

Abstract
Yeast Npl3 is homologous to SR proteins in higher eukaryotes, a family of RNA-binding proteins that play multiple essential roles in RNA metabolism. This protein competes with 3′-end processing factors for binding to the nascent RNA, protecting the transcript from premature termination and coordinating transcription termination and the packaging of the fully processed transcript for export. The NMR structure of its RNA-binding domain shows two unusually compact RNA Recognition Motifs, identifies the RNA recognition surface in Npl3. Biochemical and NMR studies identify a class of G/U-rich RNA sequences with high specificity for this protein. The protein binds to RNA by forming a single globular structure, but the two RRMs of Npl3 are not equivalent, with the second domain forming much stronger interactions with G/U-rich RNA sequences that occur independently of the interaction of the first RRM. The specific binding to G/U-rich RNAs observed for the two RRMs of Npl3 is masked in the full-length protein by a much stronger but non-sequence-specific RNA-binding activity residing outside of its RRMs. The preference of Npl3 for G/U-rich sequences supports the model for its function in regulating recognition of 3′-end processing sites through competition with the Rna15 (yeast analog of human CstF-64 protein) subunit of the processing complex.
INTRODUCTION
Coupling of mRNA processing with transcription is necessary for the production of functional transcripts [1, 2]. Conversely, the formation of mature RNA 3′-ends of eukaryotic mRNA by cleavage and polyadenylation determines where transcription is terminated [3–5]. The formation of a heterogeneous ribonucleoprotein (hnRNP) complex competent for nucleocytoplasmic transport is also coordinated with transcription and 3′-end processing [6–8]. During these events, fully processed mRNAs are directed for export while defective hnRNP assembly or 3′-end formation during transcription results in nuclear retention and degradation of the RNAs [7]. A detailed molecular understanding of how these events (i.e., transcription, mRNP assembly and 3′-end formation) are coordinated with each other and lead to RNA release and export is still lacking.
The Serine/Arginine (SR) proteins are a conserved family in higher eukaryotes that play essential roles both in co-transcriptional alternative splicing of precursor mRNA [9–12], and in RNA export and quality control [8, 13, 14]. A close relative of SR proteins in the yeast Saccharomyces cerevisiae is Npl3. At its C terminus, this protein contains a domain rich in arginine and serine residues (RS), which is essential for its role in RNA transport [6]. We have uncovered yet another similarity of Npl3 to SR proteins in the co-transcriptional processing of mRNA in yeast, more specifically in termination and 3′-end processing. We observed that certain mutations within the putative RNA-binding domain of Npl3 facilitated transcription termination [15]. This effect occurs when mutations in Npl3 cause inefficient binding to the nascent mRNA transcript, allowing the recognition of cryptic poly(A) sites by the poly(A)/termination machinery [15, 16].
According to these recent results, wild-type Npl3 competes with polyadenylation factors for binding to the nascent RNA, perhaps protecting the transcript from premature processing [17]. We sought to understand the interaction between Npl3 and RNA through a study of the structure and function of its RNA-binding domains. Here we report the NMR structure of the two RNA Recognition Motifs of Npl3 and a characterization of its interaction with various RNA oligonucleotides corresponding to yeast 3′-end processing elements. We observe that the two RRMs of Npl3 behave differently when contacting specific G/U-rich RNA sequences compared to other RNAs, but that this specificity is not observed in the full protein because of a stronger but non specific RNA-binding activity encoded by the C-terminus of the protein. A new and unexpected mode of RNA recognition by proteins containing multiple RRM domains is observed as well. The specificity of the RRM domains of the protein for G/U-rich sequences supports a functional role for the competition between Rna15 and Npl3 in the regulation of 3′-end processing and transcription termination. The difference in specificity between full length protein and its two RNA recognition motifs suggests that the specificity of the protein can be regulated by inhibiting the stronger but non-sequence-specific activity residing in the remainder of the protein.
RESULTS
Structural analysis of the RNA binding domain of Npl3
The Npl3 protein consists of 414 amino acids with three distinctive domains. The N-terminus is rich in Proline, Glutamine and Glutamic amino acid residues and is followed by two centrally located putative RRM-type RNA-binding motifs, while the C-terminus is rich in Glycine, Arginine and Serine residues [15, 16, 18]. The two central RRMs constitute the putative RNA-binding domain of Npl3 and were studied here.
The structure of the putative RNA-binding domain of Npl3 containing the two RRMs (residues Glu121 to Arg280 of Npl3) was determined by NMR as described in Materials and Methods. Experimental and structural statistics are summarized in Table 1 and a superimposition of the 20 converged lowest energy structures is displayed in Figure 1. The high quality of the structure is demonstrated by the tight clustering of the superimposed converged structures (shown on the left side of the figure) and by the excellent structural statistics of Table 1.
Table 1.
Experimental restraints and structure calculation statistics
| RRM-1 (127–192) | RRM-2 (201–272) | |
|---|---|---|
| NMR distance restraints | 1545 | 1589 |
| Intra-residue | 301 | 366 |
| Sequential | 435 | 480 |
| Medium-range(<i-i+4) | 301 | 305 |
| Long range (>i-i+4) | 508 | 438 |
| Dihedral angle constraintsa | 102 | 110 |
| Hydrogen bond constraints | 48 | 64 |
| Total number of constraints | 1695 | 1763 |
| Number of constraints per residue | 22.9 | 22.9 |
| Residual constraint violations | ||
| Distance violations > 0.25 Å | 0 | 0 |
| Van-der-Waals violation > 0.30 Å | 0 | 0 |
| Cyana target functionb | 2.03–2.35 | 1.03–1.27 |
| r.m.s.d.c | ||
| Backbone | 0.18±.04Å | 0.59±.20Å |
| All heavy | 1.02±.09Å | 1.49±.25Å |
| Ramachandran valuesd | ||
| Most favored region | 80.3% | 80.0% |
| Additionally allowed region | 19.7% | 20.0% |
| Generously allowed region | 0.0% | 0.0% |
| Disallowed region | 0.0% | 0.0% |
Obtained from Cα, Cβ and C chemical shifts using the program TALOS [59]
Minimum and maximum values for 20 converged structures
Pair-wise among the 20 converged structures
Obtained from PROCHECK-NMR
FIGURE 1.
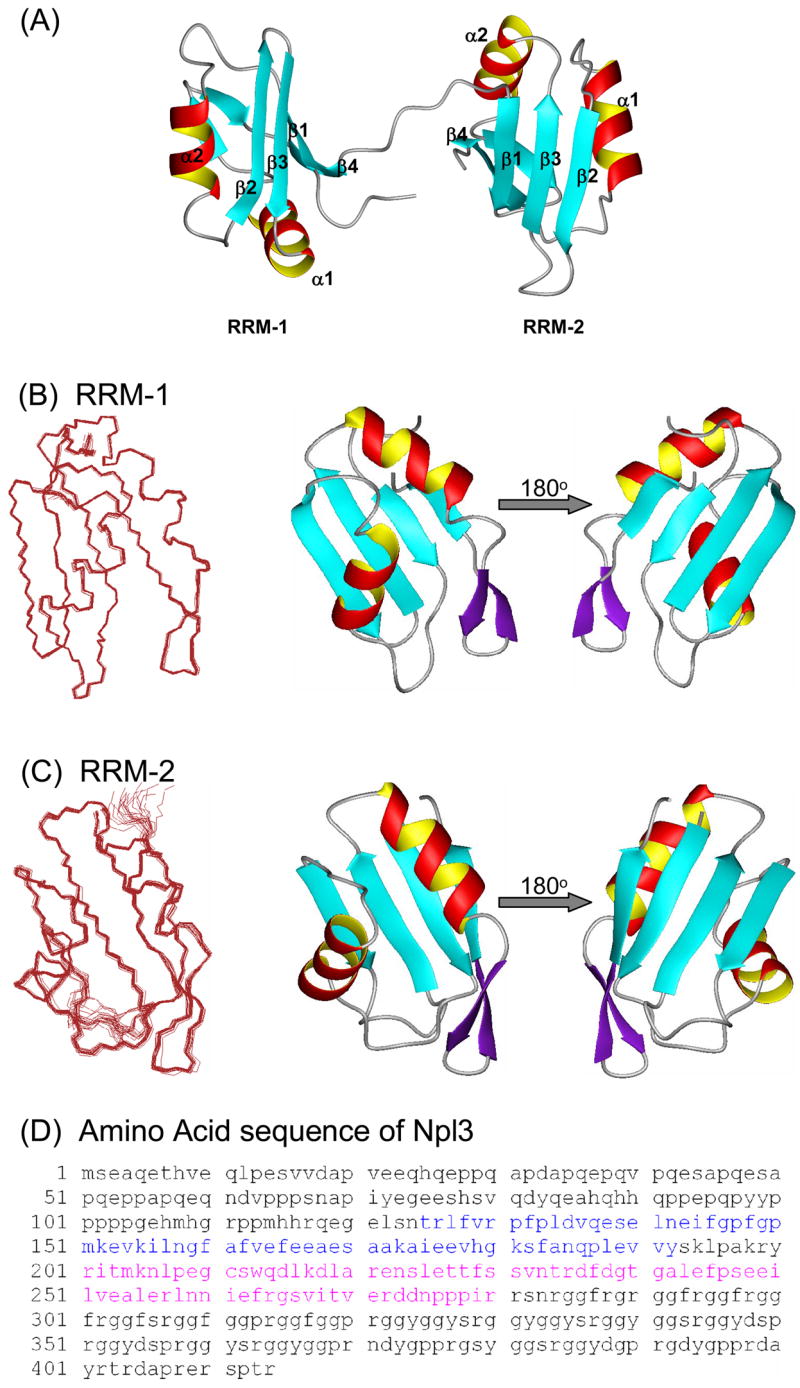
Structure of Npl3. (A) Ribbon diagrams of the two RRM domains of Npl3 as determined by NMR; the two domains are structurally independent and connected by an 8 residue flexible linker. (B) - (C) Overlays of the 20 lowest energy structures on the left, and ribbon representations of the lowest energy structure on the right. Structures for RRM-1 and RRM-2 are shown in panels (B) and (C), respectively; the two domains are shown separately here for clarity of presentation. (D) Amino acid sequence for full length Npl3 protein. Residues 121–280 were the subject of NMR structure determination; residues belonging to RRM-1 and RRM-2 are colored in blue and pink, respectively. Figures were generated using MOLMOL [61].
Most RRMs studied so far are approximately 90 amino acids long, while the Npl3 RRMs are only 70 amino acids. Despite being small, the two Npl3 domains adopt the canonical RNA Recognition Motif fold [19–21] augmented by additional features. RRM-1 extends from residues Leu127 to Tyr192; RRM-2 has roughly the same size, extending from residues Arg201 to Arg272. As often observed with proteins containing multiple RNA-binding domains [22–24], the two RRMs of Npl3 behave as independent rigid bodies in the free form of the protein. Three lines of evidence support this conclusion. First, out of ~3200 NOE restraints used in the calculation of the Npl3 structure, we were unable to find a single NOE that could be unambiguously attributed to inter-domain interactions. Second, the 8-residue linker between the two domains has chemical shift values that are close to those expected for a random coil. Third, relaxation experiments demonstrate that the two domains tumble independently of each another (see below). Accordingly, the two domains are shown as independent structures for clarity of presentation in Figure 1B and 1C. In addition to the canonical βαββαβ RRM core structure [19, 20], a short β-sheet forms in the loop between helix α2 and strand β4 in both RRM domains (Figure 1). Thus, a 2-residue β-hairpin (residues Ser182-Phe183 and Gln186-Pro187) is found in RRM-1, and a short β-hairpin forms in RRM-2 as well (Ile261-Phe263 and Ser266-Ile268).
Superposition of the two RRM domains of Npl3 shows remarkable similarity between them, with backbone RMSD of only 1.3 Å (Figure 2A). This similarity is somewhat surprising since the two RRMs have different sizes for the RNA-binding loops, making the binding surfaces in the two RRM domains in Npl3 quite different. The canonical RNA binding surface in the RRM domain is formed by the β-sheet surface, the loop between β2 and β3, and between β1 and the first α-helix [19, 21, 25]. In RRM-1, the β2- β3 loop consists of only two highly ordered residues, Asn158 and Gly159, and the Asn158 side chain folds back into the core of the protein. In RRM-2, the same loop extends from residue Asn233 to Gly239. Few NOEs are seen in this region of the protein and relaxation measurements show that the T2s in this region are lower than average, confirming the flexible nature of this loop (Figure 3). While three out of the seven residues in this loop are charged, two of the charged residues are Asp (surprisingly for an RNA-binding protein) while a Phe, seldom found in this position amongst RRMs, is also observed in this loop.
FIGURE 2.

Structural comparison of the two RRM domains of Npl3 to each other and to those of SxL protein (PDB: 1B7F). (A) Superposition of the structure of RRM-1 (gray) on RRM-2 (orange) shows that the two domains are very similar to each other, with an RMS deviation between them of only 1.3 Å despite the differences in the loop between β2 and β3. (B) Superposition of the RRM-1 domain of Npl3 (gray) with the RRM-1 domain of SxL (blue) gives an RMS deviation of only 1.1 Å, while (C) superposition of the RRM-2 domain of Npl3 (orange) with the RRM-2 domain of SxL (green) gives an RMS deviation of 1.3 Å. Figures were generated using PYMOL [62].
FIGURE 3.

Relaxation parameters versus residue number for free (red) and RNA-bound (blue) forms of Npl3. Top to bottom: 15N-T1, 15N-T2 and heteronuclear 1H-15N NOE derived from data collected at 500 MHz. Error bars represent uncertainties in the fit of the primary relaxation data to exponential decays, as described in Materials and Methods. Residues for which no result is shown correspond either to Prolines or to overlapped cross-peaks that could not be analyzed quantitatively. The secondary structure of the protein is shown on top of each chart.
The structure of the two RRM domains of Npl3 was submitted to the DALI server (http://www.ebi.ac.uk/dali/) for comparing its structure coordinates against those in the Protein Data Bank. The DALI server identified the RRM domains of Sex-Lethal (SxL) protein (PDB: 1B7F) as the closest match to Npl3, with RMSDs of only 1.11 Å and 1.29 Å for the first and second RRM, respectively (Figure 2B and C). However, unlike the majority of RRMs studied thus far, Npl3 is acidic, with 19% of its residues being negatively charged (the pI of the protein is in fact 4.58). However, the surface electrostatic potential of Npl3 shows that many of the negatively charged residues are confined to the helical face of the protein, while mostly positively charged residues cluster on the canonical RNA-binding surface (Figure 4).
FIGURE 4.

Surface electrostatics of the two RRM domains of Npl3 protein. The figure shows that the β-sheet face of the protein (shown on the left) is somewhat basic or neutral, while the other side of the protein exposing the two α-helices (shown on the right) is largely acidic.
RNA interaction studies reveal a preference of Npl3 for G/U-rich sequences
Next, we examined the interaction of Npl3 with RNA using the 414 residue full-length Npl3 protein (this section) as well as the shorter NMR-construct (in sections that follow). We had previously used primer extension (PE) to determine the locations of UV-induced protein-RNA cross-links with recombinant Npl3 bound to the GAL7 3′-UTR. We reported a preference of Npl3 for regions containing UA- or U/G-rich motifs corresponding to 3′-end processing signals [17]. We selected some of those preferred Npl3 sites and synthesized five 18-mer oligonucleotides corresponding to those that were used in the native gel electrophoretic mobility shift assay (EMSA) with recombinant Npl3 to determine approximate equilibrium dissociation constants (KD) (Table 2). A comparative gel showing binding of Npl3 to all five RNA oligonucleotides is shown in Figure 5A. When we had analyzed the binding activity of Npl3 for the GAL7-1 RNA (295-nucleotide), we observed multiple complexes indicating more than one binding site per RNA [17]. However, with the 18-mer oligonucleotides of defined sequence, we observe single RNA-protein complexes. The KD for Npl3 for the N1 RNA was 3.3 ± 0.8 nM, for the N2 RNA was 0.8 ± 0.1 nM, for the N3 RNA was 3.6 ± 0.4 nM, for the N4 RNA was 6.8 ± 0.2 nM and for the N5 RNA was 4.4 ± 0.3 nM (Figure 5 and Table 2). Thus, full length Npl3 binds strongly to all of these RNAs, and has only slightly (5-fold) higher affinity to the N2 RNA, which is notable for its lack of adenosines and its G/U-rich content (Figure 5B).
TABLE 2.
RNA sequences used in Electrophoretic Mobility Shift Assays and their dissociation constants for Npl3 and for the mutant protein Npl3-120 (Leu225Ser)
| RNA | Sequence | Dissociation constant for Npl3 (nM) | Dissociation constant for Npl3-120 (nM) |
|---|---|---|---|
| N1 | UUU UUU UUA AAU UUU UUU | 3.3 +/− 0.84 | 7.7 +/− 2.88 |
| N2 | UUG CCU GGU UGC CUG GUU | 0.8 +/− 0.08 | 2.9 +/− 0.68 |
| N3 | UAU AUA UAU AUA UAU AUA | 3.6 +/− 0.37 | 3.4 +/− 0.41 |
| N4 | UAA UAA UGA CUA UAU AUG | 6.8 +/− 1.70 | 3.8 +/− 0.20 |
| N5 | UUU CUA UUA AUU UCU AUU | 4.4 +/− 0.33 | 3.0 +/− 1.85 |
FIGURE 5.

RNA binding activity of full length Npl3 for different RNA sequences corresponding to yeast 3′-end processing signals. RNA oligonucleotides N1-N5 (Table 2) were 5′-end labeled and incubated with increasing amounts of recombinant Npl3, as indicated, under equilibrium conditions. Gels of representative mobility shifts assay for Npl3 bound to oligonucleotides N1-N5 are shown (left to right). Npl3 binds to G/U-rich sequences with slightly increased affinity. Additional dilutions of Npl3 were used to determine dissociation constants (KD) for the G/U-rich oligonucleotide N2. Binding constants for the five RNA oligonucleotides derived from the analysis of these data are shown in Table 2.
Characterization of the Npl3-RNA interaction by NMR
Having identified RNA sequences that preferentially associate with Npl3, we used NMR to study the interaction of the protein with RNA oligonucleotides corresponding to those described in the previous section (Table 3). For these NMR binding studies, we used the 2-RRM construct of Npl3 (residue Glu121 to Arg280) as described in the structural analysis section. The first class of RNAs are rich in A and U nucleotides and resemble the binding sites for the polyadenylation factors Rna15 and Hrp1 near the 3′-end processing sites to which Npl3 was also found to cross-link. These RNAs correspond to oligonucleotides N3 and N4 in Table 2. The second class of oligonucleotides is rich in Gs and Us and corresponds to oligonucleotide N2 in Table 2. The length of the shorter RNAs was chosen considering that two-domain RRM proteins typically bind 10–12 nucleotides [21]. Longer RNAs sometimes slide over the surface of the protein leading to reduced spectral quality, despite the increase in apparent affinity due to non-specific binding and avidity effects.
Table 3.
RNA sequences used for NMR binding studies and summary of their binding characteristics
| Name | RNA sequence | Affinitya |
|---|---|---|
| N2 | 5′-UUG CCU GGU UGC CUG GUU-3′ | RRM-1=Weak
RRM-2=Strong |
| N2-truncated | 5′-GCC UGG UUG C-3′ | RRM-1=Weak
RRM-2=Strong |
| AAU | 5′-AAUAUAUAUAUAUAUAUAUAAUAAU-3′ | RRM-1=Weak
RRM-2=Weak |
| AAU-truncated | 5′-AAUAAUAAU-3′ | RRM-1=Weak
RRM-2=Weak |
| AU | 5′-AAUAUAUAUAUAUAUAUAU-3′ | RRM-1=Weak
RRM-2=Weak |
| AU-truncated | 5′-AUAUAUAU-3′ | RRM-1=Weak
RRM-2=Weak |
The annotation on binding affinity is based on the NMR observations reported in the text
Titration of the four AU-rich (sequences 3 to 6 in Table 3) and the two G/U-rich RNAs (sequences 1 and 2 in Table 3) with the 15N-labeled Npl3 double RRM fragment resulted in specific changes in the resonances of residues involved in RNA binding. In fact, the chemical shift perturbation data demonstrate that all the RNAs we tested bind to the same region of the protein, corresponding to the canonical RNA-binding surface of the RRMs. However, a very significant difference was observed between the binding of AU-rich RNAs and G/U-rich RNAs to Npl3. All of the AU-rich oligonucleotides bind to both RRMs of Npl3 in the ‘fast exchange’ NMR timescale. This behavior is typical for weak affinity complexes (in the mM-μM range) and is due to the short residence time of the complex. In this regime, analysis of the chemical shift changes yield titration curves from which quantitative binding constants can be calculated, leading to KDs in the range of approximately 100 μM for the two RRM construct of Npl3 (data not shown). These results also demonstrate that AU, AU-truncated, AAU and AAU-truncated sequences (Table 3) bind to the RNA similarly to each other. The same residues on the surface of Npl3 were affected for all four sequences, with comparable changes in chemical shift (data not shown).
The second set of RNAs used in the EMSA studies was instead G/U-rich and appeared to bind the complete protein slightly more strongly compared to AU-rich RNAs (Table 2). We studied by NMR an 18-mer (N2) and a truncated version of the same RNA (N2-truncated) (Table 3). Both of these RNAs interact with the RRMs similarly to each other, but differ very significantly from the AU-rich RNAs. The interaction of both N2 and N2-truncated RNA with RRM-1 occurs on the fast time scale, as was observed for the AU and AAU RNAs. However, the interaction with RRM-2 occurs with much slower kinetics; peaks disappear during the titration due to the broadening of the NMR lines and reappear when a stoichiometric complex is formed and the line-width sharpens up again. This behavior is observed when the off-rate is in the same timescale as the NMR chemical shift, in this case corresponding to a sub-micromolar KD. Surprisingly, full titration of the protein with the shorter G/U-rich RNA sequence required a 2-fold excess of RNA, suggesting that two molecules of this RNA are required to form a stoichiometric complex (Figure 6).
FIGURE 6.

Changes in the NMR spectra of Npl3 upon RNA binding. (A) Section of the 1H–15N-HSQC spectra of free Npl3 (red) superimposed on a series of spectra of the same domain bound to N2 RNA (Table 2). Red represents the free protein spectrum while blue represents the fully bound protein (1:1 RNA:protein ratio). The peaks in orange and yellow represent spectra with RNA:protein ratio of 0.2:1 and 0.5:1, respectively. While residues Gly159 and Phe162 from RRM-1 are in fast exchange, residues Ser231 and Thr228 from RRM-2 are in intermediate exchange on the NMR timescale. (B) The weighted average chemical shift differences between the free and RNA-bound forms of Npl3 are shown. As expected, most of the residues undergoing changes can be mapped onto the β-sheet surface of the protein; a cartoon representation of the protein secondary structure is shown on top.
Because the NMR signals are relatively sharp when a stoichiometric complex is formed, broadening of the NMR signals can confidently be attributed to exchange between the free and bound forms of the protein and not to residual conformational dynamics in the protein-RNA complex. Furthermore, Figure 3 shows uniform values of T2 throughout the Npl3 protein in complex with RNA (except for the N and C terminal tails), suggesting the absence of any slower timescale motion in the protein-RNA complex that would be indicative of conformational exchange in the complex. The residues in the second RRM that are in intermediate exchange cluster in the β2 or β3 region of the protein, the canonical RNA recognition region in RRM domains (Figure 6).
These results are surprising in several ways, yet absolutely unambiguous. First, binding is significantly weaker than measured for the complete protein, strongly suggesting that other regions of Npl3 contribute to its RNA binding activity. On the other end, the two RRM construct that was studied by NMR binds with more specificity compared to the complete protein, i.e. it exhibits a much larger difference in KD between AU- and G/U-rich RNAs. Second, the two RRMs behave very differently from each other with regards to binding kinetics. If binding occurs with on rates limited by diffusion (we have no reason to think this is not the case), then broadening of residues implies a much stronger interaction (leading to slower off rates) between RRM-2 and the G/U-rich RNA, compared to RRM-1.
The RNA-bound protein forms a single globular structure
The results of the previous paragraph also imply that the interactions of RRM-1 and RRM-2 with the G/U-rich RNAs occur independently of each other. Independent binding could conceivably occur if the domains (that are connected by a flexible linker in the free protein) interacted with RNA independently of each other, leading to distinct binding affinities. In order to test this possibility, we investigated the NMR properties of the RNA-bound protein.
Analysis of NMR chemical shift perturbations of Npl3 binding to RNA demonstrates that several residues in the linker between RRM-1 and RRM-2 exhibit changes in chemical shift upon RNA binding (Figure 6). These changes suggested that the two RRM domains of Npl3, which behaved independently of one another in the free state of the protein, might adopt a fixed relative orientation with respect to one another upon RNA binding. To evaluate this possibility, we examined the NMR relaxation properties of RNA-bound Npl3 and compared them with the RNA-free protein.
The conclusion that the two domains of Npl3 act as independent units in the free protein is clearly evidenced by the NMR relaxation parameters. For the free protein, the average T1 at 500 MHz was 605.6 ms +/− 17 ms while the average T2 was 103.4 ms +/− 2.3 ms. These values are similar to those expected for a 9–10 kDa protein, the size of a single RRM. The average 15N heteronuclear-NOE was 0.88, indicative of a well folded protein, but lower values were observed for the linker between RRM-1 and RRM-2 (Lys194 to Arg199), indicating that the linker between the two RRMs is flexible (Figure 3). A quantitative analysis of the relaxation data was performed using ModelFree [26, 27]. This analysis demonstrates that each of the RRMs has a structure characterized by a rigid backbone on the ps-ns timescale, as reflected by order parameters S2 of between 0.78 and 0.85 for most residues (data not shown) and the absence of any significant conformational exchange. However, an overall correlation time of 7.71 ns was obtained, as would be expected for a protein of about 10 kDa [28, 29]; since the molecular weight of the construct of Npl3 is 17 kDa, these correlation times reflect the independent motion of each of the two RRM domains in the absence of RNA.
Upon binding to the 18mer N2 RNA, many changes in the dynamics of Npl3 protein are observed. The average T1 increases to 801.3 ± 57.1 ms while the average T2 decreases to 56.7 ± 2.5 ms (Figure 3). These values of T1 and T2 are comparable to those determined, for example, for the 23.6 kDa Adenylate Kinase protein [30], which is similar in size to our Npl3 protein-RNA complex (23 kDa). This result demonstrates very clearly that the two RRM domains of Npl3 are locked into a single globular structure upon RNA binding. The heteronuclear NOE values further add to this conclusion. While low values of the heteronuclear NOEs were observed for the linker between the two RRM domains and for the RNA binding loop in RRM-2, consistently uniform values were observed throughout the protein chain (average: 0.81 ± 0.03). These results conclusively demonstrate that the flexible linker between the two RRM domains becomes rigid upon RNA binding.
Our results thus show that the two RRM domains of Npl3 are locked into a single globular structure upon RNA binding. We also found that the G/U-rich RNA binds to RRM-1 with a ~100μM KD, while it binds to RRM-2 with a sub-micromolar KD. These results taken together suggests that each of the RRM domains of Npl3 bind to two stretches of RNA sequences that, although connected in a single phosphodiester chain, interact independently of each other with RRM-1 and RRM-2. This suggestion is strongly supported by the observation that the shorter 10-nucleotide G/U-rich sequence binds to the protein with 2:1 stoichiometry, suggesting that a 10-mer is not long enough to make all the interactions required with both domains simultaneously.
Npl3 mutants display subtle differences in RNA binding in the context of the full protein but much larger defects within the two RRM domains
We previously reported that mutations in or near the RRM-2 of Npl3 can have a profound effect on transcription termination and 3′-end processing [15, 17]. Among the most interesting alleles was the Npl3-120 mutant that differs from the wild type Npl3 by a single point mutation in RRM-2, Leu225 to Serine. Leu225 lies in the loop between α1 and β2 and, by folding into the core of the protein, participates in packing of the core itself (Figure 7B). Consistent with this structural role, a 15N-HSQC spectrum taken on the Npl3-120 protein showed that the second RRM unfolds under the conditions used for NMR spectroscopy (Figure 7C).
FIGURE 7.

Mutations within the RRM-2 of Npl3 alter the protein affinity for the G/U-rich RNAs. (A) EMSAs were performed as described in Figure 5, but the 5′ end-labeled oligonucleotides N1-N5 were incubated with increasing amounts of Npl3-120 to determine dissociation constants (KD). Representative gels are shown for RNA oligonucleotides N1 and N2 respectively; Table 2 shows the KD for the five RNAs studied. (B) The structure of the RRM-2 of Npl3 show that Leu225 participates in the formation of the protein hydrophobic core. (C) NMR data show that Npl3-120 has only one folded RRM domain. The 15N-1H HSQC spectrum of wild type Npl3 (red) is overlayed with that of Npl3-120 (blue). A closer view (on the right) shows that the peaks belonging to RRM-1 do not change in chemical shift upon mutation of L225, while peaks belonging to the RRM-2 lose their chemical shift dispersion in the mutated protein, indicating that the second domain unfolds.
We analyzed recombinant Npl3-120 in EMSAs with the 18-base RNA oligonucleotides (N1-N5) (Figure 7A); apparent equilibrium dissociation constants for Npl3-120 are shown in Table 2. We note that the standard errors for the KD determinations for Npl3-120 show increased variability, possibly due to accumulation of RNA-bound protein in the wells, probably a consequence of partial aggregation due to unfolding of RRM-2 (see below). In order to compare the RNA-bound species from wild-type and mutant, we considered only RNA-protein complexes that had migrated into the gel for KD determination. The apparent dissociation constants for four of the RNAs (N1-KD = 7.7 ± 2.9 nM, N3-KD = 3.4 ± 0.4 nM, N4-KD = 3.8 ± 0.2 nM and N5-KD = 3.0 ± 1.9 nM) are within the range (3–8 nM) of the KD determined for wild-type Npl3 (3–7 nM) (Figure 5). However, we observed a three- to four-fold difference for the N2 RNA with a KD of 2.9 ± 0.7 nM for Npl3-120 compared to a KD of 0.8 ± 0.1 nM for wild-type Npl3. Thus, the Npl3-120 mutant does not affect the non-specific RNA-binding activity of Npl3, but interferes with its ability to recognize G/U-rich sequences.
In addition to the Npl3-120 mutant, we also analyzed two other alleles of Npl3 that were isolated in the same screen, Npl3-004 (Glu244Lys) and Npl3-150 (Gly241Asn) [15]. Residue Gly241 in RRM-2 is a highly conserved Glycine in the RRM signature sequence, while Glu244 is also a conserved residue that is usually Glu/Gln or Asp/Asn in RRMs [21]. The apparent KD for the N2 RNA was 2.5 ± 0.5 nM for Npl3-004 and 2.6 ± 1.1 nM for Npl3-150. Thus, binding to the G/U-rich N2 RNA is consistently reduced for all of these mutations within RRM-2.
DISCUSSION
Transcription elongation and termination are finely balanced and controlled in a variety of ways to ensure that termination does not occur prematurely at cryptic poly(A) sites. One control mechanism is governed by the kinetics of transcriptional elongation and poly(A) complex assembly [31–34]. A second mechanism involves factors that promote transcription termination and RNA processing (e.g. Rna15 and its orthologue CstF-64) competing against those antagonizing termination (e.g. yeast Sub1 and human PC4, as well as Npl3 or Drosophila SxL) [15, 17, 35, 36]. Accordingly, we proposed that Npl3 might function to prevent premature termination by binding to weak or cryptic 3′ processing sequences. Only the high affinity binding of polyadenylation factors to “real” poly(A) sites overcomes the competition, leading to termination of transcription at proper polyadenylation sites. This model is supported by our findings that Npl3 binds to the pre-mRNA in competition with Rna15 [15, 17]. In order to understand how Npl3 functions in balancing transcription termination and anti-termination, we examined the structure of the protein and studied its interaction with RNA sequences representing yeast 3′-end processing sites.
The NMR structure of Npl3 shows two fairly canonical RRM structures, although with about 70 amino acids per domain, these are probably the smallest RRMs studied to date. The RNA-binding experiments also demonstrate that Npl3 binds to single-stranded RNAs using the classical β-sheet recognition surface. Although Npl3 is an acidic protein, most of the negatively charged residues lie on the surface of the protein opposite to the RNA-binding site.
Several observations conclusively demonstrate that the two RRM domains do not interact with each other in the RNA-free form, yet form a compact globular structure in the presence of RNA while the linker region separating the two RRMs becoming ordered as is the rest of the protein. This feature has also been seen with other multidomain RRMs, such as those in the Pab, SxL, HuD and Hrp1 proteins (PDB IDs: 1CVJ, 1FXL, 1B7F and 2CJK, respectively) [24, 37–40]. In each of these structures, the two domains combine to form a single extended RNA-binding platform that is required for the specific recognition of about 10 contiguous nucleotides. Yet the mode of RNA recognition in Npl3 is quite different from the rest of these proteins. There is a clear difference in off rates for the interaction between RRM-1 and RRM-2 and the G/U-rich RNA. This behavior suggests that the RNA binds to Npl3 through two independent recognition sequences connected by a flexible linker of a few nucleotides, and then forms a single globular structure when bound to RNA [24, 41, 42]. This conclusion is strongly supported by the observation that shorter G/U-rich RNA oligonucleotides bind to Npl3 with the same kinetic characteristics as the 18-mer, but with 2:1 stoichiometry. Thus, Npl3 is capable of forming multipartite and independent interactions with RNA sequences separated by a few or many nucleotides. Npl3 also differs from other multi-domain proteins in that while both RRMs work together to give the specificity; in Npl3, the specificity appears to be largely determined by RRM-2.
Full length Npl3 binds to various oligonucleotides that represent yeast 3′-end processing signals with nM affinity (Table 2), i.e. it has strong but non-specific RNA-binding activity. In fact full length Npl3 binds only about 5-fold more strongly to G/U-rich compared to AU-rich oligonucleotides. Although the construct of the protein containing only the two RRMs has much weaker RNA-binding activity, the difference in binding between AU- and G/U-rich sequences is much stronger. The NMR results very clearly show that the two RRMs bind weakly to AU-rich sequences, with affinity of approximately 100 μM, but very differently to G/U-rich RNAs. While binding of RRM-1 to the G/U-rich 18-mer and AU-rich oligonucleotides remains the same, RRM-2 forms a much stronger interaction with the G/U-rich RNA. Because the off rates are long, it is not possible to measure the dissociation constant directly by NMR. However, if the on rate is diffusion limited (there is no evidence to the contrary), then the NMR data demonstrate that the interaction between RRM-2 and the G/U-rich RNA is in the nM-μM range.
The structures of RRM-1 and RRM-2 of Npl3 suggest two possible reasons for this difference in affinity. First, in RRM-1 the ‘recognition’ loop connecting β2 and β3 consists of only two very highly ordered residues, a feature unique in RRMs that generally have longer flexible loops. In contrast, the length of the loop between β2 and β3 in RRM-2 is more typical, with 7 amino acids experiencing conformational exchange, representing a structure more conducive to specific RNA recognition. Second, the RNA binding β-sheet surface of RRM-2 is uniformly neutral to basic. In contrast, the β-sheet surface of RRM-1 has an acidic residue (Glu189) in the center of the RNA binding surface.
The comparison between the full-length protein and the constructs containing only the two RRM domains suggests that other parts of the protein, presumably the sequence rich in Arg, Ser and Gly near its C-terminus, contribute to the strong but non-specific RNA-binding activity of Npl3 [43, 44]. The direct interaction of RNA through the RS domain has been reported in the case of other SR proteins like the splicing factor U2AF [45, 46] and the PTB associated splicing factor (PSF) [47]. RNA recognition by Npl3 may be similar, with the RS domain contributing to the strong, but non-specific RNA binding activity of the whole protein. This non-specific binding masks the ability of RRM-2 to discriminate between G/U-rich and other RNA oligonucleotides, because the full-length protein binds with only 5-fold different affinity to G/U- and AU-rich RNAs. This result suggests that Npl3 may become a sequence-specific RNA-binding protein if the strong but non-specific RNA-binding activity of the rest of the protein is negatively regulated. Consistent with this suggestion, phosphorylation of Npl3 reduces binding to RNA 5-fold [43].
Superposition of the two RRMs of Npl3 on the SxL-protein (PDB: 1B7F) demonstrates that the two proteins are structurally very similar. We also notice that the length of the shorter G/U-rich oligonucleotide (about 10 nucleotides, Table 3) corresponds closely to the number of nucleotides that are specifically recognized by each of the two domain proteins mentioned earlier: Pab, SxL, Hrp1 and HuD. However, the binding sites for RRM-1 and RRM-2 must be separated by a flexible polynucleotide region in order to account for all the results described above. We exploited this similarity in structures and these observations to generate a model of the Npl3 protein in complex with the 18-mer RNA that incorporates this similarity as well as the independent interaction of each domain of the protein with two distinctive G/U-rich sequences separated by a flexible RNA linker (Figure 8).
FIGURE 8.

Model of Npl3 binding to N2 RNA. The pdb file was generated by using the energy minimization program MOE (version 2006.08; Chemical Computing Group, Montreal, Quebec, Canada) starting with the coordinates of the Sxl-RNA complex. Residues in Npl3 protein that are perturbed upon binding to N2 RNA are highlighted. Residues that undergo significant changes in NH chemical shift (at least 2.0-fold above average) upon RNA binding are colored in orange, while residues that are in intermediate exchange on NMR chemical shift timescale are shown in red.
The short G/U-rich sequence to which Npl3 binds (GCC UGG UUG C; Table 3) is nearly identical to the consensus sequence of another RRM binding protein, human PTB (-(A/Y) GCC UGG UGC Y-) [48]. This sequence similarity suggests that these proteins and SxL may also share a similar mechanism. Indeed, SxL and Cstf-64 compete for binding to G/U-elements in the regulation of alternative poly(A) site selection [36] and PTB competes with U2AF65 for the pyrimidine tract upstream of 3′ splice sites [49]. Thus the antagonistic interaction of these proteins functions to regulate termination/poly(A) and alternative splicing.
The NMR data strongly suggest that the RRM-2 Npl3 has a preference for sequences rich in Us and Gs that is partially masked by the non sequence-specific activity of the rest of the protein. The Rna15 consensus sequences originally identified in vitro using SELEX are also G/U-rich [50], and its vertebrate orthologue CstF-64 binds to GU or U-rich sequences as well [51]. Thus, both Npl3 and Rna15 appear to bind preferentially to G/U-rich sequences thereby providing mechanistic insight into how Npl3 prevents premature processing and transcription termination. When in complex with other CF I subunits, Rna15 contacts an A-rich sequence and Hrp1 binds specifically to an alternating AU-rich RNA upstream of the processing site [40, 52]. Consistent with the NMR results that report a much stronger interaction between RRM-2 and the G/U-rich RNAs, mutations in RRM-2 of Npl3 have significant consequences on the selection of 3′-end processing sites [17]. Presumably, reduced binding of the RRM-2 of Npl3 to G/U-rich sequences would allow for their recognition by Rna15, which could then direct binding of the rest of the 3′-end processing complex to weak or cryptic poly(A) sites. Thus, the preference of Npl3 for G/U-rich sequences is consistent with its function in regulating recognition of 3′-end processing sites via its competition with the Rna15 protein for RNA binding, and suggests that this function is encoded within RRM-2.
MATERIALS AND METHODS
Plasmids
The DNA sequence corresponding to amino acids 121 to 280 of Npl3 was amplified by PCR from the plasmid corresponding to the full-length protein and sub-cloned into pET-151 vector using the TOPO expression kit (Invitrogen). This pET-151-npl3 plasmid includes an N-terminal His-tag followed by a TEV protease cleavage site. For Npl3 mutants, the same pET-151-npl3 plasmid was used for site-directed mutagenesis (Stratagene) using appropriate mutagenic primers. Plasmids pSBEThis7-NPL3 and pSBEThis7-npl3-120 were described previously [15]. For plasmids pSBEThis7-npl3-004 and pSBEThis7-npl3-150, the ORF of NPL3 was amplified by PCR from the npl3-004 and npl3-150 alleles (oligos: 5′-CATGCCATGGCTGAAGCTCAAGAAACTCACG-3′ and 5′-CGCGGATCCGCTTACCTGGTTGGTGATCTTTCACG-3′) and cloned into BamHI/NcoI site of pSBEThis7.
Protein expression and purification
For NMR experiments, pET-151-Npl3 plasmid was transformed into E. coli Rosetta cells (Novagen) and grown in minimal media (M9) containing 100mg/l of ampicillin. 2 g/l of 13C labeled dextrose and 0.5 g/l of 15N labeled ammonium chloride were used as the sole sources of carbon and nitrogen, respectively, to prepare isotopically labeled proteins for NMR. Cells were grown at 37°C and induced at OD A600= 0.6 by using IPTG to a final concentration of 0.2 mM. After induction, the cell growth temperature was reduced to 22 °C and the cells were harvested after 12–16 hours. The cells were pelleted using a centrifuge and pellets were disrupted by sonication following re-suspension in lysis buffer, and ultracentrifuged to obtain the soluble fraction containing the protein of interest. The protein was purified by using Ni-affinity chromatography (Amersham Pharmacia). The (his)6 tag was removed using TEV protease digestion. The cleaved Npl3 protein was passed through a Q-Sepharose Fast Flow column (GE Healthcare), followed by size exclusion chromatography. No detectable impurities were found in the purified protein by polyacrylamide gel electrophoresis (PAGE) or MALDI mass spectrometry. The AKTA purification platform was used for all chromatographic steps.
For electrophoretic mobility shift assays, E. coli strain BL21 (DE3) was transformed with pSBEThis7-NPL3, pSBEThis7-npl3-120, pSBEThis7-npl3-004 or pSBEThis7-npl3-150 and recombinant Npl3 was purified as described previously [15].
RNA preparation
RNA sequences used for NMR studies were purchased as PAGE-purified oligonucleotides from IDT DNA. Samples were dissolved in water, ethanol precipitated, re-suspended and dialyzed against buffer (20 mM potassium phosphate, 20 mM KCl, pH 6.5) for 24 hours. The product was freeze-dried and re-suspended in water or 100% D2O as required for our NMR experiments.
Gel mobility shift assays
For gel electrophoretic mobility shift assays (EMSA), RNA oligos were synthesized (Invitrogen), and ATP-[32P] 5′end-labeled using T4 Polynucleotide Kinase (Invitrogen). Heat denatured [32P] RNA oligonucleotides (0.1 or 0.5 pmoles/reaction, as indicated) were mixed with recombinant Npl3, in binding buffer (1 mM magnesium acetate, 50 mM potassium acetate, 10% glycerol, 20 mM HEPES -pH 7.2, 1 mM DTT, 33 μg/ml tRNA). All reactions were incubated for 30 minutes at room temperature. Glycerol was added to 25%, and reactions were loaded on 5% polyacrylamide gels (30:0.8 acrylamide:bis) in 0.5xTBE buffer, dried and exposed to a PhosphoImager screen for quantification. All experiments were done in triplicate. The RNA-protein complexes that migrated into the gel were considered to be the RNA-bound fraction and expressed as PhosphorImager units. A least squares fit for a single binding site was obtained using the equation:
where ABmax is the maximum amount of AB complex at saturating Npl3, B is the active Npl3 concentration, and KD is the apparent equilibrium constant [53]. Reported KD values represent the average of three independent determinations.
NMR spectroscopy
All NMR samples of Npl3 were prepared in Shigemi susceptibility matched tubes. NMR experiments for structure determination were performed at a protein concentration of 1.0–1.2 mM, dissolved in a buffer containing 20 mM potassium phosphate (pH 6.5), 20 mM KCl and 4mM DTT. For RNA binding studies, the protein concentration was kept between 0.2 mM and 0.3 mM. Unless otherwise specified, all experiments were carried out at 298K. Triple resonance experiments were collected on a Bruker Avance 500 MHz spectrometer equipped with a TXI HCN triple resonance probe with triple axis gradients. 3D 15N-edited NOESY spectra and 2D NOESY spectra were recorded at Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory (PNNL) on a Varian 900MHz spectrometer. 13C-edited NOESY and HCCH-TOCSY spectra were collected on the 500MHz Bruker Avance spectrometer. All spectra were processed using NMRPipe/NMRDraw software [54]. Linear prediction and zero filling were applied in the indirect dimensions to increase the number of points in that dimension and hence to get better resolution. The resulting processed NMR spectra were analyzed using Sparky [55].
Sequence specific backbone assignments of H, N, Cα, C and side chain Cβ resonances were done using triple resonance experiments HNCO, HNCA, HN(CO)CA, NH(CO)CACB [56] along with 15N-HSQC and 13C-HSQC. 1H and 13C side chain assignments were achieved using HCCH-TOCSY, as well as 15N- and 13C-edited NOESY experiments. Side chain NH2 resonances of Asn/Gln residues were identified using 15N-edited NOESY and HSQC. Aromatic proton assignments were performed by 13C-edited NOESY experiments optimized for detection of aromatic resonances, along with a 2D proton NOESY recorded in 100% D2O. All NOESY experiments were recorded with a mixing time of 80 ms.
Structure calculation
Protein structure calculation was conducted in a semi-automated iterative manner by using CYANA version 2.0 [57, 58]. NOE data from 15N- and 13C-edited NOESY and from the NOESY collected in D2O were used for the structure calculation. The NOESY peak list used as input for CYANA analysis was generated automatically using Sparky [55] based on the chemical shift list generated in the assignment process. After the first few rounds of calculations, the spectra were analyzed again to identify additional cross-peaks consistent with the structural model and to correct misidentified NOEs.
Slowly exchanging amides were identified by lyophilizing the protein from H2O and then dissolving it in 100% D2O; hydrogen bond donors were identified by the presence of an amide peak in the HSQC recorded after 30 minutes. The corresponding acceptors were attributed by visualizing coordinates obtained from CYANA calculations without any hydrogen bonding constraint to identify carbonyl groups that were at a distance of approximately 2.0 A from slow exchanging amides. Hydrogen bonding constraints were then added at this stage of the refinement. TALOS was used to generate φ and ψ dihedral angle constraints [59].
After several rounds of refinement, the final run of structure calculations gave an average target function of 2.23 Å and 1.15 Å, respectively, for RRM-1 and RRM-2 of Npl3. No upper distance and no angle violations greater than 0.25 Å and 5°, respectively, were observed. The 20 structures with the lowest target function were selected to represent the ensemble of converged Npl3 structures. Experimental data and structural statistics are summarized in Table 1. The quality of the structure was evaluated using PROCHECK-NMR (http://rcsb-deposit.rutgers.edu).
Relaxation measurements
Standard pulse sequences were used to measure the 15N-1H T1, 15N-1H T2 and heteronuclear NOEs [60] essentially as we described recently [28]. Spectra were recorded with 112 complex points in the indirect dimension and with delays of 10, 30, 70, 120, 200, 300, 420, 550, and 700 ms for the T1 experiments and 8, 16, 24, 40, 56, 80, 104, 128, and 160 ms for T2. For the heteronuclear NOE measurements, a pair of spectra was recorded with and without proton saturation, which was achieved by application of 1H 120° pulses every 5 ms. Spectra recorded with proton saturation utilized a 2 s recycle delay followed by a 3 s period of saturation, while spectra recorded in the absence of saturation employed a recycle delay of 5 s. The NMR data was processed and analyzed essentially as we recently reported [28].
Generation of the model of the protein-RNA complex
The two domains of Npl3 protein were superimposed on the SxL protein (PDB: 1B7F), that was chosen because of its close structural homology (see text). The backbone of the G/U-rich N2 RNA (Table 2) was subsequently superimposed onto the backbone of the U-rich RNA recognized by SxL. Energy minimization was then performed in parallel with geometry optimization by using the software MOE (Version: 2006.08 Chemical Computing Group, Montreal, Quebec, Canada). Once the model was constructed, five additional nucleotides were added between the two stretches of nucleotides recognized by each of the Npl3 RRMs, in order to represent the presence of a flexible connection between the two RNAs described in the text. The conformation was finally regularized by using MOE.
Acknowledgments
P. Deka acknowledges the help received from Dr. S. Reichow in structure determination; Dr. T. Leeper in collecting NMR experiments and Dr S. Zheng in generating the model of the structure of the RNA-protein complex. M. Bucheli was supported by a fellowship from the American Cancer Society PF-03-224-01-GMC and NIH grant 1K01CA115515-01A1. This work is supported by NIH grants GM56663 to SB, GM064440 to GV, and GM68887 to CM.
Footnotes
Protein Data Bank accession numbers
Coordinates of Npl3-RRM-1 and Npl3-RRM-2 have been deposited to Protein Data Bank with accession number 2OSQ and 2OSR respectively.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hirose Y, Manley JL. RNA Polymerase II and the Integration of Nuclear Events. Genes & Develop. 2000;14:1415–1429. [PubMed] [Google Scholar]
- 2.Cho EJ, et al. mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes & Dev. 1997;11(24):3319–26. doi: 10.1101/gad.11.24.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA Processing with Transcription. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- 4.Proudfoot N. New Perspectives on Connecting messenger RNA 3′ end Formation to Transcription. Curr Op Cell Biology. 2004;16:272–278. doi: 10.1016/j.ceb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Rosonina E, Kaneko S, Manley JL. Terminating the transcript: breaking up is hard to do. Genes & Devel. 2006;20:1050–1056. doi: 10.1101/gad.1431606. [DOI] [PubMed] [Google Scholar]
- 6.Lei EP, Krebber H, Silver PA. Messenger RNAs are recruited for nuclear export during transcription. Genes & Dev. 2001;15:1771–1782. doi: 10.1101/gad.892401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen RH, et al. Early Formation of mRNP: Licence for Export or Quality Control? Mol Cell. 2003;11:1129–1138. doi: 10.1016/s1097-2765(03)00191-6. [DOI] [PubMed] [Google Scholar]
- 8.Sanford JR, et al. A novel role for shuttling SR proteins in mRNA translation. Genes & Dev. 2004;18:755–768. doi: 10.1101/gad.286404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fu XD. The Superfamily of Arginine/Serine-Rich Splicing Factors. RNA. 1995;1:663–680. [PMC free article] [PubMed] [Google Scholar]
- 10.Manley JL, Tacke R. SR Proteins and Splicing Control. Genes & Dev. 1996;10:1569–1579. doi: 10.1101/gad.10.13.1569. [DOI] [PubMed] [Google Scholar]
- 11.Valcárel J, Green MR. The SR Protein Family: Pleiotropic Functions in pre-mRNA Splicing. Trends in the Biochemical Sciences. 1996;21:296–301. [PubMed] [Google Scholar]
- 12.de la Mata M, Kornblihtt AR. RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat Struct Mol Biol. 2006;13(11):973–80. doi: 10.1038/nsmb1155. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Z, Krainer AR. Involvement of SR Proteins in mRNA Surveillance. Mol Cell. 2004;16:597–607. doi: 10.1016/j.molcel.2004.10.031. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, Steitz JA. SRprises along a Messenger’s Journey. Mol Cell. 2005;17:613–615. doi: 10.1016/j.molcel.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 15.Bucheli ME, Buratowski S. NPl3 is an Antagonist of Transcription Termination by RNA Polymerase II. EMBO J. 2005 doi: 10.1038/sj.emboj.7600687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lukasiewicz R, et al. The RGG Domain of Npl3p Recruits Sky1p through docking interactions. Journal of Molecular Biology. 2007;367(1):249–61. doi: 10.1016/j.jmb.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 17.Bucheli ME, et al. Polyadenylation Site Choice In Yeast Is Affected By Competition Between Npl3 And Polyadenylation Factor CFI. RNA. 2007 doi: 10.1261/rna.607207. under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siebel CW, Guthrie C. The essential yeast RNA binding protein Np13p is methylated. Proc Natl Acad Sci U S A. 1996;93(24):13641–6. doi: 10.1073/pnas.93.24.13641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varani G, Nagai K. RNA Recognition by RNP Proteins during RNA Processing and Maturation. Ann Rev Biophys Biomol Struct. 1998;27:407–445. doi: 10.1146/annurev.biophys.27.1.407. [DOI] [PubMed] [Google Scholar]
- 20.Nagai K, et al. The RNP Domain: a Sequence-Specific RNA-Binding Domain Involved in Processing and Transport of RNA. Trends in the Biochemical Sciences. 1995;20:235–240. doi: 10.1016/s0968-0004(00)89024-6. [DOI] [PubMed] [Google Scholar]
- 21.Maris C, Dominguez C, Allain FHT. The RNA Recognition Motif, a plastic RNA binding platform to regulate posttranscriptional gene expression. FEBS J. 2005;272:2118–2131. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 22.Crowder SM, et al. Absence of Interdomain Contacts in the Crystal Structure of the RNA Recognition Motifs of Sex-Lethal. Proc Natl Acad Sci USA. 1999;96:4892–4897. doi: 10.1073/pnas.96.9.4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dominguez C, Allain FHT. NMR Structure of the three quasi RNA Recognition Motifs (qRRMs) of human hnRNP F and interactions with Bcl-x G-tract RNA: A novel mode of RNA recognition. Nucl Acids Res. 2006 doi: 10.1093/nar/gkl488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lunde BM, Moore C, VG RNA-binding Proteins: a modular design for efficient function. Nature Rev Mol Cell Biol. 2007 doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Auwater SD, Oberstrass FC, Allain FHT. Sequence-specific Binding of Single-stranded RNA: Is There a Code for Recognition? Nucleic Acids Research. :1–47. doi: 10.1093/nar/gkl620. Nucleic Acids Res 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lipari G, Szabo A. Model-Free Approach to the Interpreptaion of Nuclear Magnetic Relaxation in Macromolecules. 1. Theory and Range of Validity. Journal of the American Chemical Society. 1982;104:4546–4559. [Google Scholar]
- 27.Lipari G, Szabo A. Model-Free Approach to the Interpreptaion of Nuclear Magnetic Relaxation in Macromolecules. 2. Analysis of Experimental Results. Journal of the American Chemical Society. 1982;104:4559–4570. [Google Scholar]
- 28.Deka P, et al. Protein and RNA Dynamics Play Key Roles in Determining the Specific Recognition of GU-rich Polydenylation Regulatory Elements by Human Cstf-64 Protein. J Mol Biol. 2005;347:719–733. doi: 10.1016/j.jmb.2005.01.046. [DOI] [PubMed] [Google Scholar]
- 29.Dobson N, et al. High-Resolution Structural Validation of the Computational Redesign of Human U1A Protein. Structure. 2006;14:847–856. doi: 10.1016/j.str.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 30.Shapiro YE, et al. Backbone Dynamics of Escherichia Coli Adenylate Kinase at the Extreme Stages of the Catalytic Cycle Studied by 15N NMR Relaxation. Biochemistry. 2000;39:6634–6644. doi: 10.1021/bi992076h. [DOI] [PubMed] [Google Scholar]
- 31.Chao L, et al. Assembly of the Cleavage and Polyadenylation Apparatus Requirs About 10 Seconds In Vivo and Is Faster for Strong than for WEak Poly(A) Sites. Molecular and Cellular Biology. 1999;19:5588–5600. doi: 10.1128/mcb.19.8.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de la Mata M, et al. A Slow RNA Polymerase II Affects Alternative Splicing in Vivo. Molecular Cell. 2003;12:525–532. doi: 10.1016/j.molcel.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 33.Park NJ, Tsao DC, Martinson HG. The Two Steps of Poly(A)-Dependent Termination, Pausing and Release, Can Be Uncoupled by Truncation of the RNA Polymerase II CTD. Molec Cell Biol. 2004 doi: 10.1128/MCB.24.10.4092-4103.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bentley DL. Rules of Engagement: co-Transcriptional Recruitment of pre-mRNA Processing Factors. Curr Op Cell Biology. 2005;17:251–256. doi: 10.1016/j.ceb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 35.Calvo O, Manley JL. Evolutionarily Conserved Interaction between CtsF-64 and PC4 Links Transcription, Polyadenylation and Termination. Mol Cell. 2001;7:1013–1023. doi: 10.1016/s1097-2765(01)00236-2. [DOI] [PubMed] [Google Scholar]
- 36.Gawande B, et al. Drosophila Sex-lethal protein mediates polyadenylation switching in the female germline. EMBO. 2006;25(6):1263–72. doi: 10.1038/sj.emboj.7601022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Handa N, et al. Structural Basis for Recognition of the tra mRNA Precursor by the Sex-Lethal Protein. Nature. 1999;398:579–585. doi: 10.1038/19242. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Tanaka-Hall T. Structural Basis for Recognition of AU-Rich Element RNA by Hu Proteins. Nature Struct Biol. 2001;8:141–146. doi: 10.1038/84131. [DOI] [PubMed] [Google Scholar]
- 39.Deo RC, et al. Recognition of Polyadenylate RNA by the Poly(A)-Binding Protein. Cell. 1999;98:835–845. doi: 10.1016/s0092-8674(00)81517-2. [DOI] [PubMed] [Google Scholar]
- 40.Perez-Canadillas JM. Grabbing the message: structural basis of mRNA 3′ UTR recognition by Hrp1. EMBO J. 2006;25:3167–3178. doi: 10.1038/sj.emboj.7601190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shamoo Y, et al. Both RNA-Binding Domains in Heterogeneous Nuclear Ribonucleoprotein A1 Contribute toward Single-Stranded-RNA Binding. Biochemistry. 1994;33:8272–8281. doi: 10.1021/bi00193a014. [DOI] [PubMed] [Google Scholar]
- 42.Shamoo Y, Abdul-Manan N, Williams KR. Multiple RNA Binding Domains (RBDs) Just Don’t Add Up. Nucleic Acids Research. 1995;23:725–728. doi: 10.1093/nar/23.5.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gilbert W, Siebel CW, Guthrie C. Phosphorylation by Sky1p promotes Npl3p shuttling and mRNA dissociation. RNA. 2001;7(2):302–13. doi: 10.1017/s1355838201002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu C, Henry MF. Nuclear export of hnRNP Hrp1p and nuclear export of hnRNP Npl3p are linked and influenced by the methylation state of Npl3p. Mol Cell Biol. 2004;24(24):10742–56. doi: 10.1128/MCB.24.24.10742-10756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen H, Green MR. A Pathway of Sequential Arginine-Serine-Rich Domain-Splicing Signal Interactions during Mammalian Spliceosome Assembly. Mol Cell. 2004;16(3):363–373. doi: 10.1016/j.molcel.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 46.Hertel KJ, Graveley BR. RS domains contact the pre-mRNA throughout spliceosome assembly. Trends Biochem Sci. 2005;30(3):115–118. doi: 10.1016/j.tibs.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 47.Huang CJ, et al. Phosphorylation by SR kinases regulates the binding of PTB-associated splicing factor (PSF) to the pre-mRNA polypyrimidine tract. FEBS Lett. 2007;581(2):223–32. doi: 10.1016/j.febslet.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh R, Valcarél J, Green MR. Distinct Binding Specificities and Functions of Higher Eukaryotes Polypirimidine Tract-Binding Proteins. Science. 1995;268:1173–1176. doi: 10.1126/science.7761834. [DOI] [PubMed] [Google Scholar]
- 49.Sauliere J, et al. The polypyrimidine tract binding protein (PTB) represses splicing of exon 6B from the beta-tropomyosin pre-mRNA by directly interfering with the binding of the U2AF65 subunit. Mol Cell Biol. 2006;26(23):8755–69. doi: 10.1128/MCB.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takagaki Y, Manley JL. RNA Recognition by the Human Polyadenylation Factor CstF. Molecular and Cellular Biology. 1997;17:3907–3914. doi: 10.1128/mcb.17.7.3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perez-Canadillas JM, Varani G. Recognition of GU-rich Polyadenylation Regulatory Elements by Human CstF-64 Protein. EMBO J. 2003;22:2821–2830. doi: 10.1093/emboj/cdg259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gross S, Moore CL. Rna15 Interaction with the A-Rich Yeast Polydenylation Signal Is an Essential Step in mRNA 3′-End Formation. Mol Cell Biol. 2001;21:8045–8055. doi: 10.1128/MCB.21.23.8045-8055.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goodrich JA, Kugel JF. Binding and Kinetics for Molecular Biologists. Cold Spring Harbor Laboratory Press; NY: 2007. [Google Scholar]
- 54.Delaglio F, et al. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. Journal of Biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 55.Goddard TD, Kneller DG. Sparky 3. University of California; San Francisco: [Google Scholar]
- 56.Sattler M, Schleucher J, Griesinger C. Heteronuclear Multidimensional NMR Experiments for the Structure Determination of Proteins in Solution Employing Pulsed Field Gradients. Prog NMR Spectr. 1999;34:93–158. [Google Scholar]
- 57.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol. 2002;319(1):209–227. doi: 10.1016/s0022-2836(02)00241-3. [DOI] [PubMed] [Google Scholar]
- 58.Guntert P. Automated NMR protein structure determination. Prog Nuclear Mag Res Spectro. 2003;(43):105–125. [Google Scholar]
- 59.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Bioml NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 60.Farrow NA, et al. Backbone Dynamics of a Free and a Phosphopeptide Complexed Src Homology 2 Domain Studied by 15N NMR Relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 61.Koradi R, Billeter M, Wuthrich K. MOLMOL: a Program for Display and Analysis of Macromolecular Structures. J Mol Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 62.De Lano WL. The PyMOL Molecular Graphics System. Delano Scientific; San Carlos USA: 2002. [Google Scholar]