206 Mb of genomic sequence orthologous to the ENCODE regions from 14 mammalian species using a targeted strategy that involved isolating89 and sequencing90 individual bacterial artificial-chromosome (BAC) clones. For an additional 14 vertebrate species, we utilised 340 Mb of orthologous genomic sequence derived from genome-wide sequencing efforts3–8, 91–93. The orthologous sequences were aligned using three alignment programs: TBA94, MAVID95, and MLAGAN96. Four independent methods that generated highly concordant results97 were then used to identify sequences under constraint (PhastCons88, GERP87, SCONE98, and BinCons86). From these analyses, we developed a high-confidence set of ‘constrained sequences’ that correspond to 4.9% of the nucleotides in the ENCODE regions. The threshold for determining constraint was set using a fixed false discovery (FDR) rate of 5% (see Margulies et al97); this level is similar to previous estimates of the fraction of the human genome under mammalian constraint4, 86–88 but the FDR rate was not chosen to fit this result. The median length of these constrained sequences is 19 bases, with the minimum being 8 bases – roughly the size of a typical transcription factor-binding site. These analyses, therefore, provide a resolution of constrained sequences that is substantially better than that currently available using only whole-genome vertebrate sequences99–102.
Intra-species variation studies mainly used single-nucleotide polymorphism (SNP) data from Phases I and II, and the 10 resequenced regions in ENCODE regions with 48 individuals of the HapMap Project103, nucleotide insertion or deletion (indel) data were from the SNP Consortium and HapMap. We also examined the ENCODE regions for the presence of overlaps with known segmental duplications104 and copy-number variants (CNVs).
Experimentally-identified functional elements and constrained sequences
We first compared the detected constrained sequences with the positions of experimentally-identified functional elements. A total of 40% of the constrained bases reside within protein-coding exons and their associated untranslated regions (Figure 10) and, in agreement with previous genome-wide estimates, the remaining constrained bases do not overlap the mature transcripts of protein-coding genes4, 5, 88, 105, 106. When we included the other experimental annotations, we found that an additional 20% of the constrained bases overlap experimentally-identified non-coding functional regions, although far fewer of these regions overlap constrained sequences compared to coding exons (see below). Most experimental annotations are significantly different from a random expectation for both base-pair or element-level overlaps (using the GSC statistic, see Supplementary Information section S1.3), with a more striking deviation when considering elements (Figure 11). The exceptions to this are pseudogenes, Un.TxFrags, and RxFrags. The increase in significance moving from base-pair measures to the element level suggests that discrete islands of constrained sequence exist within experimentally-identified functional elements, with the surrounding bases apparently not showing evolutionary constraint. This notion is discussed in greater detail in Margulies et al97.
Figure 10.
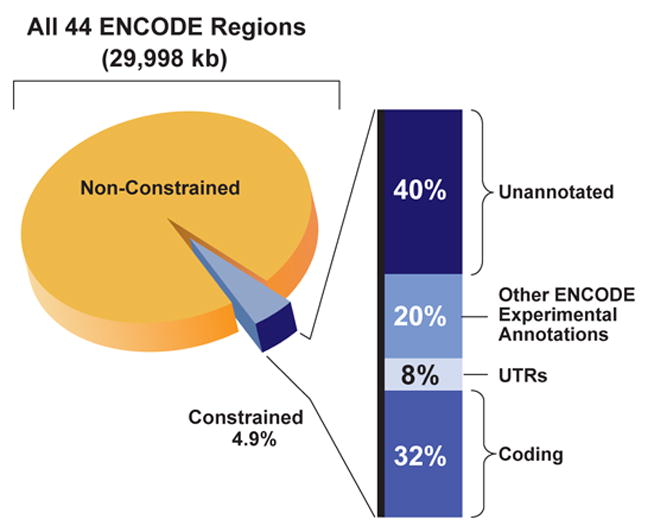
Relative proportion of different annotations among constrained sequences. The 4.9% of bases in the ENCODE regions identified as constrained is subdivided into the portions that reflect known coding regions, UTRs, other experimentally-annotated regions, and unannotated sequence.
Figure 11.
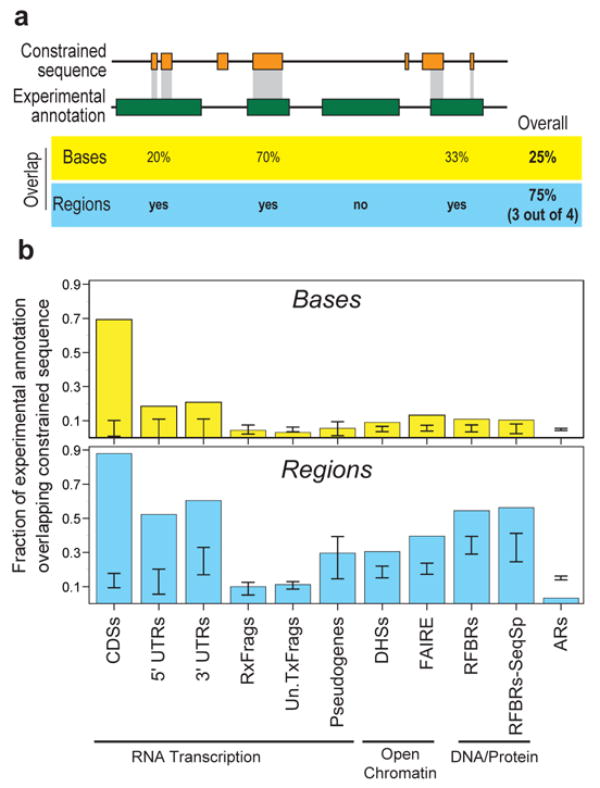
Overlap of constrained sequences and various experimental annotations. (a) A schematic depiction shows the different tests used for assessing overlap between experimental annotations and constrained sequences, both for individual bases and for entire regions. (b) Observed fraction of overlap, depicted separately for bases and regions. The results are shown for selected experimental annotations. The internal bars indicate 95% confidence intervals of randomised placement of experimental elements using the GSC methodology to account for heterogeneity in the datasets. When the bar overlaps the observed value one cannot reject the hypothesis that these overlaps are consistent with random placements.
We also examined measures of human variation (heterozygosity, derived allele-frequency spectra, and indel rates) within the sequences of the experimentally-identified functional elements (Figure 12). For these studies, ARs were used as a marker for neutrally evolving sequence. Most experimentally-identified functional elements are associated with lower heterozygosity compared to ARs, and a few have lower indel rates compared to ARs. Striking outliers are 3′ UTRs, which have dramatically increased indel rates without an obvious cause. This is discussed in more depth in Clark et al107.
Figure 12.
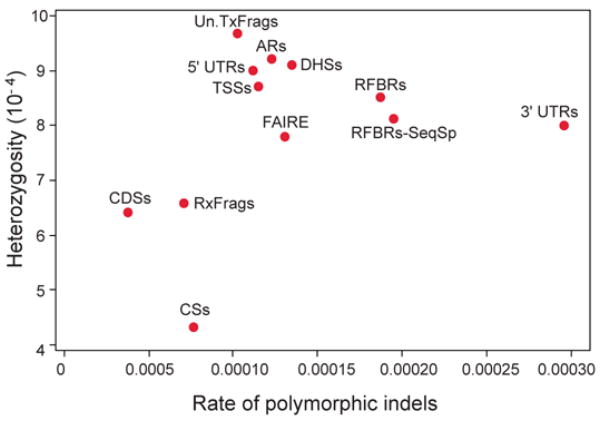
Relationship between heterozygosity and polymorphic indel rate for a variety of experimental annotations.. 3′UTRs are an expected outlier for the indel measures due to the presence of low-complexity sequence (leading to a higher indel rate).
These findings indicate that the majority of the evolutionarily-constrained, experimentally-identified functional elements show evidence of negative selection both across mammalian species and within the human population. Furthermore, we have assigned at least one molecular function to the majority (60%) of all constrained bases in the ENCODE regions.
Conservation of regulatory elements
The relationship between individual classes of regulatory elements and constrained sequences varies considerably, ranging from cases where there is strong evolutionary constraint (for example, pan-vertebrate ultraconserved regions108, 109) to examples of regulatory elements that are not conserved between orthologous human and mouse genes110. Within the ENCODE regions, 55% of RFBRs overlap the high-confidence constrained sequences. As expected, RFBRs have many unconstrained bases, presumably due to the small size of the specific binding site. We investigated whether the binding sites in RFBRs could be further delimited using information about evolutionary constraint. For 7 of 17 factors with either known TRANSFAC or Jaspar motifs, our ChIP-chip data revealed a marked enrichment of the appropriate motif within the constrained versus the unconstrained portions of the RFBRs (see Supplementary Information section S5.1). This enrichment was seen for at levels of stringency used for defining ChIP-chip-positive sites (1% and 5% FDR level), indicating that combining sequence constraint and ChIP-chip data may provide a highly sensitive means for detecting factor-binding sites in the human genome.
Genetic variation and experimentally-identified functional elements
The above studies focus on purifying (negative) selection. We used nucleotide variation to detect potential signals of adaptive (positive) selection. We modified the standard McDonald-Kreitman test (MK-test111, 112) and the Hudson-Kreitman-Aguade (HKA)113 test (Supplementary Information section S5.2.1), to examine whether an entire set of sequence elements shows an excess of polymorphisms or an excess of inter-species divergence. We found that constrained sequences and coding exons have an excess of polymorphisms (consistent with purifying selection), while 5′UTRs show evidence of an excess of divergence (with a portion likely reflecting positive selection). In general, non-coding genomic regions show more variation, with both a large number of segments that undergo purifying selection and regions that are fast evolving.
We also examined structural variation (i.e., CNVs, inversions, and translocations114; Supplementary Information section S5.2.2). Within these polymorphic regions, we encountered significant overrepresentation of CDSs, TxFrags, and intra-species constrained sequences (P<10−3, Figure 13), and also detected a statistically significant under-representation of ARs (P=10−3). A similar over-representation of CDSs and intra-species constrained sequences was found within non-polymorphic segmental duplications.
Figure 13.
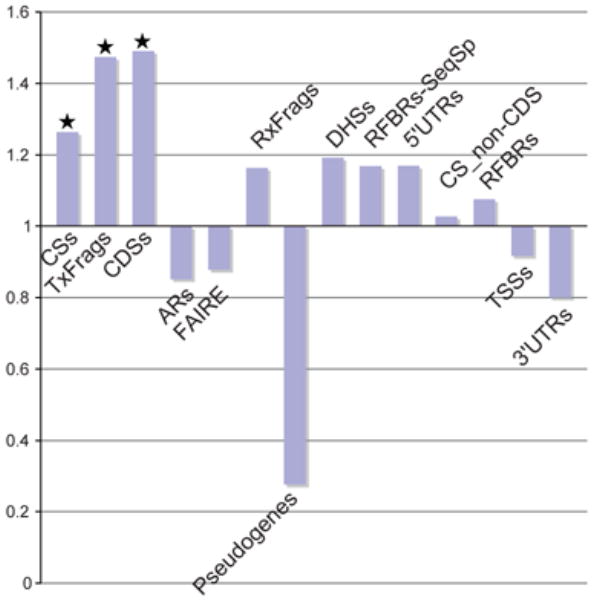
CNV enrichment. The relative enrichment of different experimental annotations in ENCODE regions associated with CNVs. CS_non-CDS are constrained sequences outside of coding regions. A value of 1 or less indicates no enrichment, and values greater than 1 show enrichment. Starred columns are cases that are significant based on this enrichment being found in less than 5% of randomisations which matched each element class for length and density of features.
Unexplained constrained sequences
Despite the wealth of complementary data, 40% of the ENCODE-region sequences identified as constrained are not associated with any experimental evidence of function. There no evidence indicating that mutational cold spots account for this constraint; they have similar measures of constraint to experimentally-identified elements and harbour equal proportions of SNPs. To further characterise the unexplained constrained sequences, we examined their clustering and phylogenetic distribution. These sequences are not uniformly distributed across most ENCODE regions, and even in most ENCODE regions the distribution is different from constrained sequences within experimentally-identified functional elements (see Supplementary Information section S5.3). The large fraction of constrained sequence that does not match any experimentally-identified elements is not surprising considering that only a limited set of transcription factors, cell lines, and biological conditions have thus far been examined.
Unconstrained experimentally-identified functional elements
In contrast, an unexpectedly large fraction of experimentally-identified functional elements show no evidence of evolutionary constraint ranging from 93% for Un.TxFrags to 12% for CDS. For most types of non-coding functional elements, roughly 50% of the individual elements appeared to be unconstrained across all mammals.
There are two methodological reasons that might explain the apparent excess of unconstrained experimentally-identified functional elements: the underestimation of sequence constraint or overestimation of experimentally-identified functional elements. We do not believe that either of these explanations fully accounts for the large and varied levels of unconstrained experimentally functional sequences. The set of constrained bases analysed here is highly accurate and complete due to the depth of the multiple alignment. Both by bulk fitting procedures and by comparison of SNP frequencies to constraint there is clearly a proportion of constrained bases not captured in the defined 4.9% of constrained sequences, but it is small (see Supplementary Information section S5.4 and S5.5). More aggressive schemes to detect constraint only marginally increase the overlap with experimentally-identified functional elements, and do so with considerably less specificity. Similarly, all experimental findings have been independently validated and, for the least constrained experimentally-identified functional elements (Un.TxFrags and binding sites of sequence-specific factors), there is both internal validation and cross-validation from different experimental techniques. This suggests that there is not likely a significant overestimation of experimentally-identified functional elements. Thus, these two explanations may contribute to the general observation about unconstrained functional elements, but cannot fully explain it.
Instead, we hypothesize five biological reasons to account for the presence of large amounts of unconstrained functional elements. The first two are particular to certain biological assays, where the elements being measured are connected to but do not perfectly coincide with the analysed region. An example of this is the parent transcript of an miRNA, where the current assays detect the exons (some of which are not under evolutionary selection), whereas the intronic miRNA actually harbours the constrained bases. Nevertheless, the transcript sequence provides the critical coupling between the regulated promoter and the miRNA. The sliding of transcription factors (which might bind a specific sequence but then migrate along the DNA) or the processivity of histone modifications across chromatin are more exotic examples of this. A related, second hypothesis is that delocalised behaviours of the genome, such a general chromatin accessibility, may be maintained by some biochemical processes (such as transcription of intergenic regions or specific factor binding) without the requirement for specific sequence elements. These two explanations of both connected components and diffuse components related to, but not coincident with, constrained sequences are particularly relevant for the considerable amount of unannotated and unconstrained transcripts.
The other three hypotheses may be more general. - the presence of neutral (or near neutral) biochemical elements, of lineage-specific functional elements, and of functionally conserved but non-orthologous elements. We believe there are a considerable proportion of neutral biochemically active elements that do not confer a selective advantage or disadvantage to the organism. This neutral pool of sequence elements may turn over during evolutionary time, emerging via certain mutations and disappearing by others. The size of the neutral pool would largely be determined by the rate of emergence and extinction via chance events; low information-content elements, such as transcription factor-binding sites110 will have larger neutral pools. Second, from this neutral pool, some elements might occasionally acquire a biological role and so come under evolutionary selection. The acquisition of a new biological role would then create a lineage-specific element. Finally, a neutral element from the general pool could also become a peer of an existing selected functional element and either of the two elements could then be removed by chance. If the older element is removed, the newer element has, in essence, been conserved without using orthologous bases, providing a conserved function in the absence of constrained sequences. For example, a common HNF4A binding site in the human and mouse genomes may not reflect orthologous human and mouse bases, though the presence of an HNF4A site in that region was evolutionarily selected for in both lineages. Note that both the neutral turnover of elements and the ‘functional peering’ of elements has been suggested for cis-acting regulatory elements in Drosophila115, 116 and mammals110. Our data support these hypotheses, and we have generalized this idea over many different functional elements. The presence of conserved function encoded by conserved orthologous bases is a commonplace assumption in comparative genomics; our findings suggest that there could be a sizable set of functionally-conserved but non-orthologous elements in the human genome, and that these seem unconstrained across mammals. Functional data akin to the ENCODE Project on other related species, such as mouse, would be critical to understanding the rate of such functionally-conserved but non-orthologous elements.
Conclusion
The generation and analyses of over 200 experimental datasets from studies examining the 44 ENCODE regions provide a rich source of functional information for 30 Mb of the human genome. The first conclusion of these efforts is that these data are remarkably informative. Although there will be on going work to enhance existing assays, invent new techniques, and develop new data-analysis methods, the generation of genome-wide experimental datasets akin to the ENCODE pilot phase would provide an impressive platform for future genome-exploration efforts. This now seems feasible in light of throughput improvements of many of the assays and the ever-declining costs of whole-genome tiling arrays and DNA sequencing. Such genome-wide functional data should be acquired and released openly, as has been done with other large-scale genome projects, to ensure its availability to as a new foundation for all biologists studying the human genome. It is these biologists who will often provide the critical link from biochemical function to biological role for the identified elements.
The scale of the pilot phase of the ENCODE Project was also sufficiently large and unbiased to reveal important principles about the organisation of functional elements in the human genome. In many cases, these principles agree with current mechanistic models. For example, trimethylation of H3K4 is enriched near active genes, which we have further refined to the ability to accurately predict gene activity based on histone modifications. However, we also uncovered some surprises that challenge the current dogma on biological mechanisms. The generation of numerous intercalated transcripts spanning the majority of the genome has been repeatedly suggested13, 14, but this phenomenon has been met with mixed opinions about the biological importance of these transcripts. Our analyses of numerous orthogonal datasets firmly establish the presence of these transcripts, and thus the simple view of the genome as having a defined set of isolated loci transcribed independently does not appear to be accurate. Perhaps the genome encodes a network of transcripts, many of which are linked to protein-coding transcripts and the majority of which we cannot (yet) assign a biological role. Our perspective of transcription and genes may have to evolve and also poses some interesting mechanistic questions. For example, how are splicing signals coordinated and used when there are so many overlapping primary transcripts? Similarly, to what extent does this reflect neutral turnover of reproducible transcripts with no biological role?
We gained subtler but equally important mechanistic findings relating to transcription, replication, and chromatin modification. Transcription factors previously thought to primarily bind promoters are more general, and those which do bind to promoters are equally likely to be downstream of a TSS as upstream. Interestingly, many elements that previously were classified as distal enhancers are, in fact, close to one of the newly-identified TSSs; only about 35% of sites showing evidence of binding by multiple transcription factors are actually distal to a TSS. This need not imply that most regulatory information is confined to classic promoters, but rather it does suggest that transcription and regulation are coordinated actions beyond just the traditional promoter sequences. Meanwhile, while distal regulatory elements could be identified in the ENCODE regions, they are currently difficult to classify, in part due to the lack of a broad set of transcription factors to use in analyzing such elements. Finally, we now have a much better appreciation about how DNA replication is coordinated with histone modifications.
At the outset of the ENCODE Project, many believed that the broad collection of experimental data would nicely dovetail with the detailed evolutionary information derived from comparing multiple mammalian sequences to provide a neat ‘dictionary’ of conserved genomic elements, each with a growing annotation about their biochemical function(s). In one sense, this was achieved; the majority of constrained bases in the ENCODE regions are now associated with at least some experimentally-derived information about function. However, we have also encountered a remarkable excess of unconstrained experimentally-identified functional elements, and these cannot be dismissed for technical reasons. This is perhaps the biggest surprise of the pilot phase of the ENCODE Project, and suggests that we take a more ‘neutral’ view of many of the functions conferred by the genome.
Methods
The methods are described in the Supplementary Information, with more technical details for each experiment often found in the references provided in Table 1. The Supplement sections are arranged in the same order as the manuscript (with similar headings to facilitate cross-referencing). The first page of the Supplement also has an index to aid navigation. Raw data are available in ArrayExpress, GEO, or EMBL/GenBank archive as appropriate, as detailed in Supplementary Information section S1.1. Processed data are also presented in a user-friendly manner at the UCSC Genome Browser’s ENCODE portal (http://genome.ucsc.edu/ENCODE/).
Supplementary Material
Table 2.
Bases detected in processed transcripts either as a GENCODE exon, a TxFrag, or as either a GENCODE exon or a TxFrag.
| GENCODE exon | TxFrag | Either GENCODE exon or TxFrag | |
|---|---|---|---|
| Total detectable transcripts | 1,776,157 bases (5.9%) | 1,369,611 bases (4.6%) | 2,519,280 bases (8.4%) |
| Transcripts detected in tiled regions of arrays | 1,447,192 bases (9.8%) | 1,369,611 bases (9.3%) | 2,163,303 bases (14.7%) |
Percentages are of total bases in ENCODE in the first row and bases tiled in arrays in the second row
Acknowledgments
We thank D. Leja for providing graphical expertise and support. Funding support is acknowledged from the following sources: National Institutes of Health, The European Union BioSapiens NoE, Affymetrix, Swiss National Science Foundation, the Spanish Ministerio de Educación y Ciencia, Spanish Ministry of Education and Science, CIBERESP, Genome Spain and Generalitat de Catalunya, Ministry of Education, Culture, Sports, Science and Technology of Japan, the NCCR Frontiers in Genetics, the Jésrôme Lejeune Foundation, the Childcare Foundation, the Novartis Foundations, the Danish Research Council, the Swedish Research Council, the Knut and Alice Wallenberg Foundation, the Wellcome Trust, the Howard Hughes Medical Institute, the Bio-X Institute, the RIKEN Institute, the US Army, National Science Foundation, the Deutsche Forschungsgemeinschaft, the Austrian Gen-AU program, the BBSRC and The European Molecular Biology Laboratory. We thank the Barcelona SuperComputing Center and the NIH Biowulf cluster for computer facilities. The Consortium thanks the ENCODE Scientific Advisory Panel for their advice on the project: G. Weinstock, M. Cherry, G. Churchill, M. Eisen, S. Elgin, J. Lis, J. Rine, M. Vidal and P. Zamore.
Analysis Coordination
Ewan Birney*,1, John A. Stamatoyannopoulos*,2, Anindya Dutta*,3, Roderic Guigó*,4, 5, Thomas R. Gingeras*,6, Elliott H. Margulies*,7, Zhiping Weng*,8, 9, Michael Snyder*,10, 11, Emmanouil T. Dermitzakis*,12;
Chromatin and Replication
John A. Stamatoyannopoulos*,2, Robert E. Thurman2, 13, Michael S. Kuehn2, 13, Christopher M. Taylor3, Shane Neph2, Christoph M. Koch12, Saurabh Asthana14, Ankit Malhotra3, Ivan Adzhubei14, Jason A. Greenbaum15, Robert M. Andrews12, Paul Flicek1, Patrick J. Boyle3, Hua Cao13, Nigel P. Carter12, Gayle K. Clelland12, Sean Davis16, Nathan Day2, Pawandeep Dhami12, Shane C. Dillon12, Michael O. Dorschner2, Heike Fiegler12, Paul G. Giresi17, Jeff Goldy2, Michael Hawrylycz18, Andrew Haydock2, Richard Humbert2, Keith D. James12, Brett E. Johnson13, Ericka M. Johnson13, Tristan T. Frum13, Elizabeth R. Rosenzweig13, Neerja Karnani3, Kirsten Lee2, Gregory C. Lefebvre12, Patrick A. Navas13, Fidencio Neri2, Stephen C. J. Parker15, Peter J. Sabo2, Richard Sandstrom2, Anthony Shafer2, David Vetrie12, Molly Weaver2, Sarah Wilcox12, Man Yu13, Francis S. Collins7, Job Dekker19, Jason D. Lieb17, Thomas D. Tullius15, Gregory E. Crawford20, Shamil Sunayev14, William S. Noble2, Ian Dunham12, Anindya Dutta*,3;
Genes and Transcripts
Roderic Guigó*,4, 5, France Denoeud5, Alexandre Reymond21, 22, Philipp Kapranov6, Joel Rozowsky11, Deyou Zheng11, Robert Castelo5, Adam Frankish12, Jennifer Harrow12, Srinka Ghosh6, Albin Sandelin23, Ivo L. Hofacker24, Robert Baertsch25, 26, Damian Keefe1, Paul Flicek1, Sujit Dike6, Jill Cheng6, Heather A. Hirsch27, Edward A. Sekinger27, Julien Lagarde5, Josep F. Abril5, 28, Atif Shahab29, Christoph Flamm24, 30, Claudia Fried30, Jörg Hackermüller31, Jana Hertel30, Manja Lindemeyer30, Kristin Missal30, 32, Andrea Tanzer24, 30, Stefan Washietl24, Jan Korbel11, Olof Emanuelsson11, Jakob S. Pedersen26, Nancy Holroyd12, Ruth Taylor12, David Swarbreck12, Nicholas Matthews12, Mark C. Dickson33, Daryl J. Thomas25, 26, Matthew T. Weirauch25, James Gilbert12, Jorg Drenkow6, Ian Bell6, XiaoDong Zhao34, K.G. Srinivasan34, Wing-Kin Sung34, Hong Sain Ooi34, Kuo Ping Chiu34, Sylvain Foissac4, Tyler Alioto4, Michael Brent35, Lior Pachter36, Michael L. Tress37, Alfonso Valencia37, Siew Woh Choo34, Chiou Yu Choo34, Catherine Ucla22, Caroline Manzano22, Carine Wyss22, Evelyn Cheung6, Taane G. Clark38, James B. Brown39, Madhavan Ganesh6, Sandeep Patel6, Hari Tammana6, Jacqueline Chrast21, Charlotte N. Henrichsen21, Chikatoshi Kai23, Jun Kawai23, 40, Ugrappa Nagalakshmi10, Jiaqian Wu10, Zheng Lian41, Jin Lian41, Peter Newburger42, Xueqing Zhang42, Peter Bickel43, John S. Mattick44, Piero Carninci40,Yoshihide Hayashizaki23, 40, Sherman Weissman41, Emmanouil T. Dermitzakis*,12, Elliott H. Margulies*,7, Tim Hubbard12, Richard M. Myers33, Jane Rogers12, Peter F. Stadler24, 30, 45, Todd M. Lowe25, Chia-Lin Wei34, Yijun Ruan34, Michael Snyder*,10, 11, Ewan Birney*,1, Kevin Struhl27, Mark Gerstein11, 46, 47, Stylianos E. Antonarakis22, Thomas R. Gingeras*,6;
Integrated Analysis and Manuscript Preparation
James B. Brown39, Paul Flicek1, Yutao Fu8, Damian Keefe1, Ewan Birney*,1, France Denoeud5, Mark Gerstein11, 46, 47, Eric D. Green7, 48, Philipp Kapranov6, Ulaf Karaöz8, Richard M. Myers33, William S. Noble2, Alexandre Reymond21, 22, Joel Rozowsky11, Kevin Struhl27, Adam Siepel25, 26, $, John A. Stamatoyannopoulos*,2, Christopher M. Taylor3, James Taylor49, 50, Robert E. Thurman2, 13, Thomas D. Tullius15, Stefan Washietl24, Deyou Zheng11;
Management Group
Laura A. Liefer51, Kris A. Wetterstrand51, Peter J. Good51, Elise A. Feingold51, Mark S. Guyer51, Francis S. Collins52;
Multi-species Sequence Analysis
Elliott H. Margulies*,7, Gregory M. Cooper33,%, George Asimenos53, Daryl J. Thomas25, 26, Colin N. Dewey54, Adam Siepel25, 26,$, Ewan Birney*,1 , Damian Keefe1, Minmei Hou49, 50, James Taylor49, 50, Sergey Nikolaev22, Juan I. Montoya-Burgos55, Ari Löytynoja1, Simon Whelan1,¶, Fabio Pardi1, Tim Massingham1, James B. Brown39, Haiyan Huang43, Nancy R. Zhang43, 56, Peter Bickel43, Ian Holmes57, James C. Mullikin7, 48, Abel Ureta-Vidal1, Benedict Paten1, Michael Seringhaus11, Deanna Church58, Kate Rosenbloom26, W. James Kent25, 26, NISC Comparative Sequencing Program‡, Baylor College of Medicine Human Genome Sequencing Center‡, Washington University Genome Sequencing Center‡, Broad Institute‡, Children’s Hospital Oakland Research Institute‡, Mark Gerstein11, 46, 47, Stylianos E. Antonarakis22, Serafim Batzoglou53, Nick Goldman1, Ross C. Hardison50, 59, David Haussler25, 26, 60, Webb Miller49, 50, 61, Lior Pachter36, Eric D. Green7, 48, Arend Sidow33, 62;
‡ A list of participants and affiliations appears below
NISC Comparative Sequencing Program
Gerard G. Bouffard7, 48, Xiaobin Guan48, Nancy F. Hansen48, Jacquelyn R. Idol7, Valerie V.B. Maduro7, Baishali Maskeri48, Jennifer C. McDowell48, Morgan Park48, Pamela J. Thomas48, Alice C. Young48, and Robert W. Blakesley7, 48;
Baylor College of Medicine Human Genome Sequencing Center
Donna M. Muzny63, Erica Sodergren63, David A. Wheeler63, Kim C. Worley63, Huaiyang Jiang63, George M. Weinstock63, and Richard A. Gibbs63;
Washington University Genome Sequencing Center
Tina Graves64, Robert Fulton64, Elaine R. Mardis64, and Richard K. Wilson64;
Broad Institute
Michele Clamp65, James Cuff65, Sante Gnerre65, David B. Jaffe65, Jean L. Chang65, Kerstin Lindblad-Toh65, and Eric S. Lander65, 66;
Children’s Hospital Oakland Research Institute
Maxim Koriabine67, Mikhail Nefedov67, Kazutoyo Osoegawa67, Yuko Yoshinaga67, Baoli Zhu67, and Pieter J. de Jong67;
Transcriptional Regulatory Elements
Zhiping Weng*,8, 9, Nathan D. Trinklein33,#, Yutao Fu8, Zhengdong D. Zhang11, Ulaf Karaöz8, Leah Barrera68, Rhona Stuart68, Deyou Zheng11, Srinka Ghosh6, Paul Flicek1, David C. King50, 59, James Taylor49, 50, Adam Ameur69, Stefan Enroth69, Mark C. Bieda70, Christoph M. Koch12, Heather A. Hirsch27, Chia-Lin Wei34, Jill Cheng6, Jonghwan Kim71, Akshay A. Bhinge71, Paul G. Giresi17, Nan Jiang72, Jun Liu34, Fei Yao34, Wing-Kin Sung34, Kuo Ping Chiu34, Vinsensius B. Vega34, Charlie W.H. Lee34, Patrick Ng34, Atif Shahab29, Edward A. Sekinger27, Annie Yang27, Zarmik Moqtaderi27, Zhou Zhu27, Xiaoqin Xu70, Sharon Squazzo70, Matthew J. Oberley73, David Inman73, Michael A. Singer72, Todd A. Richmond72, Kyle J. Munn72, 74, Alvaro Rada-Iglesias74, Ola Wallerman74, Jan Komorowski69, Gayle K. Clelland12, Sarah Wilcox12, Shane C. Dillon12, Robert M. Andrews12, Joanna C. Fowler12, Phillippe Couttet12, Keith D. James12, Gregory C. Lefebvre12, Alexander W. Bruce12, Oliver M. Dovey12, Peter D. Ellis12, Pawandeep Dhami12, Cordelia F. Langford12, Nigel P. Carter12, David Vetrie12, Philipp Kapranov6, David A. Nix6, Ian Bell6, Sandeep Patel6, Joel Rozowsky11, Ghia Euskirchen10, Stephen Hartman10, Jin Lian41, Jiaqian Wu10, Alexander E. Urban10, Peter Kraus10, Sara Van Calcar68, Nate Heintzman68, Tae Hoon Kim68, Kun Wang68, Chunxu Qu68, Gary Hon68, Rosa Luna75, Christopher K. Glass75, M. Geoff Rosenfeld75, Shelley Force Aldred33,#, Sara J. Cooper33, Anason Halees8, Jane M. Lin9, Hennady P. Shulha9, Xiaoling Zhang8, Mousheng Xu8, Jaafar N. S. Haidar9, Yong Yu9, Ewan Birney*,1, Sherman Weissman41, Yijun Ruan34, Jason D. Lieb17, Vishwanath R. Iyer71, Roland D. Green72, Thomas R. Gingeras*,6, Claes Wadelius74, Ian Dunham12, Kevin Struhl27, Ross C. Hardison50, 59, Mark Gerstein11, 46, 47, Peggy J. Farnham70, Richard M. Myers33, Bing Ren68, Michael Snyder*,10, 11;
UCSC Genome Browser
Daryl J. Thomas25, 26, Kate Rosenbloom26, Rachel A. Harte26, Angie S. Hinrichs26, Heather Trumbower26, Hiram Clawson26, Jennifer Hillman-Jackson26, Ann S. Zweig26, Kayla Smith26, Archana Thakkapallayil26, Galt Barber26, Robert M. Kuhn26, Donna Karolchik26, David Haussler25, 26, 60, W. James Kent25, 26;
Variation
Emmanouil T. Dermitzakis*,12, Lluis Armengol76, Christine P. Bird12, Taane G. Clark38, Gregory M. Cooper33,%, Paul I. W. de Bakker77, Andrew D. Kern26, Nuria Lopez-Bigas5, Joel D. Martin50, 59, Barbara E. Stranger12, Daryl J. Thomas25, 26, Abigail Woodroffe78, Serafim Batzoglou53, Eugene Davydov53, Antigone Dimas12, Eduardo Eyras5, Ingileif B. Hallgrímsdóttir79, Ross C. Hardison50, 59, Julian Huppert12, Arend Sidow33, 62, James Taylor49, 50, Heather Trumbower26, Michael C. Zody77, Roderic Guigó*,4, 5, James C. Mullikin7, Gonçalo R. Abecasis78, Xavier Estivill76, 80 and Ewan Birney*,1.
* Co-Chairs of the ENCODE analysis groups, and corresponding authors (E-mail: Ewan Birney, birney@ebi.ac.uk; John A. Stamatoyannopoulos, jstam@u.washington.edu; Anindya Dutta, ad8q@virginia.edu; Roderic Guigó, rguigo@imim.es; Thomas R. Gingeras, Tom_Gingeras@affymetrix.com; Elliott H. Margulies, elliott@nhgri.nih.gov; Zhiping Weng, zhiping@bu.edu; Michael Snyder, michael.snyder@yale.edu; Emmanouil T. Dermitzakis, md4@sanger.ac.uk)
% Current Address: Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA.
$ Current Address: Department of Biological Statistics & Computational Biology, Cornell University, Ithaca, New York 14853, USA.
¶ Current Address: Faculty of Life Sciences, University of Manchester, Michael Smith Building, Oxford Road, Manchester, M13 9PT, UK.
# Current Address: SwitchGear Genomics, 1455 Adams Drive, Suite 2015, Menlo Park, California 94025, USA.
EMBL-European Bioinformatics Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SD, UK.
Department of Genome Sciences, 1705 NE Pacific Street, Box 357730, University of Washington, Seattle, Washington 98195, USA.
Department of Biochemistry and Molecular Genetics, Jordan 1240, Box 800733, 1300 Jefferson Park Ave, University of Virginia School of Medicine, Charlottesville, Virginia 22908, USA.
Genomic Bioinformatics Program, Center for Genomic Regulation, C/Dr. Aiguader 88, Barcelona Biomedical Research Park Building, 08003 Barcelona, Catalonia, Spain.
Research Group in Biomedical Informatics, Institut Municipal d’Investigació Mèdica/Universitat Pompeu Fabra, C/Dr. Aiguader 88, Barcelona Biomedical Research Park Building, 08003 Barcelona, Catalonia, Spain.
Affymetrix, Inc., Santa Clara, California 95051, USA.
Genome Technology Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
Bioinformatics Program, Boston University, 24 Cummington St., Boston, Massachusetts 02215, USA.
Biomedical Engineering Department, Boston University, 44 Cummington St., Boston, Massachusetts 02215, USA.
Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, Connecticut 06520, USA.
Department of Molecular Biophysics and Biochemistry, Yale University, PO Box 208114, New Haven, Connecticut 06520, USA.
The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge, CB10 1SA, UK.
Division of Medical Genetics, 1705 NE Pacific Street, Box 357720, University of Washington, Seattle, Washington 98195, USA.
Division of Genetics, Brigham and Women’s Hospital and Harvard Medical School, 77 Avenue Louis Pasteur, Boston, Massachusetts 02115, USA.
Department of Chemistry and Program in Bioinformatics, Boston University, 590 Commonwealth Ave, Boston, Massachusetts 02215, USA.
Genetics Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
Department of Biology and Carolina Center for Genome Sciences, CB# 3280, 202 Fordham Hall, The University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA.
Allen Institute for Brain Sciences, 551 N. 34th Street, Seattle, Washington 98103, USA.
Program in Gene Function and Expression and Department of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, 364 Plantation Street, Worcester, Massachusetts 01605, USA.
Institute for Genome Sciences & Policy and Department of Pediatrics, 101 Science Drive, Duke University, Durham, North Carolina 27708, USA.
Center for Integrative Genomics, University of Lausanne, Genopode building, 1015 Lausanne, Switzerland.
Department of Genetic Medicine and Development, University of Geneva Medical School, 1211 Geneva, Switzerland.
Genome Exploration Research Group, RIKEN Genomic Sciences Center (GSC), RIKEN Yokohama Institute, 1-7-22, Suehiro-cho, Tsurumi-ku, Yokohama, Kanagawa, 230-0045, Japan.
Institute for Theoretical Chemistry, University of Vienna, Währingerstraße 17, A-1090 Wien, Austria.
Department of Biomolecular Engineering, University of California, Santa Cruz, 1156 High Street, Santa Cruz, California 95064, USA.
Center for Biomolecular Science and Engineering, Engineering 2, Suite 501, Mail Stop CBSE/ITI, University of California, Santa Cruz, California 95064, USA.
Department of Biological Chemistry & Molecular Pharmacology, Harvard Medical School, 240 Longwood Avenue, Boston, Massachusetts 02115, USA.
Department of Genetics, Facultat de Biologia, Universitat de Barcelona, Av Diagonal, 645, 08028, Barcelona, Catalonia, Spain.
Bioinformatics Institute, 30 Biopolis Street, #07-01 Matrix, Singapore, 138671, Singapore.
Bioinformatics Group, Department of Computer Science, University of Leipzig, Härtelstraße 16-18, D-04107 Leipzig, Germany.
Fraunhofer Institut für Zelltherapie und Immunologie - IZI, Deutscher Platz 5e, D-04103 Leipzig, Germany.
Interdisciplinary Center of Bioinformatics, University of Leipzig, Härtelstraße 16-18, D-04107 Leipzig, Germany.
Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA.
Genome Institute of Singapore, 60 Biopolis Street, Singapore 138672, Singapore.
Laboratory for Computational Genomics, Washington University, Campus Box 1045, Saint Louis, Missouri 63130, USA.
Department of Mathematics and Computer Science, University of California, Berkeley, California 94720, USA.
Spanish National Cancer Research Centre, CNIO, Madrid, Spain, and Biosapiences NoE.
Department of Epidemiology and Public Health, Imperial College, St Mary’s Campus, Norfolk Place, London W2 1PG, UK.
Department of Applied Science & Technology, University of California, Berkeley, California 94720, USA.
Genome Science Laboratory, Discovery and Research Institute, RIKEN Wako Institute, 2-1 Hirosawa, Wako, Saitama, 351-0198, Japan.
Department of Genetics, Yale University School of Medicine, 333 Cedar Street, New Haven, Connecticut 06510, USA.
Department of Pediatrics, University of Massachusetts Medical School, 55 Lake Avenue, North Worcester, Massachusetts 01605, USA.
Department of Statistics, University of California, Berkeley, California 94720, USA.
Institute for Molecular Bioscience, University of Queensland, St. Lucia, QLD 4072, Australia.
The Santa Fe Institute, 1399 Hyde Park Road, Santa Fe, New Mexico 87501, USA.
Department of Computer Science, Yale University, PO Box 208114, New Haven, Connecticut 06520-8114, USA.
Program in Computational Biology & Bioinformatics, Yale University, PO Box 208114, New Haven, Connecticut 06520-8114, USA.
NIH Intramural Sequencing Center, National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 20892, USA.
Department of Computer Science and Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA.
Center for Comparative Genomics and Bioinformatics, Huck Institutes for Life Sciences, The Pennsylvania State University, University Park, Pennsylvania 16802, USA.
Division of Extramural Research, National Human Genome Research Institute, National Institute of Health, 5635 Fishers Lane, Suite 4076, Bethesda, Maryland 20892-9305, USA.
Office of the Director, National Human Genome Research Institute, 31 Center Drive, Suite 4B09, Bethesda, Maryland 20892-2152, USA.
Department of Computer Science, Stanford University, Stanford, California 94305, USA.
Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, 6720 MSC, 1300 University Ave, Madison, Wisconsin 53706, USA.
Department of Zoology and Animal Biology, Faculty of Sciences, University of Geneva, Switzerland.
Department of Statistics, Stanford University, Stanford, California 94305, USA.
Department of Bioengineering, University of California, Berkeley, California 94720-1762, USA.
National Center for Biotechnology Information, National Institutes of Health, Bethesda, Maryland 20894, USA.
Department of Biochemistry and Molecular Biology, Huck Institutes of Life Sciences, The Pennsylvania State University, University Park, Pennsylvania 16802, USA.
Howard Hughes Medical Institute, University of California, Santa Cruz, California 95064, USA.
Department of Biology, The Pennsylvania State University, University Park, Pennsylvania 16802, USA.
Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, USA.
Human Genome Sequencing Center and Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA.
Genome Sequencing Center, Washington University School of Medicine, Campus Box 8501, 4444 Forest Park Avenue, Saint Louis, Missouri 63108, USA.
Broad Institute of Harvard University and Massachusetts Institute of Technology, 320 Charles Street, Cambridge, Massachusetts 02141, USA.
Whitehead Institute for Biomedical Research, 9 Cambridge Center, Cambridge, Massachusetts 02142, USA.
Children’s Hospital Oakland Research Institute, BACPAC Resources, 747 52nd Street, Oakland, California 94609, USA.
Ludwig Institute for Cancer Research, 9500 Gilman Drive, La Jolla, California 92093-0653, USA.
The Linnaeus Centre for Bioinformatics, Uppsala University, BMC, Box 598, SE-75124 Uppsala, Sweden.
Department of Pharmacology and the Genome Center, University of California, Davis, California 95616, USA.
Institute for Cellular & Molecular Biology, The University of Texas at Austin, 1 University Station A4800, Austin, Texas 78712, USA.
NimbleGen Systems, Inc., 1 Science Court, Madison, Wisconsin 53711, USA.
University of Wisconsin Medical School, Madison, Wisconsin 53706, USA.
Department of Genetics and Pathology, Rudbeck Laboratory, Uppsala University, SE-75185 Uppsala, Sweden.
University of California, San Diego School of Medicine, 9500 Gilman Drive, La Jolla, California 92093, USA.
Genes and Disease Program, Center for Genomic Regulation, C/Dr. Aiguader 88, Barcelona Biomedical Research Park Building, 08003 Barcelona, Catalonia, Spain.
Broad Institute of MIT and Harvard, 7 Cambridge Center, Cambridge, Massachusetts 02142, USA.
Center for Statistical Genetics, Department of Biostatistics, SPH II, 1420 Washington Heights, Ann Arbor, Michigan 48109-2029, USA.
Department of Statistics, University of Oxford, Oxford, UK.
Universitat Pompeu Fabra, C/Dr. Aiguader 88, Barcelona Biomedical Research Park Building, 08003 Barcelona, Catalonia, Spain.
Box 1: Frequently used abbreviations in this paper
- AR
Ancient Repeat. A repeat that was inserted into the early mammalian lineage and has since become dormant. The majority of ancient repeats are thought to be neutrally evolving.
- CAGE tag
A short sequence from the 5′ end of a transcript
- CDS
Coding sequence. Region of a cDNA or genome which encodes proteins
- ChIP-chip
Chromatin immunoprecipitation followed by detection of the products using a genomic tiling array
- CNV
‘Copy Number Variants’ Regions of the genome which have large duplications in some individuals in the human population
- CS
‘Constrained Sequence;’ a genomic region associated with evidence of negative selection (i.e., rejection of mutations relative to neutral regions)
- DHS
‘DNaseI Hypersensitive Site’ A region of the genome showing a sharply different sensitivity to DNaseI compared to its immediate locale
- EST
‘Expressed Sequence Tag’ A short sequence of a cDNA indicative of expression at this point
- FAIRE
‘Formaldehyde Assisted Isolation of Regulatory Elements’ A method to assay open chromatin using Formaldehyde crosslinking followed by detection of the products using a genomic tiling array
- FDR
‘False Discovery Rate’ A statistical method for setting thresholds on statistical tests to correct for multiple testing.
- GENCODE
Integrated annotation of existing cDNA and protein resources to define transcripts with both manual review and experimental testing procedures
- GSC
‘Genome Structure Correction’ A method to adapt statistical tests to make fewer assumptions about the distribution of features on the genome sequence. This provides a conservative correction to standard tests
- HMM
‘Hidden Markov Model;’ a machine-learning technique that can establish optimal parameters for a given model to explain the observed data
- Indel
An insertion or deletion; two sequences often show a length differences within alignments, but it is not always clear whether this reflects a previous insertion or a deletion
- PET tag
A short sequence that contains both the 5′ and 3′ ends of a transcript
- RACE
‘Rapid Amplification of cDNA Ends;’ a technique for amplifying cDNA sequences between a known internal position in a transcript and its 5′ end
- RFBR
‘Regulatory Factor Binding Region;’ a genomic region found by a ChIP-chip assay to be bound by a protein factor
- RFBR-Seqsp
Regulatory Factor Binding Regions which are from sequence specific binding factors
- RT-PCR
‘Reverse Transcriptase Polymerase Chain Reaction;’ a technique for amplifying a specific region of a transcript
- RxFrag
A ‘Fragment of a RACE Reaction;’ a genomic region found to be present in a RACE product via an unbiased tiling-array assay
- SNP
‘Single Nucleotide Polymorphism’ a single base pair change between two individuals in the human population
- STAGE
‘Sequence Tag Analysis of Genomic Enrichment’ A method similar to Chip/Chip for detecting protein factor binding regions but utilising short sequence determination rather than genomic tiling arrays
- SVM
‘Support Vector Machine;’ a machine-learning technique that can establish an optimal classifier based on labelled training data
- TR50
A measure of replication timing corresponding to the time in the cell cycle when 50% of the cells have replicated their DNA at a specific genomic position
- TSS
‘Transcription Start Site’
- TxFrag
A ‘Fragment of a Transcript;’ a genomic region found to be present in a transcript via an unbiased tiling-array assay
- Un.TxFrag
A TxFrag that is not associated with any other functional annotation
- UTR
Untranslated region. Part of a cDNA either at the 5′ or 3′ end which does not encode a protein sequence
Footnotes
Authors
The paper should be cited as “The ENCODE Project Consortium” as the author. The list of individual authors is divided among the six main analysis groups and five organisational groups. The groups are listed alphabetically. The corresponding authors are listed in the Analysis Coordination group. They can be collectively reached by emailing encode_chairs@ebi.ac.uk.
References
- 1.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 3.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 4.International Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 5.Rat Genome Sequencing Project Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- 6.Lindblad-Toh K, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–819. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 7.International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- 8.Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 9.ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–40. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 10.Zhang ZD, et al. Statistical analysis of the genomic distribution and correlation of regulatory elements in the ENCODE regions. Genome Res. 2006 doi: 10.1101/gr.5573107. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Euskirchen GM, et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: Comparison of array and sequencing based technologies. Genome Res. 2006 doi: 10.1101/gr.5583007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willingham AT, Gingeras TR. TUF love for “junk” DNA. Cell. 2006;125:1215–20. doi: 10.1016/j.cell.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Carninci P, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nature Genetics. 2006;38:626–35. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 14.Cheng J, et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science. 2005;308:1149–54. doi: 10.1126/science.1108625. [DOI] [PubMed] [Google Scholar]
- 15.Bertone P, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306:2242–6. doi: 10.1126/science.1103388. [DOI] [PubMed] [Google Scholar]
- 16.Guigo R, et al. EGASP: the human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006;7(Suppl 1):S21–31. doi: 10.1186/gb-2006-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denoeud F, et al. Prominent use of distal 5′ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res. 2006 doi: 10.1101/gr.5660607. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tress ML, et al. The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0700800104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rozowsky J, et al. The DART classification of unannotated transcription within ENCODE regions: Associating transcription with known and novel loci. Genome Res. 2006 doi: 10.1101/gr.5696007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kapranov P, et al. Examples of the complex architecture of the human transcriptome revealed by RACE and high-density tiling arrays. Genome Res. 2005;15:987–97. doi: 10.1101/gr.3455305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balakirev ES, Ayala FJ. Pseudogenes: are they “junk” or functional DNA? Annu Rev Genet. 2003;37:123–51. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- 22.Mighell AJ, Smith NR, Robinson PA, Markham AF. Vertebrate pseudogenes. FEBS Lett. 2000;468:109–14. doi: 10.1016/s0014-5793(00)01199-6. [DOI] [PubMed] [Google Scholar]
- 23.Zheng D, et al. Pseudogenes in the ENCODE regions: Consensus annotation, analysis of transcription and evolution. Genome Res. 2006 doi: 10.1101/gr.5586307. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng D, et al. Integrated pseudogene annotation for human chromosome 22: evidence for transcription. J Mol Biol. 2005;349:27–45. doi: 10.1016/j.jmb.2005.02.072. [DOI] [PubMed] [Google Scholar]
- 25.Harrison PM, Zheng D, Zhang Z, Carriero N, Gerstein M. Transcribed processed pseudogenes in the human genome: an intermediate form of expressed retrosequence lacking protein-coding ability. Nucleic Acids Res. 2005;33:2374–83. doi: 10.1093/nar/gki531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Washietl S, et al. Structured RNAs in the ENCODE selected regions of the human genome. Genome Res. 2007 doi: 10.1101/gr.5650707. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Carninci P, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–63. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 28.Runte M, et al. The IC-SNURF-SNRPN transcript serves as a host for multiple small nucleolar RNA species and as an antisense RNA for UBE3A. Hum Mol Genet. 2001;10:2687–700. doi: 10.1093/hmg/10.23.2687. [DOI] [PubMed] [Google Scholar]
- 29.Seidl CI, Stricker SH, Barlow DP. The imprinted Air ncRNA is an atypical RNAPII transcript that evades splicing and escapes nuclear export. Embo J. 2006;25:3565–75. doi: 10.1038/sj.emboj.7601245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parra G, et al. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res. 2006;16:37–44. doi: 10.1101/gr.4145906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maston GA, Evans SK, Green MR. Transcriptional Regulatory Elements in the Human Genome. Annu Rev Genomics Hum Genet. 2006 doi: 10.1146/annurev.genom.7.080505.115623. [DOI] [PubMed] [Google Scholar]
- 32.Trinklein ND, Aldred SJ, Saldanha AJ, Myers RM. Identification and functional analysis of human transcriptional promoters. Genome Res. 2003;13:308–12. doi: 10.1101/gr.794803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper SJ, Trinklein ND, Anton ED, Nguyen L, Myers RM. Comprehensive analysis of transcriptional promoter structure and function in 1% of the human genome. Genome Res. 2006;16:1–10. doi: 10.1101/gr.4222606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cawley S, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 35.Yelin R, et al. Widespread occurrence of antisense transcription in the human genome. Nat Biotechnol. 2003;21:379–86. doi: 10.1038/nbt808. [DOI] [PubMed] [Google Scholar]
- 36.Katayama S, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–6. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 37.Ren B, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–9. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 38.Iyer VR, et al. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature. 2001;409:533–8. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- 39.Horak CE, et al. GATA-1 binding sites mapped in the beta-globin locus by using mammalian CHIp-chip analysis. Proc Natl Acad Sci U S A. 2002;99:2924–9. doi: 10.1073/pnas.052706999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei CL, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 41.Kim J, Bhinge AA, Morgan XC, Iyer VR. Mapping DNA-protein interactions in large genomes by sequence tag analysis of genomic enrichment. Nat Methods. 2005;2:47–53. doi: 10.1038/nmeth726. [DOI] [PubMed] [Google Scholar]
- 42.Dorschner MO, et al. High-throughput localization of functional elements by quantitative chromatin profiling. Nat Methods. 2004;1:219–25. doi: 10.1038/nmeth721. [DOI] [PubMed] [Google Scholar]
- 43.Sabo PJ, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–8. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- 44.Crawford GE, et al. DNase-chip: a high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat Methods. 2006;3:503–9. doi: 10.1038/NMETH888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hogan GJ, Lee CK, Lieb JD. Cell Cycle-Specified Fluctuation of Nucleosome Occupancy at Gene Promoters. PLoS Genet. 2006;2 doi: 10.1371/journal.pgen.0020158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koch CM, et al. The Landscape of Histone Modifications across 1% of the Human Genome in Five Human Cell Lines. Genome Res. 2006 doi: 10.1101/gr.5704207. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. 2003;72:449–79. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- 48.Mito Y, Henikoff JG, Henikoff S. Genome-scale profiling of histone H3.3 replacement patterns. Nat Genet. 2005;37:1090–7. doi: 10.1038/ng1637. [DOI] [PubMed] [Google Scholar]
- 49.Heintzman ND, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- 50.Yusufzai TM, Tagami H, Nakatani Y, Felsenfeld G. CTCF tethers an insulator to subnuclear sites, suggesting shared insulator mechanisms across species. Mol Cell. 2004;13:291–8. doi: 10.1016/s1097-2765(04)00029-2. [DOI] [PubMed] [Google Scholar]
- 51.Kim TH, et al. Direct isolation and identification of promoters in the human genome. Genome Res. 2005;15:830–9. doi: 10.1101/gr.3430605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruppert S, Wang EH, Tjian R. Cloning and expression of human TAFII250: a TBP-associated factor implicated in cell-cycle regulation. Nature. 1993;362:175–9. doi: 10.1038/362175a0. [DOI] [PubMed] [Google Scholar]
- 54.Fernandez PC, et al. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–29. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z, et al. A global transcriptional regulatory role for c-Myc in Burkitt’s lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164–9. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orian A, et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101–14. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Laat W, Grosveld F. Spatial organization of gene expression: the active chromatin hub. Chromosome Res. 2003;11:447–59. doi: 10.1023/a:1024922626726. [DOI] [PubMed] [Google Scholar]
- 58.Trinklein ND, et al. Integrated analysis of experimental datasets reveals many novel promoters in 1% of the human genome. Genome Res. 2007;17:720–731. doi: 10.1101/gr.5716607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeon Y, et al. Temporal profile of replication of human chromosomes. Proc Natl Acad Sci U S A. 2005;102:6419–24. doi: 10.1073/pnas.0405088102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woodfine K, et al. Replication timing of the human genome. Hum Mol Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- 61.White EJ, et al. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc Natl Acad Sci U S A. 2004;101:17771–6. doi: 10.1073/pnas.0408170101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schubeler D, et al. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat Genet. 2002;32:438–42. doi: 10.1038/ng1005. [DOI] [PubMed] [Google Scholar]
- 63.MacAlpine DM, Rodriguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004;18:3094–105. doi: 10.1101/gad.1246404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gilbert DM. Replication timing and transcriptional control: beyond cause and effect. Curr Opin Cell Biol. 2002;14:377–83. doi: 10.1016/s0955-0674(02)00326-5. [DOI] [PubMed] [Google Scholar]
- 65.Schwaiger M, Schubeler D. A question of timing: emerging links between transcription and replication. Curr Opin Genet Dev. 2006;16:177–83. doi: 10.1016/j.gde.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 66.Hatton KS, et al. Replication program of active and inactive multigene families in mammalian cells. Mol Cell Biol. 1988;8:2149–58. doi: 10.1128/mcb.8.5.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gartler SM, Goldstein L, Tyler-Freer SE, Hansen RS. The timing of XIST replication: dominance of the domain. Hum Mol Genet. 1999;8:1085–9. doi: 10.1093/hmg/8.6.1085. [DOI] [PubMed] [Google Scholar]
- 68.Azuara V, et al. Heritable gene silencing in lymphocytes delays chromatid resolution without affecting the timing of DNA replication. Nat Cell Biol. 2003;5:668–74. doi: 10.1038/ncb1006. [DOI] [PubMed] [Google Scholar]
- 69.Cohen SM, Furey TS, Doggett NA, Kaufman DG. Genome-wide sequence and functional analysis of early replicating DNA in normal human fibroblasts. BMC Genomics. 2006;7:301. doi: 10.1186/1471-2164-7-301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao R, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 71.Muller J, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 72.Bracken AP, Dietrich N, Pasini D, Hansen KH, Helin K. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006;20:1123–36. doi: 10.1101/gad.381706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kirmizis A, et al. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 2004;18:1592–605. doi: 10.1101/gad.1200204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee TI, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karnani N, Taylor C, Malhotra A, Dutta A. Pan-S replication patterns and chromosomal domains defined by genome tiling arrays of human chromosomes. Genome Res. 2007 doi: 10.1101/gr.5427007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Delaval K, Wagschal A, Feil R. Epigenetic deregulation of imprinting in congenital diseases of aberrant growth. Bioessays. 2006;28:453–9. doi: 10.1002/bies.20407. [DOI] [PubMed] [Google Scholar]
- 77.Dillon N. Gene regulation and large-scale chromatin organization in the nucleus. Chromosome Res. 2006;14:117–26. doi: 10.1007/s10577-006-1027-8. [DOI] [PubMed] [Google Scholar]
- 78.Burkhoff AM, Tullius TD. Structural details of an adenine tract that does not cause DNA to bend. Nature. 1988;331:455–7. doi: 10.1038/331455a0. [DOI] [PubMed] [Google Scholar]
- 79.Price MA, Tullius TD. How the structure of an adenine tract depends on sequence context: a new model for the structure of TnAn DNA sequences. Biochemistry. 1993;32:127–36. doi: 10.1021/bi00052a018. [DOI] [PubMed] [Google Scholar]
- 80.Greenbaum JA, Parker SCJ, Tullius TD. Detection of DNA structural motifs in functional genomic elements. Genome Res. 2007 doi: 10.1101/gr.5602807. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thurman RE, Day N, Noble WS, Stamatoyannopoulos JA. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2006 doi: 10.1101/gr.6081407. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gilbert N, et al. Chromatin architecture of the human genome: gene-rich domains re enriched in open chromatin fibers. Cell. 2004;118:555–66. doi: 10.1016/j.cell.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 83.Nobrega MA, Ovcharenko I, Afzal V, Rubin EM. Scanning human gene deserts for long-range enhancers. Science. 2003;302:413. doi: 10.1126/science.1088328. [DOI] [PubMed] [Google Scholar]
- 84.Woolfe A, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Drake JA, et al. Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet. 2006;38:223–7. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- 86.Margulies EH, Blanchette M, Haussler D, Green ED NISC Comparative Sequencing Program. Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13:2507–18. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Thomas JW, et al. Parallel construction of orthologous sequence-ready clone contig maps in multiple species. Genome Res. 2002;12:1277–85. doi: 10.1101/gr.283202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Blakesley RW, et al. An intermediate grade of finished genomic sequence suitable for comparative analyses. Genome Res. 2004;14:2235–44. doi: 10.1101/gr.2648404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aparicio S, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–10. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- 92.Jaillon O, et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431:946–957. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- 93.Margulies EH, et al. An initial strategy for the systematic identification of functional elements in the human genome by low-redundancy comparative sequencing. Proc Natl Acad Sci U S A. 2005;102:4795–4800. doi: 10.1073/pnas.0409882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Blanchette M, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–15. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bray N, Pachter L. MAVID: constrained ancestral alignment of multiple sequences. Genome Res. 2004;14:693–699. doi: 10.1101/gr.1960404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brudno M, et al. LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003;13:721–731. doi: 10.1101/gr.926603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Margulies EH, et al. Relationship between evolutionary constraint and genome function for 1% of the human genome. Genome Res. 2006 submitted. [Google Scholar]
- 98.Asthana S, Roytberg M, Stamatoyannopoulos JA, Sunyaev S. Analysis of sequence conservation at nucleotide resolution in ENCODE regions. PLoS Comp Biol. doi: 10.1371/journal.pcbi.0030254. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cooper GM, Brudno M, Green ED, Batzoglou S, Sidow A. Quantitative estimates of sequence divergence for comparative analyses of mammalian genomes. Genome Res. 2003;13:813–20. doi: 10.1101/gr.1064503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eddy SR. A model of the statistical power of comparative genome sequence analysis. PLoS Biol. 2005;3:e10. doi: 10.1371/journal.pbio.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stone EA, Cooper GM, Sidow A. Trade-offs in detecting evolutionarily constrained sequence by comparative genomics. Annu Rev Genomics Hum Genet. 2005;6:143–164. doi: 10.1146/annurev.genom.6.080604.162146. [DOI] [PubMed] [Google Scholar]
- 102.McAuliffe JD, Jordan MI, Pachter L. Subtree power analysis and species selection for comparative genomics. Proc Natl Acad Sci U S A. 2005;102:7900–7905. doi: 10.1073/pnas.0502790102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cheng Z, et al. A genome-wide comparison of recent chimpanzee and human segmental duplications. Nature. 2005;437:88–93. doi: 10.1038/nature04000. [DOI] [PubMed] [Google Scholar]
- 105.Cooper GM, et al. Characterization of evolutionary rates and constraints in three Mammalian genomes. Genome Res. 2004;14:539–48. doi: 10.1101/gr.2034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dermitzakis ET, Reymond A, Antonarakis SE. Conserved non-genic sequences - an unexpected feature of mammalian genomes. Nat Rev Genet. 2005;6:151–7. doi: 10.1038/nrg1527. [DOI] [PubMed] [Google Scholar]
- 107.Clark TG, et al. Small insertions/deletions and functional constraint in the ENCODE regions. Genome Biology. 2007 doi: 10.1186/gb-2007-8-9-r180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bejerano G, et al. Ultraconserved elements in the human genome. Science. 2004;304:1321–5. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- 109.Woolfe A, et al. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dermitzakis ET, Clark AG. Evolution of transcription factor binding sites in Mammalian gene regulatory regions: conservation and turnover. Mol Biol Evol. 2002;19:1114–21. doi: 10.1093/oxfordjournals.molbev.a004169. [DOI] [PubMed] [Google Scholar]
- 111.McDonald JH, Kreitman M. Adaptive protein evolution at the Adh locus in Drosophila. Nature. 1991;351:652–4. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 112.Andolfatto P. Adaptive evolution of non-coding DNA in Drosophila. Nature. 2005;437:1149–52. doi: 10.1038/nature04107. [DOI] [PubMed] [Google Scholar]
- 113.Hudson RR, Kreitman M, Aguade M. A test of neutral molecular evolution based on nucleotide data. Genetics. 1987;116:153–9. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 115.Ludwig MZ, et al. Functional evolution of a cis-regulatory module. PLoS Biol. 2005;3:e93. doi: 10.1371/journal.pbio.0030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ludwig MZ, Kreitman M. Evolutionary dynamics of the enhancer region of even-skipped in Drosophila. Mol Biol Evol. 1995;12:1002–11. doi: 10.1093/oxfordjournals.molbev.a040277. [DOI] [PubMed] [Google Scholar]
- 117.Harrow J, et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7(Suppl 1):S41–9. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Emanuelsson O, et al. Assessing the performance of different high-density tiling microarray strategies for mapping transcribed regions of the human genome. Genome Res. 2006 doi: 10.1101/gr.5014606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kapranov P, et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–9. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- 120.Bhinge AA, Kim J, Euskirchen G, Snyder M, Iyer VR. Mapping the chromosomal targets of STAT1 by Sequence Tag Analysis of Genomic Enrichment (STAGE) Genome Res. 2006 doi: 10.1101/gr.5574907. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ng P, et al. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nat Methods. 2005;2:105–11. doi: 10.1038/nmeth733. [DOI] [PubMed] [Google Scholar]
- 122.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2006 doi: 10.1101/gr.5533506. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rada-Iglesias A, et al. Binding sites for metabolic disease related transcription factors inferred at base pair resolution by chromatin immunoprecipitation and genomic microarrays. Hum Mol Genet. 2005;14:3435–47. doi: 10.1093/hmg/ddi378. [DOI] [PubMed] [Google Scholar]
- 124.Kim TH, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–80. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Halees AS, Weng Z. PromoSer: improvements to the algorithm, visualization and accessibility. Nucleic Acids Res. 2004;32:W191–4. doi: 10.1093/nar/gkh433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Guigó R, et al. EGASP: The human ENCODE Genome Annotation Assessment Project. Genome Biology. 2006;7:S2.1–S2.31. doi: 10.1186/gb-2006-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bajic VB, et al. Performance assessment of promoter predictions on ENCODE regions in the EGASP experiment. Genome Biol. 2006;7(Suppl 1):S3.1–13. doi: 10.1186/gb-2006-7-s1-s3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zheng D, Gerstein MB. A computational approach for identifying pseudogenes in the ENCODE regions. Genome Biology. 2006;7:S13.1–S13.10. doi: 10.1186/gb-2006-7-s1-s13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stranger BE, et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 2005;1:e78. doi: 10.1371/journal.pgen.0010078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol. 2005;12:110–2. doi: 10.1038/nsmb0205-110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.