

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-regulated chloride channel localized primarily at the apical or luminal surfaces of epithelial cells that line the airway, gut, and exocrine glands; it is well established that CFTR plays a pivotal role in cholera toxin (CTX)-induced secretory diarrhea. Lysophosphatidic acid (LPA), a naturally occurring phospholipid present in blood and foods, has been reported to play a vital role in a variety of conditions involving gastrointestinal wound repair, apoptosis, inflammatory bowel disease, and diarrhea. Here we show, for the first time, that type 2 LPA receptors (LPA2) are expressed at the apical surface of intestinal epithelial cells, where they form a macromolecular complex with Na+/H+ exchanger regulatory factor–2 and CFTR through a PSD95/Dlg/ZO-1–based interaction. LPA inhibited CFTR-dependent iodide efflux through LPA2-mediated Gi pathway, and LPA inhibited CFTR-mediated short-circuit currents in a compartmentalized fashion. CFTR-dependent intestinal fluid secretion induced by CTX in mice was reduced substantially by LPA administration; disruption of this complex using a cell-permeant LPA2-specific peptide reversed LPA2-mediated inhibition. Thus, LPA-rich foods may represent an alternative method of treating certain forms of diarrhea.
Infectious diarrhea disease is second only to cardiovascular disease as cause of death (1) and is one of the major causes of morbidity and mortality among children. It accounts for 21% of deaths of children who are younger than 5 yr of age in the developing world, and causes 2.5 million deaths per year (2). Although incidence rates increase with a country's decreasing socioeconomic status, infectious diarrhea also is a common illness in Western industrialized societies, particularly among vulnerable groups of young children, elderly adults, and people who have underlying diseases (3). Two major organisms that cause infectious diarrhea in humans are Escherichia coli and Vibrio cholerae; their secreted toxins (heat stable or heat labile toxin, and cholera toxin [CTX], respectively) influence gastrointestinal epithelial cell function and induce diarrhea by numerous mechanisms (4). CTX is likely the most recognizable enterotoxin that causes secretory diarrhea by stimulating transepithelial Cl− secretion, thereby increasing the osmotic impetus for fluid secretion (5). Diarrhea also can result from an increased immunoinflammatory response that is triggered by cytokine or mediator (e.g., adenosine; ADO) secretion from intestinal mucosal inflammatory cells in response to luminal factors (e.g., dietary or bacterial antigens). This also is a common symptom that is associated with the major gastrointestinal disorders, such as inflammatory bowel disease (IBD) afflicting >1 million Americans (6), and irritable bowel syndrome, which affects 15 million Americans (7). Mucosal inflammation plays an essential role in the pathogenesis of irritable bowel syndrome and IBD (8, 9). ADO, an inflammatory mediator, stimulated chloride secretion in several types of secretory epithelia, including mammalian ileum and colon (10, 11).
Cystic fibrosis transmembrane conductance regulator (CFTR) is a cAMP-regulated chloride channel that is localized primarily at the apical or luminal surfaces of epithelial cells that line the airway, gut, and exocrine glands (12); it is well-established that CFTR plays a pivotal role in CTX-mediated diarrhea (13). Lysophosphatidic acid (LPA), a naturally occurring phospholipid present in blood and foods (14, 15), acts on the LPA receptors (LPA1, LPA2, and LPA3), which are G-protein coupled receptors that belong to the endothelial cell differentiation gene family (16–18). LPA was reported to play an essential role in a variety of conditions involving gastrointestinal wound repair, apoptosis, IBD, and diarrhea (19–23). In the present study, we investigated how LPA-elicited LPA2-mediated signaling might regulate CFTR function in the gut, and evaluated the usefulness of LPA in preventing CTX-induced secretory diarrhea in mice.
RESULTS
LPA2 and Na+/H+ exchanger regulatory factor–2 are expressed in intestinal epithelial cells and localized primarily at the apical cell surfaces
Using RT-PCR, we demonstrated that the transcripts for LPA1 and LPA2, but not LPA3, were present in mouse intestinal tissue (Fig. S1, A and B, available at http://www.jem.org/cgi/content/full/jem.20050421/DC1). HT29-CL19A (colonic epithelial cells) and Calu-3 (airway serous gland epithelial cells) also expressed LPA2 transcript (Fig. S1 C), and the expression of LPA2 in plasma membranes was verified using an LPA2-specific antibody (Fig. 1 A). LPA2 was localized predominantly to the apical surfaces of HT29-CL19A cells by confocal microscopy (Fig. 1 B, top) and surface biotinylation (24) (Fig. 1 C, top), where it colocalized with CFTR (Fig. 1 B, middle; Fig. 1 C, bottom). An apical membrane marker, ezrin, was used to confirm the subcellular (apical) localization of LPA2 and CFTR (Fig. 1 B, bottom).
Figure 1.

LPA2 and NHERF2 are expressed in epithelial cells and localized primarily at the apical cell surfaces. (A) LPA2 expression in crude plasma membranes prepared from HT29-CL19A and Calu-3 cells. (B) Apical localization of LPA2, CFTR, and ezrin in polarized HT29-CL19A cells by confocal microscopy. AP, apical; BL, basolateral. (C) Expression of LPA2 and CFTR in apical membranes of HT29-CL19A cells by surface biotinylation method. Smaller amounts of LPA2 also were observed in the basolateral membrane. (D) NHERF2 expression and apical localization in epithelial cells. The nucleus was stained with propidium iodide solution (bottom).
LPA2 was reported to interact specifically with Na+/H+ exchanger regulatory factor (NHERF)-2 (25, 26), a PSD95/Dlg/ZO-1 (PDZ) domain-containing protein that has been well-documented to cluster signaling molecules into supramolecular complexes (27). Here we showed that NHERF2 was expressed at various levels in epithelial cells (e.g., HT29-CL19A, Calu-3, mouse intestine epithelial cells) (Fig. 1 D, top right). This isoform-specific antibody did not cross-react with NHERF1, which shares ∼50% sequence identity with NHERF2 (Fig. 1 D, top left). NHERF2 also was localized to the apical surfaces in HT29-CL19A cells (Fig. 1 D, bottom) where CFTR and LPA2 reside (Fig. 1 B).
LPA2 binds NHERF2 with the highest affinity
The COOH-terminal tails of LPA1 and LPA2, but not LPA3, contain a consensus for the PDZ motif, the short COOH-terminal sequences that specifically interact with the PDZ-binding domains (usually ∼70–90 amino acid sequences) of PDZ proteins, including NHERF2 (28) (Fig. S2 A, boxed area, available at http://www.jem.org/cgi/content/full/jem.20050421/DC1). Thus, LPA1 and LPA2 are likely candidates with which PDZ-domain–containing proteins can interact, modulate, and mediate their signals. Pull-down assays demonstrated that LPA2 interacted directly with NHERF2 with the highest affinity (Fig. S2, B and C), as reported previously (25, 26).
LPA inhibits CFTR-dependent iodide efflux through LPA2 receptor–mediated Gi pathway
Although all three LPA receptors (LPA1, LPA2, and LPA3) respond to LPA, LPA2 has the highest affinity to the lipid (29), and leads to the activation of at least three distinct G-protein pathways: Gi, Gq, and G12/13 (17, 18). Activation of these receptors by LPA results in inhibition of the adenylate cyclase (AC) pathway, which, in turn, decreases cAMP levels (16–18). The Gi activation pathway of the receptor is summarized in Fig. S2 D.
Given that LPA2 binds NHERF2 (25, 26) (Fig. S2, B and C) and our previous demonstration that NHERF2 can form a macromolecular complex with CFTR (30), we hypothesized that LPA might regulate CFTR Cl− channel function which is regulated by cAMP. To test this, the effect of LPA on CFTR activity was monitored using iodide efflux measurements (31). Calu-3 cells, like HT29-CL19A cells, express all three proteins (CFTR, NHERF2, and LPA2) at the apical surfaces, and show a robust CFTR function because they express a higher level of CFTR compared with HT29-CL19A cells. Therefore, they represent a better model cell line for demonstrating the effects of LPA on CFTR function. LPA pretreatment (20 μM LPA 18:1 for 4 min before ADO addition) reduced the response of Calu-3 cells to cAMP-elevating ligand ADO in a dose-dependent manner (Fig. 2, A and B). CFTR activity was not inhibited by a structurally related lysophospholipid, sphingosine-1-phosphate (S1P; data not shown), whose receptors also are expressed in the gut epithelium but lack PDZ domain, nor by a related lipid—phosphatidic acid (PA) (Fig. 2 B)—which is not a ligand for LPA receptors (16). Similar results also were observed with HT29-CL19A cells (unpublished data). These results clearly demonstrate that LPA can inhibit CFTR function in epithelial cells.
Figure 2.

LPA inhibits CFTR-dependent iodide efflux through LPA2 receptor–mediated Gi pathway. (A) Representative ADO-activated CFTR-mediated iodide efflux in Calu-3 cells treated with LPA 18:1 (20 μM, BSA-complexed). (B) ADO-activated iodide efflux in LPA 18:1- or PA-pretreated Calu-3 cells. Note: the peak efflux is shown (5-min time point). (C) Calu-3 cells were pretreated with PTX (100 and 200 ng) for 24 h at 37°C and subjected to iodide efflux activated by ADO in the presence of LPA 18:1 (20 μM). (D) Peak efflux of iodide in response to ADO in Calu-3 cells that were treated with various LPA analogs (20 μM). Results are presented as mean ± SEM (n = 4–6 each group). *P < 0.05; **P < 0.01.
To test the hypothesis that the inhibition of CFTR function by LPA is receptor mediated, we pretreated cells with pertussis toxin (PTX), which catalyzes the ADP-ribosylation of the Gi α-subunit, and specifically disrupts the Gi pathway (32). PTX pretreatment reversed LPA-elicited inhibition of CFTR function in response to ADO in Calu-3 cells (Fig. 2 C) and HT29-CL19A cells in a dose-dependent manner (not depicted). Therefore, it is likely that inhibition of CFTR function by LPA is receptor mediated through the Gi pathway. Several fatty acid analogs of LPA were screened, and the rank order of inhibition was LPA 20:4 > LPA 18:2 > LPA 18:1 (Fig. 2 D), which matches their relative abundance in human serum (33).
LPA inhibits CFTR-dependent short-circuit currents in polarized epithelial cells and mouse intestinal epithelia preparation
The effect of LPA on CFTR-dependent short-circuit currents (Isc) also was monitored on polarized epithelial monolayers (30). LPA substantially inhibited CFTR-dependent Cl− currents in response to ADO activation in HT29-CL19A cells (Fig. 3 A) and Calu-3 cells (Fig. 3 B). Isc stimulated by ADO (Fig. 3 C) or 8-(4-chlorophenylthio)-cyclic AMP (cpt-cAMP) (a cell-permeant isoform of cAMP, a submaximal CFTR activating dose was used; Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20050421/DC1) also was inhibited, in a dose-dependent fashion, in LPA-pretreated epithelia that were isolated from mouse intestine (34) (Fig. S3 B). The inhibitory effect of LPA on CFTR Cl− currents in the intestinal epithelia was more prominent when CFTR was activated at lower concentrations of ADO (Fig. 3 D), whereas LPA failed to inhibit the channel function significantly when CFTR was activated maximally by a cocktail agonist mixture in intestinal epithelia (Fig. 3, C and D) and cultured epithelial cell monolayers (Fig. S3 C). The Isc in response to ADO was inhibited by CFTR blockers, diphenylamine-2-carboxylate (DPC) (Fig. 3 B) and glybenclamide (not depicted). PA and S1P did not inhibit the CFTR Cl− channel (unpublished data). ADO is produced at almost 1 μM in unstressed tissue, whereas it can be ≥100 μM in inflamed or ischemic tissues (35). Under conditions of inflammation (e.g., IBD), ADO is secreted into the gut, which activates CFTR and leads to increased chloride and fluid secretion into the gut (10, 11, 36).
Figure 3.

LPA inhibits CFTR-dependent Isc in polarized epithelial cells and isolated mouse intestinal epithelia. (A) Representative traces of CFTR-mediated Isc in response to ADO in HT29-CL19A cells without (top) or with (bottom) LPA 20:4 pretreatment. (B) Isc in response to ADO in Calu-3 cells. DPC (500 μM) was used as a CFTR blocker toward the end of the experiment. (C) Isc in response to ADO and cocktail (20 μM forskolin; 100 μM IBMX) in mouse intestinal epithelia. (D) Peak Cl− currents (% control) activated by ADO or cocktail in mouse intestine epithelia summarized from (C) (n = 4).
LPA inhibits CFTR single-channel Cl− currents in a compartmentalized fashion
The intracellular cAMP accumulation in cell extracts in response to various agonists in Calu-3 cell monolayers also was monitored. The cAMP accumulation at a lower concentration of ADO (2 μM) was almost indistinguishable from that in unstimulated cells (Fig. 4 A). However, CFTR is activated at this ligand concentration (Fig. 3 B, top), and LPA can inhibit channel activity substantially (Fig. 3 B, bottom). Our result is consistent with the concept that upon ADO stimulation, cAMP is likely to be generated in a compartmentalized manner, localized to the apical membrane (37). At a higher ADO concentration (20 μM), when the cAMP within the cell increased globally (Fig. 4 A), LPA inhibition of CFTR function became less obvious (Fig. 3 B, compare the maximal Isc response between control and LPA-treated cells). In addition, although LPA did not inhibit CFTR function in Calu-3 cells that were activated maximally by a cocktail mixture (Fig. S3 C), LPA significantly reduced the intracellular cAMP accumulation compared with the control (Fig. S3 D). This may have been because although the total intracellular cAMP level was reduced significantly by LPA, it was still high enough to activate CFTR almost maximally. Altogether, our data suggest that localized LPA signaling and cAMP accumulation within a compartmentalized domain under the apical epithelial cell membranes.
Figure 4.

LPA inhibits CFTR-dependent Cl− currents in a compartmentalized fashion. (A) Intracellular cAMP levels in Calu-3 cells under various conditions. (n = 4). (B) Single-channel recordings of CFTR in cell-attached configuration in HT29-CL19A cells under different recording conditions as indicated. C, channel closed state; FSK, forskolin; O, channel open state. (C) Summary of CFTR channel open probability (NPo). **P < 0.01; ***P < 0.001.
Cell-attached single-channel recordings also were performed; they confirmed that LPA inhibited cAMP generation in a compartmentalized fashion. ADO, forskolin, or cpt-cAMP was applied in the pipette to evoke a localized generation of cAMP or in the bath to elicit a global elevation of the second messenger (37). The CFTR channel was activated when 2 μM ADO was added in the pipette, but not when it was added to the bath (Fig. 4, B and C). In cells that were pretreated with LPA, the stimulatory effect of ADO (2 μM, applied in the pipette) on the channel activity was inhibited (Fig. 4, B and C). In contrast, when 10 μM forskolin was used in the pipette or the bath (leading to a global increase in cAMP), LPA no longer attenuated channel function (Fig. 4, B and C). Similar results were observed when cpt-cAMP was used to activate the channel. A lower concentration of cpt-cAMP (20 μM) stimulated localized CFTR activity that could be inhibited by LPA, whereas a higher concentration of cpt-cAMP (200 μM) led to global stimulation that could not be attenuated by LPA (Fig. S4, A and B, available at http://www.jem.org/cgi/content/full/jem.20050421/DC1). These single-channel recordings clearly confirmed the findings that LPA inhibits CFTR-mediated Cl− currents in a compartmentalized fashion.
LPA inhibits CFTR-dependent CTX-induced mouse intestinal fluid secretion in vivo
To provide proof of concept that LPA could be used in a therapeutic setting, we tested the effect of LPA on CTX-induced diarrhea in a mouse model (38). Binding of the cholera enterotoxin from V. cholerae to the intestinal enterocyte leads to ADP-ribosylation of the Gs α-subunit, which, in turn, activates AC of enterocytes and leads to an elevation of cAMP (39, 40). Thus, the apical membrane CFTR Cl− channel is activated, which results in a voluminous Cl− and fluid secretion that can be fatal, if untreated (5, 39). In ligated ileal loops, CTX treatment elicited fluid accumulation in a dose-dependent manner (Fig. 5 A). A linear increase in cAMP accumulation within the loop tissue was elicited by the increasing amounts of CTX (injected into the ileal loops) in the range of 0–10 μg (Fig. S5 A, available at http://www.jem.org/cgi/content/full/jem.20050421/DC1). These results are consistent with the reports that CTX can induce a dose-dependent increase in intracellular cAMP production in cultured cells (41–43) as well as a dose-dependent fluid accumulation in nonligated intestine in vivo (44). LPA treatment significantly reduced the fluid accumulation in the toxin-treated intestinal loops in a dose-dependent fashion (Fig. 5 B). PA (10–100 μM) was used as a negative control in these experiments, and showed no effect in reversing CTX action (i.e., fluid accumulation) (unpublished data).
Figure 5.
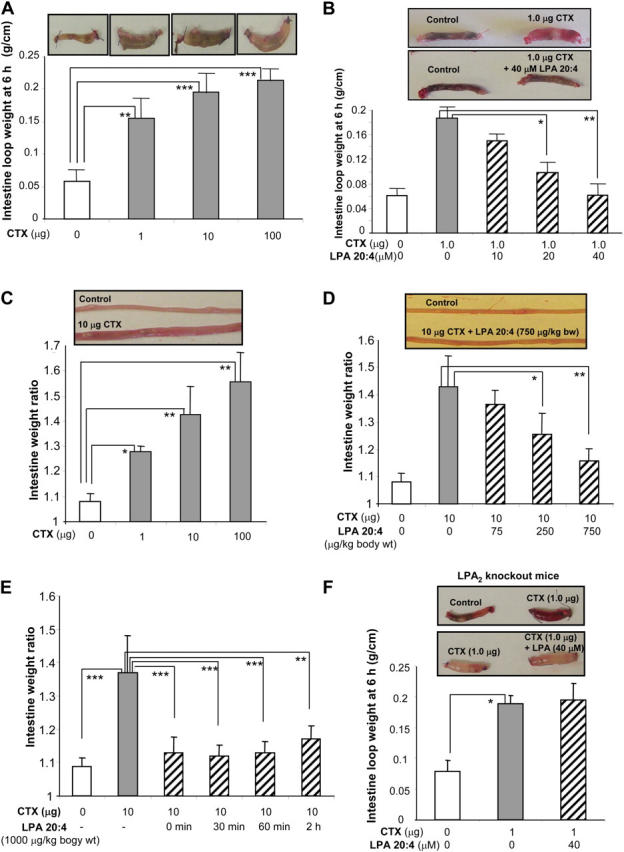
LPA inhibits CFTR-dependent intestinal fluid secretion induced by CTX. (A) Representative mouse ileal loops 6 h after luminal injection with CTX (pictures). The bar graphs (bottom) show the averaged loop weight (n = 3). (B) Representative ileal loops 6 h after luminal injection of CTX without or with LPA 20:4 (pictures). The bar graphs (bottom) show the averaged loop weight (n = 3–5). (C) Representative segments of mouse whole small intestine from open-loop model 6 h after oral gavage of CTX (pictures). Bar graphs show averaged fluid accumulation in the whole small intestine. Data shown as entire intestine weight before/intestine weight after luminal fluid removal (n = 3–4). (D) Representative segments of mouse small intestine from open-loop model 6 h after simultaneous oral gavage of CTX and LPA 20:4. Bar graphs show averaged fluid accumulation in the whole small intestine (n = 3–4). (E) CTX-induced intestinal fluid secretion after subsequent oral administration of LPA 20:4 in mouse open-loop model. Oral administration of LPA 20:4 was performed at different time points after CTX feeding (n = 5–8). (F) Representative ileal loops 6 h after luminal injection of CTX without or with LPA 20:4 in LPA2 knockout mice (pictures). The bar graphs (bottom) show the averaged loop weight (n = 5–6). *P < 0.05; **P < 0.01; ***P < 0.001.
Additionally, in an open-loop mouse model, oral administration of CTX induced a marked fluid accumulation within the whole small intestine in a dose-dependent manner (Fig. 5 C). This was attenuated substantially by simultaneous administration of LPA in a dose-dependent fashion (Fig. 5 D). Biologically effective portions of orally applied LPA remain intact in the whole intestine during the procedure (Fig. S5 B). To explore the therapeutic potential of LPA treatment of diarrhea and to exclude the possibility that LPA might interact directly with CTX or prevent binding of CTX to the ganglioside receptor (4), we also tested LPA-mediated inhibition of intestinal fluid accumulation in mice that were prestimulated by CTX. Mice that received a dose of LPA at 0–2 h after exposure to CTX also showed a significant decrease in intestinal fluid secretion (Fig. 5 E). Ileal loop experiments also were performed in LPA2 knockout mice. The inhibitory effect of LPA on CFTR-dependent fluid secretion was not observed in LPA2 knockout mice (Fig. 5 F). These results clearly demonstrate that inhibition of CFTR-mediated secretory diarrhea by LPA requires the LPA2 receptor.
LPA2 forms a macromolecular complex with CFTR mediated by way of NHERF2
We hypothesized that the interaction between CFTR and LPA2 likely was mediated by NHERF2. To test this, we assembled a macromolecular complex of LPA2, NHERF2, and CFTR in vitro, which is represented schematically in Fig. 6 A (24). A macromolecular complex was formed between maltose binding protein (MBP)-C–CFTR, glutathione S-transferase (GST)-NHERF2, and LPA2 (Fig. 6 B). MBP-C–CFTR did not bind directly to LPA2 nor did the complex form in the presence of GST or MBP alone (Fig. 6 B). The complex formation was PDZ-motif dependent. Mutating the last amino acid (aa) of LPA2 (L351A) reduced the complex formation, whereas deleting the last three aa's of LPA2 (ΔSTL) completely eliminated the macromolecular complex (Fig. 6 C).
Figure 6.

LPA2 forms a macromolecular complex with CFTR mediated through NHERF2. (A) Pictorial representation of the macromolecular complex assay (see Materials and methods for details). (B) Macromolecular complex of MBP-C–CFTR, GST–NHERF2, and Flag-tagged LPA2. (C) Macromolecular complex of MBP-C–CFTR, GST–NHERF2, and Flag-tagged LPA2, LPA2–ΔSTL, or LPA2–L351A. (D) BHK or BHK–CFTR cells transiently transfected with Flag-LPA2 were cross-linked using 1 mM DSP and coimmunoprecipitated using anti-CFTR antibody (NBD-R) and probed for LPA2. (E) Cell lysates from HT29-CL19A (left) and Calu-3 (right) were coimmunoprecipitated using anti-CFTR antibody (R1104) and probed for NHERF2 and LPA2. LPA2 blot was visualized using SuperSignal West Femto Maximum Sensitivity Substrate because of the weak signal (Pierce Chemical Co.).
The in vitro macromolecular complex assembly does not indicate whether the complex exists in native membranes; therefore, cross-linking experiments were performed to capture the complex in overexpressing cells (30). Fig. 6 D shows a cross-linked complex that contains CFTR and LPA2 in BHK cells that express endogenous NHERF2. We next immunoprecipitated CFTR from cultured epithelial cells and demonstrated that LPA2 and NHERF2 were present in the complex in HT29-CL19A and Calu-3 cells (Fig. 6 E). These studies suggest that a macromolecular complex of LPA2–NHERF2–CFTR likely is present in the apical surface of epithelial cells.
Disruption of the macromolecular complex of LPA2–NHERF2–CFTR using an LPA2-specific peptide prevents LPA2-mediated CFTR inhibition by LPA
We next determined if a physical association of LPA2 and CFTR by way of NHERF2 was essential for the LPA-elicited inhibition of the Cl− channel. To disrupt the complex, we delivered an LPA2 isoform-specific peptide containing the PDZ motif (last 11 aa's of LPA2; aa's 341–351), using the Chariot system (45), into polarized epithelial cells before Isc measurements. Isc in response to ADO in the presence of LPA was monitored. LPA2-specific peptide significantly prevented the LPA-elicited inhibition of CFTR-dependent Cl− currents in polarized HT29-CL19A (Fig. 7 A) and Calu-3 monolayers (Fig. S6 A, available at http://www.jem.org/cgi/content/full/jem.20050421/DC1). At the end of the experiment, the epithelial cells were fixed and immunostained to confirm the delivered peptide (Fig. 7 B and Fig. S6 B). Other peptides (CFTR peptide aa's 107–117) were used as a negative control (unpublished data). We also demonstrated that the LPA2 peptide could compete with MBP–LPA2 C-tail for binding to NHERF2 in a dose-dependent manner (Fig. 7 C).
Figure 7.

Disruption of the macromolecular complex of LPA2-NHERF2–CFTR using a LPA2-specific peptide prevents LPA2-mediated CFTR inhibition by LPA. (A) Isc in response to ADO in HT29-CL19A cells with or without LPA2-specific peptide delivered before Isc measurement. Cells in both groups were pretreated with LPA 20:4 (25 μM) for 30 min before activating with ADO. (B) Immunofluorescence micrographs of HT29-CL19A cells from (A) showing the LPA2-specific peptide delivered (top; green). The nucleus was stained with propidium iodide (red). HT29-CL19A cells express endogenous LPA2 (bottom). (C) NHERF2 binding to LPA2 in the presence of LPA2-specific peptide. A curve showing the averaged band density also is presented. *P < 0.05; **P < 0.01 compared with control (n = 3).
DISCUSSION
In the present study, by performing a series of in vitro, ex vivo, and in vivo experiments, we investigated the mechanism of how the macromolecular complex of CFTR, NHERF2, and LPA2 regulates the channel function. This is the first study to show that a macromolecular complex (CFTR–NHERF2–LPA2) formed in vitro is also physiologically and functionally relevant in vivo. We have made a compelling case to demonstrate that (a) there is a physical interaction among CFTR, NHERF2, and LPA2; (b) this interaction is functionally relevant in inhibiting the CFTR function; and (c) such protein–protein interactions are functionally relevant in the normal physiology of the gut. The relevance of these studies will be in trying to prevent secretory diarrhea, which is prevalent in developing countries, with 1.5 billion episodes of diarrhea and 4 million associated deaths in children annually (46).
We tested the effect of LPA on CFTR channel function under various conditions. For cultured cells or polarized cell monolayers, we used LPA of 0–40 μM, which is within the physiologic concentrations of LPA in blood serum (range, 5–50 μM) that was reported previously (47, 48). In the open-loop experiments, the maximal dose of LPA that we delivered into the mouse stomach was 1 mg/kg body weight, which is similar to the suggested dose for various dietary supplements (e.g., vitamins, minerals).
ADO is an endogenous purine nucleoside that is generated at sites of tissue stress and injury, including inflammation, ischemia, and tissue remodeling. Following its production, ADO diffuses to the surrounding cells, where it binds to the ADO receptors, and acts as a paracrine factor with diverse effects on a variety of organ systems. It was reported that in the intestinal lumen, ADO is produced in crypt abscesses during active inflammation by the interaction of neutrophils with the intestinal epithelia, whose ectonucleotidases convert neutrophil-derived ATP into ADO, which acts through the A2b ADO receptor subtype to induce vectorial chloride transport (49–51). Based on these observations, it was hypothesized that luminal ADO may be involved in secretory diarrhea that is elicited in pathophysiologic states, such as parasitic or allergic diseases, and hypereosinophilic syndromes (52, 53). ADO is produced at almost 1 μM in unstressed tissue, whereas in inflamed or ischemic tissues it can be ≤100 μM (35). In light of extensive literature support for the physiologic and pathophysiologic role of ADO, our application of 0.2–100 μM of this ligand is well within the (patho)physiologic range. In addition to ADO, we also used cpt-cAMP at submaximal CFTR activating conditions and forskolin (increases cAMP globally at 10 μM concentration) as agonists.
It was demonstrated that cAMP accumulation inside cells is inhibited by LPA treatment (54, 55). In this study, we also monitored cAMP levels under various conditions. We observed that upon stimulation with low concentration of ADO (2 μM), cAMP accumulation inside polarized epithelial cells (Calu-3) was almost indistinguishable from that of unstimulated cells (Fig. 4 A). However, CFTR is functional under these conditions and LPA can inhibit channel activity significantly (Fig. 3 B). Our data are consistent with the published results of Stutts's group (37), who demonstrated a compartmentalized signaling from the receptor (A2b) to the channel (CFTR) using electrophysiologic methods. This group reported that CFTR chloride channel function was observed upon stimulation of the cells by a low concentration of ADO (1 μM); the accumulation of cAMP inside the cell was not significantly higher than that from unstimulated control cells (37). In the present study, we found that LPA efficiently inhibits CFTR function (in response to 2 μM ADO; Fig. 3 B) without causing a decrease in the global cAMP accumulation in the cell (Fig. 4 A). Our results support the notion that cAMP likely is generated in a compartmentalized pocket upon stimulation by receptor-mediated agonists (ADO). However, when the ADO level was increased to 20 μM, we observed that the CFTR function increased slightly compared with 2 μM ADO, but there was a significant increase in cAMP accumulation inside the cell (global cAMP accumulation). Although there is a significant decrease in cAMP accumulation with LPA treatment, the Isc is not significantly different (in the presence or absence of LPA); this suggests that a global increase in cAMP may offset CFTR inhibition that is elicited by LPA.
LPA inhibition of CFTR function is most prominent when tested at a low concentration of ADO in the polarized monolayers (0.2–2 μM; Fig. 3, A and B) and in intestinal epithelia (2–40 μM; Fig. 3, C and D), or at a submaximal activating dose of cpt-cAMP (20–100 μM) in isolated intestinal epithelia (Fig. S3 A). This is because polarization of epithelia leads to the accurate targeting of the channel (CFTR) and the receptor (LPA2) to the apical surfaces, compared with nonpolarized cells. In addition, in Ussing chamber experiments, the CFTR-mediated Cl− currents (short-circuit currents) were monitored in real time, and we could titrate various concentrations of the agonists and monitor the CFTR response that shows maximum inhibition by LPA. In contrast, cells that were used for iodide efflux measurement (Fig. 2, A–D) were grown in 60-mm dishes and were not polarized. Due to the experimental limitations, we had to choose a concentration of ADO (2 μM). cpt-cAMP used in this study (Fig. S3 A) activated CFTR submaximally (<50% of maximum activating cocktail). Therefore, external addition of a low concentration of cpt-cAMP would be very similar to cAMP accumulation observed with low concentrations of agonists, such as ADO, although ADO would likely generate a more compartmentalized cAMP accumulation.
Diarrhea is the most common gastrointestinal disorder. It is well-established that CFTR plays a central role in this process (5, 13, 36). Any reagent or factor that augments CFTR activity (e.g., cAMP in CTX-induced diarrhea) at the luminal surface of the colonic epithelial cells is likely to lead to diarrhea. Conversely, factors that inhibit the CFTR Cl− channel are likely to be beneficial in cases of CFTR-induced diarrhea. Given that LPA2 is expressed in the gut, localized to the apical surface, and activates the Gi pathway and leads to decreased cAMP accumulation, we propose a novel model (Fig. 8). According to our hypothesis, LPA2 and CFTR are associated physically with NHERF2, which clusters LPA2 and CFTR into a macromolecular complex at the apical plasma membranes of epithelial cells. This macromolecular complex is the foundation of functional coupling between LPA signaling and CFTR-mediated Cl− transport. The model predicts that if the physical association is disrupted, functional coupling will be compromised. Upon LPA stimulation of the receptor, AC is inhibited through the Gi pathway, which leads to a decrease in cAMP level. This decreased local or compartmentalized accumulation of cAMP results in the reduced activation of Cl− channel in the vicinity by CFTR agonists (e.g., ADO). Our findings shed light on the detailed mechanism that underlies the transduction pathway of the LPA2 agonists, and helps to clarify how LPA inhibits CFTR-dependent Cl− channel activity; all of this will lead to the alleviation of diarrhea. Because LPA is richly available in certain forms of foods (14, 15), our proof of concept study might pave the way for the use of certain diets to control diarrhea.
Figure 8.

A schematic view of LPA inhibition on CFTR-dependent Cl− transport.
MATERIALS AND METHODS
Tissue culture and transfection.
HT29-CL19A, Calu-3, and BHK (baby hamster kidney cells) were cultured as described (30). For Ussing chamber experiments, HT29-CL19A and Calu-3 cells were grown on permeable filters (6.5-mm diameter). BHK cells were transfected transiently with various wild-type Flag-tagged lysophosphatidic acid (LPA) receptors or mutant Flag-tagged LPA2 receptors using a vaccinia virus expression system (56). Animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Tennessee Health Science Center.
Antibodies, reagents, and constructs.
Cystic fibrosis transmembrane conductance regulator (CFTR) antibodies, R1104 monoclonal mouse antibody and NBD-R polyclonal rabbit antibody, have been described previously (57). Anti-LPA2 antibody (rabbit-2143) against the last 11 amino acids (aa's 341–351) of LPA2 and anti–Na+/H+ exchanger regulatory factor (NHERF)-2 antibody (rabbit-2346) against the full-length NHERF2 protein were generated by Genemed Synthesis, CA. Anti-Flag mAb, cholera toxin (CTX), and pertussis toxin (PTX) were obtained from Sigma-Aldrich. Maltose binding protein (MBP)-fusion proteins for COOH-terminal tails of LPA1 (aa's 316–364), LPA2 (aa's 298–351), and LPA3 (aa's 298–353) were generated using pMAL vectors (New England Biolabs, Inc.). Flag-tagged full-length LPA2, LPA2-ΔSTL, and LPA2-L351A were generated using pcDNA-3 vector (Invitrogen) and QuickChange mutagenesis kit (Stratagene). The peptide delivery system (Chariot) was procured from Active Motif. LPA 18:1, sphingosine-1-phosphate (S1P), and phosphatidic acid (PA) were purchased from Avanti Polar Lipids, Inc. Other lipids, LPA (20:4) and LPA (18:2), have been described (21). Other reagents were described previously (30).
Pull-down assay.
Pull-down assay was performed as described (30). In brief, BHK or COS-7 cells expressing Flag-tagged LPA receptors (or CFTR) were lysed in lysis buffer (PBS 0.2% Triton X-100 plus protease inhibitors). Cell lysates were mixed at 4°C for 15 min followed by centrifugation at 15,000 g for 10 min at 4°C. Glutathione S-transferase (GST), GST–NHERF1, and GST–NHERF2 (0–3 μM) were added and mixed at 4°C with the clear supernatant or MBP-C–LPA2 for the direct binding between MBP-C–LPA2 and GST–fusion proteins. After 30–60 min incubation, glutathione sepharose beads (20 μl) were added and mixed for an additional 3 h. Thereafter, the mixture was spun at 800 g for 2 min, and the beads were washed three times with the same lysis buffer before the proteins were eluted from beads with Laemmli sample buffer (containing 2.5% β-mercaptoethanol). Eluates were separated on 4–15% gel, and were immunoblotted with anti-FLAG and anti-LPA2 antibodies.
Coimmunoprecipitation and immunoblotting.
Cells were harvested and processed as described previously (30). In brief, BHK cells (cross-linked with dithiobis(succinimidyl)propionate as described before [reference 30]) and epithelial cells (HT29-CL19A and Calu-3) were solubilized in RIPA buffer (plus protease inhibitors) on ice for 20 min, and lysates were spun at 15,000 g for 15 min at 4°C to pellet insoluble material. Protein concentration of the cell lysates was determined by the bicinchoninic assay (Pierce Chemical Co.). For coimmunoprecipitation, CFTR monoclonal antibody (R1104 IgG, 1.0 μg) was cross-linked to 20 μl protein A/G agarose as described previously (57). The supernatant was incubated with the cross-linked beads overnight at 4°C under a constant mixing. The beads were washed three times with RIPA buffer before the immunoprecipitated proteins were eluted with sample buffer containing 2.5% β-mercaptoethanol. The immunoprecipitated proteins were separated on 4–15% gel, and blotted using polyclonal anti-NHERF2 IgG, anti-LPA2 IgG, and anti-CFTR IgG (NBD-R IgG). For immunoblotting, cell lysates were separated on 4–15% SDS-PAGE, transferred to PVDF membranes, and immunoblotted for CFTR, NHERF2, and LPA2 using specific antibodies.
Short-circuit current measurements.
Calu-3 and HT29-CL19A polarized cell monolayers were grown to confluency on Costar Transwell permeable supports (filter area is 0.33 cm2). Filters were mounted in an Ussing chamber, and short-circuit currents (Isc) mediated through CFTR Cl− channel were performed as described (30). Epithelia were bathed in Ringer's solution (mM) (serosal/basolateral: 140 NaCl, 5 KCl, 0.36 K2HPO4, 0.44 KH2PO4, 1.3 CaCl2, 0.5 MgCl2, 4.2 NaHCO3, 10 Hepes, 10 glucose, pH 7.2, [Cl−] = 149), and low Cl− Ringer's solution (mM) (luminal/apical: 133.3 Na-gluconate, 5 K-gluconate, 2.5 NaCl, 0.36 K2HPO4, 0.44 KH2PO4, 5.7 CaCl2, 0.5 MgCl2, 4.2 NaHCO3, 10 Hepes, 10 mannitol, pH 7.2, [Cl] = 14.8) at 37°C, and gassed with 95% O2 and 5% CO2. LPA (20:4) was added into the luminal and serosal sides of the cell monolayers for 30 min before ADO was added into the luminal side. In parallel, some filters also were pretreated for 30 min with PA (0–35 μM) and S1P (0–35 μM) before ADO addition. CFTR Cl− channel inhibitor DPC (500 μM; added into luminal side) was used to inhibit the Cl− currents toward the end of the experiment.
To demonstrate the effect of LPA on Cl− transport in the mouse mucosa, the mice were killed, and the distal small intestine (∼2–4 cm proximal to the cecum) was removed and immediately placed in ice-cold, oxygenated Ringer solution, and opened along the mesenteric border. Indomethacin (10 μM) was present in the rinse and experimental Ringer solutions to prevent prostanoid generation during tissue manipulation. A patch of small intestine proximal to the cecum was stripped of serosal and smooth muscle layers (34) before being mounted in a Ussing chamber (0.112 cm2 exposed surface area). Any nerve plexus activity in the partially remaining myenteric plexus was blocked by tetrodotoxin acetate (1 μM), and local prostaglandin E production was inhibited by indomethacin (10 μM). LPA (20:4; 0–35 μM) was added into the luminal and serosal sides of the isolated mouse intestine for 30 min. cpt-cAMP (0–100 μM) or ADO (0–100 μM) were applied to both sides to elicit a CFTR-dependent Cl− current response, followed by luminal addition of diphenylamine-2-carboxylate (DPC; 500 μM) toward the end of the experiment. The epithelial integrity was monitored by passing a 3-mV pulse (for cultured cells) or 0.5-mV pulse (for isolated intestinal preparation) across the epithelia every min during the entire experiments.
Intestinal fluid secretion (in vivo) experiments.
The method of Verkman's group was followed with modifications (38). CD1 mice (body weight 20–22 g; obtained from Charles River Laboratories) were held off food for 24 h before inducing anesthesia using pentobarbital (60 mg/kg). Mouse body temperature was maintained at 36–38°C during surgery using a circulating water heating pad. A small abdominal incision was made to expose the small intestine. Ileal loops (∼20 mm) proximal to the cecum were exteriorized and isolated (two loops per mouse: 1 loop PBS, 1 loop PBS plus CTX). The closed-loops were injected with 100 μl PBS alone or PBS containing CTX (1–100 μg). The control and CTX-injected loops were tested in the presence and absence of LPA (10–40 μM). The abdominal incision and skin incision were closed with wound clips, and the mice were allowed to recover. Intestinal loops were collected in a terminal procedure 6 h later. PA (10–100 μM) and S1P (10–100 μM) also were used in parallel experiments as described above.
In the open-loop mouse model of secretory diarrhea, mice were given single doses of CTX (1, 10, or 100 μg) dissolved in 100 μl 7% NaHCO3 buffer (or buffer alone) in the presence or absence of LPA 20:4 (BSA-complexed, 75, 250, or 750 μg/kg body weight) delivered by an orogastric feeding needle. In some experiments, oral administration of LPA 20:4 (BSA-complexed, 1,000 μg/kg body weight) was performed at different time points (0–2 h) after CTX feeding. 6 h later, the mice were killed. The entire small intestine (from pylorus to cecum) was exteriorized and isolated with care to avoid tissue rupture and fluid loss. The associated mesentery and connective tissues were removed, and the weight of the intestinal tissue and the enclosed fluid was determined. The intestine was opened longitudinally to remove luminal fluid by blotting, and was weighed again. Intestinal fluid accumulation was determined from the ratio of intestinal weight before/intestinal weight after luminal fluid removal.
Macromolecular complex assembly.
This assay (24) was performed using maltose binding protein (MBP)-CFTR-C tail fusion protein (1 μM) immobilized on amylose beads (20 μl) and incubated with GST–NHERF2. This step, which is called pairwise binding, was done in 200 μl lysis buffer (PBS-0.2% Triton X-100 plus protease inhibitors) and mixed at 22°C for 2 h. The complex was washed once with the same buffer, and allowed to bind Flag-tagged LPA2 from lysates of BHK cells expressing the receptor. The binding was done at 4°C for 3 h with constant mixing. The complex was washed with lysis buffer, eluted with sample buffer, and analyzed by immunoblotting using anti-Flag mAb.
Delivery of LPA2 receptor-specific peptide and immunostaining.
Delivery of LPA2-specific peptide was performed using the Chariot system according to the manufacturer's instructions (Active Motif) (45). In brief, 1.2 μM peptide containing the PDZ motif of LPA2 (last 11 aa's, 341–351, ENGHPLMDSTL) was mixed with Chariot solution (total volume: 400 μl) at room temperature for 30 min. The Chariot-peptide complex was added to luminal and serosal sides of polarized Calu-3 and HT29-CL19A cells grown on permeable supports and incubated for 1 h at 37°C in a humidified atmosphere containing 5% CO2 before mounting in an Ussing chamber. At the end of the experiment, the epithelial cells were fixed and immunostained for the efficiency of the peptide delivery using anti-LPA2 antibody, and were subjected to immunofluorescence confocal microscopy. Other peptide (CFTR peptides, aa's 107–117) also was delivered as described before as a control.
Cell-attached single-channel recordings.
Single-channel recordings were obtained from HT29-CL19A cells using the cell-attached configuration (58). Patch-clamp pipettes were obtained using quartz class (Sutter Instrument Co.), and a Sutter model P-2000 puller and had resistance of 6–8 MΩ. The extracellular (pipette and bath) solution contained 140 mM NMDG-Cl, 2 mM MgCl2, 2 mM CaCl2, 10 mM Hepes, and 200 μM DIDS (to block non-CFTR anion channel pharmacologically) titrated to a pH of 7.4 with NMDG. CFTR channels were activated with 2 μM ADO or 10 μM forskolin included in the pipette or bath solution as indicated. Single-channel currents were recorded continuously for 5 min at a test potential of 60 mV (referenced to the cell interior) delivered from the recording electrode, and were filtered at 1 kHz and sampled at 2 kHz. All experiments were conducted at room temperature (22–24°C) using an EPC-9 patch clamp amplifier (HEKA Electronik GmbH) and the Pulse + PulseFit V 8.65 acquisition program (HEKA Electronik GmbH). Data analysis was performed using TacX4.1.5 (Bruxton Corp.).
Measurement of cAMP level in cultured cells and intestine tissue.
Calu-3 or HT29-CL19A cells were cultured (100-μl volumes) in standard 96-well microplates (tissue-culture grade), with cell concentrations of between 104–106 cells/ml. The plates were incubated overnight at 37°C (5% CO2 and 95% humidity). On the third day, the medium was decanted and 100 μl PBS (± LPA) was added and incubated for 20–30 min before the addition of agonists (ADO, or CFTR agonist cocktail) and incubation for another 5 min. The cells were lysed, and intracellular cAMP levels were measured using a cAMP Biotrak competitive enzyme immunoassay system (GE Healthcare) according to the manufacturer's instructions. For the measurement of cAMP levels in the native intestine tissue, ileal loops (∼2 cm) were exposed to 0.25–100 μg CTX. After 6 h of CTX treatment, the excised tissue sections were frozen rapidly in liquid nitrogen and samples were stored at −20°C until the cAMP assay was performed using the liquid phase extraction method according to the manufacturer's instructions (cAMP Biotrak enzyme immunoassay, GE Healthcare).
Statistical analysis.
Results are presented as mean ± SEM for the indicated number of experiments. Statistical analyses were performed using Student's t test and one-way ANOVA. A value of P < 0.05, P < 0.01, or P < 0.001 was considered to be statistically significant.
Online supplemental material.
Fig. S1 shows that LPA2 mRNA is detected in mouse intestinal tissue and various epithelial cells. Fig. S2 shows that LPA2 interacts preferentially with NHERF2. Fig. S3 shows that LPA inhibits CFTR-dependent Cl− currents in isolated intestine epithelia, but failed to inhibit Cl− currents when CFTR was activated by cocktail. Fig. S4 shows the single-channel recordings of CFTR in cell-attached configuration in HT29-CL19A cells in response to cpt-cAMP stimulation with or with LPA pretreatment. Fig. S5 shows that CTX induced a dose-dependent increase of cAMP production in gut tissue, and intact LPA is recovered 2 h later from intestine when complexed with BSA. Fig. S6 shows that LPA2-specific peptide prevented the LPA inhibition of CFTR-mediated Isc in polarized Calu-3 cells. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20050421/DC1.
Acknowledgments
We thank Dr. D.L. Armbruster for critically reading the manuscript, Dr. E.E. Strehler for NHERF2 antibody, Dr. J.A. Bobich and Dr. L.L. Clarke for technical advice, Dr. J. Jaggar for help with intestine preparation, M. McCarty for help with open-loop experiments, and Dr. K. Liliom and F. Zhou for help with data analysis.
C. Li is a recipient of the Dorothy K. and Daniel L. Gerwin graduate scholarship, and the Leonard Share Young Investigator Award from the University of Tennessee Health Science Center. This work was supported by grants from the National Institutes of Health (to A.P. Naren , G.J. Tigyi, and D.J. Nelson), and a Career Investigator Award from the American Lung Association (to A.P. Naren).
The authors have no conflicting financial interests.
Abbreviations used: aa, amino acid; AC, adenylate cyclase; ADO, adenosine; CFTR, cystic fibrosis transmembrane conductance regulator; cpt-cAMP, 8-(4-chlorophenylthio)-cyclic AMP; CTX, cholera toxin; DPC, diphenylamine-2-carboxylate; GST, glutathione S-transferase; IBD, inflammatory bowel disease; Isc, short-circuit current; LPA, lysophosphatidic acid; LPA1/2/3, type 1/2/3 LPA receptors; MBP, maltose binding protein; NHERF1/2, Na+/H+ exchanger regulatory factor–1 or –2; PA, phosphatidic acid; PDZ, PSD95/Dlg/ZO-1; PTX, pertussis toxin; S1P, sphingosine-1-phosphate.
References
- 1.Goodman, L., and J. Segreti. 1999. Infectious diarrhea. Dis. Mon. 45:268–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kosek, M., C. Bern, and R.L. Guerrant. 2003. The global burden of diarrheal disease, as estimated from studies published between 1992 and 2000. Bull. World Health Organ. 81:197–204. [PMC free article] [PubMed] [Google Scholar]
- 3.Guerrant, R.L., and D.A. Bobak. 1991. Bacterial and protozoal gastroenteritis. N. Engl. J. Med. 325:327–340. [DOI] [PubMed] [Google Scholar]
- 4.Sears, C.L., and J.B. Kaper. 1996. Enteric bacterial toxins: mechanisms of action and linkage to intestinal secretion. Microbiol. Rev. 60:167–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke, L.L., B.R. Grubb, S.E. Gabriel, O. Smithies, B.H. Koller, and R.C. Boucher. 1992. Defective epithelial chloride transport in a gene targeted mouse model of cystic fibrosis. Science. 257:1125–1128. [DOI] [PubMed] [Google Scholar]
- 6.Bassaganya-Riera, J., K. Reynolds, S. Martino-Catt, Y. Cui, L. Hennighausen, F. Gonzalez, J. Rohrer, A.U. Benninghoff, and R. Hontecillas. 2004. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 127:777–791. [DOI] [PubMed] [Google Scholar]
- 7.Sandler, R.S., J.E. Everhart, M. Donowitz, E. Adams, K. Cronin, C. Goodman, E. Gemmen, S. Shah, A. Avdic, and R. Rubin. 2002. The burden of selected digestive diseases in the United States. Gastroenterology. 122:1500–1511. [DOI] [PubMed] [Google Scholar]
- 8.Collins, S.M., T. Piche, and P. Rampal. 2001. The putative role of inflammation in the irritable bowel syndrome. Gut. 49:743–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiocchi, C. 1998. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 115:182–205. [DOI] [PubMed] [Google Scholar]
- 10.Dobbins, J.W., J.P. Laurenson, and J.N. Forrest Jr. 1984. Adenosine and adenosine analogues stimulate adenosine cyclic-3′, 5′-monophosphate-dependent chloride secretion in the mammalian ileum. J. Clin. Invest. 74:929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grasl, M., and K. Turnheim. 1984. Stimulation of electrolyte secretion in rabbit colon by adenosine. J. Physiol. 346:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cormet-Boyaka, E., A.P. Naren, and K.L. Kirk. 2003. CFTR interacting proteins. The Cystic Fibrosis Transmembrane Conductance Regulator. K.L. Kirk and D.C. Dawson, editors. Kluwer Academic/Plenum Publishers, New York. 94–118.
- 13.Gabriel, S.E., K.N. Brigman, B.H. Koller, R.C. Boucher, and M.J. Stutts. 1994. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science. 266:107–109. [DOI] [PubMed] [Google Scholar]
- 14.Tokumura, A., K. Fukuzawa, Y. Akamatsu, S. Yamada, T. Suzuki, and H. Tsukatani. 1978. Identification of vasopressor phospholipid in crude soybean lecithin. Lipids. 13:468–472. [DOI] [PubMed] [Google Scholar]
- 15.Nakane, S., A. Tokumura, K. Waku, and T. Sugiura. 2001. Hen egg yolk and white contain high amounts of lysophosphatidic acids, growth factor-like lipids: distinct molecular species compositions. Lipids. 36:413–419. [DOI] [PubMed] [Google Scholar]
- 16.Tigyi, G., and A.L. Parrill. 2003. Molecular mechanisms of lysophosphatidic acid action. Prog. Lipid Res. 42:498–526. [DOI] [PubMed] [Google Scholar]
- 17.Mills, G.B., and W.H. Moolenaar. 2003. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer. 3:582–591. [DOI] [PubMed] [Google Scholar]
- 18.Ishii, I., N. Fukushima, X. Ye, and J. Chun. 2004. Lysophospholipid receptors: signaling and biology. Annu. Rev. Biochem. 73:321–354. [DOI] [PubMed] [Google Scholar]
- 19.Sturm, A., J. Zeeh, T. Sudermann, H. Rath, G. Gerken, and A.U. Dignass. 2002. Lisofylline and lysophospholipids ameliorate experimental colitis in rats. Digestion. 66:23–29. [DOI] [PubMed] [Google Scholar]
- 20.Funk-Archuleta, M.A., M.W. Foehr, L.D. Tomei, K.L. Hennebold, and I.C. Bathurst. 1997. A soy-derived antiapoptotic fraction decreases methotrexate toxicity in the gastrointestinal tract of the rat. Nutr. Cancer. 29:217–221. [DOI] [PubMed] [Google Scholar]
- 21.Sturm, A., and A.U. Dignass. 2002. Modulation of gastrointestinal wound repair and inflammation by phospholipids. Biochim. Biophys. Acta. 1582:282–288. [DOI] [PubMed] [Google Scholar]
- 22.Deng, W., L. Balazs, D.A. Wang, L. Van Middlesworth, G. Tigyi, and L.R. Johnson. 2002. Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology. 123:206–216. [DOI] [PubMed] [Google Scholar]
- 23.Deng, W., D.A. Wang, E. Gosmanova, L.R. Johnson, and G. Tigyi. 2003. LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 284:G821–829. [DOI] [PubMed] [Google Scholar]
- 24.Naren, A.P., B. Cobb, C. Li, K. Roy, D. Nelson, G.D. Heda, J. Liao, K.L. Kirk, E.J. Sorscher, J. Hanrahan, and J.P. Clancy. 2003. A macromolecular complex of beta 2 adrenergic receptor, CFTR, and ezrin/radixin/moesin-binding phosphoprotein 50 is regulated by PKA. Proc. Natl. Acad. Sci. USA. 100:342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yun, C.C., H. Sun, D. Wang, R. Rusovici, A. Castleberry, R.A. Hall, and H. Shim. 2005. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am. J. Physiol. Cell Physiol. 289:C2–11. [DOI] [PubMed] [Google Scholar]
- 26.Oh, Y.S., N.W. Jo, J.W. Choi, H.S. Kim, S.W. Seo, K.O. Kang, J.I. Hwang, K. Heo, S.H. Kim, Y.H. Kim, et al. 2004. NHERF2 specifically interacts with LPA2 receptor and defines the specificity and efficiency of receptor-mediated phospholipase C-beta3 activation. Mol. Cell. Biol. 24:5069–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hung, A.Y., and M. Sheng. 2002. PDZ domains: structural modules for protein complex assembly. J. Biol. Chem. 277:5699–5702. [DOI] [PubMed] [Google Scholar]
- 28.Songyang, Z., A.S. Fanning, C. Fu, J. Xu, S.M. Marfatia, A.H. Chishti, A. Crompton, A.C. Chan, J.M. Anderson, and L.C. Cantley. 1997. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 275:73–77. [DOI] [PubMed] [Google Scholar]
- 29.Im, D.S., C.E. Heise, M.A. Harding, S.R. George, B.F. O'Dowd, D. Theodorescu, and K.R. Lynch. 2000. Molecular cloning and characterization of a lysophosphatidic acid receptor, Edg-7, expressed in prostate. Mol. Pharmacol. 57:753–759. [PubMed] [Google Scholar]
- 30.Li, C., K. Roy, K. Dandridge, and A.P. Naren. 2004. Molecular assembly of cystic fibrosis transmembrane conductance regulator in plasma membrane. J. Biol. Chem. 279:24673–24684. [DOI] [PubMed] [Google Scholar]
- 31.Chang, X.-B., J.A. Tabcharani, Y.X. Hou, T.J. Jensen, N. Kartner, N. Alon, J.W. Hanrahan, and J.R. Riordan. 1993. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J. Biol. Chem. 268:11304–11311. [PubMed] [Google Scholar]
- 32.Bokoch, G.M., T. Katada, J.K. Northup, E.L. Hewlett, and A.G. Gilman. 1983. Identification of the predominant substrate for ADP-ribosylation by islet activating protein. J. Biol. Chem. 258:2072–2075. [PubMed] [Google Scholar]
- 33.Sano, T., D. Baker, T. Virag, A. Wada, Y. Yatomi, T. Kobayashi, Y. Igarashi, and G. Tigyi. 2002. Multiple mechanisms linked to platelet activation result in lysophosphatidic acid and sphingosine 1-phosphate generation in blood. J. Biol. Chem. 277:21197–21206. [DOI] [PubMed] [Google Scholar]
- 34.Clarke, L.L., and M.C. Harline. 1998. Dual role of CFTR in cAMP-stimulated HCO3− secretion across murine duodenum. Am. J. Physiol. 274:G718–G726. [DOI] [PubMed] [Google Scholar]
- 35.Hasko, G., and B.N. Cronstein. 2004. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 25:33–39. [DOI] [PubMed] [Google Scholar]
- 36.Barrett, K.E., and S.J. Keely. 2000. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu. Rev. Physiol. 62:535–572. [DOI] [PubMed] [Google Scholar]
- 37.Huang, P., E.R. Lazarowski, R. Tarran, S.L. Milgram, R.C. Boucher, and M.J. Stutts. 2001. Compartmentalized autocrine signaling to cystic fibrosis transmembrane conductance regulator at the apical membrane of airway epithelial cells. Proc. Natl. Acad. Sci. USA. 98:14120–14125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma, T., J.R. Thiagarajah, H. Yang, N.D. Sonawane, C. Folli, L.J. Galietta, and A.S. Verkman. 2002. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 110:1651–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Field, M. 1971. Intestinal secretion: effect of cyclic AMP and its role in cholera. N. Engl. J. Med. 284:1137–1144. [DOI] [PubMed] [Google Scholar]
- 40.Kimberg, D.V., M. Field, J. Johnson, A. Henderson, and E. Gershon. 1971. Stimulation of intestinal mucosal adenyl cyclase by cholera enterotoxin and prostaglandins. J. Clin. Invest. 50:1218–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tilly, J.L., and A.L. Johnson. 1989. Regulation of androstenedione production by adenosine 3′, 5′-monophosphate and phorbol myristate acetate in ovarian thecal cells of the domestic hen. Endocrinology. 125:1691–1699. [DOI] [PubMed] [Google Scholar]
- 42.Tilly, J.L., and A.L. Johnson. 1990. Control of plasminogen activator activity in the thecal layer of the largest preovulatory follicle in the hen ovary. Endocrinology. 126:2079–2087. [DOI] [PubMed] [Google Scholar]
- 43.Pinot, F., H. Walti, H.P. Haagsman, B.S. Polla, and M. Bachelet. 2000. Curosurf modulates cAMP accumulation in human monocytes through a membrane-controlled mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 278:L99–104. [DOI] [PubMed] [Google Scholar]
- 44.Bowman, C.C., and J.D. Clements. 2001. Differential biological and adjuvant activities of cholera toxin and Escherichia coli heat-labile enterotoxin hybrids. Infect. Immun. 69:1528–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris, M.C., J. Depollier, J. Mery, F. Heitz, and G. Divita. 2001. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 19:1173–1176. [DOI] [PubMed] [Google Scholar]
- 46.Bern, C., J. Martines, I. de Zoysa, and R.I. Glass. 1992. The magnitude of the global problem of diarrhoeal disease: a ten-year update. Bull. World Health Organ. 70:705–714. [PMC free article] [PubMed] [Google Scholar]
- 47.Baker, D.L., D.M. Desiderio, D.D. Miller, B. Tolley, and G.J. Tigyi. 2001. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal. Biochem. 292:287–295. [DOI] [PubMed] [Google Scholar]
- 48.Aoki, J., A. Taira, Y. Takanezawa, Y. Kishi, K. Hama, T. Kishimoto, K. Mizuno, K. Saku, R. Taguchi, and H. Arai. 2002. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem. 277:48737–48744. [DOI] [PubMed] [Google Scholar]
- 49.Madara, J.L., S. Nash, and C. Parkos. 1991. Neutrophil-epithelial cell interactions in the intestine. Adv. Exp. Med. Biol. 314:329–334. [DOI] [PubMed] [Google Scholar]
- 50.Sitaraman, S.V., D. Merlin, L. Wang, M. Wong, A.T. Gewirtz, M. Si-Tahar, and J.L. Madara. 2001. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J. Clin. Invest. 107:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eltzschig, H.K., J.C. Ibla, G.T. Furuta, M.O. Leonard, K.A. Jacobson, K. Enjyoji, S.C. Robson, and S.P. Colgan. 2003. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 198:783–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choy, M.Y., J.A. Walker-Smith, C.B. Williams, and T.T. MacDonald. 1990. Activated eosinophils in chronic inflammatory bowel disease. Lancet. 336:126–127. [DOI] [PubMed] [Google Scholar]
- 53.Weller, P.F. 1991. The immunobiology of eosinophils. N. Engl. J. Med. 324:1110–1118. [DOI] [PubMed] [Google Scholar]
- 54.Tigyi, G., D.J. Fischer, A. Sebok, F. Marshall, D.L. Dyer, and R. Miledi. 1996. Lysophosphatidic acid-induced neurite retraction in PC12 cells: neurite-protective effects of cyclic AMP signaling. J. Neurochem. 66:549–558. [DOI] [PubMed] [Google Scholar]
- 55.Fischer, D.J., K. Liliom, Z. Guo, N. Nusser, T. Virag, K. Murakami-Murofushi, S. Kobayashi, J.R. Erickson, G. Sun, D.D. Miller, and G. Tigyi. 1998. Naturally occurring analogs of lysophosphatidic acid elicit different cellular responses through selective activation of multiple receptor subtypes. Mol. Pharmacol. 54:979–988. [DOI] [PubMed] [Google Scholar]
- 56.Naren, A.P., M.W. Quick, J.F. Collawn, D.J. Nelson, and K.L. Kirk. 1998. Syntaxin 1A inhibits CFTR chloride channels by means of domain-specific protein-protein interactions. Proc. Natl. Acad. Sci. USA. 95:10972–10977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naren, A.P. 2002. Methods for the study of intermolecular and intramolecular interactions regulating CFTR function. Methods Mol. Med. 70:175–186. [DOI] [PubMed] [Google Scholar]
- 58.Chang, S.Y., A. Di, A.P. Naren, H.C. Palfrey, K.L. Kirk, and D.J. Nelson. 2002. Mechanisms of CFTR regulation by syntaxin 1A and PKA. J. Cell Sci. 115:783–791. [DOI] [PubMed] [Google Scholar]