

Abstract
Insertions in HIV-1 reverse transcriptase’s fingers subdomain can enhance chain terminator excision and confer resistance to multiple nucleoside analogs. Inserts that resemble flanking sequences likely arise by local sequence duplication. However, a remarkable variety of non-repeat fingers insertions have been observed. Here, molecular epidemiology, sequence analyses and mechanistic modeling were employed to show that one Japanese isolate’s RT fingers insert likely resulted from non-homologous recombination between virus and host sequences and the transductive copying of 37 nucleotides from human chromosome 17. These findings provide evidence that human sequence transduction can, at least rarely, contribute to genetic and phenotypic variation in pandemic HIV.
Keywords: Genetic recombination, Retroviruses, Reverse transcriptase, Drug resistance
Introduction
Much of HIV-1’s genetic variation arises by stepwise – albeit at times hypermutation-accelerated – accumulation of point mutations (Harris et al., 2003; Leitner and Albert, 1999). Genetic recombination also contributes to HIV genetic diversity and occurs about 10-fold more frequently than base substitution (An and Telesnitsky, 2002; Jetzt et al., 2000). Because fewer molecular events are required to introduce panels of mutations by recombination than by serial mutation, clustered genome changes are generally believed to reflect recombination or related rearrangements (Malim and Emerman, 2001; Wain-Hobson et al., 2003). Retroviral recombination results from template switching during reverse transcription and generally occurs in regions of high sequence similarity between the two intact genomes each retrovirus co-packages (An and Telesnitsky, 2002). Non-homologous recombination, often guided by microhomology between donor and acceptor templates, can also occur (Hajjar and Linial, 1993; Zhang and Temin, 1993). Deletions and insertions can arise via non-homologous recombination between discontinuous portions of the viral genome (Parthasarathi et al., 1995; Temin, 1993). Duplications result if template switch occurs from one RNA position to a locus further downstream on the co-packaged RNA, while deletions arise when reverse transcriptase “jumping” bypasses sequences and terminates upstream of the point of template departure (Parthasarathi et al., 1995). Insertion-in-a-deletion or insertion-in-a-duplication mutations can result from a series of non-homologous crossovers (Lobato et al., 2002; Parthasarathi et al., 1995; Pathak and Temin, 1990). Either virus or host sequences can template insertions, as postulated by models for oncogene transduction (Muriaux and Rein, 2003). Although whole gene transduction is rare, incorporation of short host segments into defective viral genomes is observed fairly frequently (Dunn et al., 1992; Fang and Pincus, 1995; Hajjar and Linial, 1993; Mikkelsen and Pedersen, 2000; Pulsinelli and Temin, 1991; Sun et al., 2001). Thus, experimentally, the use of host sequences to bridge non-homologous recombination junctions and the insertion of host segments at strong stop or non-homologous crossover sites is a well-established phenomenon. Possible contributions of host sequences to HIV clinical isolates has been less clear: both because pre- and post-mutation sequence data are seldom available and because inserts maintained in replication-competent virus are generally too short to implicate a specific template (Winters and Merigan, 2005).
Results
In this report, we examined an unusually lengthy insertion mutation in an HIV-1 isolate from a Japanese child, with the goal of elucidating its likely origins (Sato et al., 2001). Epidemiologic investigation provided sequence information for the viral populations observed in the patient, referred to as NH3, during the 6 years prior to and at the onset of clinical drug resistance. These included the multi-drug-resistant isolate itself (designated 99JP-NH3-II) and several co-circulating insertion region variants (Fig. 1A). Additional sequence information was available for isolates from NH3’s father, NH1, who had been infected in Thailand, and from his mother, NH2, who had contracted HIV-1 from NH1 and subsequently infected NH3 through maternal transmission. This clinical history confirmed that the insertion arose during highly active antiretroviral treatment in NH3 (Sato et al., 2001). A comparison of virus with and without the insert demonstrated that the 11-amino-acid insertion in the reverse transcriptase of this CRF001_AE circulating recombinant form variant contributed to its high-level resistance to multiple nucleoside analogs.
Fig. 1.

Sequence analyses of the 99JP-NH3-II insertion region. (A) Deduced preinsertion sequence, sequence insertion in 99JP-NH3-II, and insertion region variants. The NH3 isolates shown are GenBank accession nos. AB053002, AB053074, AB053071, AB053005, AB053038, AB053073, AB053065, AB053035, AB053084, AB053003, AB053077, AB053068, AB053058, and AB053083. Numbers at top represent amino acid residue numbers within reverse transcriptase. Line under 99JP-NH3-II is region of greatest human match. Shaded sequences are positions that differ from 99JP-NH3-II. (B) Comparisons of the 99JP-NH3-II insertion region to other sequences in GenBank. Underlined sequences indicate greatest match to the 51-base insertion region of 99JP-NH3-II. At the top (‘All sequences’): blastn results from nr GenBank. The human match encompasses an intron/exon junction in AC135178 (Chromosome 17 open reading frame 68; also found in NC_00017); the orangutan match lies within CR857769; Bdellovibrio: BX842654, Drosophila: AY183918. Expect values at left are those assigned by blastn, as described in Materials and methods. Note that although human and chimpanzee sequences are highly related in the 3′ regions of human chromosome 17 orf 68, the orf 68 5′ region differs markedly between human and chimpanzee, and thus, no match between the 99JP-NH3-II insert and GenBank chimpanzee sequences is detectable. At the bottom of panel B: blastn results from nr GenBank limited to viruses, after manual removal of NH3 sequences. Accession number for the circovirus: AY321985; for herpesvirus: AY509253; for Kenyan HIV-1: AF457073.
The region encompassing the 33 nucleotides inserted in NH3’s RT gene was evaluated for similarity to experimentally described retroviral recombination products. The insertion did not mirror flanking sequences and thus was not a simple duplication (Parthasarathi et al., 1995). The entire sequence of the 99JP-NH3-II genome was available and none of it resembled the 33-nucleotide insert. Thus, sequential template switches among viral segments could not account for NH3’s RT insert.
Support for possible non-viral origins of the 33-base insertion came from its striking nucleotide composition. Whereas typical HIV sequences have a <40% G + C content (Berkhout et al., 2002), this insert was 67% G + C. Because abnormal G + C content is a hallmark of horizontal gene transfer (Hacker and Kaper, 2000), we explored possible human origins for the insert. If microhomology guided template switch had contributed to insert generation, viral sequences flanking the insert would be predicted to retain human sequence homology. To accommodate this, a 51-base segment comprised of the 33-base insert plus nine nucleotides from either flank was used in the database searches summarized in Fig. 1B.
When this 51-base segment was used to query unrestricted databases in GenBank, the best match was to 99JP-NH3-II itself, followed closely by the insert variants isolated from NH3 (Fig. 1A). The similarity of these NH3 isolates to one another and the differences between their inserts and all other HIV sequences (see below) makes it reasonable to assume that a single insertion event gave rise to all insert variants in patient NH3: an interpretation also supported by our previous studies on viral molecular evolution in NH3 (Sato et al., 2001).
Excluding NH3’s HIV strains, the closest GenBank match to this 51-base sequence was a 30/31-base match to a non-repetitive sequence on human chromosome 17 (Fig. 1B). This was followed by a 27/28 match to the orthologous locus in orangutan, with the remaining top matches including a 20/20 match to the genome of the predatory bacterium Bdellovibrio, followed by less extensive matches to sequences from Drosophila, mouse, dog, and rice, but notably not from any non-NH3 HIV-1 isolates.
To assess similarity of the 99JP-NH3-II insert to sequences in other HIV isolates, the 51-base segment was subsequently BLASTed against all virus entries in GenBank (‘Virus sequences’ in Fig. 1B). Besides 99JP-NH3-II and the co-circulating isolates from NH3, no viral sequences in GenBank yielded expect values of <1. Because expect values are measures of probability roughly equivalent to P values, the absence of <1 matches in any isolate of any type of virus suggests that the insert in 99JP-NH3-II is even less similar to any known virus sequence than would be predicted by random chance. Whereas BLAST had assigned the 30/31 human genome match an expect score of 0.00005, the only HIV match assigned an expect score <10 was a 15/15-base match (expect score 6.6) to a portion of env in a clinical isolate from Kenya (Fig. 1B).
That the virus–human match included a few bases downstream of the insertion was consistent with the possibility that microhomology guided recombination between HIV-1 and the BLAST-identified human sequence generated this insertion. These observations and the mutation’s structure suggested that the NH3 insertion was generated via the splinted non-homologous recombination model shown in Fig. 2. Briefly, an HIV-1 provirus was established on chromosome 17 just upstream of the putative insert-encoding sequences (Fig. 2A). Viral polyadenylation signal read-through generated a chimeric HIV-human RNA that became encapsidated. During subsequent reverse transcription, microhomology-guided template switching between portions of the RT gene and human sequences on the read-through RNA generated the observed insertion-in-a-duplication structure (Fig. 2B).
Fig. 2.
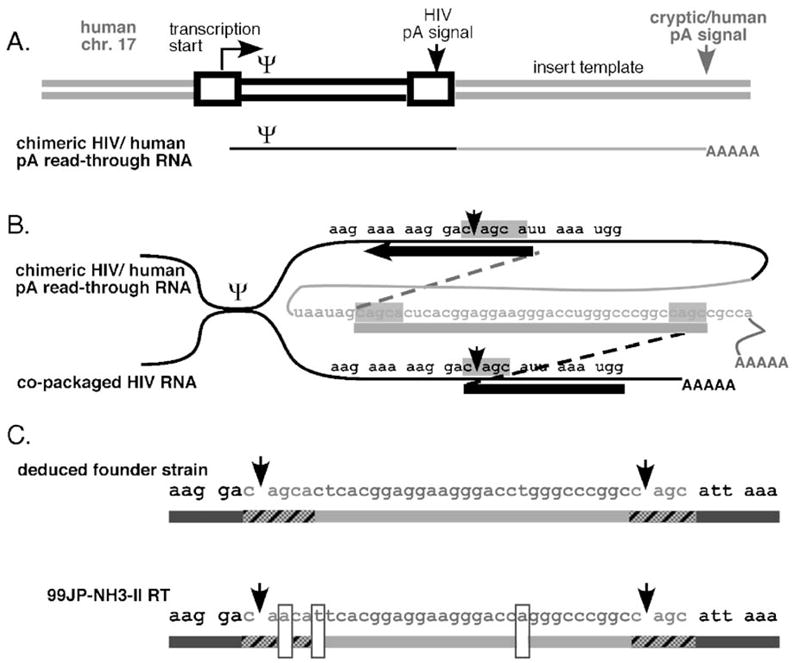
Splinted recombination model for human sequence insertion. (A) Proviral insertion and read-through RNA. Not to scale. Boxes indicate HIV-1 long terminal repeats; Ψ indicates RNA packaging/dimerization signal. Thick lines are DNA; thin lines are RNA. Black lines are viral sequences; shaded lines are human sequences. Arrows indicate direction of transcription and positions of polyadenylation signals. (B) Template switches postulated to generate 99JP-NH3-II. Shown are two co-packaged RNAs joined at Ψ, and DNA products of template switching (thick and dotted lines: shaded boxes indicate regions of microhomology between viral and human sequences). Note that experimental studies have demonstrated that polyadenylation read-through occurs during the synthesis of about 10% of all HIV RNAs and that even unlinked host sequences can become incorporated in proviruses (Olsen et al., 1990; Sun et al., 2001; An and Telesnitsky, 2004 and references therein). (C) Genetic differences between 99JP-NH3-II and the predicted founder strain. Arrowheads indicate borders of the 33 base insert; hatched sequences were templated by microhomology, boxed residues are sites of mutations evoked to generate 99JP-NH3-II from the deduced founder strain.
This model is based on experimental outcomes of non-homologous recombination, models for retroviral transduction, and properties of the putative human bridging template (An and Telesnitsky, 2002; Mikkelsen and Pedersen, 2000; Muriaux and Rein, 2003). Specifically, the putative human template straddles an intron/exon junction that lies in the antisense orientation of a mapped and verified mRNA encoding a putative protein of unknown function (Gao et al., 2005; Ota et al., 2004). Thus, although recombination between viral and unlinked host sequences has been reported previously (Sun et al., 2001), for the unspliced antisense sequences in this case, host sequences were more likely to have become encapsidated on a read-through transcript than as a free RNA (Muriaux and Rein, 2003).
Because it differs at three nucleotide positions from the founder strain predicted by our model (Fig. 2C), the postulated recombination events alone cannot explain the JP-NH3-II RT insertion mutation. However, the spectra of insert variants isolated from NH3 differed from one another at up to 4 positions within the examined sequence interval (Fig. 1A). Thus, these variants’ sequence heterogeneity demonstrates that the extent of viral diversification required to generate JP-NH3-II’s insert from the putative founder strain indisputably did occur within patient NH3 after initial insert acquisition. This supports the notion that JP-NH3-II arose via the mechanism outlined in Figs. 2A and B, followed by the introduction of point mutations at the positions boxed in Fig. 2C.
Discussion
The findings here analyzed a drug resistance-associated sequence insertion that is not closely related to sequences in any other HIV-1 isolate in GenBank. The closest match to this insert among all sequences in GenBank was to a portion of human chromosome 17. The structure of this mutation resembles insertion-within-a-duplication mutations that are well represented among defective retroviral replication products in the experimental literature. This insert and its flanking sequences are so dissimilar from one another that the alternate possibility for insert generation – local sequence duplication followed by mutation – can in large part be ruled out because the number of rare events required to generate the observed structure would far exceed those required by the postulated splinted recombination mechanism. Because RT is not known to polymerize more than a single nucleotide or two without a template, it is likely that all retroviral insertions longer than a couple of bases are synthesized using some form of template (Pathak and Temin, 1990; Preston and Dougherty, 1996). Thus, there is no precedence for de novo generation of a heteropolymeric insert of this length.
The insert examined in this study was located in RT’s β3- β4 hairpin, a region where multiple drug resistance-associated insertions are observed in roughly 1% of the viral strains in treated HIV/AIDS patients world-wide (Winters and Merigan, 2005). Although a simple duplication of flanking sequences is sufficient to enhance drug resistance and may be the most frequent cause of β3- β4 insertions, a wide variety of β3- β4 insert sequences – varying in both length and sequence composition – have been observed (Winters and Merigan, 2005).
Identifying likely human origins of the duplication-flanked insert in 99JP-NH3-II was possible because this insert was unusually lengthy. However, alternate means of searching reveal that Genbank contains additional examples of direct repeat-flanked RT insertions, albeit with shorter sequence regions with uncertain origins (Masquelier et al., 2001; van der Hoek et al., 2005) (see Materials and methods). Although these latter inserts are too short for homology searching to implicate specific sequences, it seems reasonable to postulate that their synthesis involved either virus or host bridging templates.
Not surprisingly, when the human genome is used to query all HIV-1 sequences in GenBank, the few isolates that appear genuine and contain long (>50 nt) human inserts are annotated as replication-defective (for example, GenBank accession no. AY561239; see Materials and methods). Among replication-competent HIV-1 isolates, the inserts with strongest virus–human match belong to the class of mutations called AVT codon-rich env variable region extensions (Kitrinos et al., 2003). The codon bias differences between these inserts and other HIV-1 sequences, as well as the similarity of these inserts to repetitive human micro-satellite sequences, have been described previously (An and Telesnitsky, 2004; Bosch et al., 1994; Kitrinos et al., 2003). However, most HIV AVT-rich extensions identified by BLAST using human query sequences have fewer than 30 contiguous bases of match to the human genome reference sequence. Assessing possible human origins for these AVT-rich inserts is complicated by these inserts’ genetic plasticity, the extent of microsatellite variation within the human population, and the relatively high frequency with which AVT-rich extensions are observed: leaving open the possibility that some arose via recombination between viral isolates (Kitrinos et al., 2003; Zhivotovsky et al., 2003). Nonetheless, observations such as RT β3- β4 inserts that lack genetically linked regions of sequence identity (Masquelier et al., 2001) and the de novo generation of AVT-rich extensions during virus replication in tissue culture (Kuhmann et al., 2004) suggest that on rare occasions additional instances of short patch human sequence transduction, like that reported here for 99JP-NH3-II, may contribute to HIV-1 genetic variation.
Materials and methods
Sequences and sequence analysis
Analyses shown were performed using default settings in nucleotide-nucleotide BLAST (blastn) via NCBI (http://www.ncbi.nlm.nih.gov). All sequences analyzed in this report were previously deposited in GenBank. Searches described as unrestricted databases in GenBank or “nr” were searches of (all GenBank + RefSeq Nucleotides + EMBL + DDBJ + PDB sequences). Expect values presented in Fig. 1B are those assigned by NCBI blastn using default gap and mismatch penalties.
Additional examples of β3- β4 insert splinted recombinants (GenBank accession nos. AY877315 and AF315271) mentioned in the discussion were identified using blastn by querying Genbank with an artificial sequence comprised of NH3’s preinsertion sequence (Fig. 1A) modified to contain a β3- β4 insert of arbitrary length (24 b) while applying reduced mismatch and increased gap penalty values and then by manually screening isolates for strong epidemiologic support and structures predicted by splinted recombination.
Methods and findings from querying all HIV sequences in GenBank with the human genome will be described elsewhere.
Acknowledgments
The authors thank Yuki Naito and James Gergel for their help with the sequence analyses, and David Friedman and Mary Jane Wieland for their useful discussions. This work was supported by NIH no. GM64479 to AT.
References
- An W, Telesnitsky A. HIV-1 genetic recombination: experimental approaches and observations. AIDS Rev. 2002;4:195–212. [PubMed] [Google Scholar]
- An W, Telesnitsky A. Human immunodeficiency virus type 1 transductive recombination can occur frequently and in proportion to polyadenylation signal readthrough. J Virol. 2004;78:3419–3428. doi: 10.1128/JVI.78.7.3419-3428.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B, Grigoriev A, Bakker M, Lukashov VV. Codon and amino acid usage in retroviral genomes is consistent with virus-specific nucleotide pressure. AIDS Res Hum Retroviruses. 2002;18:133–141. doi: 10.1089/08892220252779674. [DOI] [PubMed] [Google Scholar]
- Bosch ML, Andeweg AC, Schipper R, Kenter M. Insertion of N-linked glycosylation sites in the variable regions of the human immunodeficiency virus type 1 surface glycoprotein through AAT triplet reiteration. J Virol. 1994;68:7566–7569. doi: 10.1128/jvi.68.11.7566-7569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn MM, Olsen JC, Swanstrom R. Characterization of unintegrated retroviral DNA with long terminal repeat-associated cell-derived inserts. J Virol. 1992;66:5735–5743. doi: 10.1128/jvi.66.10.5735-5743.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Pincus SH. Unique insertion sequence and pattern of CD4 expression in variants selected with immunotoxins from human immunodeficiency virus type 1-infected T cells. J Virol. 1995;69:75–81. doi: 10.1128/jvi.69.1.75-81.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XS, Cheng J, Zhen Z, Guo J, Zhang LY, Tao ML. Cloning of hepatitis B virus DNAPTP1 transactivating genes by suppression subtractive hybridization technique. Shijie Huaren Xiaohua Zazhi. 2005;13:2371–2374. [Google Scholar]
- Hacker J, Kaper JB. Pathogenicity islands and the evolution of microbes. Annu Rev Microbiol. 2000;54:641–679. doi: 10.1146/annurev.micro.54.1.641. [DOI] [PubMed] [Google Scholar]
- Hajjar AM, Linial ML. A model system for nonhomologous recombination between retroviral and cellular RNA. J Virol. 1993;67:3845–3853. doi: 10.1128/jvi.67.7.3845-3853.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates Innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Jetzt AE, Yu H, Klarmann GJ, Ron Y, Preston BD, Dougherty JP. High rate of recombination throughout the human immunodeficiency virus type 1 genome. J Virol. 2000;74:1234–1240. doi: 10.1128/jvi.74.3.1234-1240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitrinos KM, Hoffman NG, Nelson JA, Swanstrom R. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J Virol. 2003;77:6811–6822. doi: 10.1128/JVI.77.12.6811-6822.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhmann SE, Pugach P, Kunstman KJ, Taylor J, Stanfield RL, Snyder A, Strizki JM, Riley J, Baroudy BM, Wilson IA, Korber BT, Wolinsky SM, Moore JP. Genetic and phenotypic analyses of human immunodeficiency virus type 1 escape from a small-molecule CCR5 inhibitor. J Virol. 2004;78:2790–2807. doi: 10.1128/JVI.78.6.2790-2807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner T, Albert J. The molecular clock of HIV-1 unveiled through analysis of a known transmission history. Proc Natl Acad Sci USA. 1999;96:10752–10757. doi: 10.1073/pnas.96.19.10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato RL, Kim EY, Kagan RM, Merigan TC. Genotypic and phenotypic analysis of a novel 15-base insertion occurring between codons 69 and 70 of HIV type 1 reverse transcriptase. AIDS Res Hum Retroviruses. 2002;18:733–736. doi: 10.1089/088922202760072375. [DOI] [PubMed] [Google Scholar]
- Malim MH, Emerman M. HIV-1 sequence variation: drift, shift, and attenuation. Cell. 2001;104 (4):469–472. doi: 10.1016/s0092-8674(01)00234-3. [DOI] [PubMed] [Google Scholar]
- Masquelier B, Race E, Tamalet C, Descamps D, Izopet J, Buffet-Janvresse C, Ruffault A, Mohammed AS, Cottalorda J, Schmuck A, Calvez V, Dam E, Fleury H, Brun-Vezinet F ANRS AC11 Resistance study group French Agence Nationale de Recherches sur le SIDA. Genotypic and phenotypic resistance patterns of human immunodeficiency virus type 1 variants with insertions or deletions in the reverse transcriptase (RT): multicenter study of patients treated with RT inhibitors. Antimicrob Agents Chemother. 2001;45:1836–1842. doi: 10.1128/AAC.45.6.1836-1842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen JG, Pedersen FS. Genetic reassortment and patch repair by recombination in retroviruses. J Biomed Sci. 2000;7:77–99. doi: 10.1007/BF02256615. [DOI] [PubMed] [Google Scholar]
- Muriaux D, Rein A. Encapsidation and transduction of cellular genes by retroviruses. Front Biosci. 2003;8:135–142. doi: 10.2741/950. [DOI] [PubMed] [Google Scholar]
- Olsen JC, Bova-Hill C, Grandgenett DP, Quinn TP, Manfredi JP, Swanstrom R. Rearrangements in unintegrated retroviral DNA are complex and are the result of multiple genetic determinants. J Virol. 1990;64 (11):5475–5484. doi: 10.1128/jvi.64.11.5475-5484.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- Parthasarathi S, Varela-Echavarria A, Ron Y, Preston BD, Dougherty JP. Genetic rearrangements occurring during a single cycle of murine leukemia virus vector replication: characterization and implications. J Virol. 1995;69:7991–8000. doi: 10.1128/jvi.69.12.7991-8000.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak VK, Temin HM. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc Natl Acad Sci USA. 1990;87:6024–6028. doi: 10.1073/pnas.87.16.6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston BD, Dougherty JP. Mechanisms of retroviral mutation. Trends Microbiol. 1996;4 (1):16–21. doi: 10.1016/0966-842x(96)81500-9. [DOI] [PubMed] [Google Scholar]
- Pulsinelli GA, Temin HM. Characterization of large deletions occurring during a single round of retrovirus vector replication: novel deletion mechanism involving errors in strand transfer. J Virol. 1991;65:4786–4797. doi: 10.1128/jvi.65.9.4786-4797.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Tomita Y, Ebisawa K, Hachiya A, Shibamura K, Shiino T, Yang R, Tatsumi M, Gushi K, Umeyama H, Oka S, Takebe Y, Nagai Y. Augmentation of human immunodeficiency virus type 1 subtype E (CRF01_AE) multiple-drug resistance by insertion of a foreign 11-aminoacid fragment into the reverse transcriptase. J Virol. 2001;75 (12):5604–5613. doi: 10.1128/JVI.75.12.5604-5613.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G, O’Neil PK, Yu H, Ron Y, Preston BD, Dougherty JP. Transduction of cellular sequence by a human immunodeficiency virus type 1-derived vector. J Virol. 2001;75:11902–11906. doi: 10.1128/JVI.75.23.11902-11906.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temin HM. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc Natl Acad Sci USA. 1993;90 (15):6900–6903. doi: 10.1073/pnas.90.15.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoek L, Back N, Jebbink MF, de Ronde A, Bakker M, Jurriaans S, Reiss P, Parkin N, Berkhout B. Increased mulitnucleoside drug resistance and decreased replicative capacity of a human immunodeficiency virus type 1 variant with an 8-amino-acid insert in the reverse transcriptase. J Virol. 2005;79:3536–3543. doi: 10.1128/JVI.79.6.3536-3543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wain-Hobson S, Renoux-Elbe C, Vartanian JP, Meyerhans A. Network analysis of human and simian immunodeficiency virus sequence sets reveals massive recombination resulting in shorter pathways. J Gen Virol. 2003;84:885–895. doi: 10.1099/vir.0.18894-0. [DOI] [PubMed] [Google Scholar]
- Winters MA, Merigan TC. Insertions in the human immunodeficiency virus type 1 protease and reverse transcriptase genes: Clinical impact and molecular mechanisms. Antimicrob Agents Chemother. 2005;49:2575–2582. doi: 10.1128/AAC.49.7.2575-2582.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Temin HM. 3′ junctions of oncogene-virus sequences and the mechanisms for formation of highly oncogenic retroviruses. J Virol. 1993;67 (4):1747–1751. doi: 10.1128/jvi.67.4.1747-1751.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky LS, Rosenberg NA, Feldman MW. Features of evolution and expansion of modern humans, inferred from genomewide microsatellite markers. Am J Hum Genet. 2003;72:1171–1186. doi: 10.1086/375120. [DOI] [PMC free article] [PubMed] [Google Scholar]