

Abstract
Interleukin (IL)-2 plays a crucial role in the maintenance of natural immunologic self-tolerance. Neutralization of circulating IL-2 by anti–IL-2 monoclonal antibody for a limited period elicits autoimmune gastritis in BALB/c mice. Similar treatment of diabetes-prone nonobese diabetic mice triggers early onset of diabetes and produces a wide spectrum of T cell–mediated autoimmune diseases, including gastritis, thyroiditis, sialadenitis, and notably, severe neuropathy. Such treatment selectively reduces the number of Foxp3-expressing CD25+ CD4+ T cells, but not CD25− CD4+ T cells, in the thymus and periphery of normal and thymectomized mice. IL-2 neutralization inhibits physiological proliferation of peripheral CD25+ CD4+ T cells that are presumably responding to normal self-antigens, whereas it is unable to inhibit their lymphopenia-induced homeostatic expansion in a T cell–deficient environment. In normal naive mice, CD25low CD4+ nonregulatory T cells actively transcribe the IL-2 gene and secrete IL-2 protein in the physiological state. IL-2 is thus indispensable for the peripheral maintenance of natural CD25+ CD4+ regulatory T cells (T reg cells). The principal physiological source of IL-2 for the maintenance of T reg cells appears to be other T cells, especially CD25low CD4+ activated T cells, which include self-reactive T cells. Furthermore, impairment of this negative feedback loop via IL-2 can be a cause and a predisposing factor for autoimmune disease.
Accumulating evidence indicates that IL-2 may contribute to the maintenance of natural immunologic self-tolerance and prevention of autoimmune disease. For example, IL-2–deficient (IL-2−/−) mice succumb to lethal immunopathology that includes autoimmune components (1, 2). Autoimmune disease, such as type 1 diabetes (T1D), can be prevented in animal models by IL-2 administration (3–5). Allelic variations of the IL-2 gene are associated with genetic susceptibility to T1D and experimental allergic encephalitis (EAE) in animal models (6). Furthermore, the majority of naturally arising CD4+ regulatory T cells (T reg cells) express CD25, the IL-2Rα chain, and abnormality in their development or function leads to various autoimmune diseases, including T1D in rodents and humans (7, 8). These findings collectively indicate a common role of IL-2 in various autoimmune diseases. In this report we have attempted to determine how IL-2 contributes to the development of autoimmune disease, in particular, whether neutralization of circulating IL-2 can cause autoimmune disease in otherwise normal animals.
Naturally arising CD25+ CD4+ T reg cells, which constitute 5–10% of peripheral CD4+ T cells in normal rodents and humans, are produced at least in part by the normal thymus as a functionally mature and distinct subpopulation of T cells (9). They play key roles not only for the maintenance of immunologic self-tolerance, but also for the control of aberrant or excessive immune responses to invading or commensal microbes and to innocuous environmental substances. For example, depletion of CD25-expressing CD4+ T cells elicits autoimmune disease in otherwise normal rodents (8). Genetic abnormality of the Foxp3 gene, which is specifically expressed in natural CD25+ CD4+ T reg cells, impairs their development and function, causing severe autoimmune diseases (in particular T1D), allergy, and inflammatory bowel disease in humans (7). Foxp3 also controls the expression of CD25 in natural T reg cells (10). These findings illustrate CD25 to be a good marker for natural T reg cells. CD25 is, on the other hand, expressed on any T cell upon activation (11, 12). Therefore, a critical issue is to determine whether CD25 is merely an indicator of the chronically activated state of natural T reg cells or a key molecule, specific and essential for their generation, maintenance, or function as an indispensable component of the high affinity IL-2R (13). If the latter is the case, it needs to be determined which cells are the physiological source of IL-2 for natural T reg cells.
Although IL-2 was initially supposed to be an essential cytokine for T cell growth in general, it was later shown that IL-2 deficiency hardly altered the development and function of T cells (14, 15). IL-2 deficiency in mice, however, produced fatal lymphoproliferative inflammatory disease, termed “IL-2 deficiency syndrome,” which is manifested by inflammatory bowel disease, autoimmune hemolytic anemia, splenomegaly, lymphadenopathy, and multi-organ lymphocytic infiltration (1, 2). CD25 or CD122 (IL-2Rβ) deficiency also produced similar lethal immunopathology (16, 17). Despite the ability of IL-2 to induce activation-induced cell death in T cells including self-reactive T cells (18), the IL-2 deficiency syndrome is not a T cell–autonomous disease. For example, IL-2 deficiency substantially reduced natural CD25+ CD4+ T reg cells (19), and transfer of CD25+ CD4+ T cells suppressed IL-2−/− T cell proliferation (20). In addition, inoculation of IL-2–replete spleen cells or thymocytes delayed the onset of multi-organ immunopathology in IL-2−/− mice (21). Furthermore, there is in vivo and in vitro evidence that IL-2 is required for functional activation of T reg cells (22–24). Despite these findings supporting the vital contribution of IL-2 to the development and function of natural T reg cells, it is still unclear whether IL-2 is required for the thymic generation or peripheral expansion/survival of natural T reg cells. It is also obscure whether IL-2 deficiency in normal animals can cause T cell–mediated autoimmune diseases, such as T1D, thyroiditis, and gastritis, similar to those produced in normal mice by depletion of natural T reg cells (8).
In this report, we have further investigated the role of IL-2 in self-tolerance and autoimmune disease, in particular, how IL-2 neutralization affects natural CD25+ CD4+ T reg cells in normal mice and whether this can cause or enhance autoimmune disease in genetically normal or autoimmune-prone mice. We show that (a) IL-2 is essential for physiological expansion and survival of natural CD25+ CD4+ T reg cells in the periphery, (b) CD25low CD4+ T cells, which appear to include self-reactive T cells, physiologically sustain this proliferation/survival of CD25+ CD4+ natural T reg cells by secreting IL-2, and (c) IL-2 neutralization for a limited period can impair this feedback control, reduce natural T reg cells, and elicit T cell–mediated autoimmune disease in normal mice. Furthermore, in genetically autoimmune-prone mice, the treatment enhances predisposed autoimmunity and produces other autoimmune diseases de novo. Thus, IL-2 is a cytokine crucially required for homeostatic maintenance of natural CD25+ CD4+ T reg cells. Genetic anomaly or environmental perturbation of IL-2 formation, and for that matter IL-2R expression and function, can be a cause and also a key predisposing factor for autoimmune disease.
Results
Neutralization of IL-2 by a specific mAb selectively reduces CD25+ CD4+ T cells in normal mice
CD25+ CD4+ CD8− cells in the thymus and spleen of normal naive mice constitutively expressed IL-2Rβ (CD122) as well as IL-2Rγc chain (CD132), whereas CD25− CD4+ CD8− cells expressed only the latter, indicating that CD25+ CD4+ T cells constitutively express the high affinity IL-2R even in the thymus (Fig. 1 A).
Figure 1.
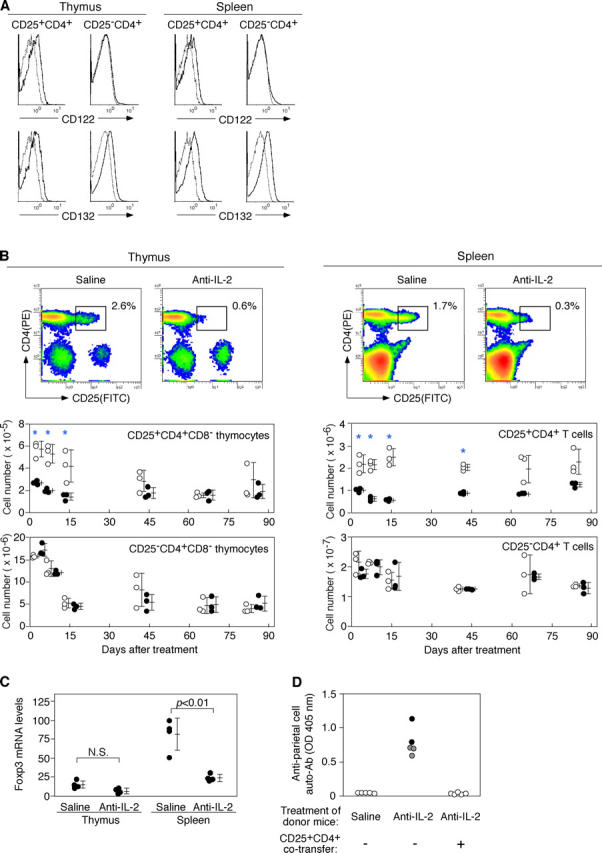
Neutralization of IL-2 by specific mAbs reduces the number of Foxp3 + CD25+ CD4+ T reg cells in normal naive mice. (A) Expression of CD122 (IL-2Rβ) and CD132 (IL-2Rγc) by CD25+ or CD25− CD4+ T cells in the thymus or spleen of normal BALB/c mice. Dotted lines represent control staining with isotype-matched mAb. (B) Representative flow cytometric profiles of thymus and spleen cells from 8-wk-old BALB/c mice 14 d after injection of anti–IL-2 mAb or saline. Kinetics of the number of CD25+ CD4+ T cells and CD25− CD4+ T cells after injection of anti–IL-2 mAb (black circles) or saline (white circles). Asterisks indicate significant differences (P < 0.05). (C) Relative Foxp3 mRNA levels assessed by real-time quantitative PCR with CD4+ CD8− thymocytes or CD4+ splenocytes from mice treated with anti–IL-2 mAbs. Quantity of Foxp3 normalized to HPRT. (D) BALB/c nude mice were i.v. transferred with spleen cells (3 × 107) from BALB/c mice injected with anti–IL-2 mAbs or saline 14 d earlier with or without CD25+ CD4+ T cells (106) from nonmanipulated mice and examined histologically and serologically 3 mo later. Titers of anti-parietal cell autoantibodies were assessed by ELISA. Black circles, macroscopically and histologically evident grade 2 gastritis; gray circles, histologically evident grade 1 gastritis; white circles, intact gastritis mucosa. See reference 55 for histological grading.
To determine the effects of IL-2 neutralization on the composition of CD25+ or CD25− CD4+ T cells in the thymus and the periphery, we i.v. administered 1 mg of neutralizing anti–IL-2 mAb to normal BALB/c mice and assessed the number of CD25+ or CD25− CD4+ CD8− thymocytes/T cells. The treatment reduced the percentages and absolute numbers of CD25+ CD4+ T cells (Fig. 1 B) and GITRhigh CD4+ T cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20041982/DC1) in the thymus, spleen, and lymph nodes (not depicted), confirming a previous result by others for splenocytes (25). Reduction was significant from 3 d after anti–IL-2 injection for nearly 2 mo, gradually recovering to normal levels by 3 mo after treatment. Recovery in the thymus was faster than in the periphery. In contrast, the treatment did not significantly alter the number of CD25− CD4+ T cells in the thymus and spleen.
Because IL-2 regulates the expression of CD25 (26), it is possible that IL-2 neutralization might impair CD25 expression by CD25+ CD4+ T cells rather than reducing their cell number. To examine this possibility, we assessed the expression levels of Foxp3 14 d after anti–IL-2 treatment (10, 27, 28). Splenic CD4+ T cells purified from anti–IL-2–treated mice exhibited significantly (3.4-fold; P < 0.01) reduced amounts of Foxp3 mRNA compared with those from control saline-treated mice (Fig. 1 C). CD4+ CD8− thymocytes from anti–IL-2–treated mice also showed reduced Foxp3 mRNA expression, but the reduction was not so marked as in splenic CD4+ T cells (2.2-fold; P < 0.06), presumably due to physiologically lower expression levels of Foxp3 in thymic CD25+ CD4+ CD8− thymocytes than in CD25+ CD4+ splenic T cells (Fig. S2 A, available at http://www.jem.org/cgi/content/full/jem.20041982/DC1).
To determine then whether anti–IL-2 treatment reduces natural T reg cells with autoimmune-preventive activity, we transferred spleen cells from BALB/c mice 14 d after treatment to syngeneic nude mice and examined possible development of autoimmune disease (8). Fig. 1 D shows that all of the recipients developed histologically and serologically evident autoimmune gastritis, whereas nude mice cell transferred from control BALB/c mice did not. In addition, cotransfer of anti–IL-2–treated spleen cells with CD25+ CD4+ T cells from untreated normal animals inhibited the development of gastritis.
Taken together, these results indicate that IL-2 neutralization can selectively reduce the number of autoimmune-preventive Foxp3-expressing natural CD25+ CD4+ T reg cells in the immune system, especially in the periphery.
IL-2 is required for the maintenance of CD25+ CD4+ T reg cells in the periphery
To determine whether IL-2 neutralization affects the thymic generation, peripheral maintenance (survival and/or growth), or both, of CD25+ CD4+ T reg cells, we thymectomized (Tx) 5–6-wk-old BALB/c mice and injected 1 mg anti–IL-2 mAb or saline 3 wk later. 14 d after anti–IL-2 mAb treatment, the percentage and the number of CD25+ CD4+ T cells decreased equally in Tx and sham-Tx mice (Fig. 2, A and B). This indicates that IL-2 neutralization reduces peripheral CD25+ CD4+ T reg cells whether there is a thymic supply of T reg cells or not.
Figure 2.

IL-2 neutralization decreases the number of CD25+ CD4+ T cells in Tx mice. Adult BALB/c mice were Tx (Tx, +) or sham operated (Tx, −). 3 wk later, animals were injected with anti–IL-2 mAb or saline and the percentages of CD25+ CD4+ T cells among the CD4+ T cells (A) and their absolute numbers (B) were determined on day 14 after antibody injection.
IL-2 is required for in vivo physiological proliferation of CD25+ CD4+ T cells, but not for their lymphopenia-driven expansion
Despite in vitro anergy of CD25+ CD4+ T reg cells, they proliferate on transfer to lymphopenic mice (29, 30). Furthermore, in normal nonlymphopenic mice, peripheral CD25+ CD4+ T cells show more active proliferation than CD25− CD4+ T cells in the physiological state (Fig. 3 A; references 9, 31, and 32). Based on these findings, we first determined whether IL-2 neutralization in normal nonlymphopenic mice blocked natural proliferation of CD25+ CD4+ T cells. Normal BALB/c mice were injected with anti–IL-2 mAb or saline and subsequently with 1 mg 5-bromo-2′-deoxyuridine (BrdU) every 12 h for 3 d. IL-2 neutralization reduced incorporation of BrdU in CD25+ CD4+ T cells to one third of control mice, with no reduction in CD25− CD4+ T cells, indicating that CD25+ CD4+ T cells require IL-2 for their physiological proliferation in the periphery (Fig. 3, A and B).
Figure 3.

Neutralization of IL-2 inhibits natural in vivo proliferation of peripheral CD25+ CD4+ T cells in normal mice. BALB/c mice received one injection of 1 mg anti–IL-2 mAb or saline and were treated with BrdU for the next 3 d. Percentage of BrdU-labeled CD25+ or CD25− CD4+ T cells from anti–IL-2–treated mice or saline-treated mice was assessed by flow cytometry using FITC-labeled anti-BrdU mAb. (A) Representative flow cytometric profiles. (B) Percent of BrdU+ cells among CD25+ or CD25− CD4+ T cells in mice treated with anti–IL-2 mAb or saline. NS, not significant.
To determine then whether IL-2 is also essential for lymphopenia-driven expansion of CD25+ CD4+ T cells, we transferred highly purified (>99%) 5- and 6-carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled CD25+ CD4+ T cells from BALB/c-Thy1.1 congenic mice to BALB/c–RAG-2−/− recipients and treated them with anti–IL-2 mAb to exclude a possible contribution of IL-2 derived from the host (for example, produced by the host DCs; reference 33) or transferred CD25+ CD4+ T cells themselves. Assessment 4 d later revealed that transferred Thy1.1+ T cells had vigorously proliferated, and this proliferation was not affected by anti–IL-2 (Fig. 4 A). This IL-2–independent proliferation was further confirmed by a similar level of proliferation of CD25+ CD4+ T cells transferred to BALB/c–RAG-2−/− IL-2−/− mice (not depicted). Furthermore, when these recipient mice were examined 4 wk later, the number of Thy1.1+ splenic T cells was not significantly different among the recipients, whether they were IL-2+/+ or IL-2−/−, cotransferred with CD25− CD4+ T cells or not, or treated with anti–IL-2 or saline (Fig. 4 B).
Figure 4.

IL-2–independent lymphopenia-driven expansion of CD25+ CD4+ T cells. (A) CD25+ CD4+ T cells (3 × 105) purified from Thy-1.1 BALB/c mice were labeled with CFSE and injected into RAG-2−/− mice that were treated with anti–IL-2 mAb or control rat IgG and assessed for the percentage of CFSE-labeled cells 4 d later. (B) CD25+ CD4+ T cells (3 × 105) highly purified from BALB/c-Thy1.1 mice were transferred to BALB/c IL-2+/+ or IL-2−/− BALB/c RAG-2−/− mice. A group of IL-2−/− RAG-2−/− BALB/c mice transferred with CD25+ CD4+ T cells were treated with anti–IL-2 mAb. Another group of IL-2−/− RAG-2−/− BALB/c mice received a cotransfer of an equal number (3 × 105) of CD25+ CD4+ T cells and CD25− CD4+ T cells sorted from the lymph nodes of normal BALB/c mice. The numbers of Thy1.1+ TCRβ+ cells recovered in the spleen of individual recipients were assessed 4 wk after transfer.
Taken together, these results indicate that IL-2 is an indispensable growth factor for physiological proliferation and survival of CD25+ CD4+ T reg cells in normal nonlymphopenic conditions but dispensable for lymphopenia-induced homeostatic expansion or survival.
IL-2 transcription is confined to CD25low CD4+ nonregulatory T cells in normal naive mice
Next, we attempted to determine which cells produce the IL-2 required for the growth/survival of CD25+ CD4+ T reg cells in normal naive nonlymphopenic mice. We quantitatively assessed IL-2 mRNA expression by real-time PCR in various cell populations prepared from the spleens of normal naive BALB/c mice. The analyzed populations included macrophages (CD11b+), DCs (CD11chigh MHCII+), B cells (CD19+), NKT cells (TCRβ+ α-GalCer/CD1d-tetramer+), NK cells (DX5+ TCRβ−), CD8+ T cells and CD4+ T cells, which were sorted to CD25−, CD25low, and CD25high cells (Fig. 5 A). High IL-2 transcription was detected in CD4+ T cells, especially CD25low CD4+ T cells, and to lesser degrees in NKT, NK, and CD8+ T cells. In accord with this, CD25low CD4+ T cells secreted IL-2 in significantly higher amounts than CD25high or CD25− CD4+ T cells during 24 h in vitro culture (Fig. 5 A). Similar results were obtained with DO11.10 TCR transgenic mice. CD25low CD4+ T cells showed an approximately five- or twofold higher transcription of IL-2 than CD25high or CD25− CD4+ T cells, respectively, whereas CD4+ T cells from RAG−/− DO11.10 mice, which lack CD25+ CD4+ T reg cells (34), scarcely transcribed the gene (Fig. 5 B). This CD25low CD4+ population was mainly composed of KJ1-26negative-low T cells similar to CD25high CD4+ T cells, indicating that CD25low CD4+ T cells expressed endogenous TCR α chains paired with transgenic TCR β chains. In contrast to IL-2 transcription, Foxp3 expression was higher (approximately threefold) in the CD25high population than in the CD25low CD4+ population in BALB/c mice and DO11.10 transgenic mice, exhibiting an inverse correlation between IL-2 and Foxp3 expression (Fig. 5 C; reference 10). Furthermore, consistent with their low Foxp3 expression status, CD25low CD4+ T cells failed to suppress the anti-CD3–driven proliferation of CD25− CD4+ T cells in vitro, contrasting with potent suppression by CD25high CD4+ T cells (Fig. 5 D). Notably, however, not only CD25high but also CD25low CD4+ T cells proliferated poorly upon anti-CD3 stimulation (Fig. 5 D). Judging from low level expression of Foxp3 by CD25low CD4+ T cells (10-fold less than CD25high cells), their hypo-responsiveness could be attributed to suppression by a small number of contaminating Foxp3 high CD25high T reg cells due to the limit of fidelity in cell sorting. Alternatively, Foxp3 low CD25low CD4+ T cells themselves might be hypoproliferative but not suppressive. Likewise, small amounts of IL-2 mRNA detected in the CD25high or CD25− fraction could be due to contaminating IL-2–secreting CD25low cells or production of a small amount of IL-2 by these populations.
Figure 5.

Active transcription of the IL-2 gene in CD25low CD4+ nonregulatory T cells in the spleens of normal BALB/c mice. (A) Splenic CD4+ T cells were sorted into CD25high, CD25low, and CD25− subpopulations as shown and compared with other cell populations in the spleen (see Results) for expression levels of IL-2 mRNA assessed by real-time quantitative RT-PCR (normalized to HPRT). The average IL-2 mRNA levels of total unseparated spleen cells was defined to be 1. The level of IL-2 mRNA in in vitro–activated CD25− CD4+ T cells (stimulated for 24 h with precoated 1 μg/ml anti-CD3 and 1 μg/ml of soluble anti-CD28) was 1,000–1,500 (not depicted). Sorted CD25high, CD25low, and CD25− CD4+ T cells were cultured for 24 h, and the IL-2 concentrations in the supernatant were determined by ELISA. The results of three independent experiments are shown. (B) CD25high, CD25low, and CD25− CD4+ populations from DO11.10 TCR transgenic mice (DO.RAG+) or the CD25− CD4+ population from RAG−/− DO11.10 transgenic mice (DO.RAG−) were assessed for the expression of IL-2 mRNA by real-time PCR and for the expression of transgenic TCR by FACS analysis using the clonotypic KJ1-26 mAb. (C) Relative Foxp3 mRNA levels in CD25high, CD25low, or CD25− CD4+ T cells from the spleens of DO.RAG+, DO.RAG−, or normal BALB/c mice. (D) In vitro responsiveness and suppressive activity of splenic CD25high or CD25low CD4+ T cells from normal BALB/c mice. These populations were stimulated for 3 d with anti-CD3 mAb and APC either alone or in the presence of an equal number of CD25− CD4+ responder T cells, and [3H]thymidine incorporation was measured. A representative of three independent experiments is shown in A–D. N.D., none detected.
CD25low CD4+ CD8− thymocytes were present in the thymus of normal naive mice and expressed Foxp3 mRNA at levels one third of thymic CD25high CD4+ T cells (Fig. S2 A). The IL-2 mRNA levels of CD25low CD4+ CD8− thymocytes were, however, much lower than the levels of the peripheral counterpart and equivalent to those of CD25high CD4+ CD8− thymocytes (Fig. S2 B). In addition, in contrast with splenic CD25low CD4+ T cells, which were higher in CD44 expression than peripheral CD25high or CD25− CD4+ T cells, CD25low CD4+ CD8− thymocytes were intermediate in CD44 expression between CD25high and CD25− CD4+ thymocytes (Fig. S2 C). These data indicate that splenic CD25low CD4+ T cells are activated in the periphery and therefore actively transcribe IL-2.
Taken together, the CD25+ CD4+ T cell population in normal naive mice is functionally heterogeneous, containing activated nonregulatory CD25low T cells, which might be the main source of IL-2 in normal naive mice. Assuming that CD25+ CD4+ T cells that express endogenous TCR α chains in TCR transgenic mice are more self-reactive than other T cells (34), such CD25low CD4+ T cells might be more self-reactive than CD25− CD4+ T cells and therefore activated by self-antigens in the physiological state and actively forming IL-2.
Induction of autoimmune gastritis in BALB/c mice by anti–IL-2 mAb treatment
Based on the result that anti–IL-2 mAb administration reduced the number of functional T reg cells (Fig. 1), we examined whether autoimmune disease would develop in normal BALB/c mice after anti–IL-2 mAb injection on days 10 and 20 after birth. Such treatment indeed induced by 3 mo of age histologically evident gastritis accompanying the appearance of anti-parietal cell autoantibodies in the circulation (Fig. 6, A and B). Furthermore, splenocytes from gastritis-bearing BALB/c mice adoptively transferred gastritis to BALB/c athymic nude mice, indicating that the lesion was a bona fide T cell–mediated autoimmune disease (Fig. 6 C).
Figure 6.

Induction of autoimmune gastritis in BALB/c mice by IL-2 neutralization. BALB/c mice were i.p. injected with 1 mg purified anti–IL-2 mAb or saline on days 10 and 20 after birth and were examined at 3 mo of age for histological and serological evidence for gastritis. (A) Hematoxylin and eosin staining of gastric mucosa of anti–IL-2–treated and saline-treated mice (a magnification of 20). (B) Titers of anti-parietal cell autoantibodies were determined by ELISA. The gastric lesion was histologically graded as described in Fig. 1 D. (C) 3 × 107 spleen cells from anti–IL-2–treated mice showing gastritis or from saline-treated control mice were i.v. transferred to BALB/c athymic nude mice, which were histologically and serologically examined 3 mo later. Gastritis was scored as in Fig. 1 D.
This anti–IL-2 mAb treatment of young mice significantly reduced the number of CD25+ CD4+ T cells for nearly 2 mo. The number gradually recovered to the control level by 3 mo after treatment (Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20041982/DC1), similar to the result with adult mice (Fig. 1 B). The recovered CD25+ CD4+ T cells were equally anergic and exhibited comparative degrees of suppressive activity and similar levels of Foxp3 mRNA expression as those from control mice (Fig. S3, B and C), indicating that the majority of them were T reg cells but not activated autoimmune T cells. Thus, IL-2 neutralization and the resulting transient T reg cell deficiency is sufficient to elicit autoimmune disease in otherwise normal mice.
Induction of a wide spectrum of organ-specific autoimmune diseases including autoimmune neuritis in nonobese diabetic (NOD) mice by IL-2 neutralization
Diabetes-prone NOD mice are known to be genetically susceptible to other organ-specific autoimmune diseases such as thyroiditis and sialadenitis (35, 36). Therefore, we examined whether neutralization of IL-2 would exacerbate diabetes and produce autoimmune disease in a wider spectrum of organs compared with nontreated NOD mice. A similar course of anti–IL-2 treatment as shown in Fig. 6 for BALB/c mice triggered an earlier onset of diabetes, resulting in a higher incidence of diabetes (Fig. 7 A) and more severe forms of insulitis at the time of examination (Fig. 7, B and C). Furthermore, the incidence and histological severity of gastritis and thyroiditis increased in these anti–IL-2–treated mice. Histologically evident gastritis and thyroiditis in the treated mice was 90.9 and 54.5%, respectively, compared with 0% in control NOD mice at 20 wk of age (Fig. 7, B, D, and E, and see Fig. 9). These mice with gastritis or thyroiditis developed high titers of anti-parietal cell or anti-thyroglobulin autoantibodies, respectively (Fig. 7, D and E). Histologically evident inflammation in lacrimal glands and salivary glands was also observed in 36.4 and 9%, respectively, of anti–IL-2–treated NOD mice but absent in control mice (Fig. 7 B and see Fig. 9).
Figure 7.

Exacerbation of diabetes and induction of other organ-specific autoimmune diseases in NOD mice by IL-2 neutralization. Female NOD mice were injected i.p. with 1 mg of purified anti–IL-2 mAbs (n = 16) or rat IgG (n = 16) on days 10 and 20 after birth. Their blood glucose levels were measured every week starting from 7 wk of age. (A) Incidence of diabetes. (B) Hematoxylin and eosin staining of pancreas, thyroid, and lachrymal glands (a magnification of 5–10). (C) Severity of insulitis. Grade 1, peri-insulitis; grade 2, moderate insulitis; grade 3, severe insulitis (reference 59). (D and E) Titers of anti-parietal cell autoantibodies (D) and anti-thyroglobulin autoantibodies (E) in the sera of anti–IL-2–treated mice (n = 11) and rat IgG-treated mice (n = 12) assessed by ELISA.
Figure 9.

Development of various autoimmune diseases in NOD mice after anti–IL-2 treatment. Individual NOD female mice that were treated as shown in Fig. 7 and individual mice that survived to 20 wk of age (11 anti–IL-2–treated mice and 12 control mice) were assessed clinically and histologically. DM, diabetes mellitus; NP, neuropathy; Neur, neuritis; Ins, insulitis; Gas, gastritis; Thyr, thyroiditis; Sial, sialadenitis; lacr, lacrimal gland adenitis.
Notably, some mice developed ataxia and paralysis of the limbs around 15 wk after anti–IL-2 treatment (Fig. 8, A and B). Histological examination of the nervous system revealed lymphocytic infiltration and demyelination in peripheral nerves (Fig. 8 C) but not in the central nervous system (not depicted). Anti–IL-2 treatment elicited neuropathy in 56.3% of NOD mice and triggered diabetes in 50% of NOD mice assessed at 3 mo of age. Interestingly, neuropathy and diabetes were apparently mutually exclusive: only 1 of 11 mice developed both diseases (Fig. 9). The peripheral neuropathy could be adoptively transferred to naive NOD.SCID mice by spleen cell suspensions from neuropathic NOD mice. Both CD4+ and CD4− T cells from neuropathic NOD mice induced histologically evident neuritis in the SCID recipients (100% by CD4+ T cells and 40% by CD4− T cells), accompanying clinical signs of neuropathy such as trembling limbs (80% by CD4+ T cells and 40% by CD4− T cells; Fig. 8, D and E).
Figure 8.

Spontaneous development of autoimmune neuropathy in NOD mice treated with anti–IL-2 mAb. Female NOD mice treated as shown in Fig. 7 were checked weekly for clinical signs of neuropathy (hind and front leg paralysis). (A) Incidence of macroscopically evident neuropathy. (B) A mouse with paralysis of hind limbs. (C) Hematoxylin and eosin staining of the sections of the peripheral nerves (left; a magnification of 20) and sciatic nerves (middle; a magnification of 40). Luxol fast blue staining of peripheral nerves (right; a magnification of 40). (D) A NOD.SCID mouse showing paralysis of hind legs after receiving CD4+ T cells from anti–IL-2–treated NOD mice with neuropathy (left). Hematoxylin and eosin staining of sciatic nerve from the NOD.SCID mouse (right; a magnification of 40). (E) Spleen cells from NOD mice with neuropathy were sorted to CD4+ and CD4− cells by MACS, and 4–6 × 106 cells of each population were transferred to NOD.SCID mice. Grade 1, lymphocytic infiltration in the peripheral nerves without clinical signs; grade 2, clinical signs of neuropathy with lymphocytic infiltration in peripheral nerves.
Thus, anti–IL-2 mAb treatment enhances diabetes and produces a wide spectrum of organ-specific autoimmune diseases and most notably, peripheral neuritis in autoimmune-prone NOD mice.
Discussion
The following findings in this report support IL-2 being critically required for the peripheral maintenance of natural CD25+ CD4+ T reg cells and consequently the immunologic self-tolerance sustained by them. First, injection of anti–IL-2 mAb to Tx mice resulted in a marked reduction of the number of peripheral CD25+ CD4+ T cells, which corresponded to Foxp3-expressing natural T reg cells. The number of CD4+ T cells expressing high levels of GITR, which is more stably expressed by Foxp3-expressing natural T reg cells than CD25 (37), was also reduced after anti–IL-2 neutralization (Fig. S1), supporting the reduction of T reg cells rather than conversion of T reg cells from CD25+ to CD25−. Second, neutralization of IL-2 in normal mice inhibited the physiological proliferation of peripheral CD25+ CD4+ T cells. Third, IL-2 neutralization in normal mice for a limited period induced autoimmune disease similar to the one produced by depletion of CD25+ CD4+ T reg cells. Furthermore, transfer of spleen cells from BALB/c mice with low numbers of CD4+ T cells expressing CD25 and Foxp3 after anti–IL-2 treatment produced in nude mice autoimmune diseases similar to those seen after the transfer of CD25+ cell–depleted BALB/c spleen cells, and cotransfer of normal CD25+ CD4+ T cells prevented the autoimmune development (8). Taken together, these findings indicate passive cell death of CD25+ CD4+ T reg cells as a major mechanism of T reg cell reduction upon IL-2 deprivation, although our results do not exclude the possibility that IL-2 neutralization may reduce CD25 expression by natural T reg cells, thereby impairing their activation or down-regulating their Foxp3 transcription, leading to the attenuation of their suppressive activity.
Previous studies with IL-2−/− or IL-2R−/− mice also demonstrated a critical role for IL-2 and IL-2R in homeostasis of peripheral CD25+ CD4+ T reg cells. For example, not only IL-2−/− mice but also IL-2Rβ−/− mice bear few CD25+ CD4+ T cells in the thymus and periphery (19, 38). In chimeric RAG-2−/− mice reconstituted with IL-2−/− and CD25−/− BW cells, the normal number of peripheral CD25+ CD4+ T cells was established solely from IL-2−/− BM cells (39). This indicates that IL-2 produced by CD25−/− cells promoted the differentiation of IL-2−/− T cells to T reg cells. In IL-2Rβ−/− mice, the number of CD25+ CD4+ T cells was restored in the thymus and the periphery by thymic expression of an IL-2Rβ transgene, indicating a critical role of the IL-2/IL-2R pathway for the thymic generation of CD25+ CD4+ T cells (38). Thus, our present results, together with these findings by others, indicate that IL-2 is crucially required for the maintenance of peripheral CD25+ CD4+ T reg cells. IL-2 may also be required for the activation of CD25+ CD4+ T reg cells to exert suppression (23). It is likely that severe inflammation/autoimmunity in IL-2−/−, CD25−/−, or CD122−/− mice could be attributed to a synergistic effect of the reduction of natural T reg cells and their impaired function and differentiation in addition to possible altered activation-induced cell death of self-reactive T cells.
In contrast to the requirement of IL-2 for physiological expansion of CD25+ CD4+ T reg cells in normal nonlymphopenic mice, homeostatic proliferation in a lymphopenic environment appears to be IL-2 independent. It has been shown that CD25+ CD4+ T cells as well as conventional CD25− CD4+ T cells undergo extensive homeostatic expansion when transferred into lymphopenic recipients (29, 30). It is therefore likely that strong TCR stimulation of CD25+ CD4+ T reg cells by self-peptide/MHC, together with costimulation via CD28, may suffice to sustain their homeostatic proliferation in a T cell–deficient environment. It may correspond to the finding that TCR and CD28 costimulation can abrogate the anergic state of CD25+ CD4+ T reg cells and evoke their proliferation in vitro, as TCR stimulation in the presence of high dose IL-2 has a similar in vitro effect (40, 41).
We have shown that splenic CD25+ CD4+ T cells are phenotypically and functionally heterogeneous and can be divided to two functionally distinct populations according to the level of CD25 expression: one is the CD25high population, which exhibits suppressive activity, expresses a high level of Foxp3, and produces little IL-2, the other being the CD25low population, which has little suppressive activity, expresses a low level of Foxp3, and produces IL-2. The CD25+ CD4+ T cell population thus contains both CD25low CD4+ activated non–T reg cells and Foxp3-expressing CD25high CD4+ T reg cells. TCR transgenic mice also harbor IL-2–transcribing Foxp3 low CD25low CD4+ T cells, whereas RAG−/− TCR transgenic mice did not. These findings collectively indicate the following. First, some of the IL-2–transcribing CD25low CD4+ T cells might be activated by self-antigens because this population in TCR transgenic mice predominantly expresses endogenous TCR α chains paired with transgenic β chains and appears to be positively selected because of high self-reactivity of such TCRs, similar to positive selection of CD25high CD4+ T reg cells (34). Second, the presence of activated CD25low CD4+ non–T reg cells within the CD25+ CD4+ T cell population explains the finding by others that CD25+ CD4+ T cells isolated from normal naive mice were able to induce colitis when transferred to SCID mice and subsequently treated with anti–IL-10R mAb (42). Such colitis-inducing CD25+ CD4+ T cells may well have contained pathogenic CD25low CD4+ T cells in addition to CD25+ CD4+ T reg cells, and the IL-10R blockade may have abrogated the suppression exerted by the latter, allowing the former to cause colitis. Third, our results substantiate the finding in humans and mice that the CD25high CD4+ population predominantly contains T reg cells (43, 44). Thus, assuming that CD25low CD4+ T cells secrete IL-2 in the physiological state by responding to self-antigens or commensal bacteria, IL-2 can be a mediator of negative feedback control by which activated CD25low CD4+ nonregulatory T cells contribute to the maintenance and activation of natural CD25+ CD4+ T reg cells, which in turn limit the expansion of the former. This may occur locally where both natural T reg cells and effector T cells are recruited to APCs presenting particular self- or nonself-antigens.
IL-2 neutralization enhanced autoimmunity in NOD mice presumably by reducing natural CD25+ CD4+ T reg cells. The treatment facilitated the onset of T1D, produced more severe insulitis with higher incidence of diabetes, and provoked other autoimmune diseases in more severe a form than control mice. Interestingly, anti–IL-2 mAb-treated NOD mice also developed T cell–mediated autoimmune neuropathy similar to the one that developed in B7-2–deficient or anti–B7-2 mAb-treated NOD mice (45). It was shown that treatment with pertussis toxin alone, which impairs blood brain barrier, evoked EAE in NOD mice (46). In addition, allelic variants of the IL-2 gene contribute to the development of T1D and EAE in NOD congenic mice (6). Taken together, it is likely in NOD mice that genetic variations of IL-2 may contribute to determining the genetic susceptibility to these autoimmune diseases by altering the development and function of natural T reg cells (47). Furthermore, such genetic variation/anomalies of IL-2 may cause different autoimmune disease in combination with other genes (47, 48), as illustrated by the development of autoimmune gastritis in BALB/c mice and autoimmune neuropathy and other autoimmune diseases in NOD mice (9).
In conclusion, IL-2 is an indispensable cytokine for the peripheral maintenance of natural CD25+ CD4+ T reg cells. Alteration in the formation of IL-2 by other T cells, structural abnormality of the IL-2 molecule itself (49), or anomalies in intracellular signal transduction through IL-2R (50–52) might be predisposing to, or even causative of, various autoimmune diseases (6, 53). Furthermore, not only genetic modification but also environmental alteration of IL-2 formation may cause autoimmune disease. For example, cyclosporin A can cause autoimmune disease similar to the one produced by IL-2 neutralization, presumably by inhibiting IL-2 formation and consequently reducing natural CD25+ CD4+ T reg cells or impairing their functions (54). On the other hand, IL-2 can be used for strengthening natural self-tolerance and preventing autoimmune disease or stabilizing transplantation tolerance by maintaining the survival/expansion and augmenting the suppressive activity of natural T reg cells.
Materials and Methods
Mice.
BALB/c, BALB/c athymic nude, C.B-17 SCID, NOD, and NOD.SCID mice were purchased from CLEA. IL-2–deficient mice were purchased from The Jackson Laboratory and backcrossed to BALB/c mice more than eight times. BALB/c-Thy1.1 congenic mice were established in our laboratory (55). DO.11.10 TCR transgenic mice and RAG-2–deficient BALB/c mice were provided by D. Loh (Washington University, St. Louis, MO) and Y. Shinkai (Kyoto University, Kyoto, Japan), respectively. All mice were maintained in our animal facility and treated in accordance with the guidelines for animal care approved by the Institute for Frontier Medical Sciences, Kyoto University.
Reagents.
The rat anti–IL-2 mAb (IgG2a, clone S4B6; reference 56) was purified from ascites using protein G columns (Amersham Biosciences). The following reagents were purchased from BD Biosciences: purified anti-CD3ɛ (145-2C11), anti-CD16/CD32 (2.4G2), and anti–IL-2Rβ (TM-β1); and FITC-labeled or biotinylated anti-CD25 (7D4), FITC–anti-CD4 (RM4-5), PE–anti-CD4 (H129.19), FITC–anti-CD19 (1D3), PE–anti-CD11c (HL3), PE–anti-CD8 (53–6.7), PE–anti-Thy1.1 (OX-7), PE–anti-CD122 (TM-β1), and PE–anti-CD132 (4G3). PE–anti-CD3ɛ (145-2C11), PE–anti-CD11b (M1/70), and PE–anti-pan NK cells (DX-5) were purchased from eBioscience. α-GalCer (KRN7000) was provided by Kirin Brewery Co. α-GalCer/CD1d tetramer was provided by H. Wakasugi and Y. Ikarashi (National Cancer Institute, Tokyo, Japan). Anti-GITR mAb (DTA-1) was produced, purified, and labeled with Alexa in our laboratory (57).
Preparation of lymphocytes, flow cytometry, and cell sorting.
Lymphocyte suspensions were prepared from thymi, spleens, and lymph nodes. Erythrocytes were lysed with ACK buffer. Thymic CD4+ CD8+ and CD8+ cells were depleted by treatment with anti-CD8 mAb (3.155) and rabbit complement (Cedarlane Laboratories). For flow cytometric analysis, 106 cells were incubated with FITC- or PE-labeled or biotinylated mAbs and then with CyChrome- or PE-streptavidin (BD Biosciences) as a secondary reagent for biotinylated antibodies. Cells were then analyzed by flow cytometry (Epics-XL; Beckman Coulter). To sort peripheral CD4+ T cell subpopulations, CD4+ T cells were first enriched by depleting B cells, CD8+ cells, and adherent cells by panning as described previously (8). After staining with fluorescent antibodies, cells were sorted by an Epics Altra cell sorter (Beckman Coulter). The purity of the resulting CD25+ or CD25− CD4+ population was ⩾99%. In some experiments, CD25+ CD4+ T cells were purified using the MACS system (Miltenyi Biotec). The enriched CD4+ T cell fraction was stained with biotinylated anti-CD25 followed by streptavidin PE. Cells were then labeled with anti-PE microbeads and passed through LS columns twice to obtain CD25+ cells. The purity of the resulting CD25+ CD4+ T cells was 90–95%.
Histology and serology.
Organs were fixed with 10% formalin and processed for hematoxylin and eosin staining. Autoantibodies specific for the gastric parietal cells and thyroglobulin were assayed by ELISA (8). Gastritis and thyroiditis were graded 0–2+ as described previously (8).
Real-time PCR.
Foxp3 or IL-2 mRNA levels were quantified by using real-time PCR as described previously (10). IL-2 primer sequences used in this study were: 5′-tccagaacatgccgcagag-3′ and 5′-cctgagcaggatggagaattaca-3′. The IL-2 probe used was 5′-FAM-actccccaggatgctcaccttcaaattt-3′. The primers were designed to hybridize on an intron–exon junction to prevent amplification of contaminating genomic DNA.
ELISA.
Purified CD25high, CD25low, and CD25− CD4+ T cells (8.5 × 105 per well in a flat-bottomed 96-well plate) were cultured in 80 μl of complete RPMI medium for 24 h. To block the binding of IL-2 to IL-2R on these populations, 10 μg/ml anti–IL-2Rβ was added in the culture. IL-2 concentration in culture supernatants was determined by sandwich ELISA (eBioscience), which has a detection limit of 2 pg/ml.
In vitro proliferation assay.
Sorted CD25− CD4+ cells (2.0 × 104 per well in a U-bottomed 96-well plate) and 4.0 × 104 irradiated APCs per well were cultured for 72 h in the presence of 1.0 μg/ml anti-CD3ɛ mAb (145-2C11; BD Biosciences). CD3+ cell–depleted spleen cells were used as APCs. [3H]thymidine (1 μCi/well; DuPont/NEN Life Science Products) was added during the last 6 h of culture. Background counts in the wells with APCs alone were always <200 cpm (40).
In vivo BrdU labeling.
Mice were injected i.p. with 1.0 mg BrdU (Sigma-Aldrich) every 12 h for 3 d. 107 pooled LN and spleen cells were surface stained with PE-CD4 and biotin-CD25 followed by RPE-Cy5-streptavidin (DakoCytomation). BrdU staining was performed as reported by others (58) using FITC-labeled anti-BrdU mAb (DakoCytomation).
Lymphocyte labeling with CFSE.
Purified CD25+ CD4+ T cells were incubated with 3 μM CFSE in PBS for 10 min at room temperature. An equal volume of normal mouse serum was then added to stop the reaction. After washing twice, 106 labeled cells were i.v. injected.
Thymectomy.
Female 5–6-wk-old mice were anesthetized by 10% nembutal and Tx or sham operated as previously described (8).
Assessment of diabetes.
Blood glucose levels were measured every week with a Medisafe reader (TERUMO). Mice were considered diabetic when blood glucose levels were >300 mg/dl after two consecutive measurements.
Statistical analysis.
All statistical analyses were performed by Student's paired t test.
Online supplemental material.
Fig. S1 shows that IL-2 neutralization decreases the number of CD4+ GITRhigh cells in the thymus and periphery. Fig. S2 shows possible activation of CD25low CD4+ T cells in the periphery. Fig. S3 shows that the size of CD25+ CD4+ T cells decreases and then recovers in BALB/c mice treated with anti–IL-2 on days 10 and 20 after birth. Figs. S1–S3 are available at http://www.jem.org/cgi/content/full/jem.20041982/DC1.
Acknowledgments
We thank Z. Fehervari for critically reading the manuscript; K. Hirota and R. Ishii for assistance with cell sorting; T. Matsushita for histology; M Kakino for maintaining mice; and H. Wakasugi, Y. Ikarashi, and Kirin Brewery Co. for reagents.
This work was supported by grants-in-aid from the Ministry of Education, Sports and Culture of Japan.
The authors have no conflicting financial interests.
Abbreviations used: BrdU, 5-bromo-29-deoxyuridine; CFSE, 5- and 6-carboxyfluorescein diacetate succinimidyl ester; EAE, experimental allergic encephalitis; NOD, nonobese diabetic; T1D, type 1 diabetes; T reg cell, regulatory T cell; Tx, thymectomized.
References
- 1.Horak, I., J. Lohler, A. Ma, and K.A. Smith. 1995. Interleukin-2 deficient mice: a new model to study autoimmunity and self-tolerance. Immunol. Rev. 148:35–44. [DOI] [PubMed] [Google Scholar]
- 2.Kramer, S., A. Schimpl, and T. Hunig. 1995. Immunopathology of interleukin (IL)-2–deficient mice: thymus dependence and suppression by thymus-dependent cells with an intact IL-2 gene. J. Exp. Med. 182:1769–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serreze, D.V., K. Hamaguchi, and E.H. Leiter. 1989. Immunostimulation circumvents diabetes in NOD/Lt mice. J. Autoimmun. 2:759–776. [DOI] [PubMed] [Google Scholar]
- 4.Zielasek, J., V. Burkart, P. Naylor, A. Goldstein, U. Kiesel, and H. Kolb. 1990. Interleukin-2-dependent control of disease development in spontaneously diabetic BB rats. Immunology. 69:209–214. [PMC free article] [PubMed] [Google Scholar]
- 5.Gutierrez-Ramos, J.C., J.L. Andreu, Y. Revilla, E. Vinuela, and C. Martinez. 1990. Recovery from autoimmunity of MRL/lpr mice after infection with an interleukin-2/vaccinia recombinant virus. Nature. 346:271–274. [DOI] [PubMed] [Google Scholar]
- 6.Encinas, J.A., L.S. Wicker, L.B. Peterson, A. Mukasa, C. Teuscher, R. Sobel, H.L. Weiner, C.E. Seidman, J.G. Seidman, and V.K. Kuchroo. 1999. QTL influencing autoimmune diabetes and encephalomyelitis map to a 0.15-cM region containing Il2. Nat. Genet. 21:158–160. [DOI] [PubMed] [Google Scholar]
- 7.Gambineri, E., T.R. Torgerson, and H.D. Ochs. 2003. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr. Opin. Rheumatol. 15:430–435. [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi, S., N. Sakaguchi, M. Asano, M. Itoh, and M. Toda. 1995. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 155:1151–1164. [PubMed] [Google Scholar]
- 9.Sakaguchi, S. 2004. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22:531–562. [DOI] [PubMed] [Google Scholar]
- 10.Hori, S., T. Nomura, and S. Sakaguchi. 2003. Control of regulatory T cell development by the transcription factor Foxp3. Science. 299:1057–1061. [DOI] [PubMed] [Google Scholar]
- 11.Uchiyama, T., S. Broder, and T.A. Waldmann. 1981. A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T cells. I. Production of anti-Tac monoclonal antibody and distribution of Tac+ cells. J. Immunol. 126:1393–1397. [PubMed] [Google Scholar]
- 12.Malek, T.R., R.J. Robb, and E.M. Shevach. 1983. Identification and initial characterization of a rat monoclonal antibody reactive with the murine interleukin 2 receptor-ligand complex. Proc. Natl. Acad. Sci. USA. 80:5694–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chastagner, P., J.L. Moreau, Y. Jacques, T. Tanaka, M. Miyasaka, M. Kondo, K. Sugamura, and J. Theze. 1996. Lack of intermediate-affinity interleukin-2 receptor in mice leads to dependence on interleukin-2 receptor alpha, beta and gamma chain expression for T cell growth. Eur. J. Immunol. 26:201–206. [DOI] [PubMed] [Google Scholar]
- 14.Ruscetti, F.W., and R.C. Gallo. 1981. Human T-lymphocyte growth factor: regulation of growth and function of T lymphocytes. Blood. 57:379–394. [PubMed] [Google Scholar]
- 15.Schorle, H., T. Holtschke, T. Hunig, A. Schimpl, and I. Horak. 1991. Development and function of T cells in mice rendered interleukin-2 deficient by gene targeting. Nature. 352:621–624. [DOI] [PubMed] [Google Scholar]
- 16.Willerford, D.M., J. Chen, J.A. Ferry, L. Davidson, A. Ma, and F.W. Alt. 1995. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 3:521–530. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki, H., T.M. Kundig, C. Furlonger, A. Wakeham, E. Timms, T. Matsuyama, R. Schmits, J.J. Simard, P.S. Ohashi, H. Griesser, et al. 1995. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor beta. Science. 268:1472–1476. [DOI] [PubMed] [Google Scholar]
- 18.Lenardo, M.J. 1991. Interleukin-2 programs mouse alpha beta T lymphocytes for apoptosis. Nature. 353:858–861. [DOI] [PubMed] [Google Scholar]
- 19.Papiernik, M., M.L. de Moraes, C. Pontoux, F. Vasseur, and C. Penit. 1998. Regulatory CD4 T cells: expression of IL-2R alpha chain, resistance to clonal deletion and IL-2 dependency. Int. Immunol. 10:371–378. [DOI] [PubMed] [Google Scholar]
- 20.Wolf, M., A. Schimpl, and T. Hunig. 2001. Control of T cell hyperactivation in IL-2-deficient mice by CD4+CD25− and CD4+CD25+ T cells: evidence for two distinct regulatory mechanisms. Eur. J. Immunol. 31:1637–1645. [DOI] [PubMed] [Google Scholar]
- 21.Klebb, G., I.B. Autenrieth, H. Haber, E. Gillert, B. Sadlack, K.A. Smith, and I. Horak. 1996. Interleukin-2 is indispensable for development of immunological self-tolerance. Clin. Immunol. Immunopathol. 81:282–286. [DOI] [PubMed] [Google Scholar]
- 22.Thornton, A.M., E.E. Donovan, C.A. Piccirillo, and E.M. Shevach. 2004. Cutting edge: IL-2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J. Immunol. 172:6519–6523. [DOI] [PubMed] [Google Scholar]
- 23.Furtado, G.C., M.A. Curotto de Lafaille, N. Kutchukhidze, and J.J. Lafaille. 2002. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J. Exp. Med. 196:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De La Rosa, M., S. Rutz, H. Dorninger, and A. Scheffold. 2004. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur. J. Immunol. 34:2480–2488. [DOI] [PubMed] [Google Scholar]
- 25.Murakami, M., A. Sakamoto, J. Bender, J. Kappler, and P. Marrack. 2002. CD25+CD4+ T cells contribute to the control of memory CD8+ T cells. Proc. Natl. Acad. Sci. USA. 99:8832–8837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim, H.P., J. Kelly, and W.J. Leonard. 2001. The basis for IL-2-induced IL-2 receptor alpha chain gene regulation: importance of two widely separated IL-2 response elements. Immunity. 15:159–172. [DOI] [PubMed] [Google Scholar]
- 27.Fontenot, J.D., M.A. Gavin, and A.Y. Rudensky. 2003. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4:330–336. [DOI] [PubMed] [Google Scholar]
- 28.Khattri, R., T. Cox, S.A. Yasayko, and F. Ramsdell. 2003. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 4:337–342. [DOI] [PubMed] [Google Scholar]
- 29.Annacker, O., R. Pimenta-Araujo, O. Burlen-Defranoux, T.C. Barbosa, A. Cumano, and A. Bandeira. 2001. CD25+ CD4+ T cells regulate the expansion of peripheral CD4 T cells through the production of IL-10. J. Immunol. 166:3008–3018. [DOI] [PubMed] [Google Scholar]
- 30.Gavin, M.A., S.R. Clarke, E. Negrou, A. Gallegos, and A. Rudensky. 2002. Homeostasis and anergy of CD4+CD25+ suppressor T cells in vivo. Nat. Immunol. 3:33–41. [DOI] [PubMed] [Google Scholar]
- 31.Hori, S., M. Haury, J.J. Lafaille, J. Demengeot, and A. Coutinho. 2002. Peripheral expansion of thymus-derived regulatory cells in anti-myelin basic protein T cell receptor transgenic mice. Eur. J. Immunol. 32:3729–3735. [DOI] [PubMed] [Google Scholar]
- 32.Fisson, S., G. Darrasse-Jeze, E. Litvinova, F. Septier, D. Klatzmann, R. Liblau, and B.L. Salomon. 2003. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J. Exp. Med. 198:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Granucci, F., C. Vizzardelli, N. Pavelka, S. Feau, M. Persico, E. Virzi, M. Rescigno, G. Moro, and P. Ricciardi-Castagnoli. 2001. Inducible IL-2 production by dendritic cells revealed by global gene expression analysis. Nat. Immunol. 2:882–888. [DOI] [PubMed] [Google Scholar]
- 34.Itoh, M., T. Takahashi, N. Sakaguchi, Y. Kuniyasu, J. Shimizu, F. Otsuka, and S. Sakaguchi. 1999. Thymus and autoimmunity: production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 162:5317–5326. [PubMed] [Google Scholar]
- 35.Bernard, N.F., F. Ertug, and H. Margolese. 1992. High incidence of thyroiditis and anti-thyroid autoantibodies in NOD mice. Diabetes. 41:40–46. [DOI] [PubMed] [Google Scholar]
- 36.Goillot, E., M. Mutin, and J.L. Touraine. 1991. Sialadenitis in nonobese diabetic mice: transfer into syngeneic healthy neonates by splenic T lymphocytes. Clin. Immunol. Immunopathol. 59:462–473. [DOI] [PubMed] [Google Scholar]
- 37.Nishimura, E., T. Sakihama, R. Setoguchi, K. Tanaka, and S. Sakaguchi. 2004. Induction of antigen-specific immunologic tolerance by in vivo and in vitro antigen-specific expansion of naturally arising Foxp3+ CD25+CD4+ regulatory T cells. Int. Immunol. 16:1189–1201. [DOI] [PubMed] [Google Scholar]
- 38.Malek, T.R., A. Yu, V. Vincek, P. Scibelli, and L. Kong. 2002. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rbeta-deficient mice. Implications for the nonredundant function of IL-2. Immunity. 17:167–178. [DOI] [PubMed] [Google Scholar]
- 39.Almeida, A.R., N. Legrand, M. Papiernik, and A.A. Freitas. 2002. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 169:4850–4860. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi, T., Y. Kuniyasu, M. Toda, N. Sakaguchi, M. Itoh, M. Iwata, J. Shimizu, and S. Sakaguchi. 1998. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10:1969–1980. [DOI] [PubMed] [Google Scholar]
- 41.Thornton, A.M., and E.M. Shevach. 1998. CD4+ CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med. 188:287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Asseman, C., S. Read, and F. Powrie. 2003. Colitogenic Th1 cells are present in the antigen-experienced T cell pool in normal mice: control by CD4+ regulatory T cells and IL-10. J. Immunol. 171:971–978. [DOI] [PubMed] [Google Scholar]
- 43.Kuniyasu, Y., T. Takahashi, M. Itoh, J. Shimizu, G. Toda, and S. Sakaguchi. 2000. Naturally anergic and suppressive CD25+CD4+ T cells as a functionally and phenotypically distinct immunoregulatory T cell subpopulation. Int. Immunol. 12:1145–1155. [DOI] [PubMed] [Google Scholar]
- 44.Baecher-Allan, C., J.A. Brown, G.J. Freeman, and D.A. Hafler. 2001. CD4+CD25high regulatory cells in human peripheral blood. J. Immunol. 167:1245–1253. [DOI] [PubMed] [Google Scholar]
- 45.Salomon, B., L. Rhee, H. Bour-Jordan, H. Hsin, A. Montag, B. Soliven, J. Arcella, A.M. Girvin, J. Padilla, S.D. Miller, and J.A. Bluestone. 2001. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2–deficient NOD mice. J. Exp. Med. 194:677–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winer, S., I. Astsaturov, R. Cheung, L. Gunaratnam, V. Kubiak, M.A. Cortez, M. Moscarello, P.W. O'Connor, C. McKerlie, D.J. Becker, and H.M. Dosch. 2001. Type I diabetes and multiple sclerosis patients target islet plus central nervous system autoantigens; nonimmunized nonobese diabetic mice can develop autoimmune encephalitis. J. Immunol. 166:2831–2841. [DOI] [PubMed] [Google Scholar]
- 47.Wicker, L.S., J.A. Todd, and L.B. Peterson. 1995. Genetic control of autoimmune diabetes in the NOD mouse. Annu. Rev. Immunol. 13:179–200. [DOI] [PubMed] [Google Scholar]
- 48.Kanagawa, O., S.M. Martin, B.A. Vaupel, E. Carrasco-Marin, and E.R. Unanue. 1998. Autoreactivity of T cells from nonobese diabetic mice: an I-Ag7-dependent reaction. Proc. Natl. Acad. Sci. USA. 95:1721–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Podolin, P.L., M.B. Wilusz, R.M. Cubbon, U. Pajvani, C.J. Lord, J.A. Todd, L.B. Peterson, L.S. Wicker, and P.A. Lyons. 2000. Differential glycosylation of interleukin 2, the molecular basis for the NOD Idd3 type 1 diabetes gene? Cytokine. 12:477–482. [DOI] [PubMed] [Google Scholar]
- 50.Snow, J.W., N. Abraham, M.C. Ma, B.G. Herndier, A.W. Pastuszak, and M.A. Goldsmith. 2003. Loss of tolerance and autoimmunity affecting multiple organs in STAT5A/5B-deficient mice. J. Immunol. 171:5042–5050. [DOI] [PubMed] [Google Scholar]
- 51.Antov, A., L. Yang, M. Vig, D. Baltimore, and L. Van Parijs. 2003. Essential role for STAT5 signaling in CD25+CD4+ regulatory T cell homeostasis and the maintenance of self-tolerance. J. Immunol. 171:3435–3441. [DOI] [PubMed] [Google Scholar]
- 52.Burchill, M.A., C.A. Goetz, M. Prlic, J.J. O'Neil, I.R. Harmon, S.J. Bensinger, L.A. Turka, P. Brennan, S.C. Jameson, and M.A. Farrar. 2003. Distinct effects of STAT5 activation on CD4+ and CD8+ T cell homeostasis: development of CD4+CD25+ regulatory T cells versus CD8+ memory T cells. J. Immunol. 171:5853–5864. [DOI] [PubMed] [Google Scholar]
- 53.Becker, K.G., R.M. Simon, J.E. Bailey-Wilson, B. Freidlin, W.E. Biddison, H.F. McFarland, and J.M. Trent. 1998. Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc. Natl. Acad. Sci. USA. 95:9979–9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sakaguchi, S., and N. Sakaguchi. 1989. Organ-specific autoimmune disease induced in mice by elimination of T cell subsets. V. Neonatal administration of cyclosporin A causes autoimmune disease. J. Immunol. 142:471–480. [PubMed] [Google Scholar]
- 55.Asano, M., M. Toda, N. Sakaguchi, and S. Sakaguchi. 1996. Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J. Exp. Med. 184:387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosmann, T.R., H. Cherwinski, M.W. Bond, M.A. Giedlin, and R.L. Coffman. 1986. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136:2348–2357. [PubMed] [Google Scholar]
- 57.Shimizu, J., S. Yamazaki, T. Takahashi, Y. Ishida, and S. Sakaguchi. 2002. Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3:135–142. [DOI] [PubMed] [Google Scholar]
- 58.Tough, D.F., and J. Sprent. 1994. Turnover of naive- and memory-phenotype T cells. J. Exp. Med. 179:1127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salomon, B., D.J. Lenschow, L. Rhee, N. Ashourian, B. Singh, A. Sharpe, and J.A. Bluestone. 2000. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12:431–440. [DOI] [PubMed] [Google Scholar]