

Abstract
One major objective of tumor immunologists is to prevent cancer development in individuals at high risk. (TG.AC × C57BL/6)F1 mice serve as a model for testing the feasibility of this objective. The mice carry in the germline a mutant ras oncogene that has an arginine at codon 12 instead of glycine present in the wild-type, and after physical (wounding) or chemical promotion, these mice have a high probability for developing papillomas that progress to cancer. Furthermore, F1 mice immunized with Arg12 mutant ras peptide in complete Freund's adjuvant (CFA) develop T cells within 10 d that proliferate in vitro on stimulation with the Arg12 mutant ras peptide. Within 14 d, these mice have delayed-type hypersensitivity to the peptide. Immunization with CFA alone or with a different Arg12 mutant ras peptide in CFA induced neither response. To determine the effect of immunization on development of tumors, mice immunized 3 wk earlier were painted on the back with phorbol 12-myristate 13-acetate every 3 d for 8 wk. The time of appearance and the number of papillomas were about the same in immunized and control mice, but the tumors grew faster and became much larger in the mice immunized with the Arg12 mutant ras peptide. Thus, the immunization failed to protect against growth of papillomas. The peptide-induced CD4+ T cells preferentially recognized the peptide but not the native mutant ras protein. On the other hand, mice immunized with Arg12 mutant ras peptide and bearing papillomas had serum antibodies that did bind native mutant ras protein. Together, these studies indicate that active immunization of cancer-prone individuals may result in immune responses that fail to eradicate mutant oncogene–expressing tumor cells, but rather induce a remarkable enhancement of tumor growth.
Keywords: primary tumor, immune stimulation, active immunization, mutant ras gene, cancer-prone mice
Introduction
More than 20 human hereditary cancer syndromes have been described, and germline mutations that predispose to development of cancer continue to be discovered 1. Furthermore, during the development of some human cancers, a predictable set of somatic mutations in known oncogenes or suppressor genes occurs. Starting from the mutant sequence of these genes, candidate peptides have been derived that bind MHC molecules and induce immune responses. Preimmunization of cancer-prone individuals with the mutant peptides might prevent the development of cancers in patients carrying such mutant oncogenes or suppressor genes. To test this possibility, a murine model of a hereditary cancer syndrome has been developed using mice that carry the Harvey ras oncogene, which has a glycine-to-arginine point mutation at codon 12 and an alanine-to-threonine point mutation at codon 59 2. Several features of this model are attractive. As observed in humans, the mutant oncogene is closely related to the normal cellular gene in that it differs from the normal cellular homologue by only two point mutations 3, and the development of tumors is influenced by nonmutagenic environmental factors, i.e., tumor development depends on chemical or biological promotion that by itself is not tumorigenic 2 4 5. The Arg12 mutant ras protein is expressed in papillomas and cancers that develop in TG.AC mice, but is barely detectable or absent in skin of TG.AC mice, even when the skin has been exposed repeatedly to chemical promoters 2 6. Expression of the mutant protein in the tumor-prone transgenic mice seems to be focal and occurs at the time tumors begin to develop 5 6 7. This late expression of the mutant protein should preclude the development of neonatal and/or peripheral tolerance, whereas the restricted expression should allow selective immunological destruction of malignant and premalignant foci without destruction of normal tissues. T cells have been reported to recognize the mutant oncoprotein 8, and peptides containing the arginine for glycine amino acid substitution induce highly specific CD4+ T cell responses 8. Furthermore CD4+ T cells can destroy MHC class II–negative tumor cells, even in the absence of CD8+ T cells 9 10, by an IFN-γ–dependent mechanism 11. As might have been predicted from these previous observations, we found that cancer-prone mice immunized with the mutant peptide had highly specific T cell responses to the peptide, but interestingly and contrary to expectation, the growth of the tumors in these specifically immunized mice was markedly enhanced. Our findings provide a cautionary note for investigators intending to prevent or treat cancers by immunizing patients against cancer antigens using mutant peptides.
Materials and Methods
Mice.
6–8-wk-old germ-free–derived specific pathogen-free C57BL/6 (H-2b) females were purchased from the National Cancer Institute, Frederick Cancer Research Facility, and FVB/N were purchased from Taconic Farms. A stock of specific pathogen-free TG.AC mice were obtained in 1995 from the National Institute of Environmental Health Sciences colony kept at Taconic Farms. As described previously 2, the TG.AC founders are of FVB (H-2q) origin and are transgenic for the viral Harvey ras (vHa-ras) oncogene under the control of the ζ-globin promoter. The vast majority (>95%), but not all TG.AC mice, develop multiple mutant ras–expressing papillomas 6–8 wk after promotion with the phorbol ester PMA (GIBCO BRL Life Technologies; reference 2). We have carried this line by brother–sister matings, and these mice continue to develop papillomas at a >95% incidence. Recently, a confounding nonresponder TG.AC genotype has been described to have arisen in the Taconic Farms colony 12 13. The genotype becomes apparent in the hemizygous TG.AC mice (F1 between homozygous TG.AC and the parental FVB) that were sold by Taconic Farms 12 13. These hemizygous nonresponder mice (even though they are homozygous for the promotion-sensitive FVB background) develop no papillomas (90% of the mice) or only one papilloma (10% of the mice) upon promotion. We have no evidence that a nonresponder genotype developed in our TG.AC colony, as our hemizygous F1 mice have an up to 100% papilloma response rate when properly promoted. (TG.AC × C57BL/6)F1 (TGB6F1) mice that carry one allele of the mutant ras transgene and (FVB × C57BL/6)F1 (FVB6F1) mice were bred and housed in a specific pathogen-free barrier facility at The University of Chicago 14. C57BL/6 mice are virtually nonresponders to chemical promotion 15. Therefore, the TGB6F1 mice we generated are less susceptible to tumor induction than homozygous TG.AC mice, in that with shorter length of promotion some of these mice may not develop tumors. Though we have no evidence whatsoever that a nonresponsive genotype was present in our hemizygous TGB6F1 mice, we have reevaluated our results as though this was the case, and we find that there is no noticeable difference in the results (data not shown).
Cell Lines.
Tumor cell lines were passed in DMEM supplemented with glutamine (GIBCO BRL Life Technologies) and 10% FCS (HyClone Laboratories). T cell hybridoma cell lines and CTLL cells were passed as described 16. An anti-Arg12 mutant ras peptide–specific T cell line was generated from lymph node cells (LNCs) obtained from a C57BL/6 mouse immunized with Arg12 mutant ras peptide in CFA. The LNCs were cultured using IL-2 and antigen. 4 d after passage, 2 × 107 T cells and 2 × 107 BW5147 cells were fused as described 16 to generate anti-Arg12 ras T cell hybridoma no. 1. A second anti-Arg12 mutant ras peptide–specific CD4+ T cell line has been described as 2F9 8; T cells of this line were fused to generate anti-Arg12 ras T cell hybridoma no. 2.
Ras Peptides and Proteins.
The wild-type ras peptide consisted of amino acids 5–17 (KLVVVGAGGVGKS). The Arg12 mutant ras peptide also consisted of amino acids 5-17 (KLVVVGARGVGKS), but had a G to R substitution at codon 12. The Leu61 mutant ras peptide consisted of amino acids 54–67 (DILDTAGLEEYSAM) and had a Q to L substitution at codon 61. Peptides were obtained from either The University of Chicago Protein Core Facility or from Chiron Mimotopes or SynPep. Peptides prepared to at least 70% purity as verified by mass spectroscopy were resuspended before use in double-distilled H2O to a final concentration of 10 mg/ml and stored at −80°C until used.
To generate recombinant ras proteins at highest purity, we developed a new ras vector using the glutathione S-transferase (GST) Gene Fusion System (Amersham Pharmacia Biotech). The vHa-ras gene was amplified by PCR from the PA9 plasmid 2 using a 5′ primer containing a BamHI site (CGTGGATCCATGACAGAATACAAGCTTGT) and a 3′ primer containing an EcoRI site (CGATGAATTCAGGACAGCACACACTTGCAGCT). 10-min initial denaturation was followed by 35 cycles of 55°C annealing (30 sec), 72°C extension (45 sec), and 94°C denaturation (30 sec). 600 base pair fragments containing the vHa-ras gene were amplified, purified using Qiaquick PCR columns (Qiagen), and cloned into the pGEX-6P-1 vector using the BamHI and EcoRI restriction sites that in previous studies was used to generate GST fusion proteins (Amersham Pharmacia Biotech). The fusion protein was prepared and purified according to the manufacturer's detailed instructions. In brief, the recombinant fusion protein made in BL21 strain of Escherichia coli was bound to glutathione Sepharose 4B (Amersham Pharmacia Biotech), washed three times with large volumes of PBS, and then eluted with glutathione elution buffer. Free fusion protein was evaluated by Western blot assay using ras-specific antibodies 17. The fusion protein was subsequently cut with Precision Protease (Amersham Pharmacia Biotech) and repurified with glutathione Sepharose 4B to remove the GST protein. The Precision Protease is a GST fusion protein that also binds to glutathione Sepharose 4B. The resulting highly purified recombinant ras protein retains only five amino acids (GPLGS) of GST. After final purification, the GST ras tumor protein appears to be 99% pure as assessed by silver-stained gels. In some experiments, mutant ras protein was digested using endoproteinase Glu-C (Boehringer); the protease was added to a final volume of 2% (vol/vol), and the protein was digested at 37°C overnight. The enzyme in the mixture was then inactivated by boiling. As control antigen, the ribosomal protein L26 was made as a recombinant fusion protein using the same procedures for purification. After final purification, the GST L26 fusion protein appears to be >90% pure as assessed by Coomassie silver-stained gels. All proteins were stored in aliquots at −80°C. All mutant ras proteins were stored in aliquots at −80°C. In some experiments, we used a recombinant Arg12 ras protein provided by Dr. R.G. Fenton (National Cancer Institute, Frederick Cancer Research Facility, Frederick, MD). This protein had been purified by ion exchange chromatography and gel filtration.
Immunizations and Promotion.
Each hind footpad of naive animals was injected with 50–75 μg of the mutant Arg12 ras or the mutant Leu61 ras peptide (total dose 100–150 μg) emulsified in CFA. 3 wk after immunization, the backs of mice were shaved using electric clippers (Wahl Clipper Corp.) without nicking the skin. 200 μl containing 2.5 μg of PMA in acetone (99.5% pure ACS spectrometric grade; Sigma-Aldrich) was distributed evenly over the shaved back using an Eppendorf pipettor and a 200-μl yellow plastic pipette tip with 2 mm of the tip cut off. PMA was applied every 3 d for 20 applications. Hair was shaved several times during promotion as required by hair growth. Individual papillomas were measured in three orthogonal dimensions with a caliper. Tumor measurements usually continued for 16–20 wk after the start of promotion. Tumor volume was estimated by π × abc/6, where a, b, and c are three orthogonal tumor diameters recorded in millimeters.
Proliferation, IL-2 Release, and Delayed-type Hypersensitivity Assays.
Draining popliteal or paraaortic LNs were harvested 7 d after immunization. Suspensions of the LNCs were cultured in duplicate or triplicate with 106 cells per well in 96-well flat-bottomed plates. Unless otherwise indicated, each culture contained 100 μg/ml antigen and 1% normal mouse serum. Wells were pulsed on day 2–3 of culture [methyl-3H]thymidine (Amersham Pharmacia Biotech) as described 16. 24 h later, cells were harvested and the radioactivity was measured in a liquid scintillation counter as described 16. Proliferative responses of the T cell lines to the antigens were measured by culturing 1–2 × 105 T cells, 106 irradiated syngeneic spleen cells as APCs, and 10 μg/ml of Arg12 mutant ras peptide for 2–3 d, pulsing with 3H-TdR, and assaying 24 h later. The hybridomas were used to evaluate whether mutant ras protein could be processed and presented by APCs. In this assay, 105 hybridoma cells and 7.5 × 105 irradiated syngeneic spleen cells were combined and cultured for 24 h with either no antigen, 40 μg/ml peptide, or 40 μg/ml protein. Supernatants were removed after 24 h and analyzed for IL-2. IL-2 released by the T cell hybridoma was measured by the growth of IL-2–dependent CTLL cells using 3-(4,5-dimethylthiazol-2-yl),-2, 5 diphenyltetrazolium bromide (MTT; absorbance at 570 nm and absorbance at 650 nm) as described 16 18.
Delayed-type hypersensitivity (DTH) was measured 14 d after immunization. The dorsal surface of each ear was injected with 10 μl containing 10–20 μg of peptide or saline alone using a 30-gauge needle. Ear thickness was measured with precision spring-loaded dial calipers (Mitutoyo; no. 7326, Precision Gage Co.) 24 h after challenge. The averages of triplicate measurements were compared with baseline measurements made immediately before challenge injections.
ELISA for Measuring Anti-Ras Serum Antibody Titers.
The titers of anti-ras antibody in the serum of mice immunized with Arg12 ras peptide were measured using a modification of an ELISA described previously 19. Mutant ras peptides (Arg12 and Leu61) coupled to OVA or OVA alone and diluted in carbonate buffer (pH 9.6) to a concentration of 3 μg/ml were immobilized in the wells of 96-well microtiter plates (no. 442404; Nalge Nunc International) by overnight incubation at 4°C. Recombinant full-length ras protein or control proteins (30 μg/ml) were similarly immoblized. Microwells were washed once with distilled water followed by three washes with PBS containing 0.05% Triton X-100 (Sigma-Aldrich). Sera from control mice or mice immunized with ras peptide were diluted in PBS containing 1% BSA (Sigma-Aldrich; no. A7030). 50 μl were added to microwells containing test or control antigens. Sera were incubated for 2 h at room temperature, and the wells of the plates were washed as described above. 50 μl of alkaline phosphatase–conjugated goat anti–mouse antisera (BD PharMingen; no. 12063E) diluted 1:1,000 was added to each well, and plates were incubated at room temperature for 1 h. Plates were washed as described above, and 100 μl of p-nitrophenyl phosphate substrate (Sigma-Aldrich; no. N9389, 1 mg/ml dissolved in diethanolamine buffer) was added to each well. The reaction was allowed to develop for 60 min at room temperature before reading at dual wavelength (405 nm minus 650 nm) using an ELISA reader (Molecular Devices).
Results
TGB6F1 Mice Respond to Immunization with the Arg12 Mutant Ras Peptide.
FVB mice, FVB6F1 mice, and the Arg12 mutant ras transgenic TGB6F1 mice were immunized in the footpads with the Arg12 mutant ras peptide in CFA. LNCs from FVB6F1 mice immunized with Arg12 mutant ras peptide responded when restimulated with the Arg12 mutant ras peptide, whereas FVB mice, the strain of origin of the TG.AC mice, did not respond (Fig. 1 A). The magnitude of responses of cells from FVB6F1 mice was similar to that of cells from C57BL/6 mice immunized with the Arg12 mutant ras peptide (data not shown). LNCs from mice immunized with adjuvant alone or with an unrelated antigen did not mount Arg12 mutant ras peptide–specific proliferative responses (data not shown). Fig. 1 A also shows that cells from TGB6F1 mice immunized with Arg12 mutant ras peptide mounted specific proliferative responses to the peptide, indicating that tumor-free transgenic mice remained responsive to the immunogenic mutant ras peptide sequence encoded by the inherited oncogene. Consistent with this finding is the fact that immunized TGB6F1 mice also developed DTH to the Arg12 mutant ras peptide (Fig. 1 B). Most importantly, TGB6F1 mice developed papillomas after promotion with PMA. Thus, TGB6F1 mice were immunologically responsive to Arg12 mutant ras peptide and were appropriate for determining the effect of immunization with Arg12 mutant ras peptide on development of papillomas after promotion.
Figure 1.

FVB6F1 mice and cancer-prone TGB6F1 mice carrying a mutant ras transgene mount a specific DTH response when immunized against the mutant ras peptide, and lymphocytes from these mice mount a specific proliferative response to the mutant peptide. (A) The proliferative response of lymphocytes from immunized FVB, FVB6F1, and TGB6F1 mice was analyzed by harvesting draining LNCs 7–10 d after immunization and pulsing them with 3H-TdR after 2 d of culture with or without antigen, as indicated. Three representative experiments using cells taken from a single or a pool of animals are shown. (B) DTH reactions were assayed in TGB6F1 9-wk -old (Group 1)and 13-wk-old (Group 2) mice that were naive or had been immunized with the Arg12 mutant ras peptide in CFA or with saline in CFA. 14 d after the immunization, mice were challenged with Arg12 mutant ras peptide in one ear and saline in the other. The ear swelling was measured 24 h later. Bars represent SEM of groups consisting of 10 mice.
Preimmunization of TGB6F1 Mice with the Arg12 Mutant Ras Peptide Enhances the Growth of Papillomas.
12-wk-old TGB6F1 mice (five per group) were immunized with the Arg12 ras peptide in CFA, the Leu61 ras peptide in CFA, or with CFA alone (time 0). Painting with the promoter began 3 wk after and ended 12 wk after the immunization. Tumors began to appear near the end of PMA treatment, and increased thereafter (Fig. 2 A). By week 13, four of the five Arg12 mutant ras peptide–immunized mice had larger tumors than mice immunized with Leu61 mutant ras peptide in CFA or with saline in CFA (Fig. 3). Tumors in the Arg12 mutant ras–immunized mice grew larger in the following weeks (Fig. 2 A), and differences between groups increased (Fig. 2 B, left). This remarkable difference between the Arg12 mutant ras peptide–immunized mice and the control groups was accounted for primarily by the much larger average volume of the papillomas in the Arg12 mutant ras–immunized mice (Fig. 2 B, right), though the number of papillomas was also marginally greater in the Arg12 mutant ras–immunized group. The tumor volume per mouse in the Arg12 mutant ras peptide–immunized group increased more than fivefold during the following 5 wk (Fig. 2 A). By contrast, the papillomas in the control groups remained about the same size or became smaller during this time. The experiment was terminated at 20 wk because of the large size of the tumors in four of the five Arg12 mutant–immunized mice. Histologically, there was no apparent difference between tumors from the Arg12 mutant ras peptide–immunized mice and tumors in the control groups at the time of killing. Clearly, the larger tumors in the immunized groups were not due to increased inflammatory infiltrates (Fig. 4). The volumes of the 10 largest tumors of the Arg12 mutant immunized group compared with the volumes of the 10 largest tumors in either of the control groups at 14, 17, or 20 wk were significantly larger (P < 0.001) at each of the three time points. There were no significant differences between the control groups.
Figure 2.


Immunization against a mutant region of the ras oncoprotein results in enhanced growth of primary tumors in mice carrying the mutant ras gene in the germline. (A) Kinetics of tumor development in relation to immunization, DTH analysis, and promotion. Chemical promotion with PMA once every 3 d (20 treatments) was begun 3 wk after and ended 11.7 wk after immunization. Five out of five Arg12 mutant ras–immunized mice, five out of five Leu61 mutant ras–immunized mice, and three out of five mice immunized with CFA developed two or more papillomas. (B) The average total volume of tumors per mouse and the average volume of papillomas within each group of mice. Tumor volumes in B were measured 2.5 wk after the end of PMA treatment. The numbers on top of the bars at left represent the average number of papillomas per mouse within that group. Five mice per group were used. (C and D) The design of the experiment and results were similar, except the experiment was done ∼1 yr later with 10 mice per group. Tumor volumes in D were measured 12.1 wk after the end of the PMA treatment. 8/10 Arg12 mutant ras–immunized mice, 1/10 Leu61 ras–immunized mice, and 3/10 mice immunized with CFA alone developed two or more papillomas. The data in Fig. 2A–D remain virtually unchanged when all mice that developed no or only one papilloma after promotion are excluded as potential “nonresponders” (references 12, 13; described in Materials and Methods).
Figure 3.
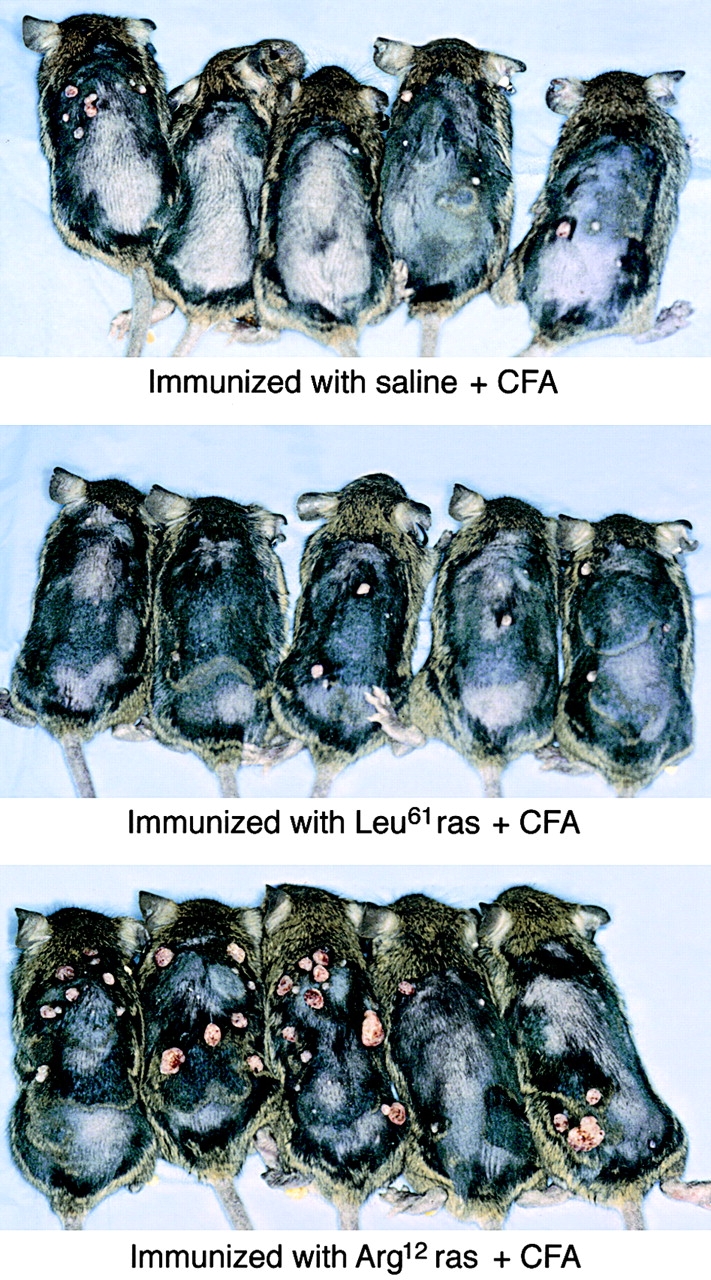
Enhanced tumor development in TGB6F1 mice with the Arg12 mutant ras peptide. Five mice per group were immunized with the Arg12 mutant ras peptide in CFA, the Leu61 mutant ras peptide in CFA, or with saline in CFA. Pictures were taken 1.5 wk after the end of promotion (mice anesthetized for photography). Although the difference between the specifically immunized and the control groups is apparent, the bottom panel shows that papillomas developed more slowly in one of the five Arg12 mutant ras peptide–immunized mice, and some of the papillomas in the Arg12 mutant ras of the other four similarly immunized group remained small. The results shown here and in Fig. 2A and Fig. B are derived from the same experiment; pictures shown here were taken 1 wk earlier than results shown for the same mice in Fig. 2 B.
Figure 4.

Photomicrograph illustrating the appearance of a papilloma on the back skin of an Arg12 mutant ras–immunized mouse at time of sacrifice. The lower magnification (original magnification: ×20) shows the keratinization of the papilloma. At the higher magnification (original magnifications: ×100 and ×200), the dysplastic epithelial changes and the neutrophilic inflammatory infiltrate at the epidermal–dermal interphase become evident. These inflammatory infiltrates do not contribute significantly to the tumor volume and are similarly found in papillomas of control mice (not shown). Hematoxylin and eosin staining.
The experiment was repeated using newly synthesized batches of peptide reagents. Because some older mice spontaneously develop jaw and other non-skin tumors 2 6, separate groups of 13-wk-old and 9-wk-old mice (10 per group) were compared. Both groups of mice immunized with the Arg12 mutant ras peptide and challenged with the Arg12 mutant ras peptide had DTH reactions, whereas mice immunized with CFA alone did not, showing as in previous experiments that the mice were not tolerant to the peptide. Mice immunized with Leu61 mutant ras peptide and challenged with the Leu61 mutant ras peptide had weak responses that were less specific for the Leu61 mutant ras peptide (data not shown). 1 wk after testing for DTH, promotion began. Tumors started to appear after cessation of promotion (Fig. 2 C), and from week 14 on, tumor volumes per mouse were much larger (>18-fold) in the Arg12 mutant ras–immunized group than in the control groups (Fig. 2 D, left). As observed earlier, this difference was due to an increased average volume of the papillomas in the Arg12 mutant ras–immunized mice (Fig. 2 D, right). The results for older and younger mice were comparable (data not shown).
Mice Immunized with the Arg12 Mutant Ras Peptide Respond Preferentially to the Arg12 Peptide and Not to the Intact Arg12 Mutant Ras Oncoprotein.
We next sought evidence that CD4+ T cells specific for Arg12 mutant ras and induced by immunization with this peptide indeed responded to the intact protein produced by tumor cells. Thus, we immunized mice with the Arg12 mutant ras peptide and analyzed the response of these T cells to peptide or the intact protein. Fig. 5 (top) shows that the LNCs from these mice responded preferentially to the peptide in repeated experiments. This predominant response pattern is also reflected in T cell hybridomas derived from T cells of Arg12 mutant ras peptide–immunized mice by fusion with BW5417 cells (Fig. 5, middle and bottom). However, lysates of several Arg12 mutant ras protein–expressing tumor cells failed to stimulate specifically the hybridoma in the presence of APCs. More surprisingly, the T cell line and the hybridoma even failed to respond specifically to lysates of cells that had been infected with Arg12 mutant ras vaccinia virus, and were expressing large amounts of the Arg12 mutant ras protein (data not shown). We then generated large amounts of affinity-purified recombinant Arg12 mutant ras protein. However, the hybridoma also did not respond to this protein in the presence of APCs (Fig. 4, right). These results suggested that when the intact Arg12 mutant ras protein was available for exogenous presentation, the antigen was not recognized by the Arg12 mutant ras–specific CD4+ T cells 19.
Figure 5.



Arg12 mutant ras peptide is preferentially recognized by T cells from mice immunized with the Arg12 mutant ras peptide or by T hybridoma cells derived from these T cells. Top: proliferative response of LNCs in vitro from mice immunized with Arg12 mutant ras peptide, and with CFA 9 d earlier. Three representative experiments using cells taken from a single or pool of animals are shown. Mutant L26 (MutL26) protein is used as a control. Middle: proliferation of an anti-Arg12 mutant ras T cell line when cultured with the Arg12 mutant ras peptide (left). T cell hybridoma derived from this T cell line remains Arg12 mutant ras peptide specific, but does not respond to the Arg12 mutant ras protein (right). Cells from the T cell line or T cell hybridoma were cultured with antigen, and spleen cells were provided as APCs. The antigen-specific stimulation of the T cells caused increased 3H-TdR incorporation, whereas the antigen-specific stimulation of the T cell hybridoma caused IL-2 release, which was analyzed using growth stimulation of the IL-2–dependent CTLL cells as determined by a colorimetric assay (described in Materials and Methods). Bottom: same as top, except a different T cell line 2F9 8 was used. This T cell line 2F9 is also specific for the Arg12 mutant ras peptide, and the T cell hybridoma derived from this T cell line again only recognizes the mutant peptide and not the intact protein. Affinity-purified (Affin.) Arg12 mutant ras protein was prepared from a recombinant GST fusion protein as described. The ion exchange (Ion-ex.) matrix–purified Arg12 and Leu61 mutant ras proteins were a gift from Dr. R.G. Fenton. The average of duplicate samples are shown. Wt, wild-type.
This response pattern was confirmed with a second hybridoma derived from another T cell line, 2F9 8, which also originated from LNCs of Arg12 mutant ras peptide–immunized mice. Fig. 5 (middle and bottom) shows that the 2F9-derived hybridoma also failed to respond to the purified protein we had made. Interestingly, we observed a response to a recombinant Arg12 mutant ras protein that had been purified in another laboratory by gel filtration 20. These differences are likely due to the substantially different purification procedures. We used affinity purification: the NH2-terminal immunogenic portion of the ras protein will only be found in the purified fraction if it was part of the complete protein that contained the COOH-terminal end that binds to the affinity column (discussed in Materials and Methods). This is important because recombinant protein preparations from bacteria are likely to contain protein fragments and unfolded proteins because of the action of bacterial endopeptidases. Indeed, Fig. 6 shows that the affinity-purified Arg12 mutant ras protein, when cut with the bacterial endopeptidase (Glu-C), was as stimulatory of the anti-Arg12 mutant ras hybridoma as the mutant peptide (using equimolar concentrations). Undigested highly purified protein failed to show any stimulation, even at the highest concentrations used. The bacterial endoproteinase, Glu-C, would be expected to cut the mutant ras protein at glutamic acid residues at positions 3 and 31, thereby releasing an immunostimulatory 28-mer oligopeptide. Together, our results suggest that the CD4+ T cells predominantly induced by immunization with the Arg12 mutant ras peptide were specific for this peptide and could not recognize the intact mutant protein that is produced by tumor cells in the presence of APCs.
Figure 6.

The mutant Arg12 ras peptide and digests of the Arg12 mutant ras protein but not the intact protein stimulate Arg12 mutant ras peptide–specific T hybridoma cells. Equimolar concentrations of either the Arg12 mutant ras peptide, the undigested protein, or protein digested by exposure to the bacterial endoprotease Glu-C were tested for antigen-specific stimulation of hybridoma R12-H2 derived from the 2F9 T cell line. Antigen recognition by the hybridoma cells resulted in IL-2 release, which was analyzed using growth stimulation of the IL-2–dependent CTLL cells in a colorimetric assay. Proteins were prepared and digested as described in Materials and Methods.
Mice Immunized with Arg12 Mutant Ras Peptide Produce Antibodies that Bind Arg12 Mutant Ras Protein.
In preliminary experiments, C57BL/6 mice immunized with the Arg12 mutant ras peptide in CFA followed by multiple boosts with the peptide in IFA produced significant titers of Arg12 mutant ras peptide–specific antibodies. There was no measurable cross-reactivity with the Leu61 mutant ras peptide (data not shown). We then immunized four TGB6F1 mice with the Arg12 mutant ras peptide in CFA followed by two booster immunizations with the peptide in IFA. All four F1 mice produced antibodies that bound the Arg12 mutant ras peptide (Fig. 7, left). Importantly, sera from all four of the TGB6F1 mice immunized with the Arg12 mutant ras peptide also contained antibodies that bound the intact Arg12 mutant ras protein (Fig. 7, middle). None of the antisera contained antibodies that bound more effectively than control serum to L26, an unrelated control protein purified by the same procedure and used at similar concentrations for coating the ELISA plates (Fig. 7, right).
Figure 7.

TGB6F1 mice immunized repeatedly with the Arg12 mutant ras peptide produce Arg12 mutant ras peptide–specific antibodies, and some of these antibodies also recognize the intact protein specifically. Mice were immunized intraperitoneally with 100 μg of the Arg12 or Leu61 mutant ras peptide in CFA and boosted twice at 2-wk intervals with the peptide in IFA. 2 wk after the last boost, sera from immunized mice were tested for antibodies specific for mutant ras peptide or protein. Sera of all four mice have antibody that binds mutant ras peptide and also the mutant ras protein, but the antibodies did not bind a control protein. For comparison, the serum of mouse no. 2 (•), which was immunized only once (1x) with the Arg12 mutant ras peptide in CFA and then promoted with PMA followed by development of papillomas, was included. This serum is identical to that of mouse no. 2 (•) shown in the middle panel. 3x, three times.
These results were confirmed and extended by immunizing TGB6F1 mice once (without boost) with the Arg12 mutant ras peptide or Leu61 mutant ras peptide (four mice per group) followed by promotion (Fig. 8 A). Sera from all four Arg12 mutant ras peptide–immunized mice taken 7 wk after the end of promotion contained Arg12 mutant peptide–specific antibodies (Fig. 8 B, left panels), whereas none of the four Leu61 mutant peptide–immunized mice had antibodies that bound either peptide (data not shown). Sera from two of the four Arg12 mutant ras peptide–immunized mice had high titers of antibody against intact Arg12 mutant ras protein, but none of the sera contained antibodies against an unrelated protein purified by the same procedures (Fig. 8 B, right panels). Remarkably, the two mice that had serum antibodies against the protein also had large tumors, whereas the two mice in the group that had failed to develop Arg12 mutant ras protein–binding antibodies failed to develop a significant tumor load (Fig. 8 B, right panels). Furthermore, the two mice immunized only once followed by promotion and tumor development had higher titers against the mutant protein than the mice in the previous experiment, which had been immunized multiple times but not promoted, and which remained tumor free. (Compare titers of serum of the singly immunized, promoted, tumor-bearing mouse no. 2 with titers of the other four sera obtained from three-times immunized, not promoted, tumor-free mice in Fig. 6).
Figure 8.
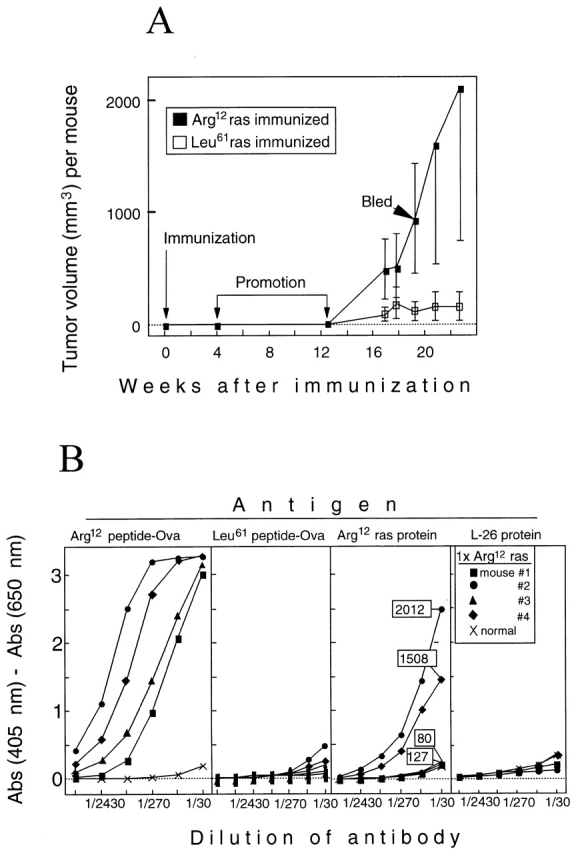
TGB6F1 mice immunized only once but then promoted with PMA can produce antibody specific for Arg12 mutant ras peptide, and antibody titers correlate directly with tumor burden. (A) Experimental design and kinetics of tumor growth. TGB6F1 mice were immunized with 150 μg of either Arg12 or Leu61 mutant ras peptide in CFA (four mice per group) and then promoted with PMA as described in Materials and Methods. Kinetics of tumor development and time of serum sampling in this group is indicated. (B.) Sera of all of the four TGB6F1 mice singly (1x) immunized with the Arg12 mutant ras peptide and then promoted with PMA have antibody that binds Arg12 mutant ras peptide and not the Leu61 mutant ras peptide, but only antibodies in sera of mice nos. 2 and 4 also bind mutant ras protein. These antibodies were Arg12 mutant ras protein specific because they did not bind to the L26 control protein. Protein binding antibody titers correlated with the host tumor burden. Numbers in the rectangles represent total tumor volume in mm3 for the indicated mouse at the time the serum was taken. Sera from Leu61 mutant peptide–immunized mice do not have antibodies that bind either ras peptide (data not shown).
Discussion
Immunizing cancer-prone mice with peptide corresponding to the mutant region of an oncoprotein led to markedly enhanced tumor growth in most mice. Immunization with the Arg12 mutant ras peptide did not induce T cell tolerance, as T cells from immunized mice proliferated in vitro when stimulated by the mutant peptide, and immunized mice had DTH to the peptide in vivo. Although T cells induced by immunization with the mutant ras peptide responded predominantly to the peptide and not to the intact mutant ras oncoprotein, this immunization did stimulate the production of antibodies that bound the mutant ras protein. In a single small experiment, the serum titers corresponded directly to the tumor burden.
Many years ago, Peyton Rous and his coworkers recognized that tumors develop from “subthreshold neoplastic states” (21 22; now referred to as the initiation stage) and that wounding could trigger tumorigenesis (now referred to as promotion). Subsequent work showed that various chemicals and inflammatory states can promote tumor development (for a review, see reference 23). This appears to be the first published report that active immunization can cause the enhanced growth of primary tumors.
Because the Arg12 mutant ras peptide–immunized mice developed large tumor burdens, they were killed before obvious malignant lesions developed. In an earlier study using the parental cancer-prone transgenic FVB strain, about one third of mice with persistent papillomas developed malignant skin cancers 6–12 mo after cessation of the PMA treatment 2, a much longer period than one could have reasonably kept the immunized mice with large papillomas. Very few papillomas regressed in the Arg12 mutant ras peptide–immunized mice, whereas papillomas in the control groups remained very small. Papillomas that regress after promotion are called promoter dependent, whereas papillomas that persist are called promoter independent 24. The papillomas of specifically immunized as well as control mice had persistent, i.e., promoter-independent papillomas. Presumably because of the much more rapid growth of papillomas in the specifically immunized group, the risk of these papillomas becoming malignant is increased because of the continued clonal expansion of the initiated cell population with the increased probability of additional mutations necessary for progression to malignancy. However, it is unclear how immunization against the mutant peptide led to increased proliferation and large papillomas. But regardless of the mechanism, increased proliferation appears to be the important and common mechanism whereby a wide variety of injuries, infections, hormones, growth factors, and chronic inflammation enhance the development of cancers in various organs such as bowel, liver, esophagus, breast, gall bladder, oral cavity, and skin (for a review, see reference 23).
We have not found published evidence that active immunization against an antigen expressed by initiated cells can lead to enhanced growth of primary tumors, but an immune stimulation of tumor growth has been postulated for decades 25. It has been postulated that vaccination leading to a “weak” immune reaction might stimulate rather than inhibit the growth of primary tumors 26. In the case of hepatitis B virus–mediated hepatocarcinogenesis, it is postulated that a strong T cell response can eradicate the virus from the host, whereas a response too weak to terminate the infection is procarcinogenic 27. Presumably a particular type of chronic inflammation caused by a weak response stimulates hepatocellular proliferation and the enhanced development of hepatocellular cancers. In our model, vaccination of cancer-prone mice with the Arg12 mutant ras peptide 26 failed to induce detectable destructive T cell responses, but did induce antibody responses to the mutant protein. Whether the immunological mechanism(s) responsible for the enhanced tumor growth is related to antibody itself and/or to alterations (e.g., cytokine milieu) inherent in hosts mounting an antibody response is not known. Interestingly, the level of specific antibody corresponded with the total volume of the papillomas in the four mice we tested, but a much larger group will have to be studied, and it will be interesting to determine the subclasses of the mutant ras protein–binding antibodies in mice developing tumors. We are in the process of backcrossing the TG.AC mice to mice lacking mature B cells to determine whether B cells and/or antibody play a central role in the observed enhancement.
Induction of a humoral response against the extracellular domain of the HER2/neu growth factor receptor can reduce primary mammary tumor development in transgenic mice, but antibody-mediated downregulation of the receptor appears to be the mechanism 28. In previous studies, Ig actively produced or passively given enhanced growth of tumor grafts 29 30 31, and malignant tumors grew more slowly or were rejected by mice unable to produce Ig 32 33. Also, mice with low Ig responses are more resistant to chemically induced tumorigenesis 34 35. Several mechanisms have been postulated by which Ig or B cells secreting Ig may enhance tumor growth: (a) by blocking epitopes that are required for tumor rejection 36, (b) by suppressing activation and/or proliferation of tumor-specific CTLs by TGF-β carried by IgG 37 38 (consistent with such a mechanism is the finding that conditions leading to strong antibody responses appear to inhibit CTL induction; reference 39), and (c) by antitumor antibody–tumor antigen complexes formed in and around tumors, which may alter inflammation and angiogenesis as well as have other effects 23. Also, actively growing papillomas in the tumor-prone mice are surrounded by conspicuous cellular inflammatory infiltrates 40, and B cells in such infiltrates may downregulate the production of IFN-γ and IL-12 41. This is of interest because mice deficient in IFN-γ signaling are sensitive to tumor development 42 43, particularly when the mice carry a mutant Harvey ras gene (as is the case here) or are nullizygous for p53. We have preliminary data indicating that neutralizing endogenous IFN-γ in vivo in our model enhances papilloma growth (Schreiber, K., R.D. Schreiber, and H. Schreiber, unpublished data). Thus, we are currently exploring whether active immunization may result in reduced endogenous IFN-γ production.
Mutant ras proteins should be ideal candidate antigens that are not only cancer-specific but are also shared by cancers from different individuals. For example, one of three to four different single amino acid substitutions in codon 12 of the cellular Kirsten ras gene is found in >90% of pancreatic cancers 44. Based upon the amino acid sequences encoded by these mutant ras genes, candidate peptides for immunotherapeutic trials have been designed that bind to MHC molecules and induce T cell responses, a strategy for selecting epitopes referred to as “reverse immunology” 45. CD4+ and CD8+ T lymphocytes that recognize point mutations in ras have been described in mice and humans after immunization in vitro and in vivo. In one model using mice immunized with mutant ras oncoprotein, preventive as well as therapeutic effects against transplanted tumor cells transfected to overexpress ras have been reported 20 46. Thus, certain mutant ras oncoproteins may be useful as shared yet tumor-specific antigens.
Nevertheless, these studies had certain problems. (a) MCA-induced tumors have antigens that can lead to the rejection of transplanted tumor cells; however, the tumor-rejection antigens appear to be unrelated to the mutant ras protein that these tumors express 47. (b) Studies in mice that reported the induction of CD8+ T cells and protective or therapeutic effects used malignant cell lines that had been transduced to overexpress the mutant ras gene rather than using unmanipulated tumors expressing mutant ras 20 48 49 50 51. At present, there is no convincing evidence that unmanipulated tumor cells spontaneously expressing mutant ras at transforming levels would be susceptible targets for destruction by ras-specific immune responses. (c) Studies in humans also failed to demonstrate antitumor effects against unmanipulated (untransfected) tumor cells 50 52, although in one recent study, almost 20% specific lysis of tumor cells expressing the appropriate mutant ras protein was achieved; even this low level of lysis required that the tumor targets be pretreated with IFN-γ 53. (d) Although numerous studies in humans and mice have demonstrated the induction of CD4+ T cells specific for mutant ras peptides 53 54 55 56 57 58 59 60 61 62 63 64 65, none of these studies have demonstrated restimulation of these T cells by unmanipulated lysates of untransfected tumor cells expressing an endogenous mutant ras protein. Insufficient amounts of antigen or inefficient antigen presentation may be a problem, as a recent study showed restimulation of CD4+ T cells by detergent lysates of human tumor cells extracted and enriched for the mutant ras protein by antibody-coated plates 53. (e) None of the previous studies 8 54 56 57 58 used affinity-purified protein for the induction or restimulation of mutant ras–specific CD4+ T cells, and the preparations of recombinant proteins purified by ion exchange and gel filtration are likely to have contained peptide fragments because of contamination with bacterial endopeptidases. At present, we do not know whether simply unfolding of the protein by nonionic detergent 50 or whether complexes of antibody and antigen 53 can allow mutant ras protein to be presented more effectively on MHC class II molecules by the endogenous lysosomal pathway. However, the endogenous antigen-presenting pathway does not seem to provide the Arg12 mutant ras peptide, as tumor cells expressing the mutant ras protein as well as the restricting MHC class II molecule after transfection were very poor targets for mutant ras peptide–specific CD4+ T cells in a 51Cr-release assay, unless the appropriate mutant ras peptide had been added exogenously 66. Similarly, L cells expressing the restricting MHC class II molecule after transfection and the mutant ras protein after infection with recombinant mutant ras vaccinia did not stimulate the CD4+ T cell hybridoma in vitro unless the mutant ras peptide had been added exogenously (Siegel, C., and H. Schreiber, unpublished results). It would be interesting to determine the peptides that are presented by the MHC class II molecules of APCs after processing of the intact mutant ras protein. The peptide-induced T cells must recognize conformations of the peptide that are not or are deficiently produced by APCs processing the intact protein. In any case, immunization with the mutant ras protein may induce a T cell response capable of recognizing the protein more effectively 67.
Stimulating immunity that protects against the development of cancer has many difficulties. Somatic or germline mutations in oncogenes or suppressor genes that lead to cancer usually consist of single amino acid substitutions or fusions of two different proteins. Because the gene products are, except for the fusion point or point mutation, identical to normal cellular self-proteins, neonatal and/or peripheral tolerance to the nonmutant portions of these proteins must limit the number of new epitopes produced. Therefore, an immune response is likely to be limited to the small region containing the point mutation or the junction of fused self-proteins. By contrast, in the case of viral or xenogeneic proteins, using conditions at which no normal cellular homologues of the transgenes are expressed during and after ontogeny, the entire protein can potentially serve as a source for antigenic peptides. Such strong immunogenicity may explain why active immunization or adoptively transferred T cells are effective in preventing development of primary cancers expressing the simian virus 40 T antigen as a transgene 5 68 69.
Our data showing that immunization with a mutant ras peptide led to increased tumor growth raise several intriguing but as yet unanswered questions. It is not known whether immunization with intact ras protein would have conferred protection against tumor growth in these animals. The potential role of antibody in promoting tumor growth in the mutant ras peptide–immunized animals also needs to be elucidated further. As discussed above, B cells and/or Ig may be involved in tumor/graft enhancement, but it is not clear whether the underlying mechanism is mediated by B cells and/or Ig itself, by B cells and/or Ig suppressing T cell responses, and/or by alterations in host cells and cytokine production which accompany B cell responses. Nevertheless, whatever the answers may be, our result raise an important cautionary note. Clinical trials have begun in patients bearing mutant ras–expressing cancers by active immunization with mutant ras peptides 59 70. Our results suggest that, in the absence of an effective T cell response to the mutant proteins, active immunization of cancer-prone or cancer-bearing individuals may enhance development and/or growth of cancers, and therefore immunization by certain procedures may be contraindicated. However, our results also show that an immune response to a cancer-specific peptide can profoundly and substantially perturbate the development of primary tumors in mice. Inducing a different type of immunity might confer protection against tumor development.
Acknowledgments
We deeply appreciate the generous support of several colleagues whose help was critically important for our studies. Drs. Raymond W. Tennant and Judson W. Spalding were essential and extremely helpful in establishing the TG.AC mouse model in our laboratory. We also appreciate the help of Dr. Aya Leder in the early stages of establishing the TG.AC model. We are deeply indebted to Dr. David Peace and Dr. Martin Cheever for making available to us the 2F9 T cell line specific for the Arg12 mutant ras peptide. The availability of this cell line helped us to compare the reactivity of our T cell line to one that had previously been studied carefully. Ms. Sherry Wanderling and Dr. Farley Yang were instrumental in developing the vector for the mutant Arg12 ras–GST fusion protein, the affinity purification of this protein, and the analysis of this protein by the T cell lines or hybridomas. We thank Dr. Andrea Sant for important suggestions. Dr. R.G. Fenton kindly provided us with recombinant ion exchange/column-purified mutant ras proteins. Mr. Gordon Bowie provided us with excellent photography.
This work was supported by National Institutes of Health grants RO1-CA-22677, RO1-CA-37516, and PO1-CA74182, by University of Chicago Cancer Center Core grant CA-14599, and by the Corinne Kreissl Foundation grant of the American Cancer Society IM773. C.A. Lazarski was supported by National Institutes of Health training grant 5T32-CA09594 (Cancer Biology Training Grant). C.T. Siegel was supported by National Institutes of Health training grant HL-07665 (Surgical Scientist Training Grant). The authors also gratefully acknowledge support by a gift from the Passis family.
Footnotes
Abbreviations used in this paper: DTH, delayed-type hypersensitivity; FVB6F1, (FVB × C57BL/6)F1 hybrid; GST, glutathione S-transferase; LNC, lymph node cell; TGB6F1, (TG.AC × C57BL/6)F1 hybrid; vHa-ras, viral Harvey ras.
References
- Fearon E.R. Human cancer syndromesclues to the origin and nature of cancer. Science. 1997;278:1043–1050. doi: 10.1126/science.278.5340.1043. [DOI] [PubMed] [Google Scholar]
- Leder A., Kuo A., Cardiff R.D., Sinn E., Leder P. v-Ha-ras transgene abrogates the initiation step in mouse skin tumorigenesiseffects of phorbol esters and retinoic acid. Proc. Natl. Acad. Sci. USA. 1990;87:9178–9182. doi: 10.1073/pnas.87.23.9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbacid M. ras genes. Annu. Rev. Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- Germolec D.R., Spalding J., Yu H.S., Chen G.S., Simeonova P.P., Humble M.C., Bruccoleri A., Boorman G.A., Foley J.F., Yoshida T., Luster M.I. Arsenic enhancement of skin neoplasia by chronic stimulation of growth factors. Am. J. Pathol. 1998;153:1775–1785. doi: 10.1016/S0002-9440(10)65692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon R.E., Spalding J.W., Trempus C.S., Szczesniak C.J., Virgil K.M., Humble M.C., Tennant R.W. Kinetics of wound-induced v-Ha-ras transgene expression and papilloma development in transgenic Tg.AC mice. Mol. Carcinog. 1997;20:108–114. doi: 10.1002/(sici)1098-2744(199709)20:1<108::aid-mc12>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Hansen L.A., Tennant R. Focal transgene expression associated with papilloma development in v- Ha-ras-transgenic TG.AC mice. Mol. Carcinog. 1994;9:143–154. doi: 10.1002/mc.2940090306. [DOI] [PubMed] [Google Scholar]
- Cannon R.E., Spalding J.W., Virgil K.M., Faircloth R.S., Humble M.C., Lacks G.D., Tennant R.W. Induction of transgene expression in Tg.AC(v-Ha-ras) transgenic mice concomitant with DNA hypomethylation. Mol. Carcinog. 1998;21:244–250. doi: 10.1002/(sici)1098-2744(199804)21:4<244::aid-mc3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Peace D.J., Chen W., Nelson H., Cheever M.A. T cell recognition of transforming proteins encoded by mutated ras proto-oncogenes. J. Immunol. 1991;146:2059–2065. [PubMed] [Google Scholar]
- Fujiwara H., Fukuzawa M., Yoshioka T., Nakajima H., Hamaoka T. The role of tumor-specific Lyt-1+2− T cells in eradicating tumor cells in vivo. I. Lyt-1+2− T cells do not necessarily require recruitment of host's cytotoxic T cell precursors for implementation of in vivo immunity. J. Immunol. 1984;133:1671–1676. [PubMed] [Google Scholar]
- Greenberg P.D., Kern D.E., Cheever M.A. Therapy of disseminated murine leukemia with cyclophosphamide and immune Lyt-1+,2− T cells. J. Exp. Med. 1985;161:1122–1134. doi: 10.1084/jem.161.5.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D., Monach P.A., Wanderling S., Philip M., Toledano A.Y., Schreiber R.D., Schreiber H. CD4+ T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-γ. Proc. Natl. Acad. Sci. USA. 1999;96:8633–8638. doi: 10.1073/pnas.96.15.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver J.L., Contrera J.F., Rosenzweig B.A., Thompson K.L., Faustino P.J., Strong J.M., Ellison C.D., Anderson L.W., Prasanna H.R., Long-Bradley P.E. An evaluation of the hemizygous transgenic Tg.AC mouse for carcinogenicity testing of pharmaceuticals. I. Evidence for a confounding nonresponder phenotype. Toxicol. Pathol. 1998;26:532–540. doi: 10.1177/019262339802600409. [DOI] [PubMed] [Google Scholar]
- Thompson K.L., Rosenzweig B.A., Sistare F.D. An evaluation of the hemizygous transgenic TG.AC mouse for carcinogenicity testing of pharmaceuticals. II. A genotypic marker that predicts tumorigenic responsiveness. Toxicol. Pathol. 1998;26:548–555. doi: 10.1177/019262339802600411. [DOI] [PubMed] [Google Scholar]
- Ward P.L., Koeppen H., Hurteau T., Schreiber H. Tumor antigens defined by cloned immunological probes are highly polymorphic and are not detected on autologous normal cells. J. Exp. Med. 1989;170:217–232. doi: 10.1084/jem.170.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M., Chenicek K.J., Naito Y., DiGiovanni J. Susceptibility to phorbol ester skin tumor promotion in (C57BL/6 × DBA/2) F1 mice is inherited as an incomplete dominant traitevidence for multi-locus involvement. Carcinogenesis. 1988;9:639–645. doi: 10.1093/carcin/9.4.639. [DOI] [PubMed] [Google Scholar]
- Monach P.A., Meredith S.C., Siegel C.T., Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity. 1995;2:45–59. doi: 10.1016/1074-7613(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Lacal J.C., Aaronson S.A. ras p21 deletion mutants and monoclonal antibodies as tools for localization of regions relevant to p21 function. Proc. Natl. Acad. Sci. USA. 1986;83:5400–5404. doi: 10.1073/pnas.83.15.5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survivalapplication to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Engvall E., Perlman P. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry. 1971;8:871–874. doi: 10.1016/0019-2791(71)90454-x. [DOI] [PubMed] [Google Scholar]
- Fenton R.G., Taub D.D., Kwak L.W., Smith M.R., Longo D.L. Cytotoxic T-cell response and in vivo protection against tumor cells harboring activated ras proto-oncogenes. J. Natl. Cancer Inst. 1993;85:1294–1302. doi: 10.1093/jnci/85.16.1294. [DOI] [PubMed] [Google Scholar]
- Rous P., Kidd J.G. Conditional neoplasms and subthreshold neoplastic states. A study of the tar tumors of rabbits. J. Exp. Med. 1941;73:365–390. doi: 10.1084/jem.73.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie I., Rous P. The experimental disclosure of latent neoplastic changes in tarred skin. J. Exp. Med. 1941;73:391–415. doi: 10.1084/jem.73.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber H., Rowley D.A. Inflammation and cancer. In: Gallin J.I., Snyderman R., editors. InflammationBasic Principles and Clinical Correlates. 3rd ed. Lippincott Williams and Wilkins; Philadelphia: 1999. pp. 1117–1129. [Google Scholar]
- Yuspa S.H. The pathogenesis of squamous cell cancerlessons learned from studies of skin carcinogenesis—thirty-third G.H.A. Clowes Memorial Award Lecture. Cancer Res. 1994;54:1178–1189. [PubMed] [Google Scholar]
- Prehn R.T. The immune reaction as a stimulator of tumor growth. Science. 1972;176:170–171. doi: 10.1126/science.176.4031.170. [DOI] [PubMed] [Google Scholar]
- Prehn R.T. Stimulatory effects of immune reactions upon the growths of untransplanted tumors. Cancer Res. 1994;54:908–914. [PubMed] [Google Scholar]
- Nakamoto Y., Guidotti L.G., Kuhlen C.V., Fowler P., Chisari F.V. Immune pathogenesis of hepatocellular carcinoma. J. Exp. Med. 1998;188:341–350. doi: 10.1084/jem.188.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esserman L.J., Lopez T., Montes R., Bald L.N., Fendly B.M., Campbell M.J. Vaccination with the extracellular domain of p185neu prevents mammary tumor development in neu transgenic mice. Cancer Immunol. Immunother. 1999;47:337–342. doi: 10.1007/s002620050539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell G.D. Incompatibility reactions to tumor homotransplants with particular reference to the role of the tumor. Cancer Res. 1957;17:2–10. [PubMed] [Google Scholar]
- Kaliss N. Immunological enhancement of tumor homografts in mice. A review. Cancer Res. 1957;18:992–1003. [PubMed] [Google Scholar]
- Bubenik J., Turano A. Enhancing effect on tumour growth of humoral antibodies against tumour specific transplantation antigens in tumours induced by murine sarcoma virus (Harvey) Nature. 1968;220:928–930. doi: 10.1038/220928a0. [DOI] [PubMed] [Google Scholar]
- Monach P.A., Schreiber H., Rowley D.A. CD4+ and B lymphocytes in transplantation immunity. II. Augmented rejection of tumor allografts by mice lacking B cells. Transplantation. 1993;55:1356–1361. [PubMed] [Google Scholar]
- Qin Z., Richter G., Schuler T., Ibe S., Cao X., Blankenstein T. B cells inhibit induction of T cell-dependent tumor immunity. Nat. Med. 1998;4:627–630. doi: 10.1038/nm0598-627. [DOI] [PubMed] [Google Scholar]
- Ibanez O.M., Mouton D., Ribeiro O.G., Bouthillier Y., De Franco M., Cabrera W.H., Siqueira M., Biozzi G. Low antibody responsiveness is found to be associated with resistance to chemical skin tumorigenesis in several lines of Biozzi mice. Cancer Lett. 1999;136:153–158. doi: 10.1016/s0304-3835(98)00317-6. [DOI] [PubMed] [Google Scholar]
- Brodt P., Gordon J. Natural resistance mechanisms may play a role in protection against chemical carcinogenesis. Cancer Immunol. Immunother. 1982;13:125–127. doi: 10.1007/BF00205312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson L.A. Anti-tumor immune responses of the tumor-bearing hostthe case for antibody-mediated immunologic enhancement. Clin. Immunol. Immunopathol. 1994;72:1–8. doi: 10.1006/clin.1994.1099. [DOI] [PubMed] [Google Scholar]
- Stach R.M., Rowley D.A. A first or dominant immunization. II. Induced immunoglobulin carries transforming growth factor beta and suppresses cytolytic T cell responses to unrelated alloantigens. J. Exp. Med. 1993;178:841–852. doi: 10.1084/jem.178.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley D.A., Stach R.M. B lymphocytes secreting IgG linked to latent transforming growth factor-beta prevent primary cytolytic T lymphocyte responses. Int. Immunol. 1998;10:355–363. doi: 10.1093/intimm/10.3.355. [DOI] [PubMed] [Google Scholar]
- Speidel K., Osen W., Faath S., Hilgert I., Obst R., Braspenning J., Momburg F., Hammerling G.J., Rammensee H.G. Priming of cytotoxic T lymphocytes by five heat-aggregated antigens in vivoconditions, efficiency, and relation to antibody responses. Eur. J. Immunol. 1997;27:2391–2399. doi: 10.1002/eji.1830270938. [DOI] [PubMed] [Google Scholar]
- Cardiff R.D., Leder A., Kuo A., Pattengale P.K., Leder P. Multiple tumor types appear in a transgenic mouse with the ras oncogene. Am. J. Pathol. 1993;142:1199–1207. [PMC free article] [PubMed] [Google Scholar]
- Wijesuriya R., Maruo S., Zou J.P., Ogawa M., Umehara K., Yamashita M., Ono S., Fujiwara H., Hamaoka T. B cell-mediated down-regulation of IFN-gamma and IL-12 production induced during anti-tumor immune responses in the tumor-bearing state. Int. Immunol. 1998;10:1057–1065. doi: 10.1093/intimm/10.8.1057. [DOI] [PubMed] [Google Scholar]
- Kaplan D.H., Shankaran V., Dighe A.S., Stockert E., Aguet M., Old L.J., Schreiber R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA. 1998;95:7556–7561. doi: 10.1073/pnas.95.13.7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa H., Oda E., Nakao K., Ishihara M., Ueda S., Yokochi T., Ogasawara K., Nakatsuru Y., Shimizu S., Ohira Y. Loss of transcription factor IRF-1 affects tumor susceptibility in mice carrying the Ha-ras transgene or nullizygosity for p53. Genes Dev. 1999;13:1240–1245. doi: 10.1101/gad.13.10.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruban R.H., van Mansfeld A.D., Offerhaus G.J., van Weering D.H., Allison D.C., Goodman S.N., Kensler T.W., Bose K.K., Cameron J.L., Bos J.L. K-ras oncogene activation in adenocarcinoma of the human pancreas. A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele-specific oligonucleotide hybridization. Am. J. Pathol. 1993;143:545–554. [PMC free article] [PubMed] [Google Scholar]
- Boon T., van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J. Exp. Med. 1996;183:725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton R.G., Keller C.J., Hanna N., Taub D.D. Induction of T-cell immunity against Ras oncoproteins by soluble protein or Ras-expressing Escherichia coli . J. Natl. Cancer Inst. 1995;87:1853–1861. doi: 10.1093/jnci/87.24.1853. [DOI] [PubMed] [Google Scholar]
- Carbone G., Borrello M.G., Molla A., Rizzetti M.G., Pierotti M.A., Della Porta G., Parmiani G. Activation of ras oncogenes and expression of tumor-specific transplantation antigens in methylcholanthrene-induced murine fibrosarcomas. Int. J. Cancer. 1991;47:619–625. doi: 10.1002/ijc.2910470423. [DOI] [PubMed] [Google Scholar]
- Bristol J.A., Schlom J., Abrams S.I. Persistence, immune specificity, and functional ability of murine mutant ras epitope-specific CD4+ and CD8+ T lymphocytes following in vivo adoptive transfer. Cell. Immunol. 1999;194:78–89. doi: 10.1006/cimm.1999.1489. [DOI] [PubMed] [Google Scholar]
- Peace D.J., Smith J.W., Chen W., You S.G., Cosand W.L., Blake J., Cheever M.A. Lysis of ras oncogene–transformed cells by specific cytotoxic T lymphocytes elicited by primary in vitro immunization with mutated ras peptide. J. Exp. Med. 1994;179:473–479. doi: 10.1084/jem.179.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristol J.A., Schlom J., Abrams S.I. Development of a murine mutant Ras CD8+ CTL peptide epitope variant that possesses enhanced MHC class I binding and immunogenic properties. J. Immunol. 1998;160:2433–2441. [PubMed] [Google Scholar]
- Toes R.E., Offringa R., Blom R.J., Melief C.J., Kast W.M. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc. Natl. Acad. Sci. USA. 1996;93:7855–7860. doi: 10.1073/pnas.93.15.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Elsas A., Nijman H.W., Van der Minne C.E., Mourer J.S., Kast W.M., Melief C.J., Schrier P.I. Induction and characterization of cytotoxic T-lymphocytes recognizing a mutated p21ras peptide presented by HLA-A*0201. Int. J. Cancer. 1995;61:389–396. doi: 10.1002/ijc.2910610319. [DOI] [PubMed] [Google Scholar]
- Abrams S.I., Khleif S.N., Bergmann-Leitner E.S., Kantor J.A., Chung Y., Hamilton J.M., Schlom J. Generation of stable CD4+ and CD8+ T cell lines from patients immunized with ras oncogene-derived peptides reflecting codon 12 mutations. Cell. Immunol. 1997;182:137–151. doi: 10.1006/cimm.1997.1224. [DOI] [PubMed] [Google Scholar]
- Yokomizo H., Matsushita S., Fujisao S., Murakami S., Fujita H., Shirouzu M., Yokoyama S., Ogawa M., Nishimura Y. Augmentation of immune response by an analog of the antigenic peptide in a human T-cell clone recognizing mutated Ras-derived peptides. Hum. Immunol. 1997;52:22–32. doi: 10.1016/S0198-8859(96)00254-6. [DOI] [PubMed] [Google Scholar]
- Ngo-Giang-Huong N., Kayibanda M., Deprez B., Levy J.P., Guillet J.G., Tilkin A.F. Mutations in residue 61 of H-Ras p21 protein influence MHC class II presentation. Int. Immunol. 1995;7:269–275. doi: 10.1093/intimm/7.2.269. [DOI] [PubMed] [Google Scholar]
- Gedde-Dahl T., III, Spurkland A., Fossum B., Wittinghofer A., Thorsby E., Gaudernack G. T cell epitopes encompassing the mutational hot spot position 61 of p21 ras. Promiscuity in ras peptide binding to HLA. Eur. J. Immunol. 1994;24:410–414. doi: 10.1002/eji.1830240221. [DOI] [PubMed] [Google Scholar]
- Peace D.J., Smith J.W., Disis M.L., Chen W., Cheever M.A. Induction of T cells specific for the mutated segment of oncogenic P21ras protein by immunization in vivo with the oncogenic protein. J. Immunother. 1993;14:110–114. doi: 10.1097/00002371-199308000-00005. [DOI] [PubMed] [Google Scholar]
- Qin H., Chen W., Takahashi M., Disis M.L., Byrd D.R., McCahill L., Bertram K.A., Fenton R.G., Peace D.J., Cheever M.A. CD4+ T-cell immunity to mutated ras protein in pancreatic and colon cancer patients. Cancer Res. 1995;55:2984–2987. [PubMed] [Google Scholar]
- Gjertsen M.K., Bakka A., Breivik J., Saeterdal I., Gedde-Dahl T., III, Stokke K.T., Solheim B.G., Egge T.S., Soreide O., Thorsby E., Gaudernack G. Ex vivo ras peptide vaccination in patients with advanced pancreatic cancerresults of a phase I/II study. Int. J. Cancer. 1996;65:450–453. doi: 10.1002/(SICI)1097-0215(19960208)65:4<450::AID-IJC10>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Gedde-Dahl T., III, Eriksen J.A., Thorsby E., Gaudernack G. T-cell responses against products of oncogenesgeneration and characterization of human T-cell clones specific for p21 ras-derived synthetic peptides. Hum. Immunol. 1992;33:266–274. doi: 10.1016/0198-8859(92)90334-j. [DOI] [PubMed] [Google Scholar]
- Fossum B., Gedde-Dahl T., III, Hansen T., Eriksen J.A., Thorsby E., Gaudernack G. Overlapping epitopes encompassing a point mutation (12 Gly--> Arg) in p21 ras can be recognized by HLA-DR, -DP and -DQ restricted T cells. Eur. J. Immunol. 1993;23:2687–2691. doi: 10.1002/eji.1830231045. [DOI] [PubMed] [Google Scholar]
- Johansen B.H., Gedde-Dahl T., III, Sollid L.M., Vartdal F., Thorsby E., Gaudernack G. Binding of ras oncogene peptides to purified HLA-DQ(alpha 1*0102,beta 1*0602) and -DR(alpha,beta 1*0101) molecules Scand. J. Immunol. 39 1994. 607 612[published erratum at 40:468] [DOI] [PubMed] [Google Scholar]
- Fossum B., Breivik J., Meling G.I., Gedde-Dahl T., III, Hansen T., Knutsen I., Rognum T.O., Thorsby E., Gaudernack G. A K-ras 13Gly-- > Asp mutation is recognized by HLA-DQ7 restricted T cells in a patient with colorectal cancer. Modifying effect of DQ7 on established cancers harbouring this mutation? Int. J. Cancer. 1994;58:506–511. doi: 10.1002/ijc.2910580409. [DOI] [PubMed] [Google Scholar]
- Gedde-Dahl T., III, Nilsen E., Thorsby E., Gaudernack G. Growth inhibition of a colonic adenocarcinoma cell line (HT29) by T cells specific for mutant p21 ras. Cancer Immunol. Immunother. 1994;38:127–134. [PubMed] [Google Scholar]
- Jung S., Schluesener H.J. Human T lymphocytes recognize a peptide of single point–mutated, oncogenic ras proteins. J. Exp. Med. 1991;173:273–276. doi: 10.1084/jem.173.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrams S.I., Dobrzanski M.J., Wells D.T., Stanziale S.F., Zaremba S., Masuelli L., Kantor J.A., Schlom J., Masuelle L. Peptide-specific activation of cytolytic CD4+ T lymphocytes against tumor cells bearing mutated epitopes of K-ras p21 Eur. J. Immunol. 25 1995. 2588 2597[published erratum at 25:3525] [DOI] [PubMed] [Google Scholar]
- Viner N.J., Nelson C.A., Unanue E.R. Identification of a major I-Ek-restricted determinant of hen egg lysozymelimitations of lymph node proliferation studies in defining immunodominance and crypticity. Proc. Natl. Acad. Sci. USA. 1995;92:2214–2218. doi: 10.1073/pnas.92.6.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X., McCarrick J., Jewett L., Knowles B.B. Timely immunization subverts the development of peripheral nonresponsiveness and suppresses tumor development in simian virus 40 tumor antigen-transgenic mice. Proc. Natl. Acad. Sci. USA. 1994;91:3916–3920. doi: 10.1073/pnas.91.9.3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granziero L., Krajewski S., Farness P., Yuan L., Courtney M.K., Jackson M.R., Peterson P.A., Vitiello A. Adoptive immunotherapy prevents prostate cancer in a transgenic animal model. Eur. J. Immnol. 1999;29:1127–1138. doi: 10.1002/(SICI)1521-4141(199904)29:04<1127::AID-IMMU1127>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Khleif S.N., Abrams S.I., Hamilton J.M., Bergmann-Leitner E., Chen A., Bastian A., Bernstein S., Chung Y., Allegra C.J., Schlom J. A phase I vaccine trial with peptides reflecting ras oncogene mutations of solid tumors. J. Immunother. 1999;22:155–165. doi: 10.1097/00002371-199903000-00007. [DOI] [PubMed] [Google Scholar]