

Abstract
Heat shock proteins (HSPs) like glycoprotein (gp)96 (glucose-regulated protein 94 [grp94]) are able to induce specific cytotoxic T lymphocyte (CTL) responses against cells from which they originate. Here, we demonstrate that for CTL activation by gp96-chaperoned peptides, specific receptor-mediated uptake of gp96 by antigen-presenting cells (APCs) is required. Moreover, we show that in both humans and mice, only professional APCs like dendritic cells (DCs), macrophages, and B cells, but not T cells, are able to bind gp96. The binding is saturable and can be inhibited using unlabeled gp96 molecules. Receptor binding by APCs leads to a rapid internalization of gp96, which colocalizes with endocytosed major histocompatibility complex (MHC) class I and class II molecules in endosomal compartments. Incubation of gp96 molecules isolated from cells expressing an adenovirus type 5 E1B epitope with the DC line D1 results in the activation of E1B-specific CTLs. This CTL activation can be specifically inhibited by the addition of irrelevant gp96 molecules not associated with E1B peptides. Our results demonstrate that only receptor-mediated endocytosis of gp96 molecules leads to MHC class I–restricted re-presentation of gp96-associated peptides and CTL activation; non–receptor-mediated, nonspecific endocytosis is not able to do so. Thus, we provide evidence on the mechanisms by which gp96 is participating in the cross-presentation of antigens from cellular origin.
Keywords: heat shock protein–peptide complex, cross-priming, receptor-mediated endocytosis, cytotoxic T lymphocyte activation, dendritic cell
Introduction
Activation of CTLs with exogenous cell-associated antigens requires efficient uptake and presentation of these antigens by bone marrow–derived APCs. This phenomenon was first observed by Bevan 1 2 3 for the induction of CTLs against minor H antigens. Because the antigens were expressed in foreign donor cells with different MHC molecules, this process was termed “cross-priming.” Since then, it has been shown that soluble protein antigens 4 5, antigens expressed in MHC matched cells 6 7 8, or antigens encoded by naked DNA 9 also require uptake and re-presentation by MHC molecules expressed on the surface of professional APCs. Therefore, the term “cross-presentation” was introduced to describe the general re-presentation of exogenous cell-associated antigens by MHC class I 6 and MHC class II molecules 10. In addition to CTL activation, cross-presentation can also lead to the induction of CTL tolerance 11 12.
The nature of the APCs that are able to take up and re-present cell-associated antigens on MHC class I molecules remains elusive in vivo. However, in vitro studies suggest that dendritic cells (DCs) 11 12 13, macrophages 14 15 16, or B cells 17 might be involved.
Several pathways for antigen uptake have been described, ranging from nonspecific mechanisms such as phagocytosis, pinocytosis, or macropinocytosis 18 19 20 21 22 23 to specific, receptor-operated mechanisms that include mannose- and scavenger-type receptors 22. Depending on the nature of the antigens, and consequently on the mode of uptake, antigens might be targeted to different processing compartments and be able to gain access to different antigen presentation pathways. CTL activation can be mediated by macropinocytosis or phagocytosis of exogenous soluble antigens 24 25 26. However, these pathways require high antigen concentrations and might therefore be of limited relevance in providing a mechanism for cross-presentation in vivo 23.
More recently, apoptotic bodies were shown to be phagocytosed by immature DCs, resulting in the activation of MHC class I–restricted T cells 13 27. This uptake involves CD36 and the integrin receptor αvβ5 28, which explains the high efficiency.
An additional pathway with potential relevance for cross-presentation became evident when the induction of tumor immunity and CTL activation through the injection of heat shock proteins (HSPs) such as glycoprotein (gp)96, HSP70, and HSP90 was discovered (for a review, see reference 29). The specificities of the CTL response were directed against the cells from which the HSPs were isolated. This can be explained by the association of the HSPs with peptides of cellular origin. Immune responses against several cellular antigens including minor H and tumor and viral antigens were induced (for a review, see reference 30) by using as little as 1–2 ng HSP–peptide complex in one particular case 31. It was postulated that the extremely efficient MHC presentation of HSP-associated peptides is accomplished by the receptor-mediated uptake of HSPs by professional APCs 32. Recently, binding of HSP70 and gp96 to a macrophage- and dendritic-like cell line was observed 33. This observation provides a possible explanation for the high immunogenic potential of HSPs in situations in which they are injected into mice or released from dying cells, in that they shuttle antigenic peptides to APCs 32. Receptor-mediated endocytosis of HSPs by professional APCs will lead to the accumulation of these peptide chaperones in cells crucially involved in the activation of CTLs.
We therefore decided to characterize the cell populations involved in receptor-mediated endocytosis of HSPs in detail, to follow the fate of endocytosed HSPs, and to test whether or not receptor-mediated endocytosis of HSPs indeed results in the re-presentation of HSP-associated peptides and subsequent activation of CTLs. The latter issue in particular is of crucial importance for the understanding of HSP-mediated cross-presentation, as antigen uptake by APCs does not necessarily correlate with the ability to cross-present antigens. Despite the fact that macrophages and DCs phagocytose apoptotic cells, only immature DCs are able to cross-present antigens and to activate CTLs 28.
Here, we demonstrate that members of the family of professional APCs such as macrophages, DCs, and B cells are able to bind the endoplasmic reticulum (ER)-resident HSP gp96 specifically. The binding was saturable and could be competed for with unlabeled gp96 molecules. The uptake of gp96 isolated from cells expressing the adenovirus type 5 (Ad5)-E1B epitope by the DC line D1 resulted in the activation of E1B-specific CTLs. More importantly, activation of Ad5-E1B–specific CTLs could be inhibited by competition with gp96 not associated with E1B peptide. This result clearly demonstrates that CTL activation is the consequence of receptor-mediated endocytosis of gp96 molecules followed by the class I–restricted re-presentation of associated peptides, and supports the participation of HSPs in cross-presentation of cell-associated antigens.
Materials and Methods
Mice, Cells, Antibodies, and Proteins.
The DEC-205 knockout mice were provided by Michel Nussenzweig and Ralph Steinman (The Rockefeller University, New York, NY). BALB/c and C57BL/6 mice were obtained from Charles River Laboratories. MHC class II–deficient mice ABBN5 34 and littermate ABBN6 were obtained from Taconic Farms. P388D1, RMA, and RMA-S mouse cell lines (American Type Culture Collection) were cultured in α-MEM. The cell line D2SC/1, representing an early progenitor of mouse splenic DC, and D1, a nontransformed, growth factor–dependent, long-term DC culture 35, were cultured in IMDM. All tissue culture media were supplemented with 10% FCS, 0.3 mg/ml l-glutamin, 100 U/ml penicillin/streptomycin, and 50 μM β-mercaptoethanol. To grow D1 cells, medium was additionally supplemented with 30% conditioned medium from the fibroblast cell line R1. Antibody to gp96 (SPA-850) was obtained from StressGen Biotechnologies. The following labeled antibodies to mouse and human antigens were obtained from BD PharMingen: H2-Kb–biotin, H2-Ab–biotin, CD8-FITC, IFN-γ–PE, CD16/CD32 (Fc block), CD45R/B220-PE, CD19-PE, CD14-PE, CD90.2 (Thy1.2)-PE, CD86 (B7.2)-PE, CD11c-PE, Mac-3–PE, CD1a-PE, CD83-PE, and IgG1-PE and IgG2a-PE isotype controls. Goat anti–rabbit–Alexa™ 546 and streptavidin-Alexa™ 546 (Molecular Probes) were used as secondary reagents. BSA, biotinylated BSA, OVA, and FITC were obtained from Sigma-Aldrich. Streptavidin-PE was purchased from Jackson Laboratories. BSA and OVA were labeled with FITC or biotin according to standard protocols. Free FITC molecules were removed by reaction with Tris and gel filtration through a Sephadex G-25 (Sigma-Aldrich) column. gp96 and gp96-FITC from the mouse cell line IGELa2 were provided by Immunosome. All animal studies were performed according to our institutional guidelines and approved by our Institutional Review Board.
Purification of gp96.
The transporter associated with antigen processing (TAP)-deficient RMA-S SigE1B cell line has been generated by transfection of RMA-S with the adenovirus early region 1 H2-Db–restricted E1B epitope (VNIRNCCYI) targeted to the ER in a TAP-independent fashion 36. gp96 was purified from RMA, RMA-S, and RMA-S SigE1B cell lines as described 37. The approximate concentrations were determined by measuring the OD at 280 nm using an extinction coefficient of 1.0.
Cytometry (FACS®) Binding Assay.
105 cells were incubated for 30 min on ice in 100 μl IMDM and 10% FCS containing 30 μg/ml gp96-FITC or OVA-FITC, washed three times, and fixed in 1% paraformaldehyde. For competition experiments, a given excess of unlabeled gp96 was added together with 50 μg/ml gp96-FITC simultaneously. For staining of mouse spleen cells (including erythrocytes) and human PBLs, PE-conjugated antibodies were added as markers for different cell types. Immature DCs were prepared from bone marrow of C57BL/6 mice 38 and human blood monocytes 39 as described. Cytometry measurements were performed on a FACSCalibur™ (Becton Dickinson).
Internalization Studies in Confocal Microscopy.
Immature bone marrow–derived DCs (BMDCs) were prepared from C57BL/6 mice as described 38. On day 6 of their preparation, the BMDCs were tested for CD11c, CD86, and MHC class II expression and were seeded on cover slips, precooled, and incubated for 30 min on ice with IMDM containing 10% FCS and 50 μg/ml gp96-FITC (“pulse”). The coverslips were washed twice and incubated in IMDM medium for 15 min or longer at 37°C (“chase”), washed, and fixed in 3.7% paraformaldehyde in PBS. For the colocalization experiments, cells were pre-incubated with Fc block (α-CD16/CD32) followed by biotinylated antibodies to H2-Kb or H2-Ab and 50 μg/ml gp96-FITC together with streptavidin-Alexa™ 546. For staining of lysosomes, cells were fixed with methanol/aceton (1:1, −20°C) and incubated with α–Lamp-1 (provided by M. Fukuda, La Jolla Research Center, La Jolla, CA) and goat anti–rabbit–Alexa™ 546. For microscopy, a ZEISS LSM 510 laser scanning microscope was used. “Bleeding” of emission into other detection channels was excluded using the multitracking modus of the LSM 510. Thickness of the optical plane was adjusted by the pinhole to be <1 μm.
Immunization of Mice with gp96.
C57BL/6 mice were immunized intraperitoneally with 30 μg gp96 purified from RMA-S SigE1B cells. After 10 d, mice were killed and the spleen cells were restimulated with E1B-expressing XC3 cells or Ad5-E1B peptide (50 ng/ml). Specific lysis of RMA-S SigE1B cells by CTLs contained in the spleen culture was determined by a standard chromium release assay 5 d after restimulation and after a second restimulation with XC3 cells or Ad5-E1B peptide (50 ng/ml).
CTL Cross-Presentation Assay.
The CTL clones 100B6, 0.1C2, and LN5 were described previously 36 40. CTL clones were restimulated on a weekly basis by incubation with the Ad5-E1B/E1A–expressing tumor cell line XC3. The E1B peptide was synthesized on a ABI 432 A peptide snythesizer (Applied Biosystems) applying Fmoc strategy.
Activation of CTL clones was assessed by measurement of intracellular IFN-γ production. 2.5 × 104 D1 cells were incubated with 20 μg/ml gp96 purified from RMA-S SigE1B, RMA, or RMA-S cells for 2 h at 37°C. For competition experiments, an excess of gp96 from RMA or RMA-S was added, washed four times, and incubated with 2.5 × 105 CTLs for 12 h at 37°C. 10 μg/ml Brefeldin A was added for an additional 5 h at 37°C. Cells were washed, fixed, and perforated with saponin. The fixed cells were stained with PE-labeled anti–IFN-γ or isotype control and FITC-labeled anti-CD8 antibodies, and were measured by flow cytometry.
Results
gp96 Binds Specifically to APC Lines.
Recent experiments demonstrated that HSPs are able to interact specifically with a macrophage- and a DC-like cell line 33. Therefore, we further characterized the cell types able to interact with gp96 in a specific manner. For this purpose, we incubated several cell lines with FITC-labeled gp96, always at 4°C to exclude endocytosis. We only observed a specific interaction of gp96 with APC lines like P388D1, D2SC/1, and D1, but not with the lymphoma cell lines RMA, EG.7, and T1 (Fig. 1A and Fig. B). Increasing the total concentration of gp96-FITC, the binding displayed saturation at a total concentration of 30 μg/ml (Fig. 1 C) and could only be competed for by unlabeled gp96, but not by OVA (Fig. 1 A) or BSA (not shown). A onefold excess of unlabeled gp96 resulted in a 50% reduction, and a fivefold excess resulted in an ∼75% reduction of gp96-FITC binding at saturation point, which could be inhibited completely using an excess of up to a 100-fold (Fig. 1 D). These data correspond to the theoretical values of 50% and 83% (1:1 and 1:5 dilution of gp96-FITC with unlabeled gp96), demonstrating that the FITC labeling of gp96 did not significantly affect the binding characteristics to its putative receptor. No inhibition was observed using an excess of up to 400-fold of OVA or BSA (data not shown). These data demonstrate the presence of a specific gp96 receptor that is expressed on APCs but not on other cell lines (Fig. 1A–D).
Figure 1.
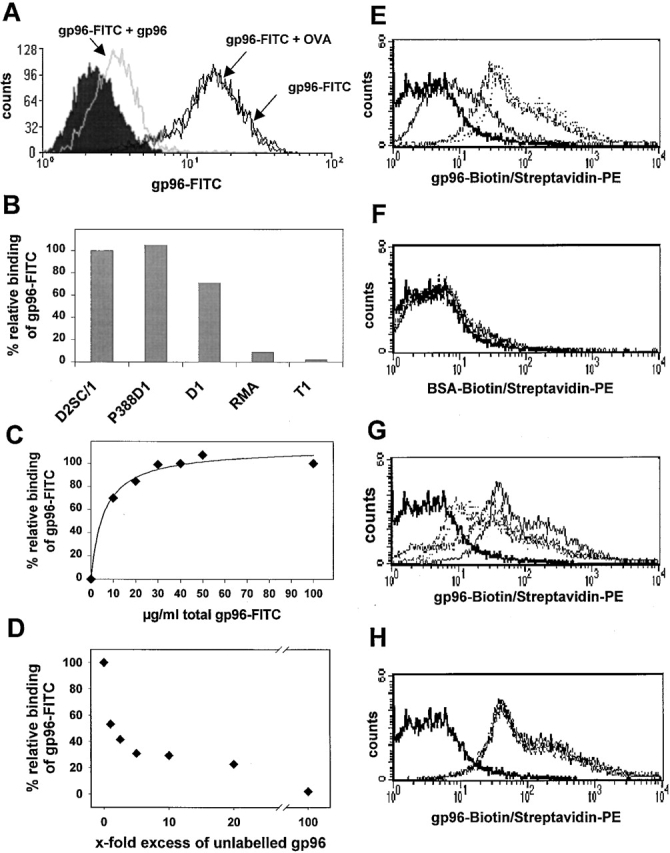
Specific binding of gp96-FITC to APC cell lines and BMDCs. Binding of 3 μg gp96-FITC to 105 D2SC/1 cells was performed, always at 4°C, in 100 μl IMDM containing 10% FCS. This binding could be competed by a 10-fold excess of unlabeled gp96, but not OVA. (A) Specific binding of gp96-FITC was observed on D2SC/1 (DC progenitor), P388D1 (macrophage), and D1 (DC), but not on RMA and T1 cells. (B) Binding could be saturated at ∼30 μg/ml for 105 D2SC/1 cells (C) and competed almost completely by a 100-fold excess of unlabeled gp96. (D) Binding is given as relative values, where 100% represents maximum binding of gp96-FITC. The concentration values shown give total concentration of gp96-FITC added to the cells. (E) Binding of 1 μg (bold line), 5 μg (broken line), and 10 μg (dotted line) gp96-biotin/streptavidin-PE to immature BMDCs from C57BL/6 mice. (F) No binding was observed for BSA-biotin/streptavidin-PE. Binding of 10 μg gp96-biotin (bold line) to BMDC is competed in a similar fashion to D by unlabeled gp96 (G), but not by unlabeled BSA (H).
gp96 Interacts Specifically with Primary APCs in Mice and Humans.
More importantly, gp96 (Fig. 1 E) but not BSA (Fig. 1 F) bound efficiently to immature BMDCs prepared 38 from C57BL/6 mice, and could be competed for by increasing amounts of unlabeled gp96 (Fig. 1 G) but not by BSA (Fig. 1 H). Specific binding was also observed when mouse spleen cells from BALB/c mice were incubated with gp96-FITC (Fig. 2). gp96 interacted specifically with cells that stained positive for MHC class II and CD45 (B220), but not with cells positive for CD90 (Thy-1) molecules. Setting the forward and side scatter gate on the bigger cells, including cells of the myeloid lineage, CD11c– and Mac-3–positive cells were also positive for gp96-FITC, indicating that the expression of the gp96 receptor is restricted to professional APCs. No staining was observed using OVA-FITC (Fig. 2, left) or BSA-FITC (data not shown). The identical outcome was observed using spleen cells from C57BL/6 mice (not shown). A similar gp96-FITC staining pattern was obtained for human PBLs. HLA-DR–, CD86-, CD19-, and CD14-positive but not CD2- or CD3-positive cells interacted specifically with gp96-FITC (Fig. 3 A). Again, no staining was observed using OVA-FITC. gp96 binding to monocytes was slightly better than to B cells in human PBLs. As expected, DCs expressing CD1a and CD83 were not detected. To determine gp96-FITC binding to this cell type, we differentiated DCs from human PBLs by the application of GM-CSF and IL-4. The whole DC population generated stained positive with gp96 but not BSA (Fig. 3 B).
Figure 2.
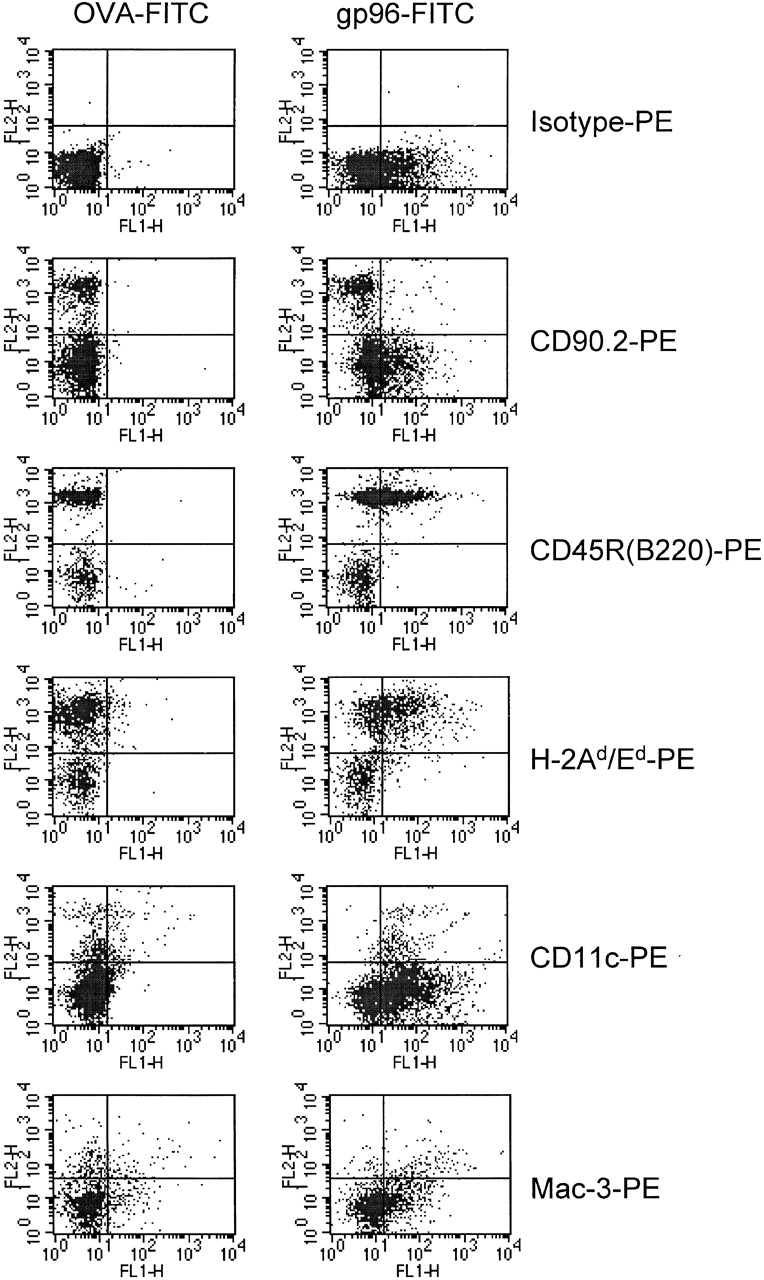
Specific binding of gp96-FITC to B cells, macrophages, and DCs, but not to T cells of a spleen cell culture. 105 fresh BALB/c spleen cells were stained with 5 μg OVA-FITC (left) or gp96-FITC (right), and different PE-labeled cell type marker antibodies to CD90.2 (Thy-1.2, T cells), CD45R/B220 (B cells), I-Ad/Ed, CD11c (DCs), and Mac-3 (monocytes and macrophages). Macrophages and DCs were counted in a different gate than lymphocytes with a higher forward scatter value.
Figure 3.
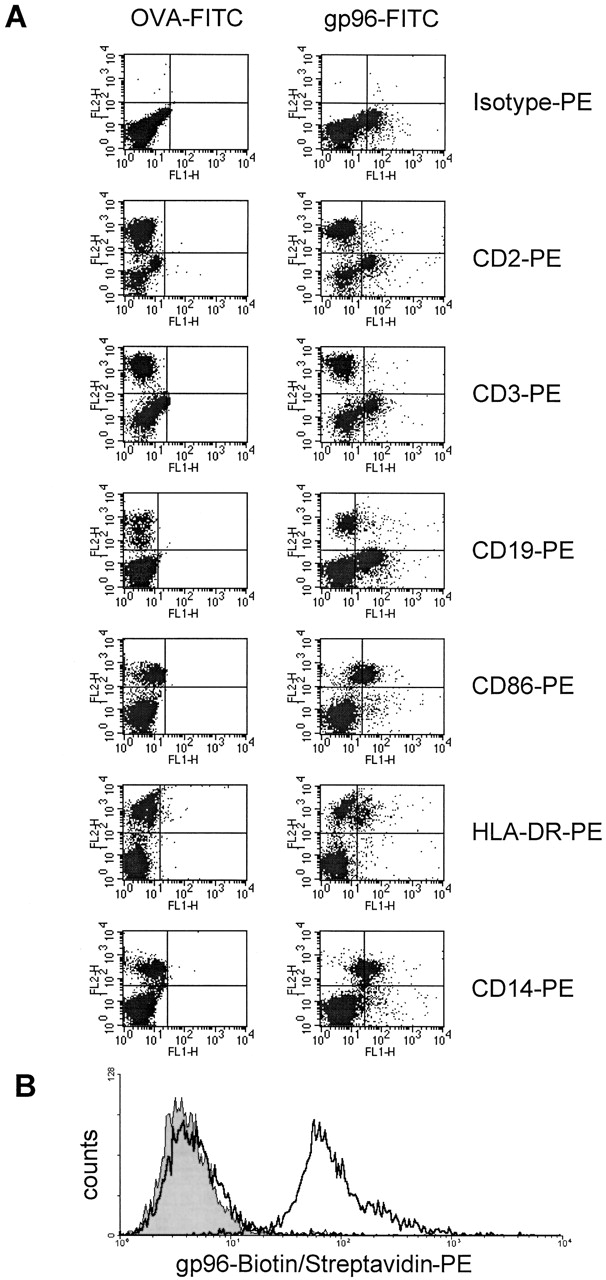
gp96-FITC binds to APCs in human PBL culture, but not to T cells. (A) 105 fresh human PBLs were stained with 5 μg OVA-FITC (left) or gp96-FITC (right) together with PE-labeled antibodies to the following cell surface antigens: CD2 (T and NK cells), CD3 (T cells), CD19 (B cells), CD86, HLA-DR, and CD14 (monocytes). The gate was set on all living cells. Therefore monocytes appear as a population with a slightly higher autofluorescence than lymphocytes in both fluorescence channels. Comparing the shifts of each population, monocytes showed slightly better binding of gp96 than B cells. (B) Binding of 0 μg (gray shaded) and 5 μg (bold line) gp96-biotin to immature DCs prepared from human PBLs. 10 μg of BSA-biotin (bold line) did not display binding.
DEC-205 and MHC Class II Molecules Are Not Required for gp96 Binding.
Because gp96 molecules contain a single high-mannose oligosaccharide 41 42, we addressed the question of whether this might allow the uptake by the DEC-205 receptor. DEC-205 is expressed on DCs and thymic epithelial cells and is capable of directing captured soluble exogenous antigens to a specialized antigen processing compartment 43. DCs were prepared from bone marrow of wild-type and DEC-205−/− mice (provided by Michel Nussenzweig and Ralph Steinman) and incubated with increasing amounts of gp96-FITC. FACS® analysis revealed identical staining (Fig. 4 A), suggesting that the DEC-205 receptor is not involved in the binding of gp96 molecules by DCs.
Figure 4.


DEC-205 and MHC class II do not function as receptors for gp96. Binding of gp96-biotin to BMDCs from wild-type and DEC-205−/− mice (A) as well as binding of gp96-FITC to spleen cells from MHC class II−/− mice and their littermate (B) showed identical staining. For staining of spleen cells in B, different cell surface markers were used (shown in Fig. 2). Only antibody to MHC class II is shown.
We further speculated whether MHC class II might function as a receptor for gp96 because gp96 showed binding to all MHC class II–positive mouse spleen cells and human PBLs. Binding together with marker antibodies to spleen cells from MHC class II knockout mice and their littermates did not reveal any difference (shown for MHC class II antibody in Fig. 4 B; other markers not shown), indicating that MHC class II molecules do not function as gp96 receptors.
gp96 is Endocytosed Efficiently and Colocalizes with Recycled MHC Class I and Class II Molecules.
Recently it has been suggested that gp96-FITC bound to peritoneal macrophages is endocytosed into early endosomes but does not reach the stage of lysosomes 44. We also attempted to determine the faith of receptor-bound gp96 at the cell surface of APCs by confocal microscopy (Fig. 5) using authentic DCs (BMDCs from C57BL/6 mice). Initial binding of FITC-labeled gp96 to the cell surface at 4°C to prevent endocytosis revealed a patched pattern. Further incubation at 37°C led to efficient endocytosis of gp96. Colocalization with lysosomes labeled with Lamp-1 antibody 45 was not observed after 15, 30, 45, 60, and 90 min of endocytosis (shown for 60 min in Fig. 5). Recently, it has been reported that internalized cell surface MHC class I molecules, like class II molecules, are able to bind their antigen in endosomal compartments, suggesting these vesicles to be putative MHC class I and class II loading compartments for exogenously derived antigen 46. We therefore attempted to determine whether gp96 taken up by receptor-mediated endocytosis can be found in compartments containing recycled MHC class I and class II molecules. Indeed, after 15 min of endocytosis, nearly all of the endocytosed H2-Kb and H2-Ab molecules colocalized with gp96. Similar results were obtained using gp96-FITC bound to the cell surface of the D2SC/1 cell line, where after 15 min of endocytosis, gp96 colocalized with transferrin Texas red (as marker for early endosomes) and endocytosed H2-Kd molecules, but were excluded from lysosomes after 30 min (data not shown).
Figure 5.

gp96-FITC is endocytosed by BMDCs efficiently and colocalizes with endocytosed MHC class I and class II molecules, but does not target to lysosomes. Internalization of gp96-FITC was followed by confocal microscopy. Representative sections are displayed. Coverslip-grown BMDCs were incubated with 50 μg/ml gp96-FITC (shown in false color green) on ice, washed, chased for 15 min or longer at 37°C, and fixed in paraformaldehyde. To follow the fate of gp96-FITC after 15, 30, 45, 60, and 90 min (only 60 min is shown) of endocytosis, cells were fixed and permeabilized with methanol/aceton and stained with antibody to the Lamp-1 and secondary Alexa™ 546-coupled antibody to visualize lysosomes (shown in false color red). No colocalization of gp96 and Lamp-1 was observed. Furthermore, cells were stained with biotinylated antibodies to MHC class I (H2-Kb) and class II (H2-Ab) and secondary streptavidin-Alexa™ 546 (both shown in red) as well as gp96-FITC (green) on ice, washed, and chased at 37°C for 15 min. After 15 min of endocytosis, nearly all vesicles containing endocytosed gp96 and MHC class I and class II molecules colocalize (shown in yellow as result of overlapping green and red).
gp96-associated Peptides Are Loaded onto MHC Class I Molecules as a Result of Receptor-mediated Endocytosis.
gp96 molecules have been observed to enter APCs by receptor-mediated endocytosis as well as by non–receptor-mediated, nonspecific endocytosis or macropinocytosis 33 44. The latter nonspecific pathways have been described many times before to introduce exogenous proteins into the MHC class I–restricted antigen pathway, but unlike receptor-mediated endocytosis require high concentrations of antigens (for a review, see reference 22).
To investigate whether receptor-mediated endocytosis can lead to cross-presentation of gp96-associated antigens, we have isolated gp96 from RMA-S SigE1B cells that stably express the H2-Db–restricted E1B epitope of Ad5, fused with an ER-targeting signal sequence. C57BL/6 mice immunized with these gp96 molecules generated CTLs that recognized efficiently RMA-S SigE1B and RMA cells pulsed with the Ad5-E1B peptide, but not RMA cells, demonstrating the presence of the Ad5-E1B epitope on gp96 molecules. Immunization with control gp96 molecules from RMA-S cells did not induce Ad5-E1B–specific CTL responses (Fig. 6).
Figure 6.

gp96–E1B complexes generate a CTL response in vivo. gp96 was purified from RMA-S SigE1B and RMA-S cells. 30 μg of gp96 from either cell type was injected into C57BL/6 mice intraperitoneally. The specificity of the generated CTLs was assayed by 51Cr-release of RMA-S SigE1B cells (▴), RMA cells incubated with 100 ng/ml Ad5-E1B peptide (□), or RMA cells (•). The figure shows one representative of three independent experiments.
To test whether the E1B epitope attached to gp96 was re-presented to CTLs after uptake by APCs, gp96 isolated from RMA-S SigE1B (or control gp96 from RMA-S cells) was incubated with the DC cell line D1 for 2 h at 37°C. The D1 cells were further incubated overnight with the Ad5-E1B–specific CTL clones 100B6 and 0.1C2 or control CTL clone LN5, specific for the Ad5-E1A epitope. Intracellular IFN-γ production was measured to determine CTL activation via the re-presentation of the Ad5-E1B peptide. As shown in Fig. 7 A, incubation of 0.1C2 CTLs with D1 cells pulsed with RMA-S SigE1B gp96 resulted in the activation of T cells. This activation was not observed if control gp96 isolated from RMA-S cells was used or if gp96 isolated from RMA-S SigE1B cells was incubated with the CTLs in the absence of D1 cells. The latter experiment clearly demonstrates that D1 cells, which efficiently bind gp96 molecules (shown in Fig. 1 B), are required for the re-presentation. The T cells themselves are not able to bind gp96 (shown in Fig. 2 and Fig. 3), and consequently do not stimulate each other.
Figure 7.

Specific activation of CTLs by DC-mediated cross-presentation of gp96-associated antigen requires receptor-mediated endocytosis of gp96–antigen complexes. (A) Activation of Ad5-E1B–specific CTL clone 0.1C2 was assayed by intracellular IFN-γ staining in flow cytometry. D1 DCs as APCs could activate the CTLs after prior incubation of D1 with gp96–E1B complexes purified from RMA-S SigE1B (top left), but not with irrelevant gp96 isolated from RMA-S (top right) or RMA cells (data not shown) or in the absence of D1 cells (bottom right). Moreover, activation by gp96–E1B complexes could be competed with a twofold excess of gp96 from RMA-S (bottom left) or RMA (not shown), but not with the same excess of BSA (not shown), indicating the presence of a receptor-mediated pathway responsible for processing of gp96 by D1 cells. PE-labeled isotype control antibody was always negative (data not shown). Results are representative for at least three experiments. (B) Summary of the activation of Ad5-E1B–specific CTL clones 0.1C2 and 100B6 as well as control CTL clone LN5 specific for Ad5 E1A. Graph shows the percentage of activated CTLs present in the gate shown in A. Addition of Ad5–E1B peptide to D1 cells resulted in the activation of ∼25% of CTL clones 0.1C2 and 100B6 (data not shown).
Most importanly, however, the activation of Ad5-E1B–specific CTLs by gp96 from RMA-S SigE1B cells could be inhibited by the addition of a twofold excess of irrelevant gp96 molecules from RMA-S and RMA cells. This excess of gp96 was able to reduce the binding of gp96-FITC by 60% (Fig. 1 D), and eliminated the activation of 0.1C2 CTLs by gp96 molecules from RMA-S SigE1B cells almost completely. The identical scenario was observed for a different Ad5-E1B–specific CTL clone, 100B6, but not for LN5 CTLs, which are specific for the control Ad5-E1A CTL epitope (Fig. 7 B). No competition was observed by using a twofold excess of BSA as control (data not shown).
Discussion
HSPs have been shown previously to induce specific immune responses against tumor, minor H, and viral antigens (for reviews, see references 29 and 30). This feature is based on peptides that are associated with HSPs and on the fact that HSPs, by an unknown mechanism, can interact very efficiently with APCs to result in the re-presentation of HSP-associated peptides and subsequent activation of T cells 31 47. We have now shown in this study that specific binding of low amounts of gp96 to a receptor present on mouse and human professional APCs is indeed required for the MHC class I–restricted re-presentation of gp96-associated peptides.
The nature of the gp96 receptor still remains unclear. We reported earlier that gp96 binding to the macrophage line P388D1 cannot be inhibited by mannan, thus arguing against the participation of the mannose receptor. Using DCs from DEC-205−/− mice, we show here that this receptor as well, which displays strong homology to the mannose receptor present on macrophages 43, is unlikely to be involved, because gp96-FITC binding is indistinguishable from that observed for DCs of wild-type mice (Fig. 4 A). Because DnaK and HSP73 molecules have been reported to bind to certain allelic products of MHC class II 48 49, they could represent another potential receptor for HSPs on the surface of APCs. The observation that gp96 binds to all MHC class II–positive cells could indicate that gp96 also uses MHC class II molecules as a receptor. However, anti–MHC class II antibodies were not able to inhibit the binding of gp96-FITC molecules (data not shown), and cells from MHC class II−/− mice showed identical gp96 binding compared with wild-type mice, thus arguing against MHC class II molecules being the receptor for gp96 (Fig. 4 B).
We further demonstrate in this study that the specific interaction of gp96 molecules with DCs results in re-presentation of associated peptides and specific activation of CTLs. gp96 molecules isolated from RMA-S SigE1B cells that carry the Ad5-E1B CTL epitope (Fig. 6) are able to activate Ad5-E1B–specific CTLs after incubation with the DC line D1, as visualized by intracellular IFN-γ staining. The control CTL line LN5 is not activated by any of the gp96 preparations tested (Fig. 7). More importantly, we are able to show here for the first time that receptor-mediated endocytosis of gp96 is indeed required for the re-presentation and subsequent activation of CTLs. By inhibiting the specific binding of RMA-S SigE1B–derived gp96 with a twofold excess of unrelated gp96 molecules that have been shown to reduce gp96-FITC binding by 60% (Fig. 1 D), we completely abolish the activation of Ad5-E1B–specific CTLs (Fig. 5). This low excess of unrelated gp96 was chosen on purpose to exclude potential toxic effects of a high gp96 concentration. Using synthetic E1B peptide, ∼25% of CTLs could be activated (data not shown), compared with 6–7% activated CTLs, as shown in Fig. 7. Therefore, the amount of RMA-S SigE1B gp96 was not able to activate all possible CTLs, most likely because of limiting amounts of peptide. As the activation of CTLs requires the activating signal to be above a certain threshold, the amount of antigen presented by MHC class I molecules in the presence of competitor could easily be below this threshold, explaining the lack of a CTL response with a twofold excess of irrelevant gp96 not associated with E1B peptide.
Because only receptor-mediated endocytosis of labeled gp96 but not nonspecific, non–receptor-mediated uptake such as pinocytosis or macropinocytosis can be inhibited by an excess of unlabeled gp96 33, our results clearly demonstrate that receptor-mediated endocytosis of gp96 molecules is the cellular pathway responsible for re-presentation of gp96-associated peptides by MHC class I molecules. Therefore, our results provide evidence for the hypothesis that professional APCs possess receptors that are able to interact specifically with HSPs 32 and direct HSP-associated peptides into the MHC class I–restricted antigen presentation pathway. This now explains why very small amounts of gp96–peptide complexes can activate T cells.
The exact intracellular pathway for the re-presentation of gp96-associated peptides requires further clarification. Confocal microscopy data point in the direction that gp96 heads for early endosomes but does not enter lysosomes. We could show that gp96 after receptor-mediated uptake enters compartments containing MHC class I and class II molecules. It can be speculated that these compartments function as putative loading compartments where antigen could be transferred to MHC class I and class II molecules 46, but it cannot be excluded that gp96–antigen complexes enter the cytosol specifically, as recently suggested for immunoglobulin–antigen complexes after endocytosis by Fc receptors in DCs 50.
Further identification of the pathway responsible for the re-presentation of gp96-associated peptides will also contribute to the understanding of the phenomenon termed cross-presentation. Until now, cross-presentation of MHC class I–restricted antigens has been shown to be induced by receptor-mediated phagocytosis of apoptotic bodies 27 28, exosomes 51, bacteria 52, and proteins, either denatured or immobilized 26 by phagocytic or nonphagocytic mechanisms 22. Unlike the latter two pathways, which in most require cases high concentrations of the antigens, receptor-mediated endocytosis of HSPs operates efficiently at antigen concentrations of ∼1–2 ng per mouse 31, and might be as efficient as receptor-mediated phagocytosis of apoptotic cells or receptor-mediated endocytosis of proteins by surface immunoglobulins on B cells. One can envisage that HSPs, released from dying cells, bind to HSP receptors of professional APCs and are endocytosed before the associated peptides are re-presented by MHC class I molecules.
The antigen carriers in apoptotic cells or exosomes are unknown, but one interesting possibility is that HSPs chaperone the antigenic peptides, thus protecting them from further degradation and directing them to the correct intracellular loading compartment. In line with this speculation is the observation that HSP70 is one of the proteins found in close association with the transferrin receptor in exosomes derived from reticulocytes 53. Whether or not the induction of apoptosis leads to a general increase of HSPs is still controversial and might depend on factors that are still to be determined. For tumor cells, it was reported that apoptotic death was associated with low HSP expression levels 54, whereas for PMNs, increased apoptosis coincided with induction of Hsp72 55. Nevertheless, an increase of HSP expression levels generally seems to correlate with increased immunogenicity 54 56, supporting the above-mentioned hypothesis.
The finding that cells deficient in TAP are still able to cross-prime as efficiently as wild-type cells 57 does not contradict the involvement of HSPs in cross-presentation. It shows that the ER-resident HSP gp96 alone is not essential for cross-priming, but it also does not exclude the participation of other HSPs such as HSP70 or HSP90 that might compensate for the absence of immunogenic gp96–peptide complexes. Another argument formulated against the participation of HSPs in cross-presentation of cellular antigens is based on an experiment performed by Carbone and Bevan 58, in which splenocytes were incubated with OVA or β-galactosidase, washed, and injected into mice. Because of the nonspecific coating of cells with the soluble proteins, an association with HSPs might be difficult to imagine. However, the incubation conditions (37°C, 10 mg/ml protein, 10 min), do not exclude the uptake and processing of proteins and the subsequent loading of antigenic peptides onto HSPs. In addition, several different pathways for cross-presentation, including apoptotic cells, exosomes, and receptor-mediated endocytosis of HSPs, might exist in parallel, each one able to induce the cross-presentation of different types of antigens.
More detailed knowledge about the gp96 receptor, its intracellular transport, and the regulation of expression in different cell types will deepen our understanding of the role of gp96 and possibly HSPs in general in cross-presentation, and could greatly improve the application of gp96 for the induction of specific immune responses in vivo.
Acknowledgments
We would like to thank Sylvia Bayertz and Ellen van der Voort for excellent technical assistance, and Hans-Ulrich Scherer, Cécile Gouttefangeas, Arnaud Moris, Volker Teichgräber, Mar Fernandez-Borja, Lauran Oomen, Eric Nooteboom, Danita Schuurhuis, Roland Brock, and Mirjam Schmid for help with various experiments. We thank Hans Bisswanger for discussion of binding kinetics, Lynne Yakes for reading the manuscript, and Ralph Steinman for discussion and for carefully reviewing the manuscript.
This work was supported by grants of the Deutsche Forschungsgemeinschaft (Leibnizprogram Ra369/4-1 to H.G. Rammensee, Sonderforschungsbereich 510, C1 to H. Schild) and the European Union (Biomed 95-1627). The research of R.E.M. Toes has been made possible by a fellowship of the Royal Netherlands Academy of Arts and Sciences.
Footnotes
Abbreviations used in this paper: Ad5, adenovirus type 5; BMDC, bone marrow–derived dendritic cell; DC, dendritic cell; ER, endoplasmic reticulum; gp, glycoprotein; HSP, heat shock protein; TAP, transporter associated with antigen processing.
H. Singh-Jasuja, R.E.M. Toes, and P. Spee contributed equally to this paper.
References
- Bevan M.J. Priming for a cytotoxic response to minor histocompatibility antigensantigen specificity and failure to demonstrate a carrier effect. J. Immunol. 1977;118:1370–1374. [PubMed] [Google Scholar]
- Bevan M.J. Minor H antigens introduced on H-2 different stimulating cells cross-react at the cytotoxic T cell level during in vivo priming. J. Immunol. 1976;117:2233–2238. [PubMed] [Google Scholar]
- Bevan M.J. Cross-priming for a secondary cytotoxic response to minor H antigens with H-2 congenic cells which do not cross-react in the cytotoxic assay. J. Exp. Med. 1976;143:1283–1288. doi: 10.1084/jem.143.5.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staerz U.D., Karasuyama H., Garner A.M. Cytotoxic T lymphocytes against a soluble protein. Nature. 1987;329:449–451. doi: 10.1038/329449a0. [DOI] [PubMed] [Google Scholar]
- Rock K.L., Gamble S., Rothstein L. Presentation of exogenous antigen with class I major histocompatibility complex molecules. Science. 1990;249:918–921. doi: 10.1126/science.2392683. [DOI] [PubMed] [Google Scholar]
- Kurts C., Heath W.R., Carbone F.R., Allison J., Miller J.F., Kosaka H. Constitutive class I–restricted exogenous presentation of self-antigens in vivo. J. Exp. Med. 1996;184:923–930. doi: 10.1084/jem.184.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toes R.E., Blom R.J., van der Voort E., Offringa R., Melief C.J., Kast W.M. Protective antitumor immunity induced by immunization with completely allogeneic tumor cells. Cancer Res. 1996;56:3782–3787. [PubMed] [Google Scholar]
- Huang A.Y., Golumbek P., Ahmadzadeh M., Jaffee E., Pardoll D., Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- Corr M., Lee D.J., Carson D.A., Tighe H. Gene vaccination with naked plasmid DNAmechanism of CTL priming. J. Exp. Med. 1996;184:1555–1560. doi: 10.1084/jem.184.4.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler A.J., Marsh D.W., Yochum G.S., Guzzo J.L., Nigam A., Nelson W.G., Pardoll D.M. CD4+ T cell tolerance to parenchymal self-antigens requires presentation by bone marrow–derived antigen-presenting cells. J. Exp. Med. 1998;187:1555–1564. doi: 10.1084/jem.187.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C., Kosaka H., Carbone F.R., Miller J.F., Heath W.R. Class I–restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 1997;186:239–245. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C., Carbone F.R., Barnden M., Blanas E., Allison J., Heath W.R., Miller J.F. CD4+ T cell help impairs CD8+ T cell deletion induced by cross-presentation of self-antigens and favors autoimmunity. J. Exp. Med. 1997;186:2057–2062. doi: 10.1084/jem.186.12.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert M.L., Sauter B., Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- Kovacsovics-Bankowski M., Clark K., Benacerraf B., Rock K.L. Efficient major histocompatibility complex class I presentation of exogenous antigen upon phagocytosis by macrophages. Proc. Natl. Acad. Sci. USA. 1993;90:4942–4946. doi: 10.1073/pnas.90.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debrick J.E., Campbell P.A., Staerz U.D. Macrophages as accessory cells for class I MHC-restricted immune responses. J. Immunol. 1991;147:2846–2851. [PubMed] [Google Scholar]
- Norbury C.C., Hewlett L.J., Prescott A.R., Shastri N., Watts C. Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity. 1995;3:783–791. doi: 10.1016/1074-7613(95)90067-5. [DOI] [PubMed] [Google Scholar]
- Ke Y., Kapp J.A. Exogenous antigens gain access to the major histocompatibility complex class I processing pathway in B cells by receptor-mediated uptake. J. Exp. Med. 1996;184:1179–1184. doi: 10.1084/jem.184.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991;9:271–296. doi: 10.1146/annurev.iy.09.040191.001415. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C., Stahl P.D., Austyn J.M. Phagocytosis of antigens by Langerhans cells in vitro. J. Exp. Med. 1993;178:509–519. doi: 10.1084/jem.178.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F., Cella M., Danieli C., Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartmentdownregulation by cytokines and bacterial products. J. Exp. Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock K.L. A new foreign policyMHC class I molecules monitor the outside world. Immunol. Today. 1996;17:131–137. doi: 10.1016/0167-5699(96)80605-0. [DOI] [PubMed] [Google Scholar]
- Jondal M., Schirmbeck R., Reimann J. MHC class I-restricted CTL responses to exogenous antigens. Immunity. 1996;5:295–302. doi: 10.1016/s1074-7613(00)80255-1. [DOI] [PubMed] [Google Scholar]
- Heath W.R., Carbone F.R. Cytotoxic T lymphocyte activation by cross-priming. Curr. Opin. Immunol. 1999;11:314–318. doi: 10.1016/s0952-7915(99)80050-8. [DOI] [PubMed] [Google Scholar]
- Norbury C.C., Chambers B.J., Prescott A.R., Ljunggren H.G., Watts C. Constitutive macropinocytosis allows TAP-dependent major histocompatibility complex class I presentation of exogenous soluble antigen by bone marrow-derived dendritic cells. Eur. J. Immunol. 1997;27:280–288. doi: 10.1002/eji.1830270141. [DOI] [PubMed] [Google Scholar]
- Kovacsovics-Bankowski M., Rock K.L. Presentation of exogenous antigens by macrophagesanalysis of major histocompatibility complex class I and II presentation and regulation by cytokines. Eur. J. Immunol. 1994;24:2421–2428. doi: 10.1002/eji.1830241024. [DOI] [PubMed] [Google Scholar]
- Bachmann M.F., Lutz M.B., Layton G.T., Harris S.J., Fehr T., Rescigno M., Ricciardi-Castagnoli P. Dendritic cells process exogenous viral proteins and virus-like particles for class I presentation to CD8+ cytotoxic T lymphocytes. Eur. J. Immunol. 1996;26:2595–2600. doi: 10.1002/eji.1830261109. [DOI] [PubMed] [Google Scholar]
- Rovere P., Vallinoto C., Bondanza A., Crosti M.C., Rescigno M., Ricciardi-Castagnoli P., Rugarli C., Manfredi A.A. Bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J. Immunol. 1998;161:4467–4471. [PubMed] [Google Scholar]
- Albert M.L., Pearce S.F., Francisco L.M., Sauter B., Roy P., Silverstein R.L., Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998;188:1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P.K., Menoret A., Basu S., Binder R.J., McQuade K.L. Heat shock proteins come of ageprimitive functions acquire new roles in an adaptive world. Immunity. 1998;8:657–665. doi: 10.1016/s1074-7613(00)80570-1. [DOI] [PubMed] [Google Scholar]
- Schild H., Arnold-Schild D., Lammert E., Rammensee H.G. Stress proteins and immunity mediated by cytotoxic T lymphocytes. Curr. Opin. Immunol. 1999;11:109–113. doi: 10.1016/s0952-7915(99)80019-3. [DOI] [PubMed] [Google Scholar]
- Blachere N.E., Li Z., Chandawarkar R.Y., Suto R., Jaikaria N.S., Basu S., Udono H., Srivastava P.K. Heat shock protein–peptide complexes, reconstituted in vitro, elicit peptide-specific cytotoxic T lymphocyte response and tumor immunity. J. Exp. Med. 1997;186:1315–1322. doi: 10.1084/jem.186.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P.K., Udono H., Blachere N.E., Li Z. Heat shock proteins transfer peptides during antigen processing and CTL priming. Immunogenetics. 1994;39:93–98. doi: 10.1007/BF00188611. [DOI] [PubMed] [Google Scholar]
- Arnold-Schild D., Hanau D., Spehner D., Schmid C., Rammensee H.G., de la Salle H., Schild H. Cutting edgereceptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J. Immunol. 1999;162:3757–3760. [PubMed] [Google Scholar]
- Grusby M.J., Johnson R.S., Papaioannou V.E., Glimcher L.H. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science. 1991;253:1417–1420. doi: 10.1126/science.1910207. [DOI] [PubMed] [Google Scholar]
- Winzler C., Rovere P., Rescigno M., Granucci F., Penna G., Adorini L., Zimmermann V.S., Davoust J., Ricciardi-Castagnoli P. Maturation stages of mouse dendritic cells in growth factor–dependent long-term cultures. J. Exp. Med. 1997;185:317–328. doi: 10.1084/jem.185.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toes R.E., Offringa R., Blom R.J., Brandt R.M., van der Eb A.J., Melief C.J., Kast W.M. An adenovirus type 5 early region 1B-encoded CTL epitope-mediating tumor eradication by CTL clones is down-modulated by an activated ras oncogene. J. Immunol. 1995;154:3396–3405. [PubMed] [Google Scholar]
- Arnold D., Faath S., Rammensee H., Schild H. Cross-priming of minor histocompatibility antigen-specific cytotoxic T cells upon immunization with the heat shock protein gp96. J. Exp. Med. 1995;182:885–889. doi: 10.1084/jem.182.3.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R.M. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A., Sapp M., Schuler G., Steinman R.M., Bhardwaj N. Improved methods for the generation of dendritic cells from nonproliferating progenitors in human blood. J. Immunol. Methods. 1996;196:121–135. doi: 10.1016/0022-1759(96)00079-8. [DOI] [PubMed] [Google Scholar]
- Kast W.M., Offringa R., Peters P.J., Voordouw A.C., Meloen R.H., van der Eb A.J., Melief C.J. Eradication of adenovirus E1-induced tumors by E1A-specific cytotoxic T lymphocytes. Cell. 1989;59:603–614. doi: 10.1016/0092-8674(89)90006-8. [DOI] [PubMed] [Google Scholar]
- Welch W.J., Garrels J.I., Thomas G.P., Lin J.J., Feramisco J.R. Biochemical characterization of the mammalian stress proteins and identification of two stress proteins as glucose- and Ca2+-ionophore-regulated proteins. J. Biol. Chem. 1983;258:7102–7111. [PubMed] [Google Scholar]
- Wearsch P.A., Nicchitta C.V. Endoplasmic reticulum chaperone GRP94 subunit assembly is regulated through a defined oligomerization domain. Biochemistry. 1996;35:16760–16769. doi: 10.1021/bi962068q. [DOI] [PubMed] [Google Scholar]
- Swiggard W.J., Mirza A., Nussenzweig M.C., Steinman R.M. DEC-205, a 205-kDa protein abundant on mouse dendritic cells and thymic epithelium that is detected by the monoclonal antibody NLDC-145purification, characterization, and N-terminal amino acid sequence. Cell. Immunol. 1995;165:302–311. doi: 10.1006/cimm.1995.1218. [DOI] [PubMed] [Google Scholar]
- Wassenberg J.J., Dezfulian C., Nicchitta C.V. Receptor mediated and fluid phase pathways for internalization of the ER Hsp90 chaperone GRP94 in murine macrophages. J. Cell. Sci. 1999;112:2167–2175. doi: 10.1242/jcs.112.13.2167. [DOI] [PubMed] [Google Scholar]
- Carlsson S.R., Roth J., Piller F., Fukuda M. Isolation and characterization of human lysosomal membrane glycoproteins, h-lamp-1 and h-lamp-2. Major sialoglycoproteins carrying polylactosaminoglycan. J. Biol. Chem. 1988;263:18911–18919. [PubMed] [Google Scholar]
- Gromme M., Uytdehaag F.G., Janssen H., Calafat J., van Binnendijk R.S., Kenter M.J., Tulp A., Verwoerd D., Neefjes J. Recycling MHC class I molecules and endosomal peptide loading. Proc. Natl. Acad. Sci. USA. 1999;96:10326–10331. doi: 10.1073/pnas.96.18.10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto R., Srivastava P.K. A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides. Science. 1995;269:1585–1588. doi: 10.1126/science.7545313. [DOI] [PubMed] [Google Scholar]
- Auger I., Roudier J. A function for the QKRAA amino acid motifmediating binding of DnaJ to DnaK. Implications for the association of rheumatoid arthritis with HLA-DR4. J. Clin. Invest. 1997;99:1818–1822. doi: 10.1172/JCI119348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich T., Gruneberg U., Trowsdale J. Heat shock proteins, HLA-DR and rheumatoid arthritis. Nat. Med. 1998;4:1210–1211. doi: 10.1038/3172. [DOI] [PubMed] [Google Scholar]
- Rodriguez A., Regnault A., Kleijmeer M., Ricciardi-Castagnoli P., Amigorena S. Selective transport of internalized antigens to the cytosol for MHC class I presentation in dendritic cells. Nat. Cell. Biol. 1999;1:362–368. doi: 10.1038/14058. [DOI] [PubMed] [Google Scholar]
- Zitvogel L., Regnault A., Lozier A., Wolfers J., Flament C., Tenza D., Ricciardi-Castagnoli P., Raposo G., Amigorena S. Eradication of established murine tumors using a novel cell-free vaccinedendritic cell-derived exosomes. Nat. Med. 1998;4:594–600. doi: 10.1038/nm0598-594. [DOI] [PubMed] [Google Scholar]
- Rescigno M., Citterio S., Thery C., Rittig M., Medaglini D., Pozzi G., Amigorena S., Ricciardi-Castagnoli P. Bacteria-induced neo-biosynthesis, stabilization, and surface expression of functional class I molecules in mouse dendritic cells. Proc. Natl. Acad. Sci. USA. 1998;95:5229–5234. doi: 10.1073/pnas.95.9.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew A., Bell A., Johnstone R.M. Hsp-70 is closely associated with the transferrin receptor in exosomes from maturing reticulocytes. Biochem. J. 1995;308:823–830. doi: 10.1042/bj3080823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher A., Todryk A., Hardwick N., Ford M., Jacobson M., Vile R.G. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat. Med. 1998;4:581–587. doi: 10.1038/nm0598-581. [DOI] [PubMed] [Google Scholar]
- Hennigan S.M., Wang J.H., Redmond H.P., Bouchier-Hayes D. Neutrophil heat shock protein expression and activation correlate with increased apoptosis following transmigration through the endothelial barrier. Shock. 1999;12:32–38. doi: 10.1097/00024382-199907000-00005. [DOI] [PubMed] [Google Scholar]
- Menoret A., Patry Y., Burg C., Le Pendu J. Co-segregation of tumor immunogenicity with expression of inducible but not constitutive hsp70 in rat colon carcinomas. J. Immunol. 1995;155:740–747. [PubMed] [Google Scholar]
- Schoenberger S.P., van der Voort E.I., Krietemeijer G.M., Offringa R., Melief C.J., Toes R.E. Cross-priming of CTL responses in vivo does not require antigenic peptides in the endoplasmic reticulum of immunizing cells. J. Immunol. 1998;161:3808–3812. [PubMed] [Google Scholar]
- Carbone F.R., Bevan M.J. Class I–restricted processing and presentation of exogenous cell–associated antigen in vivo. J. Exp. Med. 1990;171:377–387. doi: 10.1084/jem.171.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]