

Abstract
The molecular basis of thymocyte negative selection, a crucial mechanism in establishing central tolerance, is not yet resolved. Histone deacetylases (HDACs) have emerged as key transcriptional regulators in several major developmental programs. Recently, we showed that the class IIa member, HDAC7, regulates negative selection by repressing expression of Nur77, an orphan nuclear receptor involved in antigen-induced apoptosis of thymocytes. Engagement of the T cell receptor (TCR) alleviates this repression through phosphorylation-dependent nuclear exclusion of HDAC7. However, the identity of the TCR-activated kinase that phosphorylates and inactivates HDAC7 was still unknown. Here, we demonstrate that TCR-induced nuclear export of HDAC7 and Nur77 expression is mediated by activation of protein kinase D (PKD). Indeed, active PKD stimulates HDAC7 nuclear export and Nur77 expression. In contrast, inhibition of PKD prevents TCR-mediated nuclear exclusion of HDAC7 and associated Nur77 activation. Furthermore, we show that HDAC7 is an interaction partner and a substrate for PKD. We identify four serine residues in the NH2 terminus of HDAC7 as targets for PKD. More importantly, a mutant of HDAC7 specifically deficient in phosphorylation by PKD, inhibits TCR-mediated apoptosis of T cell hybridomas. These findings indicate that PKD is likely to play a key role in the signaling pathways controlling negative selection.
For many cell surface receptors, ligation initiates a cascade of intracellular events that can result in multiple cellular outcomes, depending on the extent of the ligation and the cell context. Nowhere is this more relevant than during T cell development in the thymus. Mature CD4+ and CD8+ T lymphocytes develop in the thymus from a pool of CD4+ CD8+ double positive (DP) precursors. These precursors harbor diverse clonally distributed receptors with negligible, intermediate, or high affinity for self-peptide MHC complexes (1). Most DP die “by neglect” as they fail to receive a signal from their TCR. Cells receiving strong intracellular signaling through engagement of their TCR are eliminated by an activation-induced apoptotic program termed negative selection, which allows for deletion of autoreactive T cells and establishment of central immune tolerance. In contrast, the small proportion of DP thymocytes that display intermediate reactivity for MHC ligands will receive an appropriate signal allowing for survival and further differentiation, a process named positive selection (2). As a first step toward the comprehension of these differentiative steps, a key issue is to unravel TCR signaling pathways.
Engagement of the TCR results in the activation of several downstream cascades, involving protein tyrosine kinases, protein kinase C (PKC), and mitogen-activated protein kinases (3). These signaling pathways ultimately induce the activation and/or synthesis of transcription factors, leading to changes in gene expression and activation of specific developmental programs (4). The orphan nuclear receptor Nur77 (also called TR3, NGFI-B, NAK1, Nr4a1, and several other aliases) is a potent transcription factor that plays a key role in negative selection (5). Thymocytes from Nur77-expressing transgenic mice show massive cell death in vivo (6–8). In contrast, loss of Nur77 function through overexpression of a dominant negative mutant inhibits negative selection (9). Although basal levels of Nur77 are extremely low in unstimulated T cells, engagement of the TCR induces rapid induction of Nur77. The two major signaling pathways downstream of the TCR (the PKC pathway and the calcium pathway) converge on the Nur77 promoter. Indeed, both phorbol esters (for PKC) and ionomycin (for the Ca2+ pathway) can activate Nur77 expression independently or in combination (7). Transcriptional regulation of Nur77 is mediated by two MEF2-binding sites located in the Nur77 promoter. Cabin-1, a MEF2-interacting partner, was previously shown to inhibit Nur77 expression and confer Ca2+ inducibility to the Nur77 promoter (10). We have recently reported that histone deacetylase (HDAC)7, a class IIa HDAC highly expressed in DP thymocytes, associates with MEF2D and represses Nur77 expression via its deacetylase activity (11).
By modulating the accessibility of the transcriptional machinery to DNA, acetylation and deacetylation of nucleosomal histones have emerged as key regulatory mechanisms of gene transcription. Deacetylation of histones by HDACs results in a compact chromatin structure associated with transcriptional repression. The 18 human HDACs identified to date are grouped into four distinct classes, with members of class II further subdivided into two subclasses, IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and IIb (HDAC6 and HDAC10; reference 12).
Class IIa HDACs are restricted to a limited number of cell types, suggesting a role for these proteins in specific biological processes. The class IIa HDACs contain two domains: a conserved catalytic region at their COOH terminus and an NH2-terminal extension showing limited sequence homology amongst the different members. The NH2-terminal domain of class IIa HDACs contains a conserved amino acid motif implicated in interactions with members of the MEF2 family of transcription factors. This interaction leads to repression of MEF2-targeted promoters via recruitment of class IIa–associated HDAC activity (13). In addition, the NH2-terminal region of class IIa HDACs contains serine residues important for their regulation (14). Sequence analysis previously identified these serine residues as putative targets for Ca2+/calmodulin-dependent kinases (CaMKs). Phosphorylation of these residues by CaMK signaling leads to the dissociation of MEF2 interactions and binding of 14-3-3 proteins, which induce class IIa HDAC nuclear export and derepression of their target promoters (14–17). These findings have been illustrated for HDAC5 and HDAC9 in the context of the gene expression programs associated with muscle differentiation (18) and cardiac hypertrophy (19). In developing T cells, we recently reported that HDAC7 inhibits negative selection of DP thymocytes by repressing the transcriptional activity of the Nur77 promoter (11). Upon stimulation of the TCR, the inhibitory effect of HDAC7 is relieved through its nuclear exclusion, allowing for Nur77 expression and engagement of negative selection programs.
In this report, we have examined the signaling pathways leading to the nuclear exclusion and inactivation of HDAC7 during negative selection. We show that Ca2+ signaling is not involved in TCR-mediated nuclear exclusion of HDAC7. In contrast, we demonstrate that protein kinase D (PKD; also known as PKCμ), which is activated upon engagement of the TCR, stimulates HDAC7 nuclear export by direct phosphorylation on four serine residues. Conversely, selective PKD inhibition blocks TCR-induced HDAC7 nuclear export and Nur77 expression. In addition, an HDAC7 mutant specifically deficient in phosphorylation by PKD blocks TCR-mediated apoptosis. These findings establish a key role for PKD in the signaling pathway mediating TCR-induced apoptosis in developing thymocytes and indicate that PKD is likely to play an important role in the control of central immune tolerance.
Results
TCR-dependent nuclear export of HDAC7 is regulated by a PMA-stimulated, Gö6976-sensitive kinase
Our previous results have shown that TCR stimulation of T cell hybridomas, a convenient model to study the molecular aspects of negative selection in vitro (6, 10), induces rapid and sustained relocalization of HDAC7 to the cytoplasm (11). The signals downstream of the TCR consist of two intracellular signaling pathways: the PKC pathway, which can be mimicked by the addition of PMA, and the Ca2+ pathway, which can be mimicked by the addition of calcium ionophore (ionomycin). As a step toward the identification of the HDAC7-specific kinase that is activated in thymocytes in response to TCR signaling, we assessed the subcellular localization of HDAC7 after various treatments. For this purpose, we stably transduced T cell hybridoma cells (DO11.10) with a HDAC7-GFP construct. In compliance with our previous results, we observed that unstimulated DO11.10 cells showed predominantly nuclear HDAC7, whereas treatment with anti-CD3 antibodies or a combination of PMA and ionomycin induced exclusion of HDAC7 from the nucleus (Fig. 1 A). Surprisingly, upon treatment with ionomycin alone, HDAC7 remained nuclear in most cells (Fig. 1 A). On the other hand, treatment of cells with PMA alone led to the complete exclusion of HDAC7 from the nucleus in the vast majority of cells (Fig. 1 A). These results indicate that the phosphorylation-dependent nuclear exclusion of HDAC7 upon TCR stimulation does not involve the CaMKs previously implicated in class IIa shuttling and suggest a role for a PKC-dependent pathway.
Figure 1.

TCR-induced nuclear export of HDAC7 is mediated by a PMA-stimulated and Gö6976-sensitive kinase. (A) Polyclonal DO11.10 cells stably expressing HDAC7-GFP were untreated or treated with anti-CD3 antibodies, PMA and ionomycin, ionomycin, or PMA. Subcellular localization of GFP-HDAC7 was monitored by confocal microscopy after 2 h of treatment. Bar histograms represent the mean percentages of cells showing nuclear exclusion of HDAC7-GFP in each condition. (B) Polyclonal DO11.10 cells stably expressing HDAC7-GFP were treated with PMA with or without previous incubation with the indicated inhibitors. After 2 h of PMA treatment, localization of HDAC7 was then examined using confocal microscopy. Bar histograms represent the mean percentages of cells showing nuclear exclusion of HDAC7-GFP in each condition.
To further characterize the TCR-induced HDAC7 kinase, we tested the ability of various inhibitors to interfere with the nuclear exclusion of HDAC7 upon PMA treatment (Fig. 1 B). Staurosporine, a general serine/threonine kinase inhibitor, abolished the nuclear exclusion of HDAC7 by PMA (Fig. 1 B). Surprisingly, two PKCs inhibitors selective for classical and novel isoforms, GF109203x and Ro-31-8220, were ineffective at blocking PMA-induced cytoplasmic translocation of HDAC7. On the other hand, treatment with Gö6976, an inhibitor of classical PKCs and PKD, totally prevented the nuclear exclusion of HDAC7 by PMA treatment (Fig. 1 B).
All together, these results suggest that PKD could be involved in the TCR-induced nuclear exclusion of HDAC7.
PKD is activated by TCR signaling in thymocytes
If PKD is involved in the nuclear exclusion of HDAC7, then TCR signaling would be expected to activate PKD in DO11.10 cells. Autophosphorylation of serine 916 follows activation of PKD and can thus be used as a marker for PKD activity (20). We used an antibody specific for phosphorylated serine 916 of PKD to monitor the activation of PKD in DO11.10 cells after various stimuli. As shown in Fig. 2 A, unstimulated cells exhibited low basal activity of PKD that was dramatically enhanced upon triggering of the TCR with an anti-CD3 antibody. Stimulation of cells with a combination of PMA and ionomycin or PMA alone enhanced even further the activity of PKD.
Figure 2.

PKD is expressed and activated by TCR in thymocytes. (A) DO11.10 cells were left unstimulated (n/s) or stimulated for 15 min with anti-CD3 antibodies, PMA and ionomycin, PMA alone, or ionomycin alone. Total protein extracts were analyzed by Western blotting with an antibody specific for phosphorylated serine 916 of PKD1 (pPKD1, which recognizes active PKD1). As a control, the same membrane was stripped and probed with an antiserum against total PKD1. (B) DO11.10 cells were cultured in the presence of 10 ng/ml PMA, and activation of PKD1 was examined at 0, 2, 5, 15, 30, 120, and 240 min by Western blot using the phosphorylated serine 916–specific antibody. The same blot was probed with an antibody for total PKD1 as control.
Translocation of HDAC7 from the nucleus to the cytoplasm occurs within minutes after TCR signaling (11). To monitor the kinetic of PKD activation, we stimulated DO11.10 cells with PMA and detected active PKD at different time points (Fig. 2 B). As expected, PMA induced a marked increase in PKD activation compared with basal levels. This activation was maximal within 2 min of stimulation and was sustained for up to 2 h.
These results indicate that PKD is activated by the same extracellular signals that induce nuclear exclusion of HDAC7 and suggest that PKD could be the kinase regulating this process in T cells.
PKD associates with the amino terminus of HDAC7
Subcellular localization of HDAC7 is controlled by phosphorylation of three serine residues (S155, S318, and S449) located in its amino-terminal domain (11, 17). In an attempt to identify the HDAC7 kinase(s), we expressed the NH2 terminus of HDAC7 (1–490) as a glutathione S-transferase (GST) fusion protein in Escherichia coli. As expected, no kinase activity could be detected in the GST-HDAC7Nter or GST alone recombinant protein preparations (Fig. 3 A, − Extract). To test whether the NH2 terminus of HDAC7 might recruit kinase activity, we bound the GST-HDAC7Nter to glutathione agarose beads and incubated them with extract from DO11.10 cells. After extensive washing, the bound material was assessed for kinase activity by in vitro kinase (IVK) assay. High kinase activity was found associated with GST-HDAC7Nter, but not with the GST control (Fig. 3 A, + Extract). To further refine these observations, IVK reactions were resolved by SDS-PAGE and analyzed by autoradiography. As shown in Fig. 3 A, a radioactive band corresponding to GST-HDAC7Nter was readily detected (Fig. 3 A, + Extract, bottom). These results demonstrate that a kinase that can specifically associate with and phosphorylate the amino terminus of HDAC7 is present in extracts from DO11.10 cells.
Figure 3.
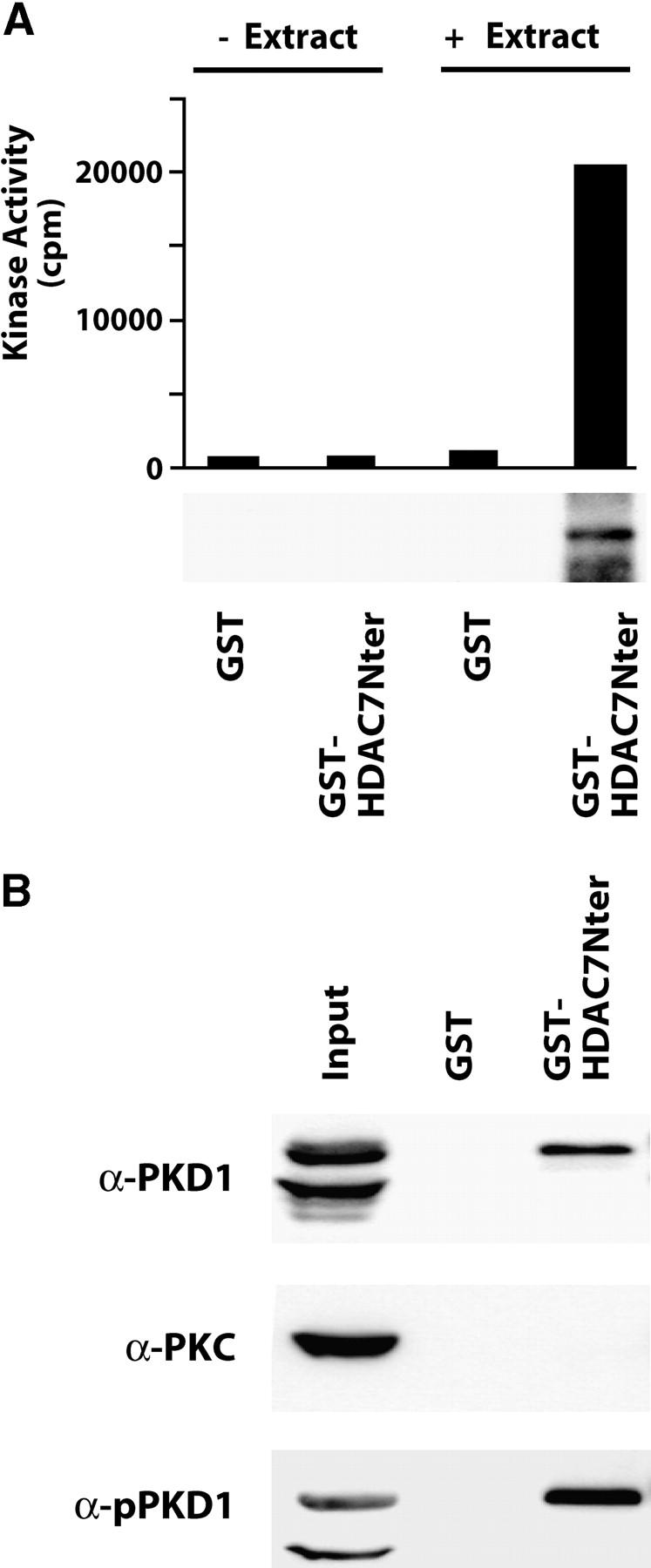
PKD associates with the NH2 terminus of HDAC7. (A) The NH2 terminus of HDAC7 (aa 1–490) was expressed as a GST fusion protein and immobilized on glutathione agarose beads. GST protein alone was used as control. Before (− Extract) and after (+ Extract) incubation of equal amounts of the fusion proteins with total cellular extracts from unstimulated, actively growing DO11.10 cells, associated kinase activity was revealed by IVK. Total incorporated radioactivity was quantified by scintillation counting. Results representative of three independent experiments are shown as a histogram (top). In parallel, IVK assays were analyzed by SDS-PAGE and autoradiography (bottom). (B) Pull-downs were analyzed for the presence of endogenous PKD1, PKC family members (PKC), and activated PKD1 (pPKD1) by Western blot.
Because our observations so far suggested a role for PKD in the TCR-induced nuclear exclusion of HDAC7, we tested if PKD could participate in the kinase activity associated with HDAC7Nter. Western blot analysis of the bound material revealed that endogenous PKD1 was specifically present in the fraction associated with the NH2 terminus of HDAC7 but not with GST alone (Fig. 3 B, α-PKD1). As a control for specificity, no association with PKC family members could be detected (Fig. 3 B, α-PKC). To demonstrate that PKD could contribute to the HDAC7Nter-specific kinase activity, we next tested the presence of active PKD in the material associated with GST-HDAC7Nter. Western blot analysis with the antibody against active PKD1 revealed that GST-HDAC7Nter was associated with endogenous active PKD1 (Fig. 3 B, α-pPKD1).
PKD phosphorylates the amino-terminal domain of HDAC7
Next, we examined whether PKD could directly phosphorylate the NH2 terminus of HDAC7. The N- (amino acids [aa] 1–490) or the COOH-terminal (aa 490–915) domains of HDAC7 were expressed as GST fusion proteins and incubated with active recombinant PKD1 in an IVK assay. SDS-PAGE analysis showed incorporation of high levels of the γ[32P] label on GST-HDAC7Nter, indicating that it was very efficiently phosphorylated by PKD in vitro (Fig. 4 A, GST-HDAC7Nter). In contrast, PKD did not efficiently phosphorylate GST-HDAC7Cter in a similar assay (Fig. 4 A, GST-HDAC7Cter).
Figure 4.

PKD1 phosphorylates S155, S181, S321, and S449 of HDAC7 in vitro. (A) The C- (aa 490–915) and N- (aa 1–490) terminal domains of HDAC7 were produced as GST fusion proteins (GST-HDAC7Cter and GST-HDAC7Nter, respectively) and immobilized on glutathione beads. Equal amounts of purified recombinant proteins were used in IVK assays with constitutively active recombinant PKD1. IVK reactions were analyzed by SDS-PAGE and Coomassie staining (left) before autoradiography (right). (B) Sequences around the four identified serines in HDAC7 match with the canonical PKD recognition motif. (C) GST fusion proteins corresponding to the sequences surrounding the four putative PKD phosphorylation sites in HDAC7 were used as substrates in independent IVK assays with constitutively active recombinant PKD1. Mutant fusion proteins harboring a serine to alanine substitution were used as controls. Reactions were resolved by SDS-PAGE and gel was stained with Coomassie, dried, and analyzed by autoradiography. The arrow indicates the radioactive band corresponding to autophosphorylated PKD1 (rPKD1).
A peptide library study previously revealed that PKD prefers a basic residue at position −3 and is highly selective for a leucine at position −5 with respect for the phosphorylated serine (21). In silico analysis of the sequences corresponding to the NH2 terminus of HDAC7 revealed four putative PKD phosphorylation sites located at S155, S181, S321, and S449 (Fig. 4 B). Remarkably, we had previously identified three of these serines, S155, S321 and S449, as crucial for the TCR-induced nuclear exclusion of HDAC7 (11). In addition, considering that the nucleo-cytoplasmic shuttling of class IIa HDACs is dependent on the binding of 14-3-3 proteins, it is very striking to note that the identified PKD sites also match the consensus for 14-3-3 binding R/KXXpSXP, in which pS means phosphoserine (17). Together, these findings strongly suggest that PKD could phosphorylate HDAC7 on S155, S181, S321, and S449. To test this hypothesis, we expressed the HDAC7 sequences surrounding each putative PKD site as GST fusion proteins (GST-S155, GST-S181, GST-S321, and GST-S449, respectively) and examined their ability to be phosphorylated by PKD1 in vitro. Mutant fusion proteins with respective serine to alanine substitutions were used as controls. As shown in Fig. 4 C, wild-type fusion proteins were very efficiently phosphorylated by recombinant active PKD (Fig. 4 C, top, GST-S155, GST-S181, GST-S321, and GST-S449). In addition, substitutions to alanine abolished phosphorylation completely, indicating that phosphorylation occurred specifically on Ser155, Ser181, Ser321, and Ser449 (Fig. 4 C, top, GST-A155, GST-A181, GST-A321, and GST-A449).
Taken together, these data strongly suggest that HDAC7 might be a physiologically relevant substrate for PKD1.
PKD overcomes HDAC7-mediated repression of Nur77 expression
If the phosphorylation sites we have identified in HDAC7 are relevant targets for PKD, then PKD would be expected to alter the subcellular localization of HDAC7. To test this possibility, we cotransfected GFP-HDAC7 and a constitutively active mutant of PKD in DO11.10 cells and examined the localization of HDAC7 by confocal microscopy (Fig. 5 A). As expected, HDAC7 was located mainly in the nucleus in the absence of active PKD1. In contrast, coexpression of active PKD1 induced nuclear export of HDAC7, similar to TCR signaling (Fig. 5 A).
Figure 5.
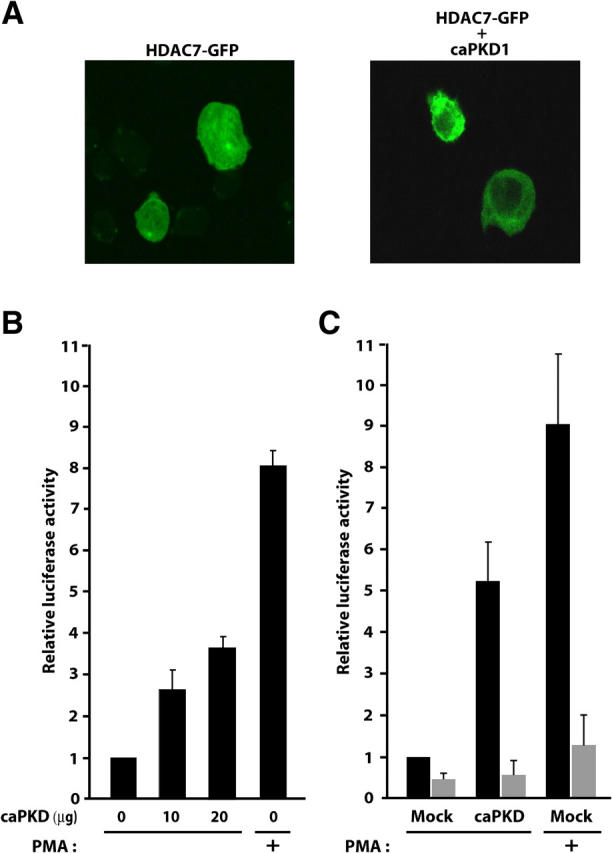
PKD activation is associated with HDAC7 nuclear export and derepression of the Nur77 promoter. (A) DO11.10 cells were transfected with an expression vector encoding GFP-HDAC7 in the absence or presence of a plasmid for activated PKD1. Subcellular localization of HDAC7 was examined by confocal microscopy. (B) Increasing amounts of an expression vector for activated PKD1 were cotransfected with a luciferase reporter plasmid driven by the Nur77 promoter (pNur77-Luc) into DO11.10 cells. The activity of the Nur77 promoter is shown relative to its basal activity, measured in the absence of activated PKD1 using dual luciferase assay. Luciferase activities were measured in untreated (−) cells or cells treated with PMA for 4 h (+). Results are from at least three independent experiments each performed in triplicate. (C) DO11.10 cells were cotransfected with Nur77-responsive reporter constructs pNBREwt-Luc (black bars) or pNBREmut-Luc (gray bars) along with an expression vector for active PKD1. Luciferase activities are presented relative to the basal luciferase activity of pNBREwt-Luc, measured in the absence of active PKD1 using the dual luciferase assay. Luciferase activities were measured in untreated (−) cells or cells treated with PMA for 4 h (+). Results are from at least three independent experiments each performed in triplicate.
We have previously shown that HDAC7 inhibits Nur77 expression and that this inhibitory action is controlled by its subcellular localization (11). As PKD promotes nuclear export of HDAC7, we investigated whether PKD would overcome the inhibitory activity of endogenous HDAC7 on the Nur77 promoter and activate its transcription in the absence of TCR signaling. We first examined the effect of PKD1 on the transcriptional activity of the isolated Nur77 promoter in a reporter assay. Constitutively active PKD1 led to a dose-dependent activation of the transfected Nur77 promoter up to about fourfold (Fig. 5 B). For comparison, the PMA-mediated transcriptional activation of the Nur77 promoter in the absence of constitutively active PKD1 was eightfold. To confirm these results, we next evaluated the ability of PKD to activate expression of endogenous Nur77. We observed that constitutively active PKD1 induced a fivefold induction of a Nur77-responsive promoter (pNBRE-Luc) after transfection (Fig. 5 C, black bars). Control treatment with PMA alone led to a ninefold induction of endogenous Nur77 activity. In addition, an NBRE-luc construct containing mutated Nur77 binding sites was only marginally affected by active PKD or PMA treatment (Fig. 5 C, gray bars).
To further demonstrate that PKD is involved in the control of Nur77 expression, we examined the capacity of chemicals inhibitors of PKD to interfere with the TCR-mediated activation of the endogenous Nur77 promoter. The Nur77 protein is not expressed in DO.11.10 cells but was highly induced by PMA treatment (Fig. 6 A, middle). PKD1 activation was observed concomitantly, confirming our previous observations (Fig. 6 A, top). Strikingly, treatment of cells with staurosporine or Gö6976 markedly impaired activation of PKD1 and concomitantly noticeably reduced Nur77 induction. In contrast, Ro-318220 and GF109203x, which have no effect on PKD, did not interfere with Nur77 activation.
Figure 6.
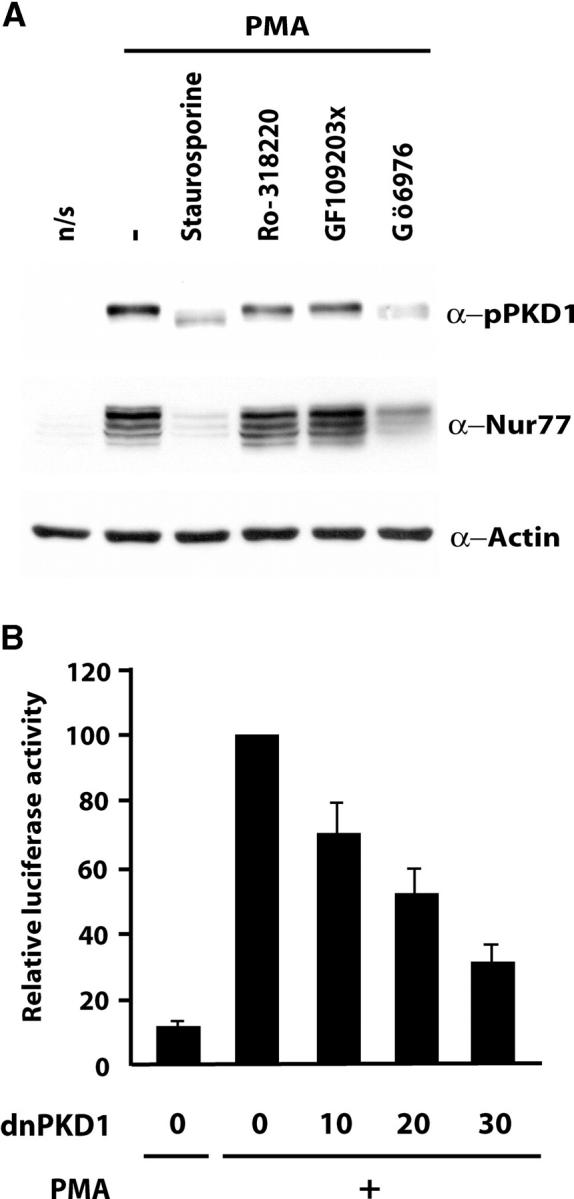
Repression of PKD is associated with reduced Nur77 promoter activity. (A) DO11.10 cells were left unstimulated (n/s) or treated with PMA with or without (−) previous incubation with the indicated inhibitors. After 2 h of PMA treatment, total cellular extracts were prepared and analyzed by Western blotting with antisera for activated PKD1 (pPKD1), Nur77, and actin. (B) DO11.10 cells were transfected with pNur77-Luc and increasing amounts of an expression vector coding for a dominant negative mutant of PKD1. 40 h after transfection, cells were left unstimulated (−) or activated with PMA for 4 h (+). The activity of the Nur77 promoter is shown relative to its activity after PMA treatment in the absence of dominant negative PKD1 measured with the dual luciferase assay. Results are from three independent experiments each performed in triplicate.
To independently demonstrate that PKD1 is involved in TCR-induced activation of Nur77, we assessed the effect of specific PKD1 inhibition on the transcriptional activity of the Nur77 promoter. As expected, PMA treatment led to a 10-fold increase in the activity of the transfected Nur77 promoter. This activation was inhibited in a dose-dependent manner by expression of a dominant negative mutant of PKD1 (Fig. 6 B).
Together, these data suggest that PKD participates in TCR-induced Nur77 expression by interfering with the inhibitory activity of HDAC7.
PKD mediates TCR-induced Nur77 expression and apoptosis
PKD is uniquely selective for substrates with a leucine residue at position −5 relative to the phosphorylated serine (21). To demonstrate that PKD is the specific TCR-activated kinase of HDAC7, we first examined the role of these critical leucine residues in the in vitro phosphorylation of HDAC7 by PKD. HDAC7 sequences centered on each phosphorylatable serine, S155, S181, S321, and S449, were expressed as GST fusion proteins (GST-L150, GST-L176, GST-L316, and GST-L444, respectively) along with the corresponding mutant fusion proteins, in which the −5 leucine was substituted with an alanine (GST-A150, GST-A176, GST-A316, and GST-A444, respectively). As expected, wild-type fusion proteins were efficiently phosphorylated by recombinant active PKD (Fig. 7 A, top). Single substitution of the −5 leucine to alanine dramatically decreased phosphorylation by PKD of S181, S321, and S449 (Fig. 7 A, top). Surprisingly, mutation of L150 into A150 had little to no effect on phosphorylation of S155 by PKD.
Figure 7.

PKD regulates Nur77 expression in response to TCR stimulation. (A) GST fusion proteins corresponding to the sequences surrounding the four putative PKD phosphorylation sites in HDAC7 were used independently as substrates in IVK assays with constitutively active recombinant PKD1. Corresponding mutant fusion proteins, harboring a leucine to alanine substitution at position −5 relative to the phosphorylated serine, were tested in parallel. IVK reactions were resolved by SDS-PAGE. The gel was stained with Coomassie, dried, and analyzed by autoradiography. The arrow indicates the radioactive band corresponding to autophosphorylated PKD1. (B) Polyclonal DO11.10 cells stably expressing wild-type HDAC7 (HDAC7wt) or the L150/176/316/444A HDAC7 mutant (HDAC7ΔL), both fused to GFP, were treated with PMA. Subcellular localization of fluorescent proteins was monitored by confocal microscopy after 2 h of treatment. Bar histograms represent the mean percentages of cells showing nuclear exclusion. (C) Polyclonal DO11.10 cell lines, stably expressing FLAG-tagged versions of HDAC7 (wt) and the HDAC7 mutant (HDAC7ΔL) or the empty vector (Empty), were left unstimulated (−) or stimulated with PMA (+) for 1. 5 h. Total cellular extracts were prepared and analyzed by Western blotting with antisera specific for FLAG, Nur77, and actin. (D) DO11.10 cells were transfected with the pNur77-Luc reporter construct and the same quantity of empty vector (Mock), wild-type HDAC7 (WT), or the ΔL mutant (ΔL). Luciferase activity was measured in cells treated with PMA (left, +) or ionomycin (right, +) and untreated cells (−). The activity of the Nur77 promoter is expressed relative to its activity in the mock-transfected cells after PMA (left) or ionomycin (right) treatment as measured by dual luciferase assay. Results are from three independent experiments each performed in triplicate.
Based on these results, we generated an HDAC7 mutant specifically deficient for phosphorylation by PKD (HDAC7ΔL) in which the leucine residues at position −5 of each phosphorylated serines (namely L150, L176, L316, and L444) were mutated into alanines. To then assess the subcellular localization of the ΔL mutant protein upon PMA activation, we generated polyclonal DO11.10 cell lines stably expressing a HDAC7ΔL-GFP construct. In unstimulated cells, HDAC7ΔL localized mainly in the nucleus (not depicted), similar to wild-type HDAC7. Although HDAC7 was efficiently exported out of the nucleus upon treatment with PMA, the HDAC7ΔL mutant remained nuclear in response to the same treatment (Fig. 7 B).
Nuclear exclusion of HDAC7 is a prerequisite for Nur77 expression (11). Therefore, we would expect the HDAC7ΔL mutant to exhibit stronger repression over the Nur77 promoter. To verify this hypothesis, we established polyclonal cell lines by infecting DO11.10 cells with a retrovirus expressing wild-type HDAC7 or the ΔL mutant. Endogenous Nur77 expression was analyzed in these cell lines after PMA treatment. As expected, Nur77 induction was weakly reduced in cells stably expressing wild-type HDAC7 (Fig. 7 C, middle). Despite being expressed at significantly lower levels (Fig. 7 C, top), the ΔL mutant was associated with a significantly stronger inhibition of Nur77 expression (Fig. 7 C, middle). These observations were confirmed using a Nur77 promoter reporter construct (Fig. 7 D, left). Compared with wild-type HDAC7, the ΔL mutant consistently exhibited a threefold stronger repression effect over PMA-induced activity of the Nur77 promoter activity (Fig. 7 D, left). Interestingly, when the Nur77 promoter activity was induced with ionomycin treatment, the export deficient mutant HDAC7-ΔL did not show any additional repressive effect over wild-type HDAC7 (Fig. 7 D, right).
Next, we reasoned that impairment of HDAC7 phosphorylation by PKD should lead to a stronger repression of TCR-induced apoptosis. We used polyclonal cell lines stably expressing wild-type HDAC7 or the ΔL mutant and measured their apoptotic rates in response to PMA/ionomycin or anti-CD3 antibodies (Fig. 8 A). Overexpression of wild-type HDAC7 weakly inhibited apoptosis induced by PMA/ionomycin or anti-CD3 antibodies (Fig. 8 A, WT). Consistent with our hypothesis, overexpression of the ΔL mutant was associated with a significantly stronger inhibition of apoptosis (Fig. 8, A and B, ΔL). Importantly, this protective effect was not observed when cell death was induced with cycloheximide (Fig. 8 A). Taken together, these data demonstrate that HDAC7 is a physiologically relevant substrate for PKD1 and strongly support a role for PKD1 in TCR-induced apoptosis of thymocytes.
Figure 8.

PKD regulates TCR-mediated apoptosis. (A) Polyclonal DO11.10 cells lines, stably expressing wild-type HDAC7 (WT), HDAC7ΔL (ΔL), or the empty vector (Mock) were left untreated or treated with PMA/ionomycin, anti-CD3 antibodies, or cycloheximide (CHX) for 20 h. Apoptotic cells were detected by PE–annexin V staining and flow cytometry. Flow histograms illustrate the percentages of apoptotic cells detected in one representative experiment. (B) Mean apoptotic rates from nine independent experiments (as described in A) performed from three independently established set of polyclonal cell lines are shown. Results are expressed relative to the apoptotic rate observed in the mock-transduced cell line and are shown as histograms.
Discussion
During thymic education, negative selection of T cells is associated with the activation of a specific apoptotic program. One of the crucial mediators of negative selection in thymocytes and T cell hybridomas is the orphan nuclear receptor Nur77, whose expression is rapidly induced during TCR-mediated apoptosis. Expression of Nur77 is tightly controlled by several proteins, amongst which the transcription factor MEF2D plays a central role by alternatively recruiting repressors or activators. We recently identified HDAC7, a class IIa HDAC, as a major MEF2D-associated transcriptional repressor of Nur77 in unstimulated thymocytes (11). Derepression of Nur77 and induction of apoptosis occurs through TCR-mediated phosphorylation of HDAC7 and subsequent nuclear exclusion. To investigate the involvement of protein kinases in the TCR-induced nuclear exclusion of HDAC7, we used several serine/threonine protein kinase inhibitors. However, kinase inhibitor studies should always be regarded with a degree of caution. Indeed, Gö6976 was also reported to be an efficient inhibitor of MAPKAP-K1b, MSK1, PKCα, and CHK1, although at a slightly higher concentration (1 μM; reference 22). Thus, we resorted to a more specific approach based on the use of a dominant negative mutant of PKD1. Our results provide strong evidence that PKD1 contributes to Nur77 expression by phosphorylating HDAC7 in response to TCR signaling during negative selection of thymocytes. However, additional experiments will be required to confirm this model.
Previous work identified KidIns220 (kinase D–interacting substrate of 220 kD), a lipid raft–associated protein selectively expressed in brain and neuroendocrine cells, as an interacting partner and a substrate for PKD (23). However, the true biological effect of KidIns220 phosphorylation remains to be elucidated. PKD was also shown to phosphorylate S351 of the RAS effector RIN1 (24). Interestingly, the authors suggested that phosphorylation of serine 351 by PKD might create a 14-3-3 binding site in RIN1. Remarkably, the sequence around S351 of RIN1 (SL346LRSMSAAF354) perfectly fits with the LXR/KXXS consensus motif that is also present in the four PKD sites identified in this study in HDAC7. We and others had previously associated phosphorylation of S155, S321, and S449 of HDAC7 with 14-3-3 binding and inactivation through nuclear export (11, 17). Thus, our findings provide additional evidence that PKD may function as a regulator of 14-3-3 binding to several cellular proteins. Identification of additional endogenous substrates of PKD should confirm this hypothesis.
It has been proposed that PKD function may also be dependent on the intracellular localization of the kinase (25). In B cells, antigen receptor stimulation rapidly activates PKD, which transiently translocates to the plasma membrane and then redistributes to the cytosol (26). Based on the B cell model, it is thought that PKD would localize first at the plasma membrane and then in the cytosol during TCR activation of T cells. Here, we show that stimulation of the TCR induces PKD-dependent phosphorylation of a nuclear protein, HDAC7, which then shuttles from the nucleus to the cytoplasm. Thus, our results imply that active PKD would be found, at least in some conditions, in the nucleus of TCR-stimulated thymocytes. This would be reminiscent of the nucleo-cytoplasmic shuttling observed for PKD in response to G protein–coupled receptor agonists or oxidative stress (27–29). These mechanisms await further studies to be fully defined in T cells.
In fibroblast and epithelial cells, PKD has been implicated in many important cellular processes, including function and organization of the Golgi apparatus (30–32), normal and tumor cell proliferation (33, 34), cancer cell invasion (35), and apoptosis (36, 37). Several interesting observations initially suggested a key role for PKD in lymphoid cells. B cell receptor or TCR engagement rapidly induces activation of PKD in B cells, peripheral blood T lymphoblasts, and Jurkat cells (38). However, very little information is available for the true biological function of PKD in T cells. In this study, we provide direct experimental evidence supporting a role for PKD in T cells, where it regulates Nur77 expression and the rate of apoptosis in response to TCR engagement. Recently, an elegant study used the CD2 promoter and locus control region to express a membrane- or cytosol-targeted active PKD in transgenic mice T cells (39). Expression of active PKD in the cytosol had a dramatic effect on thymocyte cellularity, with transgenic mice having reduced cell numbers and lacking DP and single positive (SP) compared with control littermates. This was partially explained by the capacity of cytosolic PKD to suppress rearrangement and expression of TCRβ subunits, a mechanism reminiscent of allelic exclusion. However, even in the presence of a functional TCR complex, cytosolic PKD prevented development of DP and SP thymocytes (39). This suggests that expression of constitutively active PKD in the cytoplasm could perturb thymocyte maturation downstream of TCR rearrangement. In light of our new findings, this perturbation could be partially due to an early derepression of the Nur77 promoter and premature engagement of negative selection programs in early DP thymocytes. Indeed, the cytosolic-targeted PKD used in the transgenic mice study was generated by deletion of the DAG-binding domain, which targets the protein to the membrane. One could imagine that this mutant would retain its capacity to locate to the nucleus and induces expression of Nur77 in the absence of TCR stimulation. Interestingly, decreased thymic cellularity, absence of DP and SP thymocytes, and reduced TCR αβ expression were amongst the phenotypes observed in thymi from Nur77-transgenic mice (8).
Multiple studies have demonstrated the role of Ca2+ signaling in TCR-induced transcriptional activation of Nur77 (7, 10, 40). However, published data indicate that Ca2+-independent pathways also contribute significantly to Nur77 expression (41). At moderate levels, Nur77 mRNA and protein are rapidly induced in thymocytes stimulated with phorbol ester (7). This observation can now be interpreted in light of our current results. Indeed, activation of PKD by phorbol ester would induce phosphorylation-dependent nuclear export of HDAC7. Subsequently, this would lead to the establishment of a chromatin environment favorable to Nur77 transcription and transactivation by other Ca2+-independent coactivators such as ERK5 (42). Maximal induction of Nur77 would require Ca2+-dependent inactivation of additional repressors, such as Cabin-1 (10), and recruitment of Ca2+-dependent (43) coactivators, such as NFAT. This coordinated control through release of repressors and recruitment of coactivators would constitute a tightly regulated mechanism whereby T cells can activate Nur77 transcription to a high level in a relatively short time.
The ability of recombinant CaMK to induce class IIa HDAC nuclear export in transfected cells suggested that CaMK might be involved in the transduction of extracellular stimuli through HDAC inactivation (15). CaMKI has been shown to induce nuclear export of HDAC7 when transfected into Hela cells (17). However, here we provide in vitro and in vivo experimental evidence that signal-responsive kinases other than CaMKs are involved in this process. We identified PKD1 as the kinase responsible for the direct phosphorylation and nuclear exclusion of HDAC7 after TCR stimulation of thymocytes. Interestingly, in these cells, activation of the CaMKs with ionomycin did not alter the subcellular localization of HDAC7, suggesting that HDAC7 is not responsive to the CaMK pathway in T cell hybridomas. This further suggests that class IIa HDACs might be regulated by different signaling pathways depending on the cellular context and/or the extracellular stimuli.
Previous data from us and others supported a role for S155, S321, and S449 in the nuclear exclusion mechanism of HDAC7 (11, 17). Data presented in this study identified S181 as an additional phosphorylation site in the amino terminus of HDAC7. Although the role of S181 as a true 14-3-3 binding site remains to be directly tested, sequences flanking S181 are very similar to those around S321 and S449 and match the optimal binding motif for 14-3-3. Interestingly, although S155, S321, and S449 are conserved amongst other members of the class IIa, S181 is uniquely present in HDAC7. This could support a model in which each member of the class IIa HDAC might be regulated differentially through phosphorylation.
Intriguingly, all four phosphorylation sites within HDAC7 are not equivalent. We and others have already shown that S155 was the most critical for HDAC7 nucleo-cytoplasmic regulation (11, 17). Sequence comparison showed that S155 is the most divergent of the four phosphorylation sites (refer to Fig. 4). In addition, here we show that S155 in vitro phosphorylation by PKD1 is uniquely insensitive to an L150A substitution. The identification of multiple phosphorylation sites in HDAC7 and the fact that they present specific properties raise the interesting possibility that combinatorial phosphorylation of these sites would differentially impact HDAC7 function and regulation.
Class IIa HDACs regulate myogenesis and cardiac hypertrophy (18, 19). As PKD had previously been implicated in cardiac activity (44 and unpublished data) and hypertrophy (45), it is intriguing to speculate that it could identically control these biological processes by promoting nuclear export of class IIa HDACs.
materials and methods
Plasmids, antibodies, and chemicals
C-terminal GFP proteins of human HDAC7 were constructed by inserting the full-length HDAC7 ORF (46) into peGFP-N1 (CLONTECH Laboratories, Inc.) as described previously (11). The NH2-terminal GST fusion proteins, corresponding to the NH2 (aa 1–490) and COOH terminus (aa 490–915) of HDAC7 have been described elsewhere (46). GST-S155, GST-S181, GST-S321, and GST-S449 constructs contain aa 130–180, 156–216, 267–345, and 396–490, respectively, of HDAC7 cloned into pGEX4T1 (Amersham Biosciences). Serine to alanine and leucine to alanine substitutions were introduced by PCR, and mutations were verified by direct DNA sequencing according to standard methods. The luciferase reporter plasmids driven by the Nur77 promoter (pNur77-Luc) and the wild-type and mutant Nur77-responsive promoters (pNBREwt-Luc and pNBREmut-Luc) have already been described (7, 11). Mammalian expression vectors encoding constitutively active (S744E/S748E mutant) and dominant negative (K628N) mutants of PKD1 have been described (20).
Anti-FLAG, anti-panPKC, and anti-actin antibodies were purchased from Santa Cruz Biotechnology, Inc. Anti–mouse Nur77 was from BD Biosciences. Rabbit polyclonal antibodies against PKD1 or PKD1 phosphorylated at serine 916 were generated by immunization with the KLH-linked peptides CEEREMKALSERVSIL and CERVpS916IL, respectively.
Phorbol 12-myristate 13-acetate (PMA), ionomycin, and cycloheximide were purchased from Sigma-Aldrich. Staurosporine, GF109203x, Ro-31-8220, and Gö6976 were obtained from Qbiogene. Kinase inhibitors were used at the sublethal concentrations of 200 nM for staurosporine, GF109203x, and Ro-31-8220 and 400 nM for Gö6976. Cycloheximide was used at 20 μg/ml.
Cell culture, transfections, and reporter assays
DO11.10 T cell hybridomas were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, and 50 U/ml streptomycin/penicillin. Transfections were performed by the DEAE-dextran-chloroquine method. When specified, 10 ng/ml PMA and 0.5 μM ionomycin were added separately or in combination 40 h after transfection. For anti-CD3 stimulation, monoclonal antibody 5002A was used as described previously (11). Protein kinase inhibitors were added 30 min before the stimulation. For reporter assays, all transfections were performed in triplicate and are represented as the mean of at least three independent experiments. Cells were stimulated 4 h before harvesting, and reporter assays were performed according to the dual luciferase reporter assay's recommendations (Promega).
Immunofluorescence
For immunofluorescence experiments, HDAC7-GFP constructs were transiently transfected or stably transduced into DO11.10 cells. GFP-expressing cells were left unstimulated or stimulated with PMA, ionomycin, PMA/ionomycin, or anti-CD3 antibodies in the presence or absence of the indicated protein kinase inhibitor. Localization of the fluorescent proteins was assessed by confocal microscopy (Axivert 200 with LSM 510; Carl Zeiss MicroImaging, Inc.). When indicated, the average percentage of cells showing nuclear exclusion of the GFP-tagged protein was assessed by examining at least three independent fields each containing >50 cells.
GST fusion proteins: expression, purification, and pull-downs
Fusion proteins were expressed in BL21 RIP (Stratagene) and purified according to protocols described elsewhere (47). Recombinant, active GST-PKD1 was produced and purified as described previously (20).
For pull-down reactions, GST fusion proteins were incubated with total extracts from unstimulated, actively growing DO11.10 cells. After rocking for 3 h at 4°C, reactions were washed three times in IPLS buffer (47), three times in IPHS buffer (47), and two times in IPLS buffer. Pull-down reactions were then processed for Western blotting analysis.
IVK assays
Purified GST fusion proteins were incubated with a dilution of 0.5 μg/ml of recombinant active PKD1 in 30 μl of phosphorylation mix containing 10 μM ATP, 10 μCi [γ-32P]ATP, 25 mM Tris, pH 7.5, and 10 mM MgCl2. After 30 min at 30°C, reactions were terminated by adding an equal amount of 2× SDS-PAGE sample buffer, and phosphorylated proteins were visualized by SDS-PAGE analysis and autoradiography.
SDS-PAGE and Western blotting
SDS-PAGE and Western blot analysis were performed according to standard procedures (48). Western blots were developed with the ECL detection kit (Amersham Biosciences).
Polyclonal cell lines
HDAC7 constructs were cloned into MSCV-based retroviral vectors without (for HDAC7-GFP constructs) or with (for HDAC7-FLAG–tagged constructs) an internal ribosome entry site allowing for simultaneous expression of HDAC7 and enhanced GFP (49). Production of recombinant infectious particles and infection of DO.11.10 cells were performed as described previously (11). After 48 h, GFP-expressing cells were sorted by flow cytometry using a FACS Vantage (Becton Dickinson) and expanded. Typically, the polyclonal cell lines contained >99% of GFP+ cells.
For apoptosis assay, we used PE-coupled annexin V (BD Biosciences) according to the manufacturer's instructions. Cell viability was assessed by flow cytometry using a FACScan cytometer (Becton Dickinson).
Acknowledgments
We thank Jean-Claude Twizere, Sabine Ruidant, and Wolfgang Fischle for helpful discussion.
This work was supported by the Fond National de la Recherche Scientific (F.N.R.S), the Belgian Federation against Cancer, the “Fonds voor Wetenschappelijk Onderzoek-Vlaanderen,” the Belgian Government (grant no. IUAP P5/12), the Association for International Cancer Research, INTAS (grant no. 2118), the Flemish Government (grant nos. GOA and BWTS), and the Deutsche Forschungsgemeinschaft (grant nos. SFB 518/B3 and SE 676/5-1). F. Dequiedt and R. Wattiez are Research Associates, and R. Kettmann is Research Director of the F.N.R.S.
The authors have no conflicting financial interests.
Abbreviations used: aa, amino acids; CaMK, Ca2+/calmodulin-dependent kinase; DP, double positive; GST, glutathione S-transferase; HDAC, histone deacetylase; IVK, in vitro kinase; PKC, protein kinase C; PKD, protein kinase D; SP, single positive.
F. Dequiedt and J. Van Lint contributed equally to this work.
References
- 1.Sprent, J., and H. Kishimoto. 2002. The thymus and negative selection. Immunol. Rev. 185: 126–135. [DOI] [PubMed] [Google Scholar]
- 2.Hogquist, K.A. 2001. Signal strength in thymic selection and lineage commitment. Curr. Opin. Immunol. 13:225–231. [DOI] [PubMed] [Google Scholar]
- 3.Starr, T.K., S.C. Jameson, and K.A. Hogquist. 2003. Positive and negative selection of T cells. Annu. Rev. Immunol. 21:139–176. [DOI] [PubMed] [Google Scholar]
- 4.Berg, L.J., and J. Kang. 2001. Molecular determinants of TCR expression and selection. Curr. Opin. Immunol. 13:232–241. [DOI] [PubMed] [Google Scholar]
- 5.Winoto, A. 1997. Genes involved in T-cell receptor-mediated apoptosis of thymocytes and T-cell hybridomas. Semin. Immunol. 9:51–58. [DOI] [PubMed] [Google Scholar]
- 6.Liu, Z.G., S.W. Smith, K.A. McLaughlin, L.M. Schwartz, and B.A. Osborne. 1994. Apoptotic signals delivered through the T-cell receptor of a T-cell hybrid require the immediate-early gene nur77. Nature. 367:281–284. [DOI] [PubMed] [Google Scholar]
- 7.Woronicz, J.D., A. Lina, B.J. Calnan, S. Szychowski, L. Cheng, and A. Winoto. 1995. Regulation of the Nur77 orphan steroid receptor in activation-induced apoptosis. Mol. Cell. Biol. 15:6364–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weih, F., R. Ryseck, L. Chen, and R. Bravo. 1996. Apoptosis of nur77/N10-transgenic thymocytes involves the Fas/Fas ligand pathway. Proc. Natl. Acad. Sci. USA. 93:5533–5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calnan, B.J., S. Szychowski, F.K. Chan, D. Cado, and A. Winoto. 1995. A role for the orphan steroid receptor Nur77 in apoptosis accompanying antigen-induced negative selection. Immunity. 3:273–282. [DOI] [PubMed] [Google Scholar]
- 10.Youn, H.-D., L. Sun, R. Prywes, and J.O. Liu. 1999. Apoptosis of T cells mediated by Ca2+-induced release of the transcription factor MEF2. Science. 286:790–793. [DOI] [PubMed] [Google Scholar]
- 11.Dequiedt, F., H. Kasler, W. Fischle, V. Kiermer, M. Weinstein, B.G. Herndier, and E. Verdin. 2003. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity. 18:687–698. [DOI] [PubMed] [Google Scholar]
- 12.Verdin, E., F. Dequiedt, and H.G. Kasler. 2003. Class II histone deacetylases: versatile regulators. Trends Genet. 19:286–293. [DOI] [PubMed] [Google Scholar]
- 13.Miska, E.A., C. Karlsson, E. Langley, S.J. Nielsen, J. Pines, and T. Kouzarides. 1999. HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J. 18:5099–5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grozinger, C.M., and S.L. Schreiber. 2000. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA. 97:7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu, J., T.A. McKinsey, R.L. Nicol, and E.N. Olson. 2000. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA. 97:4070–4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang, A.H., M.J. Kruhlak, J. Wu, N.R. Bertos, M. Vezmar, B.I. Posner, D.P. Bazett-Jones, and X.J. Yang. 2000. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 20:6904–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kao, H.-Y., A. Verdel, C.-C. Tsai, C. Simon, H. Juguilon, and S. Khochbin. 2001. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J. Biol. Chem. 276:47496–47507. [DOI] [PubMed] [Google Scholar]
- 18.McKinsey, T.A., C.L. Zhang, J. Lu, and E.N. Olson. 2000. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 408:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang, C.L., T.A. McKinsey, S. Chang, C.L. Antos, J.A. Hill, and E.N. Olson. 2002. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 110:479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vertommen, D., M. Rider, Y. Ni, E. Waelkens, W. Merlevede, J.R. Vandenheede, and J. Van Lint. 2000. Regulation of protein kinase D by multisite phosphorylation. J. Biol. Chem. 275:19567–19576. [DOI] [PubMed] [Google Scholar]
- 21.Nishikawa, K., A. Toker, F.-J. Johannes, Z. Songyang, and L.C. Cantley. 1997. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J. Biol. Chem. 272:952–960. [DOI] [PubMed] [Google Scholar]
- 22.Davies, S.P., H. Reddy, M. Caivano, and P. Cohen. 2000. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 351:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iglesias, T., N. Cabrera-Poch, M.P. Mitchell, T.J.P. Naven, E. Rozengurt, and G. Schiavo. 2000. Identification and cloning of Kidins220, a novel neuronal substrate of protein kinase D. J. Biol. Chem. 275:40048–40056. [DOI] [PubMed] [Google Scholar]
- 24.Wang, Y., R.T. Waldron, A. Dhaka, A. Patel, M.M. Riley, E. Rozengurt, and J. Colicelli. 2002. The RAS effector RIN1 directly competes with RAF and is regulated by 14-3-3 proteins. Mol. Cell. Biol. 22:916–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Lint, J., A. Rykx, Y. Maeda, T. Vantus, S. Sturany, V. Malhotra, J.R. Vandenheede, and T. Seufferlein. 2002. Protein kinase D: an intracellular traffic regulator on the move. Trends Cell Biol. 12:193–200. [DOI] [PubMed] [Google Scholar]
- 26.Matthews, S.A., T. Iglesias, E. Rozengurt, and D. Cantrell. 2000. Spatial and temporal regulation of protein kinase D (PKD). EMBO J. 19:2935–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Waldron, R.T., O. Rey, E. Zhukova, and E. Rozengurt. 2004. Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J. Biol. Chem. 279:27482–27493. [DOI] [PubMed] [Google Scholar]
- 28.Rey, O., J. Sinnett-Smith, E. Zhukova, and E. Rozengurt. 2001. Regulated nucleocytoplasmic transport of protein kinase D in response to G protein-coupled receptor activation. J. Biol. Chem. 276:49228–49235. [DOI] [PubMed] [Google Scholar]
- 29.Rey, O., J. Yuan, S.H. Young, and E. Rozengurt. 2003. Protein kinase C nu/protein kinase D3 nuclear localization, catalytic activation, and intracellular redistribution in response to G protein-coupled receptor agonists. J. Biol. Chem. 278:23773–23785. [DOI] [PubMed] [Google Scholar]
- 30.Liljedahl, M., Y. Maeda, A. Colanzi, I. Ayala, J. Van Lint, and V. Malhotra. 2001. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 104:409–420. [DOI] [PubMed] [Google Scholar]
- 31.Prigozhina, N.L., and C.M. Waterman-Storer. 2004. Protein kinase D-mediated anterograde membrane trafficking is required for fibroblast motility. Curr. Biol. 14:88–98. [DOI] [PubMed] [Google Scholar]
- 32.Yeaman, C., M.I. Ayala, J.R. Wright, F. Bard, C. Bossard, A. Ang, Y. Maeda, T. Seufferlein, I. Mellman, W.J. Nelson, and V. Malhotra. 2004. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat. Cell Biol. 6:106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rennecke, J., P.A. Rehberger, G. Furstenberger, F.J. Johannes, M. Stohr, F. Marks, and K.H. Richter. 1999. Protein-kinase-Cmu expression correlates with enhanced keratinocyte proliferation in normal and neoplastic mouse epidermis and in cell culture. Int. J. Cancer. 80:98–103. [DOI] [PubMed] [Google Scholar]
- 34.Zhukova, E., J. Sinnett-Smith, and E. Rozengurt. 2001. Protein kinase D potentiates DNA synthesis and cell proliferation induced by bombesin, vasopressin, or phorbol esters in Swiss 3T3 cells. J. Biol. Chem. 276:40298–40305. [DOI] [PubMed] [Google Scholar]
- 35.Bowden, E.T., M. Barth, D. Thomas, R.I. Glazer, and S.C. Mueller. 1999. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 18:4440–4449. [DOI] [PubMed] [Google Scholar]
- 36.Waldron, R.T., and E. Rozengurt. 2000. Oxidative stress induces protein kinase D activation in intact cells. J. Biol. Chem. 275:17114–17121. [DOI] [PubMed] [Google Scholar]
- 37.Storz, P., and A. Toker. 2003. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J. 22:109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matthews, S.A., E. Rozengurt, and D. Cantrell. 2000. Protein kinase D: a selective target for antigen receptors and a downstream target for protein kinase C in lymphocytes. J. Exp. Med. 191:2075–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marklund, U., K. Lightfoot, and D. Cantrell. 2003. Intracellular location and cell context-dependent function of protein kinase D. Immunity. 19:491–501. [DOI] [PubMed] [Google Scholar]
- 40.Youn, H.D., and J.O. Liu. 2000. Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity. 13:85–94. [DOI] [PubMed] [Google Scholar]
- 41.Winoto, A., and D.R. Littman. 2002. Nuclear hormone receptors in T lymphocytes. Cell. 109:S57–S66. [DOI] [PubMed] [Google Scholar]
- 42.Kasler, H.G., J. Victoria, O. Duramad, and A. Winoto. 2000. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 20:8382–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blaeser, F., N. Ho, R. Prywes, and T.A. Chatila. 2000. Ca2+-dependent gene expression mediated by MEF2 transcription factors. J. Biol. Chem. 275:197–209. [DOI] [PubMed] [Google Scholar]
- 44.Haworth, R.S., M.W. Goss, E. Rozengurt, and M. Avkiran. 2000. Expression and activity of protein kinase D/protein kinase C mu in myocardium: evidence for alpha1-adrenergic receptor- and protein kinase C-mediated regulation. J. Mol. Cell. Cardiol. 32:1013–1023. [DOI] [PubMed] [Google Scholar]
- 45.Tsybouleva, N., L. Zhang, S. Chen, R. Patel, S. Lutucuta, S. Nemoto, G. DeFreitas, M. Entman, B.A. Carabello, R. Roberts, and A.J. Marian. 2004. Aldosterone, through novel signaling proteins, is a fundamental molecular bridge between the genetic defect and the cardiac phenotype of hypertrophic cardiomyopathy. Circulation. 109:1284–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischle, W., S. Emiliani, M.J. Hendzel, T. Nagase, N. Nomura, W. Voelter, and E. Verdin. 1999. A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J. Biol. Chem. 274:11713–11720. [DOI] [PubMed] [Google Scholar]
- 47.Fischle, W., F. Dequiedt, M.J. Hendzel, M.G. Guenther, M.A. Lazar, W. Voelter, and E. Verdin. 2002. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell. 9:45–57. [DOI] [PubMed] [Google Scholar]
- 48.Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning: A Laboratory Manual. Second Edition. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 1626 pp.
- 49.Hawley, R.G., F.H. Lieu, A.Z. Fong, and T.S. Hawley. 1994. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1:136–138. [PubMed] [Google Scholar]