

Abstract
Methods are described to determine the structures of viral membrane fusion domains in detergent micelles by NMR and in lipid bilayers by site-directed spin labeling and EPR spectroscopy. Since in favorable cases, the lower-resolution spin label data obtained in lipid bilayers fully support the higher-resolution structures obtained by solution NMR, it is possible to graft the NMR structural coordinates into membranes using the EPR-derived distance restraints to the lipid bilayer. Electron paramagnetic dynamics and distance measurements in bilayers support conclusions drawn from NMR in detergent micelles. When these methods are applied to a structure determination of the influenza virus fusion domain and four point mutations with different functional phenotypes, it is evident that a fixed-angle boomerang structure with a glycine edge on the outside of the N-terminal arm is both necessary and sufficient to support membrane fusion. The human immunodeficiency virus fusion domain forms a straight helix with a flexible C-terminus. While EPR data for this fusion domain are not yet available, it is tentatively speculated that, because of its higher hydrophobicity, a critically tilted insertion may occur even in the absence of a kinked boomerang structure in this case.
1. Introduction
Determining structures of membrane proteins still presents a formidable challenge to all current methods of macromolecular structure determination. However, like other methods, magnetic resonance spectroscopy has made great strides in recent years and has become a major asset in the field of structural biology of membrane proteins. We can distinguish between three major classes of magnetic resonance techniques that have been applied to membrane protein structure determination with ever increasing success. They include solid-state NMR, solution NMR, and EPR spectroscopy. The current state of affairs of solid-state NMR spectroscopy for membrane proteins is expertly covered in other articles of this special issue. Solution NMR spectroscopy has also gained a respectable position for obtaining structures of membrane proteins. Two recent reviews on this subject give good current overviews on this topic [1, 2]. The third method, EPR spectroscopy combined with site-directed spin-labeling, provides lower-resolution information that is nevertheless extremely valuable because this information can be obtained with small amounts of sample in nearly physiological membrane environments. Excellent recent reviews on this topic can be found in the literature [3, 4].
The three methods all have their strengths and weaknesses and as such are complementary. Apart from the different spectroscopic techniques that are used in each case, sample preparation is also fundamentally different for solid-state NMR, solution NMR, and site-directed spin-label (SDSL) EPR spectroscopy. As is true so often in science, the outcome of a particular experiment does not only depend on the experimental technique employed, but also on how the samples are prepared. Solid-state NMR has the advantage that membrane protein samples are reconstituted in lipid bilayer model membranes. However, quite often these membranes have to be aligned to a high degree relative to the magnetic field, which in some cases requires stacked model membranes with relatively small amounts of water in between. These methods of sample preparation are adequate for relatively small membrane proteins with the majority of their mass embedded in the lipid bilayer. Membrane proteins with large water-exposed portions are likely affected by such preparation methods. Other solid-state NMR methods require micro-crystalline samples for optimal spectral resolution, which again does not take full advantage of NMR methods that ideally should bypass the need for difficult crystallization steps. Solution NMR by definition requires membrane protein samples that are solubilized in detergent micelles. Protein-detergent complexes that are much larger than 100 kDa tumble slowly in the magnetic field and, therefore, cause severe resonance line-broadening and spectral overlap that limit the usefulness of this method. Even TROSY and other relaxation-suppression methods cannot handle membrane proteins that are embedded in even the smallest lipid bilayer vesicles. Although high-quality solution NMR spectra of quite large membrane proteins (up to 30–40 kDa proteins in mixed micelles of up to 100 kDa) have been obtained and used to determine their structures, the question arises to what extent these micellar systems represent the lipid bilayer of a biological membrane. The answer to this question is complex and depends on the particular detergents as well as the proteins that are investigated. A good method to further investigate this question is to use SDSL EPR spectroscopy, which is well suited for structural and dynamic studies of membrane proteins in lipid micelles and bilayers in small and large unilamellar vesicles. Therefore, SDSL EPR can be used to cross-validate higher resolution NMR results that have been obtained in less physiological environments. In this review, we demonstrate how high-resolution structural data can be obtained by solution NMR in lipid micelles and then transferred to lipid bilayers by complementary EPR studies in lipid bilayers.
Since our laboratory has been interested for several years in developing a structural framework for the action of membrane fusion proteins on lipid bilayers, we demonstrate these methods by using several examples of membrane-bound viral fusion domains. All membrane-enveloped viruses enter cells by undergoing a large conformational change of their surface-glycoproteins upon binding to appropriate target membranes. As part of this conformational change, they expose quite hydrophobic fusion domains (also called “fusion peptides”), which then become inserted into the target membrane in preparation for membrane fusion. These fusion domains can be N-terminal, internal, or composed of several sequence parts of the parent fusion protein. Here, we will discuss two specific cases, namely the fusion domains of influenza A virus and of human immunodeficiency virus 1 (HIV-1). In the case of influenza virus, the membrane-embedded fusion protein on the viral surface is hemagglutinin (HA) and in the case of HIV it is gp41/gp120. The fusion activities are primarily carried out by subunits HA2 and gp41, in influenza and HIV, respectively. Both have N-terminal fusion domains and their sequences are shown in Figure 1. Highly conserved residues (between different strains of the same virus) are shown in bold face. Although there are similarities between these two sequences, there are also differences. Both are rich in glycines and HIV is also rich in alanines. Both have repeated pairings of aromatic residues with either leucines or isoleucines. The HIV fusion domain is overall more hydrophobic, whereas the flu fusion domain has a few negatively charged residues in the middle and towards the end of the sequence. This latter difference may be related to the different mechanisms of how fusion is triggered in these two viruses. HIV binds to the surface of susceptible cells (some lymphocytes and astrocytes) and then fuses at pH 7 directly at the cell surface after receptor binding. By contrast, the flu virus binds to sialic residue receptors and is first internalized into the endosomes of epithelial cells (typically in the lung). This low pH of 5 that prevails in the endosome then triggers the conformational change and fusion with the endosomal membrane in this case. For these reasons, most structural and lipid interaction studies that will be summarized in this review have been conducted at pH 5 for flu and at pH 7 for HIV. More detail on mechanisms of viral membrane fusion and how structural transformations of viral fusion fusion proteins might be coupled to transformations of lipid bilayer structures that lead to membrane fusion can be found in several recent reviews [5–9].
Figure 1.

Sequences of influenza hemagglutinin and HIV gp41 fusion domains. They are located at the extreme N-terminus of the transmembrane subunit of each viral envelope spike glycoprotein. Highly conserved (between multiple strains of each virus) residues are shown in bold face. Very conservative mutations (e.g. leucine to isoleucine) may occasionally occur at positions shown in smaller bold face, whereas residues shown in larger bold face are absolutely conserved. Frequently observed clusters of (phenylalanine, aliphatic residue, glycine) are underlined.
Fusion domains are intermediate in hydrophobicity, i.e. their solubility is between that of constitutive bona fide integral membrane proteins and that of many antimicrobial peptides, which can exist in soluble and membrane-bound forms. Fusion domains by themselves are typically not soluble in water, but often soluble in moderately polar solvents like dimethylsulfoxide or acetonitrile/water or trifluoroethanol/water mixtures. Although NMR data of fusion peptides can be obtained in these solvents, such information is probably not very informative about their conformation in membranes. When solvent-solubilized fusion domains are combined with solvent-solubilized or aqueous dispersions of detergents or lipids and the solvent is subsequently removed, these peptides often do not adopt the same conformation as they would when reconstituted in purely aqueous systems. They often self-associate in a non-specific fashion when going through such solvent reconstitution procedures and the resulting aggregated forms appear not to disperse back to monomeric forms in the final protein/lipid or protein/detergent mixtures. At least in our experience, we have not been able to obtain highly resolved NMR spectra that would be adequate for structural studies using solvent-based reconstitution procedures. Instead, we have taken a different approach for studying apolar fusion domains by NMR and other techniques. We have added a solubility tag to the C-terminus of the fusion domains via a flexible glycine linker. The solubility tag consists of 4 lysines in the case of the flu fusion domain and 5 lysines in the case of the HIV fusion domain [10, 11]. These solubility tags basically replace the hydrophilic ectodomains of the full-length fusion proteins. The flu and HIV fusion domains become soluble up to concentrations of a few hundred micromolar upon adding these solubility tags. Therefore, the modified fusion domains can be bound to lipid model membranes or detergent micelles from aqueous solutions. This reconstitution procedure, which more closely follows the biological membrane insertion process, was essential in our hands to obtain high-resolution NMR data for structural studies as described in the next section. As discussed in more detail in the primary literature, the oligolysine tails do not appear to influence the structures of the fusion domains in membranes because they are uncoupled from the fusion domains with a flexible linker. However, they do enhance the binding to negatively charged membranes. This enhancement can be quantified and the binding energies of the fusion domains themselves (i.e., without tails) can be determined by subtraction of appropriate controls [10, 12].
2. Solution NMR of Micelle-Bound Fusion Domains
The flu fusion domain was chemically synthesized with its solubility tag and bound to perdeuterated dodecylphosphocholine (DPC) micelles. This fusion domain, as well as all others described in this report, is unfolded in solution as determined by CD spectroscopy. However, when lipid bilayer membranes or micelles are present, they bind with high affinity and fold. All NMR studies described here were carried out under conditions where virtually all fusion domains are micelle-bound and folded. Homonuclear TOCSY and NOESY spectra were recorded at 600 MHz. Backbone and side-chain protons could be completely assigned from these spectra. The N-terminal half of the fusion domain (residues 2 to 11) had many medium range backbone αN(i,i+3) and αβ(i,i+3) NOEs that indicated the presence of an α-helix. Medium range αN(i,i+3) and αβ(i,i+3) NOEs indicative of a second short helix were also present from residues 14 to 18. These patterns of helical secondary structure were confirmed by positive HN and negative Hα chemical shift indices. In addtition, several NOEs and chemical shifts defined a kink in the region from residues 11 to 14. The structure that was calculated based on the experimental distance and chemical shift derived angle restraints features a kinked boomerang-shaped amphipathic domain with a hydrophobic pocket on the bottom and a neutral glycine-rich N-terminal and partially charged C-terminal outer surface on top (Figure 2) [13].
Figure 2.

Structures of the influenza and HIV fusion domains determined by solution NMR in DPC micelles. The angle of the flu fusion domain “boomerang” structure at Asn 12 and Gly 13 is relatively fixed by hydrophobic interactions between Phe 9, Ile 10, and Trp 14 and the micelle, and possibly some weak hydrogen bonds in the kink region (PDB code: 1IBN; from [13]. The C-terminal tail of the HIV fusion domain from Ala 15 is flexible (PDB code: 2PJV; from [11]). However, in SDS micelles the helix of the HIV fusion domain extends out to Met 19 [18].
Subsequently, we targeted several mutant flu fusion domains for structural studies (Table 1). These were selected based on functional fusion assays with corresponding mutations in the full-length HA proteins expressed on CV-1 cells. The cells were incubated with red blood cells (RBCs) that had been fluorescently double-labeled with the lipid octadecyl-rhodamine and the soluble compound carboxyfluorescein. After brief incubation of the cell couples with pH 5 buffer, wild-type HA expressing cells fused completely with the RBCs as evidenced by flow of the green carboxyfluorescein and red rhodamine dye into the CV-1 cells. However, some mutant HAs elicited only hemifusion, i.e. lipid connectivity without forming a fusion pore. In these cases, only the red rhodamine dye moved from the RBC to the CV-1 cell membrane and carboxyfluorescein stayed confined in the RBCs. The mutants G1S [14] and E11A [15] are hemifusion mutants. In other cases neither dye moved to the CV-1 cell upon exposure to pH 5, indicating that fusion was blocked even before the hemifusion intermediate. G1V [14], F9A/I10A [16], and W14A [15] are non-fusion mutants. Yet another class of mutations had no effect on fusion. They include F9A [15], I10A [16], and N12A [15]. Finally, G13A had an unusual phenotype. It showed lipid connectivity like the hemifusion mutants, but the contents dye carboxyfluorescein leaked out into the medium rather than into the coupled CV-1 cells, indicating a laterally porous fusion connection between the cell couples (A.L. Lai and L.K. Tamm, unpublished results).
Table 1.
Structural, dynamical, and functional features of influenza hemagglutinin fusion domain mutants.
| Mutant | Phenotypea | Structure (NMR)b | Dynamics (EPR)c | End-End Distance (DEER)d |
|---|---|---|---|---|
| Wild-type | full fusion | boomerang | low | one major at 24 Å |
| G1S | hemifusion | boomerang | n.d.e | n.d. |
| G1V | no fusion | linear | n.d. | n.d. |
| F9A | full fusion | boomerang | low | one major at 24 Å |
| I10A | full fusion | n.d. | n.d. | n.d. |
| F9A/I10A | no fusion | n.d. | high | distribution 21 – 33 Å |
| E11A | hemifusion | work in progress | n.d. | n.d. |
| N12A | full fusion | n.d. | n.d. | n.d. |
| G13A | contents leakage | work in progress | n.d. | n.d. |
| W14A | no fusion | flexible angle | high | distribution 25 – 33 Å |
from cell-cell fusion experiments
in DPC micelles
in DPC micelles and lipid bilayers
in lipid bilayers
not determined
The glycine-1 mutations had been found empirically before the structure of the wild-type fusion domain was known. However, the other mutations from phenylalanine-9 to tryptophane-14 were specifically targeted to determine their role in stabilizing the kink in the boomerang structure. The wild-type structure at pH 5 indicated that this kink is stabilized by the following hydrogen bonds: NH(Glu 11)→CO(Gly8), NH(Asn 12)→CO(Phe 9), NH(Trp 14)→CO(Phe9), and γNH(Asn 12)→CO(Gly 8). Additionally, the kink might also be stabilized by hydrophobic anchors formed by Phe 9 and Trp 14.
The structure of the hemifusion mutant G1S was relatively unperturbed. It still formed a stable boomerang in DPC micelles (Figure 3) [17]. The major difference was the serine residue in position 1, which disrupted the continuous smooth glycine edge on the outer surface of the N-terminal arm. The structural and functional effect of replacing glycine-1 with a valine was much more dramatic. The structure was linear with no evidence for a kink around asparagine-12 (Figure 3) [17]. This result was very surprising. How could a single amino acid change at the extreme N-terminus have such a long-range effect on the structure of the entire domain? Closer inspection of the structure provides interesting clues to this question. First of all, this, as well as all the other structures described in this report, is only stable in micelles or lipid bilayers, i.e. the domains are always in full contact to amphiphilic interfaces (see also EPR measurements described below). Valine-1 of G1V is hydrophobic enough so that this residue turns into the hydrophobic core of the micelle or lipid bilayers. This hydrophobic interaction apparently is strong enough to unwind the first turn of the helix in the wild-type structure. In addition, it positions the peptide differently in the interface so that the kink in the boomerang straightens out. This result also suggests that the kink in the wild-type structure, although quite fixed according to the NMR structure calculations, is not energetically very stable, but could be dynamic as demonstrated below by EPR spectroscopy. The dramatic structural change observed with the G1V mutation explains its completely aborted fusion function. This structural change also dramatically lowers the enthalpy and free energy of binding of the mutant fusion domain to lipid bilayers [12].
Figure 3.
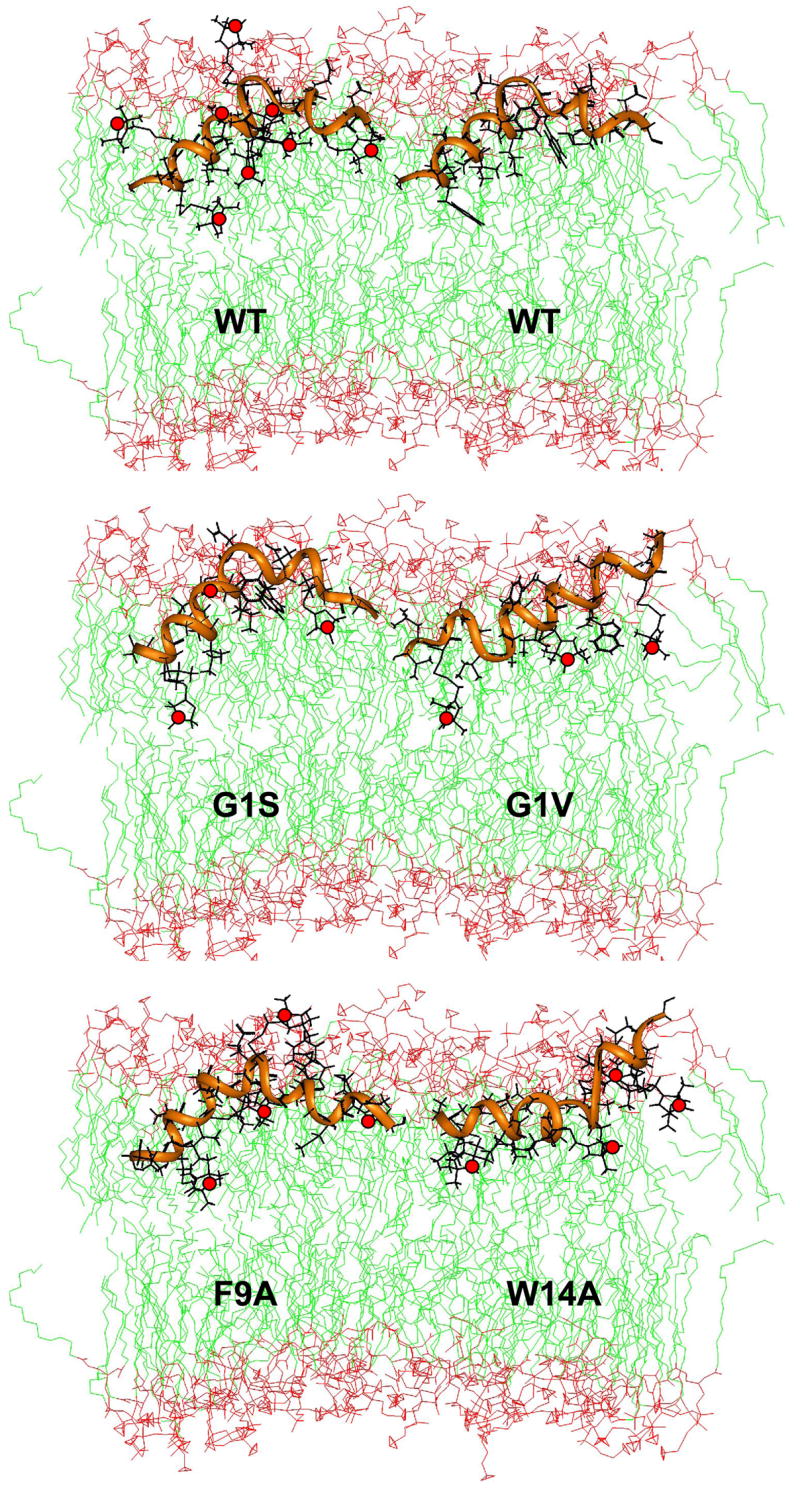
Docking of influenza hemagglutinin fusion domain structures determined by solution NMR in DPC micelles to lipid bilayers using distances from SDSL power saturation EPR spectroscopy. Top bilayer: wild-type (PDB code: 1IBN; left: with MTSL in residue positions 3, 5, 7, 9, 11, 12, 14, 18; right: all native side chains; from [13]); middle bilayer: G1S (PDB code: 1XOO; with MTSL in residue positions 3, 11, 18) and G1V (PDB code: 1XOP; with MTSL in residue positions 3, 11, 18; both from [17]); bottom bilayer: F9A (PDB code: 2JRD; with MTSL in residue positions 3, 7, 12, 18; from [16]) and W14A (PDB code: 2DCI; with MTSL in residue positions 3, 9, 12, 18; from [15]). The positions of the paramagnetic sites (unpaired electrons on nitroxide nitrogens) are shown with red dots. The POPC bilayer simulation “popc128a.pdb” available at http://moose.bio.ucalgary.ca/index.php?page=Structures_and_Topologies was used to represent the membranes.
The structure of the fully active mutant F9A was solved to serve as a positive control. As expected, it forms a relatively stable boomerang structure in DPC micelles as does the wild-type fusion domain [16] (Figure 3).
The other aromatic replacement mutant W14A produces a non-fusion phenotype, i.e. it is functionally similar to G1V. Therefore, it was interesting to solve its structure in DPC micelles by NMR as well. Interestingly, the outcome was different than for G1V. The structure had a hinge around asparagine-12, but unlike the boomerang structures of wild-type, F9A, and G1S, the hinge of W14A was very flexible and the C-terminal arm pointed in multiple directions that were all very different from those of the functional and partially functional fusion domains [15] (Figure 3).
As mentioned in the Introduction, the fusion domain of HIV gp41 is longer and significantly more hydrophobic than that of influenza HA. This required a slightly longer solubility tag and posed more difficulties with the resonance assignments from homonuclear NMR. Therefore, this fusion protein was expressed and uniformly 15N-labeled [11]. All backbone and side chain resonances were then assigned by a combination of homonuclear and heteronuclear strategies. The structure of the fusion domain bound to DPC micelles was then calculated based on NOE derived distance restraints and chemical shift and J-coupling derived dihedral angle restraints (Figure 2) [11]. The N-terminal half of structure was helical from about isoleucine-4 to alanine-14, but the C-terminal half from about alanine-15 to the end was disordered. The structure of a slightly longer construct was also solved in SDS micelles [18]. In this case the N-terminal helix extended from isoleucine-4 to methionine-19, followed by an unstable helix of three more residues and a flexible C-terminus. Otherwise, the structures in SDS and DPC were very similar although different chemical shifts were reported and reproduced in the two environments in both studies [11, 18]. Although the structure in SDS, but not in DPC was refined by the additional measurements of residual dipolar couplings, which led to a higher precision of the SDS compared to the DPC structure, most likely the longer construct and/or different detergent was responsible for the elongated helix in SDS.
3. Power-Saturation EPR to Determine Penetration Depths in Membranes
Although solution NMR provided a wealth of high-resolution information on fusion domains bound to lipid micelles with headgroups and chains that resemble those of phospholipids, these structures may not provide accurate representations of the structures of these domains in lipid bilayers. EPR spectroscopy is not limited by the size of the particles in the system as is solution NMR spectroscopy and, therefore, provides a nice tool to check at lower resolution whether the general conformational features of the NMR structures are still retained in lipid bilayers. To this end, a series of single cysteine mutants of each fusion domain are made and each molecule is labeled with a sulfhydro-reactive nitroxide spin label. The methane thiosulfonate spin label MTSL is quite small and has proven to be relatively un-perturbing to the structure of many soluble and membrane proteins. Control experiments with many fusion domains in our laboratory have shown that most (but not all) MTSL labeled sites had no effects on their CD and FTIR spectra in lipid micelles or bilayers. EPR spectra of the spin labeled domains bound to lipid bilayers are recorded under N2, O2, and nickel containing solution conditions. O2 and Ni are paramagnetic and engage in Heisenberg spin exchange when they collide with the nitroxide spin. Since O2 preferentially partitions into the bilayer and the Ni-chelate used in these studies is highly water soluble, these two exchange reagents probe different spin label environments. N2 is diamagnetic and therefore does not undergo spin exchange in any environment so that the N2 measurement can be used as a reference. To accurately measure the environment of a protein-bound spin label, the microwave power in the EPR experiment is gradually increased through saturation. Since the signal of spins undergoing Heisenberg exchange saturates at much higher power, the saturation profile can be used to measure the polarity of the environment. An experimental depth parameter is then obtained by ratioing the signals from measurements in Ni and O2 [19]. The accuracy of this method is quite high. Average penetration depths of the spin labels from the level of the lipid phosphate groups can be determined to a resolution of ±2 Å.
The wild-type fusion domain of influenza HA was most extensively studied by SDSL EPR spectroscopy. It was individually labeled with MTSL at 18 sites. Power saturation data were acquired for each site under N2, O2, and nickel conditions, and membrane penetration depths were measured for all 18 sites. It was evident from these data that the kinked boomerang structure was retained in lipid bilayers [13]. The penetration depths of the first 11 residues showed a periodic pattern with a helical periodicity of about 3.6 residues per turn. The helical period was furthermore inclined at 38° from the membrane surface. A kink was present at around asparagine-12 and another single helical “period” appeared between residues 13–18, i.e. in the same place where a helical turn had been observed by NMR in DPC micelles. The depths of the spin-labeled side chains ranged from 15 Å below (residue 3) to 6 Å above (residue 12) the level of the lipid phosphate groups. Therefore, although lower in resolution than NMR, the power saturation EPR method provided very useful information about many details of the conformation of the fusion domain in lipid bilayers, and in addition, allowed us to accurately place this domain at the appropriate depth in the lipid bilayer.
4. Docking of Solution NMR Structures to EPR Distance Restraints in Membranes
Given the close similarity of the NMR and EPR conformations of the fusion domain in DPC micelles and lipid bilayers, it is possible to use the NMR structure and dock it to the lipid bilayer membrane using the distance restraints obtained from the EPR power saturation experiments. To do so, the nitroxide spin labels are incorporated into the NMR structural model at the measured positions. The cysteine-attached MTSL has five side chain bonds with dihedral angles χ1 through χ5. In proteins, χ1 and χ2 are typically fixed in gauche conformations, and χ3 (the S–S bond) can assume a +90° or a −90° dihedral angle [20]. Therefore, this dihedral angle is optimized to either +90° or −90° by energy minimization at each site. χ4 and χ5 may assume different rotamer conformations depending on the side chain context. For simplicity, all rotamers are allowed in the docking simulations, which places the nitroxides in ranges described by cones. Since χ1 through χ3 are fixed, the accessible nitroxide ranges are quite limited. The structural model with the grafted and χ3-optimized spin labels is then docked into the lipid bilayer by minimizing the geometric differences with the experimental EPR distance restraints. The outcome of this docking experiment with the influenza wild-type fusion domain with 8 of the 18 spin-labels depicted is shown in at the top left of Figure 3. The top right panel of Figure 3 shows the same position of this domain in a lipid bilayer, but with the native side chains reinserted.
EPR power saturation experiments and subsequent in silico docking experiments were also conducted with the four mutant flu fusion domains whose NMR structures have been described above. However, in these cases only three or four spin label derivatives were made for each mutant. The results of the respective structures docked into lipid bilayers are shown in the middle and bottom rows of Figure 3. As expected, F9A docks in a similar position in lipid bilayers as wild-type. The hemifusion mutant G1S also inserts into bilayers at about the same level as the fully fusion active wild-type and F9A mutant fusion domains. Given the similar shape and position of G1S in bilayers, one concludes that the reason of G1S for blocking fusion at the hemifusion to full fusion transition must be a consequence of specific sequence contacts and perhaps interactions with other parts of the fusion protein rather than a gross conformational change or a general lipid bilayer insertion defect of G1S. We have suggested that the wild-type fusion domain may interact with the transmembrane domain of HA2 during completion of membrane fusion, i.e. when the final fusion pore opens [21]. The mutants G1V and W14A that block fusion completely do not have the typical boomerang structure and also insert differently into lipid bilayers. The linear structure of G1V inserts into the bilayer at a shallower angle of 18° (confirmed by polarized ATR-FTIR spectroscopy as 10° [12]) and the N-terminal arm of W14A orients almost parallel to the membrane surface. However, in the case of W14A, the flexible hinge directs the C-terminal arm out of the bilayer. The lack of a fixed-angle boomerang structure or the different angle of the N-terminal arm relative to the lipid bilayer could be responsible for blocking early hemifusion events in physiological cell fusion experiments with this mutation.
5. Peptide Dynamics in Micelles and Bilayers
EPR spectroscopy of spin-labeled proteins provides a useful tool to examine the backbone dynamics at the labeled site [3]. Although located eight bonds away from Cα, the motion of the nitroxide still reports on backbone fluctuations because the first three dihedral angles of the side chain are constrained by a weak hydrogen bond between Hα and Sδ [20], which leads to restricted anisotropic motions of the terminal pyrroline ring around χ4 and χ5. These motions are characterized by a side chain order parameter and correlation time, which are thought to be intrinsic side chain properties at sites that are not interacting with other side chains. Therefore, any deviation from this intrinsic side chain mobility may be interpreted as resulting from backbone motions at the site of spin label attachment. Although backbone order parameters and correlation times have been extracted by other authors in other systems by quantitative modeling of the EPR line-shapes, we applied this approach to our studies of fusion domains in only a qualitative manner by comparing the central line-widths of different sites in different mutants and different environments. The inverse of this line-width is a function of both the order parameter and the correlation time and therefore provides a qualitative measure of the involved motions. Residues 3 and 18 were chosen as representatives to probe the motions of the N- and C-terminal arms, respectively, of the flu fusion domains. The arm motions of wild-type and the F9A, F9A/I10A, and W14A mutants were then compared in DPC micelles and in small and large unilamellar vesicle (SUV and LUV) bilayers. The largest line-width, i.e. the slowest and/or most restricted motion was observed for residue 3 in wild-type in LUVs. We assign this residue a relative mobility (inverse line-width) of 1. The relative mobilities of all other situations are shown in Table 2. The most striking result is that all relative mobilities are at least 50 % larger in DPC micelles than in lipid bilayers and that the differences between LUVs and SUVs are relatively minor. This is not due to the faster overall tumbling rate of micelles compared to vesicles. Both can be considered as very large objects on the EPR time-scale. Instead these differences reflect differences of internal backbone motions in the two environments. Therefore, the peptide backbones of our fusion domains are more dynamic in micelles, which themselves are internally more dynamic than the acyl chains in lipid bilayers. For all fusion domains in all environments, the C-terminal labels are relatively more mobile than the N-terminal labels. Very interestingly, W14A, which was found by NMR to have a flexible hinge in DPC micelles, is also more mobile than wild-type and F9A in lipid bilayers. The mobility of the double mutant F9A/I10A, whose NMR structure has not been determined, is intermediate between the functional fixed-angle and non-functional flexible fusion domains. When this mutation was engineered into full-length HA and fusion was measured, it exhibited a non-fusion phenotype (Table 1). This agrees with expectations from its higher relative mobility in LUVs. The functional fusion domains are clearly more rigid than the non-functional ones, which are more mobile in lipid bilayer model membranes and detergent micelles.
Table 2.
Relative mobilities of nitroxide side chains of wild-type and mutant influenza hemagglutinin fusion domains derived from EPR line-widths in different environments.a
| LUV | SUV | DPC | ||||
|---|---|---|---|---|---|---|
| R3b | R18c | R3b | R18c | R3b | R18c | |
| Wild-type | 1.00 | 1.04 | 1.07 | 1.08 | 1.59 | 1.72 |
| F9A | 1.04 | 1.08 | 1.08 | 1.07 | 1.66 | 1.76 |
| F9A/I10A | 1.10 | 1.16 | 1.04 | 1.14 | 1.59 | 1.73 |
| W14A | 1.10 | 1.19 | 1.18 | 1.27 | 1.76 | 1.93 |
width of central EPR line of R3 of wild-type in LUV divided by width of central EPR line of given residue and environment. Data from ref [10].
MTSL at residue 3
MTSL at residue 18
6. EPR Methods to Determine Distances in Membranes
The dipole-dipole coupling between two electron spins depends on the inverse cube of the distance between them and is on the order of 25 MHz for two nitroxide spins that are separated by ~20 Å. The angular frequency of the splitting ωdd is
where, g1, g2, and ge are the g-values of the two spins and the free electron, respectively, θ is the angle between the spin-spin vector and the magnetic field, and r is the distance between the two spins. Therefore, if ωdd can be measured and 3cos2θ − 1 can be estimated, the distance r can be determined. Two methods have been used to measure electron dipolar interactions. The first uses continuous wave (CW) X-band EPR spectroscopy [22]. In this case, the spectra of the double-labeled proteins are compared with the sum of the spectra of the two single-labeled proteins and the dipolar interaction is estimated from the difference between the two. The CW EPR method, which can be applied to samples at room temperature, yields accurate measurements of distances in the 8 – 20 Å range [23]. The other method uses pulsed double electron electron resonance (DEER) spectroscopy and is useful for measuring weaker couplings arising from distances longer than 20 Å in frozen samples. A four-pulse DEER experiment produces a spin echo that is modulated by the frequency of the dipolar interaction [24]. DEER time evolution data of double-labeled samples are Fourier transformed, which for isotropically distributed proteins leads to Pake powder patterns. (Lipid vesicles and even micelles and soluble proteins tumble slowly on the EPR time scale.) The splitting of the peaks in the powder pattern corresponds to ωdd with θ = 90°, and therefore, is a direct measure of the distance r. Since the CW and DEER methods cover different distance ranges and typically use different measuring temperatures, they provide complementary information.
We used both methods to measure the distances between residues 3 and 18 on the N- and C-terminal arms of the wild-type and some mutant fusion domains in LUV lipid bilayers [16]. The purpose of these measurements was (i) to assess whether the distances measured in lipid bilayers were consistent with the solution NMR structures determined in DPC micelles and (ii) to assess whether the flexibilities of the hinge in the fusion domain structures in DPC were also manifest in lipid bilayers by showing larger distance distributions for mutants with flexible hinges than for fixed-angle boomerang structures. The distances between the Cα’s of residues 3 and 18 in the wild-type NMR structural ensemble are in the range 17.7–18.7 Å. Therefore, we expected that both the CW EPR and DEER experiments would be informative. Indeed, the CW method yielded distances greater than 20 Å between the nitroxides attached to residues 3 and 18 of wild-type and the F9A mutant fusion domains. This was outside the normal range of distance measurements by this method. However, the flexible mutant fusion domains W14A and F9A/I10A had distance components shorter than 20 Å (down to ~18 Å) as well as longer ones (up to ~30 Å). These results were further confirmed by DEER experiments with all four fusion domains (Table 1). The major distances measured between the two nitroxides in wild-type and F9A were 24 Å, whereas much wider distributions reaching from 21 Å up to 33 Å were observed for F9A/I10A and W14A. The distances observed by EPR are a little larger than the Cα distances of the corresponding sites in the NMR structures. This is not surprising given the extensions of the spin labeled side chains from the backbone and the variations in the previously described cone that is bounded by motions around the χ4 and χ5 bonds of the spin label. The fact, that we observed distance distributions extending to slightly lower values by CW EPR than by DEER spectroscopy could be due to experimental limitations of the two methods or due to the different sample temperatures that were required for the two types of measurements. Regardless of this minor uncertainty regarding precise absolute values of the measured distances, it is clear that qualitatively, a fixed-angle boomerang structure was maintained in lipid bilayers in the case of the fully functional wild-type and F9A fusion domains, while the non-functional F9A/I10A and W14A fusion domains had more flexible structures in lipid bilayers as was also observed by NMR for W14A in lipid micelles.
7. Conclusion
The described examples of several membrane fusion domains show that it is possible to combine high-resolution NMR data with lower-resolution SDSL EPR data to obtain reasonable structural models of membrane-embedded proteins in lipid bilayers. Despite the power of the combination of these techniques, one must also remain cautious about their limitations. There are two caveats that need to be carefully examined in each case. First, the structures that membrane proteins or their domains assume in lipid bilayers may not always correspond exactly to their structures in lipid bilayers. Although, in many cases, there is good first order agreement between structures in the two environments, there are also cases known where second order details are different. Among the examples that were described here, the wild-type flu fusion domain has been best characterized in this regard. The eighteen spin labels produced a picture of this fusion domain in membranes that was remarkably similar to the model produced by NMR in DPC micelles. Therefore, we can be quite confident that the transfer of these NMR coordinates into the model lipid bilayer is reasonable and gives a good first order approximation of the structure in a membrane. In the other cases discussed in this account, where for practical reasons fewer sites have been spin labeled, the uncertainty may be greater. However, as pointed out in the individual original research papers, the sites of labeling were strategically selected so that deviations from the kinked boomerang structure should become evident from the power saturation data of just a few critical labels. The second limitation to the resolution of the EPR method is the size and flexibility of the spin label itself. As can be seen in Figure 3, the spin labels and their paramagnetic centers extend quite far from their point of Cα backbone attachment. Therefore, it is not always straightforward to extrapolate information from the paramagnetic center back to the backbone. However, as was discussed in earlier sections, the conformations of many side chain dihedral angles are restricted so that the situation is not as bad as it might appear at first sight. Still, different rotamers around the χ4 and χ5 bonds are possible, which account for a few Å of uncertainty to the distances measured by EPR and DEER spectroscopy. There are methyl derivatives of MTSL whose χ4 and χ5 angles are more restricted. However, in several cases, the same results were obtained with these derivatives, which validates the use of the standard MTSL. The problem of side chain flexibility may be further alleviated by more sophisticated future structure calculations, in which the electron distances are incorporated as additional restraints. In such procedures, the specific spin label side chain conformations would be calculated in each case by global energy minimizations. It may also be possible to improve the structures and determine the spin label conformations by measuring paramagnetic relaxation enhancements (PREs) of the nuclear spins, which has become a valuable tool to get additional restraints in large proteins including membrane proteins. In summary, we believe that the combination of NMR and EPR techniques holds much promise for future structure determinations of membrane-bound proteins. At this time, we are probably witnessing just the beginning of many new technological developments in this area and the emergence of many interesting new biological insights that will be derived from these new methods.
The combined NMR and EPR approach has allowed us to better understand the structural biology of membrane fusion. Influenza HA has long served as the prototypical membrane fusion protein because it was the first fusion protein whose ectodomain structure had been solved. This paradigmatic role continues with the structure-function studies on its fusion domain in membranes that are described here. From the collective studies on this domain, it becomes clear that the kinked boomerang structure with a fixed-angle is essential for its function. The several mutant structures that have been solved and the correlation with their fusion activities in cellular fusion models provide strong evidence that the boomerang structure is both necessary and sufficient for function. The boomerang structure also appears to be required for hemifusion because the G1S mutant has a fixed-angle boomerang structure in membranes that closely resembles that of wild-type. Therefore, we think that specific protein-protein interactions, perhaps mediated by glycines in the membrane, may be necessary to progress from hemifusion to full fusion. Finally, the examples of the G1V and W14A mutants show that the fusion activity can be aborted in different ways, i.e., by straightening the kink or by making the kink flexible, respectively. A key consequence of the fixed-angle boomerang structure may be that it drives the N-terminal helical arm into the membrane at a steeper angle than in the non-fusogenic mutants. This feature may also be the answer to why the HIV fusion domain does not assume a boomerang structure. Its helical structure is much more hydrophobic than that of flu and it does not need to be regulated by pH for insertion. Although, the position of this helix has not yet been measured in membranes, it is possible that it inserts at a relatively steep tilt angle into lipid bilayers even in the absence of a C-terminal interfacial second helix that is needed for the proper membrane positioning and pH regulation of the flu fusion domain. Early polarized FTIR experiments have indicated that this is indeed the case [25]. Future experiments as described here for flu would help to further address the precise location of the HIV and possibly other viral and non-viral fusion domains in lipid bilayers.
Acknowledgments
We thank Drs. John Bushweller and David Cafiso for advice and many helpful discussions. We are also grateful to members of the Tamm group for discussions and critically reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sanders CR, Sonnichsen F. Solution NMR of membrane proteins: practice and challenges. Magn Reson Chem. 2006;44(Spec):S24–S40. doi: 10.1002/mrc.1816. [DOI] [PubMed] [Google Scholar]
- 2.Tamm LK, Liang B. NMR of membrane proteins in solution. Prog NMR Spect. 2006;48:201–210. [Google Scholar]
- 3.Columbus L, Hubbell WL. A new spin on protein dynamics. Trends Biochem Sci. 2002;27:288–295. doi: 10.1016/s0968-0004(02)02095-9. [DOI] [PubMed] [Google Scholar]
- 4.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Blumenthal R, Clague MJ, Durell SR, Epand RM. Membrane fusion. Chem Rev. 2003;103:53–69. doi: 10.1021/cr000036+. [DOI] [PubMed] [Google Scholar]
- 6.Tamm LK, Crane J, Kiessling V. Membrane fusion: a structural perspective on the interplay of lipids and proteins. Curr Opin Struct Biol. 2003;13:453–466. doi: 10.1016/s0959-440x(03)00107-6. [DOI] [PubMed] [Google Scholar]
- 7.Chernomordik LV, Kozlov MM. Protein-lipid interplay in fusion and fission of biological membranes. Annu Rev Biochem. 2003;72:175–207. doi: 10.1146/annurev.biochem.72.121801.161504. [DOI] [PubMed] [Google Scholar]
- 8.Lai AL, Li Y, Tamm LK. Interplay of proteins and lipids in virus entry by membrane fusion. In: Tamm LK, editor. Protein-Lipid Interactions. Wiley-VCH, Weinheim; Germany: 2005. pp. 279–305. [Google Scholar]
- 9.Tamm LK. Cell entry of influenza virus by membrane fusion. In: Schmidt MFG, editor. Influenza Viruses - Facts and Perspectives. Grosse; Berlin: 2007. pp. 48–55. [Google Scholar]
- 10.Han X, Tamm LK. A host-guest system to study structure-function relationships of membrane fusion peptides. Proc Natl Acad Sci USA. 2000;97:13097–13102. doi: 10.1073/pnas.230212097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Tamm LK. Structure and Plasticity of the Human Immunodeficiency Virus gp41 Fusion Domain in Lipid Micelles and Bilayers. Biophys J. 2007;93:876–885. doi: 10.1529/biophysj.106.102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Han X, Tamm LK. Thermodynamics of fusion peptide-membrane interactions. Biochemistry. 2003;42:7245–7251. doi: 10.1021/bi0341760. [DOI] [PubMed] [Google Scholar]
- 13.Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat Struct Biol. 2001;8:715–720. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 14.Qiao H, Armstrong RT, Melikyan GB, Cohen FS, White JM. A specific point mutant at position 1 of the influenza hemagglutinin fusion peptide displays a hemifusion phenotype. Mol Biol Cell. 1999;10:2759–2769. doi: 10.1091/mbc.10.8.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lai AL, Park H, White JM, Tamm LK. Fusion peptide of influenza hemagglutinin requires a fixed angle boomerang structure for activity. J Biol Chem. 2006;281:5760–5770. doi: 10.1074/jbc.M512280200. [DOI] [PubMed] [Google Scholar]
- 16.Lai AL, Tamm LK. Locking the kink in the influenza hemagglutinin fusion domain structure. J Biol Chem. 2007;282:23946–23956. doi: 10.1074/jbc.M704008200. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Han X, Lai AL, Bushweller JH, Cafiso DS, Tamm LK. Membrane structures of the hemi-fusion-inducing fusion peptide mutant G1S and the fusion-blocking mutant G1V of influenza hemagglutinin suggest a mechanism for pore opening in membrane fusion. J Virol. 2005;79:12065–12076. doi: 10.1128/JVI.79.18.12065-12076.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaroniec CP, Kaufman JD, Stahl SJ, Viard M, Blumenthal R, Wingfield PT, Bax A. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005;44:16167–16180. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- 19.Altenbach C, Greenhalgh DA, Khorana HG, Hubbell WL. A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc Natl Acad Sci U S A. 1994;91:1667–1671. doi: 10.1073/pnas.91.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langen R, Oh KJ, Cascio D, Hubbell WL. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 21.Tamm LK. Hypothesis: spring-loaded boomerang mechanism of influenza hemagglutinin-mediated membrane fusion. Biochim Biophys Acta. 2003;1614:14–23. doi: 10.1016/s0005-2736(03)00159-7. [DOI] [PubMed] [Google Scholar]
- 22.Rabenstein MD, Shin YK. Determination of the distance between two spin labels attached to a macromolecule. Proc Natl Acad Sci U S A. 1995;92:8239–8243. doi: 10.1073/pnas.92.18.8239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–82. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- 24.Jeschke G. Distance measurements in the nanometer range by pulse EPR. Chemphyschem. 2002;3:927–932. doi: 10.1002/1439-7641(20021115)3:11<927::AID-CPHC927>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 25.Martin I, Defrise-Quertain F, Decroly E, Vandenbranden M, Brasseur R, Ruysschaert JM. Orientation and structure of the NH2-terminal HIV-1 gp41 peptide in fused and aggregated liposomes. Biochim Biophys Acta. 1993;1145:124–133. doi: 10.1016/0005-2736(93)90389-h. [DOI] [PubMed] [Google Scholar]