

Abstract
Prostate cancer progresses from an androgen-dependent to androgen-independent stage after androgen ablation therapy. Mitochondrial DNA plays a role in cell death and metastatic competence. Further, heteroplasmic large-deletion mitochondrial DNA is verycommon in prostate cancer. To investigate the role of mitochondrial DNA in androgen dependence of prostate cancers, we tested the changes of normal and deleted mitochondrial DNA in accordance with the progression of prostate cancer. We demonstrated that the androgen-independent cell line C4-2, established byinoculation of the androgen-dependent LNCaP cell line into castrated mice, has a greatlyreduced amount of normal mitochondrial DNA and an accumulation of large-deletion DNA. Strikingly, the depletion of mitochondrial DNA from androgen-dependent LNCaP resulted in a loss of androgen dependence. Reconstitution of normal mitochondrial DNA to the mitochondrial DNA-depleted clone restored androgen dependence. These results indicate that mitochondrial DNA determines androgen dependence of prostate cancer cell lines. Further, mitochondrial DNA-deficient cells formed tumors in castrated athymic mice, whereas LNCaP did not. The accumulation of large deletion and depletion of mitochondrial DNA maythus playa role in the development of androgen independence, leading to progression of prostate cancers.
Keywords: mitochondrial DNA, prostate cancer, hormone dependence
Introduction
Prostate cancer is a significant health problem, representing the leading cancer diagnosis and the second leading cause of cancer death in North American men (Jemal et al., 2005). Prostate cancer begins as an androgen-dependent cancer that undergoes clinical regression in response to pharmacological or surgical strategies that reduce testosterone concentration. Despite this treatment, the cancer frequently recurs as an androgen- or hormone-independent cancer. Therefore, elucidation of the mechanisms involved in the development of androgen independence is a critical topic for the development of novel effective treatments for prostate cancer. In the absence of androgen, androgen-dependent prostate cancer cannot proliferate and enter an apoptotic pathway (Kimura et al., 2001). Therefore, androgen-independent prostate cancer cells must have acquired antiapoptotic activity and/or growth stimulatory activity in the absence (or low concentration) of androgen.
The androgen-independent human prostate cancer cell line C4-2 was established by the coinoculation of androgen-dependent LNCaP cells and nontumorigenic fibroblasts from osteosarcoma into a castrated mouse (Wu et al., 1994). C4-2 cells are androgen-independent and highly tumorigenic and have a proclivity to metastasize to the bone (Wu et al., 1994). This system is considered an appropriate model for investigating the change in androgen dependence following androgen ablation therapy in prostate cancer for the following reasons: (1) we can compare the cells before and after androgen ablation treatment and, (2) most of the prostate cancer cells in vivo still express androgen receptors after the cells become androgen-independent (Diamond and Barrack, 1984; Sommer and Haendler, 2003). In using the term ‘androgen-independent’, one should consider that the cells could survive in the absence (or low concentration) of androgen, although other specific biological functions of androgen may or may not be induced by androgen. C4-2 is the only cell line available that expresses androgen receptor and is androgen-independent. Other androgen-independent cell lines such as DU-145 do not express androgen receptor (Culig et al., 1993). We believe that changes in nuclear DNA or mitochondrial DNA (mtDNA) induced by androgen ablation altered the status of androgen-dependent LNCaP to create the androgen-independent C4-2.
Mitochondria are essential organelles that generate cellular energy and control oxidative phosphorylation. The mutation rate of mtDNA is high and can contribute to mitochondrial respiratory chain (MRC) defects. The high mtDNA sequence change rate is caused by both high rates of mutation and ‘fixing’ mutated DNA. The high mutation rate results in part from the mtDNA’s lack of protective histones, an inefficient DNA repair system, and continuous exposure to the mutagenic effect of reactive oxygen species (ROS) generated from MRC (Wallace, 1992). The high mutation fixation rate is due to the efficient intracellular sorting of mutant mtDNA and the rapid genetic drift of mtDNA in the general population (Michikawa et al., 1999).
Heteroplasmic deletion of mutant mtDNA is very common in prostate cancer (Jessie et al., 2001) and has been observed in renal carcinoma (Horton et al., 1996) and gastric adenocarcinoma (Burgart et al., 1995). Jeronimo et al. (2001) sequenced D-loop region, 16S ribosomal RNS, and NADH subunits of complex I to identify mtDNA point mutations in 16 matched PIN lesions and primary cancers. They found 20 somatic mutations detected in three of 16 patients. These mutations were homoplasmic or near homoplasmic. Another report (Jessie et al., 2001) demonstrates the accumulation of large-deletion heteroplasmic mutant mtDNA in most of prostate cancer by long range PCR. They also demonstrate the possible roles of mtDNA mutations in prostate cancer tumorigenesis (Petros et al., 2005). Chen et al. (2002) reported an extremely high incidence of somatic mutation (90%) in prostate cancer specimens found by sequencing the D-loop region. Taken together, both heteroplasmic deletion mutant mtDNA, and homoplasmic and heteroplasmic point mutant mtDNA were observed in most prostate cancer. Point mutations in mtDNA might influence the activity of certain proteins coded in mtDNA, depending on the site of mutation. If the mutation is in transfer RNA or ribosomal RNA, it may affect the activity of all or some of the proteins coded in mtDNA. If the mutation is in the D-loop region, it may affect duplication and transcription. In contrast, increases in large-deletion mutant mtDNA together with the depletion of normal mtDNA will reduce the levels of some or all of the 13 enzymes required for oxidative phosphorylation (Anderson et al., 1981) and might affect oxidative phosphorylation, redox regulation of the cells, and Ca2+ homeostasis. Cells with a high proportion of large-deletion mutant mtDNA might in fact, have characteristics similar to those with depleted mtDNA.
We previously showed that tumor necrosis factor (TNF) and serum starvation could not induce apoptosis in mtDNA-depleted cells, whereas they induced apoptosis in parental cells and cells reconstituted with normal mtDNA (Higuchi et al., 1997). These results indicate that mitochondrial respiratory function and mtDNA are critically important in apoptosis. Amuthan et al. (2001) demonstrated that mtDNA-depleted murine skeletal myoblasts C2C12 cells showed invasive phenotypes and overexpression of the tumor-specific markers cathepsin L and transforming growth factor-β, indicating that the loss of mtDNA could contribute to tumor progression and metastasis. Simonnet et al. (2002) showed a decrease in mitochondrial respiratory function in accordance with increased invasiveness of cancer. We therefore investigated whether androgen ablation affects mtDNA and mitochondrial respiratory function.
Cytoplast fusion provides a novel approach to investigate the role of mtDNA in diseases. In this fusion method, mtDNA in the platelets or cytoplasts is transferred into mtDNA-deficient cells via the formation of cybrids; once inside the cell, the mtDNA can complement the mtDNA deficiency (King and Attardi, 1988). To investigate the role of mtDNA in the role of androgen-dependence of prostate cancer, we established the mitochondrial respiration-deficient cells (LNρ0) from androgen-dependent LNCaP and reconstituted clones (LNCyb4) by the fusion of respiration-deficient cells with platelets from normal donors.
Results
Reduction of normal mtDNA and accumulation of large-deletion mutant mtDNA by androgen ablation
To evaluate the roles of mutant mtDNA in the shift from androgen dependence to androgen independence in prostate cancer, we focused on normal mtDNA and heteroplasmic large-deletion mutant mtDNA, which is present at high levels in prostate cancer (Jessie et al., 2001). We first investigated whether we could detect mtDNA deletions in LNCaP (androgen dependent) and C4-2 (androgen independent) derived by inoculation of LNCaP into castrated mice. We extracted total DNA from both of the cell lines and assessed mtDNA levels by long range PCR. The normal mtDNA band (approximately 16 kb) was readily detected in LNCaP but was greatly reduced in C4-2 (Figure 1a). In contrast, large-deletion mutant mtDNA was highly abundant in C4-2 but not in LNCaP (Figure 1a). Reduction of normal mtDNA and the increase in deleted form of mtDNA in C4-2 cells compared with LNCaP were observed in two independent experiments. To compare the amount of normal mtDNA in LNCaP and C4-2, we then ran long range PCR from the indicated amount of DNA (100–6.25 ng, two times dilution) extracted from LNCaP and C4-2 and investigated the detectable PCR band of normal mtDNA. We could detect a PCR band of normal mtDNA (16 kb) when we used more than 12.5 ng DNA from LNCaP, but we could detect the normal mtDNA band only when we used 100 ng DNA from C4-2 (Figure 1b) in two independent experiments. Normal mtDNA PCR band intensity from 100 ng total cellular DNA template in Figure 1b in experiment 1 was greatly reduced as compared with the full-length PCR band intensity in Figure 1a and experiment 2 in Figure 1b. This is possibly because of the difference in the polymerase activity of different batches of the enzymes, although we are not sure. These results indicate that the amount of normal mtDNA in C4-2 was reduced approximately eight times relative to that in LNCaP. To further confirm this result, we performed Southern blotting analysis of 10 μg DNA derived from LNCaP and C4-2 (Figure 1c). We could detect greatly reduced normal mtDNA bands in C4-2 as compared with LNCaP. We could also see faint bands below 16.5 kb in the C4-2 lane but these bands were nearly completely covered by smear bands. To investigate further, we extracted total DNA from 3 × 107 C4-2 cells by Qiagen kit and performed Southern blotting using 100 μg DNA. Although we used ten times more DNA, we could see a smear band with no bands that might represent deleted form of mtDNA (data not shown). To explore the possibility that this smear band was derived from nuclear DNA and hid the existing bands of deleted form of mtDNA, we extracted and purified mtDNA from C4-2 using Wako kit and ran Southern blotting. As shown in Figure 1d, we demonstrated one deleted form of mtDNA with approximately 7800–8800 bp. In Figure 1a in experiment 1, we observed very intense PCR band with 8800 bp (largest MW band in bands with deleted form of mtDNA), which might represent the highest molecular weight deleted form of mtDNA in C4-2. From the estimation of the molecular weight, we believe that we could detect this deleted form of mtDNA by Southern blotting in Figure 1d. We could detect bands representing other forms of deletion mtDNA by long range PCR but not by Southern blotting, possibly because of the high efficiency of PCR products of low molecular weight DNA. As shown in Figure 1a, we always detected strong PCR bands from LNCaP and C4-2 below 3054 bases like previously reported by Jessie et al. (2001). We could observe this band even when we ran PCR from mtDNA from purified mitochondria (data not shown), indicating that this band was derived from mtDNA not from nuclear DNA. However, we could not detect this band with Southern blotting, possibly because of the small copy number of this mtDNA with good efficiency in each PCR cycle.
Figure 1.

The effect of androgen ablation on mtDNA and mitochondrial respiratory function. (a) Total DNA was extracted from LNCaP and C4-2 cells and 100 ng DNA from each cell line was subjected to long range PCR as described in Materials and methods. PCR band at 16 kb indicates normal mtDNA and other bands below 16 kb indicate large deletion mutant mtDNA. (b) Indicated amounts of DNA from LNCaP and C4-2 were subjected to PCR and PCR bands (16 kb) were shown as described in Materials and methods. (c) Total DNA (10 μg) from LNCaP, C4-2 and LNρ0-8 was subjected to Southern blotting as described in Materials and methods. (d) mtDNA from 3×106 cells from C4-2 was subjected to Southern blotting as described in Materials and methods. (e) Oxygen consumption in permeabilized LNCaP and C4-2 was detected using succinate as a substrate as described in Materials and methods.
The changes in mtDNA observed in C4-2 cells may inhibit mitochondrial oxidative phosphorylation. Mitochondrial respiratory function was detected by measuring oxygen consumption using succinate as substrate. The oxygen consumption is correlated with the electron transfer from complexes II, III, IV to oxygen. Mitochondrial respiratory function was approximately 75% lower in C4-2 than in parental LNCaP (Figure 1e). These results indicate that androgen ablation reduced normal mtDNA and accumulated large-deletion mutant mtDNA, thus inhibiting mitochondrial respiratory function.
Inhibition of androgen dependence by the depletion of mtDNA from androgen-dependent LNCaP
To evaluate the roles of mtDNA in androgen dependence of prostate cancers, we investigated the effect of mtDNA depletion from LNCaP cells on androgen dependence. We established respiration-deficient and normal mtDNA-deficient LNρ0 cells by long-term treatment of LNCaP cells in low concentrations of ethidium bromide (King and Attardi, 1989). Ethidium bromide at low concentrations inhibits mtDNA synthesis but not host DNA synthesis, reduces mtDNA, and thus reduces mitochondrial respiratory function. Normal mtDNA was not detectable in LNρ0 by long range PCR, and LNρ0 could not survive in the absence of uridine and pyruvate, possibly because of the lack of respiratory function (data not shown). R1881, a synthetic androgen, had a growth stimulatory effect on LNCaP cells (Figure 2a). In contrast, the growth promoting effect of R1881 was absent in both C4-2 and LNρ0 (Figure 2a). Therefore, depletion of mtDNA from androgen-dependent LNCaP cells abrogated androgen dependence.
Figure 2.
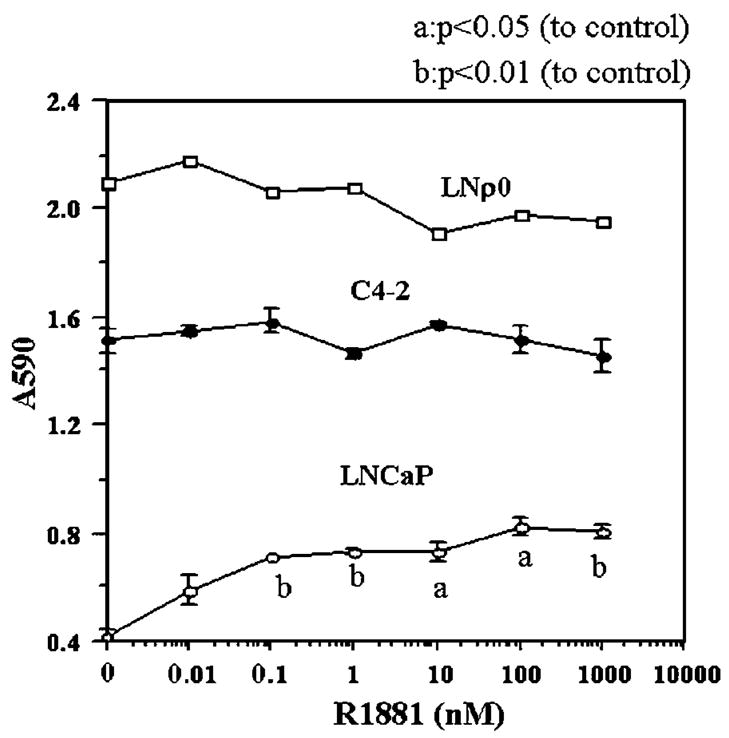
The effect of synthetic androgen R1881 on the growth of LNCaP, C4-2 and LNρ0. LNCaP, C4-2 and LNρ0 were incubated in 96 wells in the indicated concentration of R1881 in DMEM medium supplemented with 1% FCS and 5% charcoal stripped bovine serum. Five days later, relative cell number was assayed by crystal violet staining as described in Materials and methods.
We also investigated the growth of these cell lines in the absence (or low concentration) of androgen (Figure 3). C4-2 and LNρ0 cells proliferated even in the absence of R1881. In the absence of androgen, LNCaP cells did not begin to grow until 48 h in culture (Figure 3). These results indicate that C4-2 and LNρ0 cells had acquired the ability to proliferate without androgen. Interestingly, the growth of LNρ0 cells in the absence of androgen was more rapid than that of either LNCaP or C4-2. This is particularly surprising since the growth of most mtDNA-deficient cells is much slower than that of their parental cell lines because of loss of energy-generation through normal respiratory function. This might suggest that the growth-inhibitory mechanisms specific to prostate cells induced in the absence of androgen were abrogated in LNρ0 cells by depletion of mtDNA.
Figure 3.
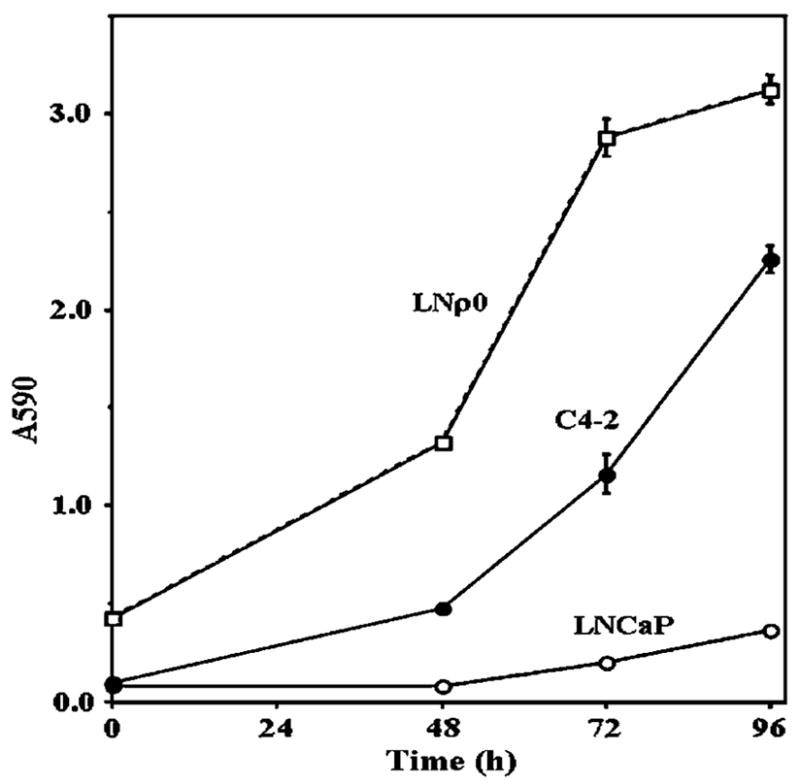
The growth of LNCaP, C4-2 and LNρ0 in reduced androgen condition. LNCaP, C4-2 and LNρ0 were incubated in 96 wells in DMEM medium supplemented with 1% FCS and 5% charcoal stripped bovine serum overnight. Cells were then incubated for indicated times. Relative cell number was assayed by crystal violet staining as described in Materials and methods.
Establishment of a cybrid that had nuclear DNA from LNCaP with normal mtDNA
To confirm that ethidium bromide treatment of LNCaP cells affected only mtDNA for androgen dependence, we reintroduced normal mtDNA into mtDNA-deficient prostate cancer cells. We first tried to establish cybrids by the fusion of LNρ0 cells with platelets from a healthy donor (MH). After fusion, cells were incubated in DMEM plus 10% dialysed FCS. Dialysed FCS is used basically to remove uridine and pyruvate completely from the medium (King and Attardi, 1988). LNρ0 died in a week in the absence of uridine and pyruvate. Cybrids continued to survive as long as 2 weeks but eventually died, potentially because dialysed serum lacks specific essential supplements. Our preliminary results indicate that even androgen-independent C4-2 cells could not grow for a long time in this condition. In contrast, other cell lines such as HeLa and ML-1a cells grew in the same condition. These results suggest that even androgen-independent prostate cancer cell lines might require specific supplements lacking in dialysed FCS. The purpose for using dialysed FCS was to remove uridine and pyruvate to eliminate the growth of ρ0 cells and select cybrids. Instead of using dialysed FCS, we planned to use FCS. However, LNρ0 cells were not appropriate since they can survive up to 3 weeks in DMEM plus 10% FCS. To solve this problem, we cloned LNρ0 cells, and obtained 20 clones. We incubated these 20 clones in DMEM medium plus 10% FCS in which prostate cancer cell lines such as LNCaP, C4-2, PC3 and DU145 can survive. Two clones died in 1 week (clone 6 and 8), 15 clones (clone 1, 2, 3, 4, 5, 9 and 12–20) died in 2 weeks, and the other three clones (clone 7, 10 and 11) could survive up to 3 weeks. Then we decided to evaluate clone LNρ0-8. We confirmed that LNρ0-8 has no mtDNA by Southern blotting (Figure 1c). As LNρ0 continued to survive for as long as 3 weeks in these conditions, LNρ0-8 might require higher concentration of uridine and/or pyruvate than is present in DMEM plus 10% FCS. At 2 weeks after fusion of LNρ0-8 and platelets from a healthy donor (MH), all LNρ0-8 cells died and cybrids (LNCyb4) started to grow. Theoretically, LNCyb4 has nuclei from LNCaP and mtDNA from platelets, but to completely rule out the possibility that LNCyb4 were hybrid cells between LNρ0-8 and mononuclear cells contaminated in platelets fraction, we performed a kariotyping analysis. Cell lines LNp08 and LNCyb4 showed identical structural chromosome aberrations and similar modal chromosome numbers in the range of 59–62.
Recovery of androgen dependence by the reconstitution of mtDNA to LNρ0-8 cells
We investigated the effect of R1881 on the cell growth of LNρ0-8 and LNCyb4. As expected, R1881 did not stimulate the proliferation of LNρ0-8 cells (Figure 4). R1881 did have a growth-stimulatory effect on LNCyb4 (Figure 4), indicating that reconstitution of normal mtDNA restored androgen dependence.
Figure 4.
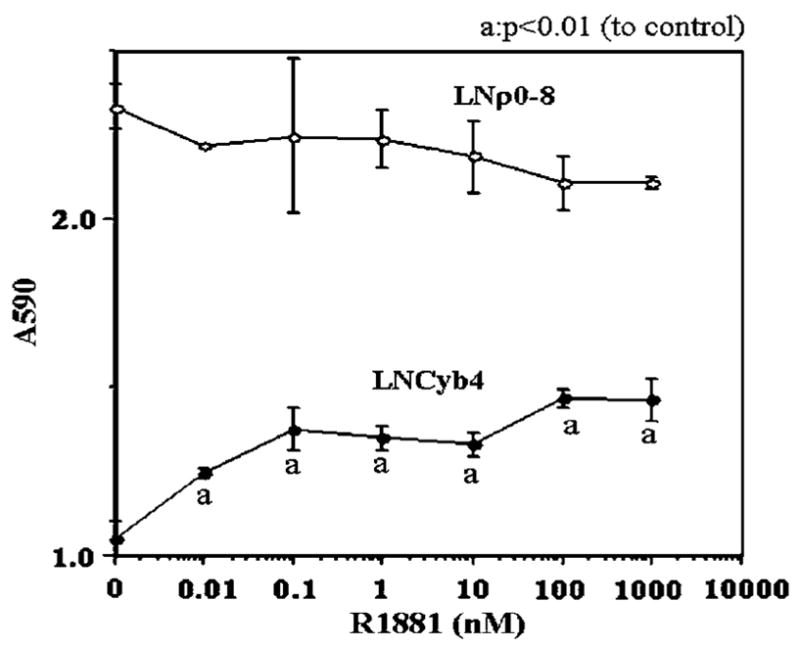
The effect of synthetic androgen R1881 on the growth of LNρ0-8 and LNCyb4. LNρ0-8 and LNCyb4 were incubated in 96 wells with the indicated concentration of R1881 in DMEM medium supplemented with 1% FCS and 5% charcoal stripped bovine serum. After 5 days, relative cell number was assayed by crystal violet staining as described in Materials and methods.
The effect of depletion of mtDNA on androgen dependence in vivo
In order to investigate the role of mtDNA in vivo, we inoculated LNCaP and LNρ0-8 cells into athymic nude mice in three groups; (1) castrated, (2) uncastrated, and (3) castrated 28 days after tumor inoculation (postcastrated). Both LNCaP and LNρ0-8 grew rapidly in uncastrated mice (Figure 5). However, LNρ0-8 but not LNCaP grew in castrated mice (Figure 5). Furthermore, castration of the mice induced tumor regression in LNCaP but not in LNρ0-8 tumors (Figure 5). Thus, depletion of mtDNA was sufficient to render cells androgen independent and resistant to the effects of androgen ablation by castration.
Figure 5.
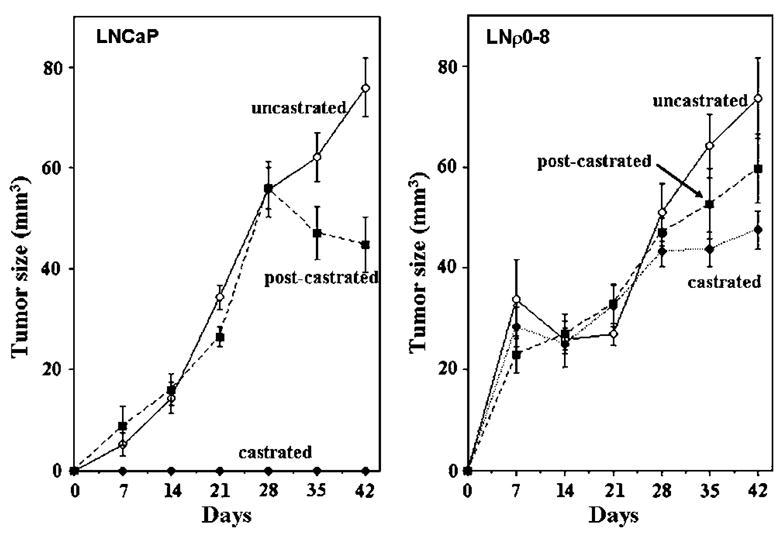
Tumor formations of LNCaP and LNρ0-8 in athymic nude mice with or without castration. Tumor formations of LNCaP and LNρ0-8 in uncastrated athymic (closed circle) nude mice, mice castrated before inoculation (open circle) and mice castrated 28 days after inoculation (closed square) were assayed as described in Materials and methods.
To determine the induction of cell apoptosis, the tumor tissues were immunostained with an antibody against single-stranded DNA. Large numbers of apoptotic LNCaP cells were present in mice castrated after inoculation (Figure 6b), whereas apoptotic cells were rarely seen in LNCaP cells in uncastrated mice (Figure 6a). In contrast, LNρ0-8 did not undergo apoptosis in either uncastrated or castrated mice (Figure 6c and d). Thus, depletion of mtDNA inhibits apoptosis induced by androgen ablation in prostate cancer cells in vivo and may contribute to the continued growth of tumors in castrated mice.
Figure 6.
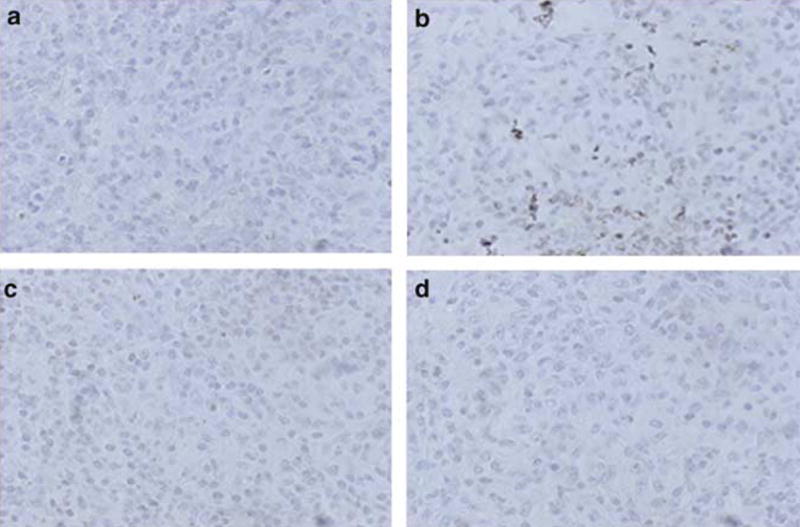
Castration induced apoptotic cell death in LNCaP but not in LNρ0-8. Tumor tissues of LNCaP from uncastrated (a) and postcastrated (b) athymic nude mice and those of LNρ0-8 from uncastrated (c) and postcastrated (d) athymic nude mice were immunostained for the single-stranded DNA as described in Materials and methods.
Discussion
Our data are the first to establish a significant role for mtDNA in the loss of androgen dependence and progression of prostate cancer. Androgen ablation inhibited cell growth in androgen-dependent prostate cancer and at the same time reduced normal mtDNA, with a concomitant accumulation of deletion mutant mtDNA. These changes directly reduced mitochondrial respiratory function and finally led to the conversion of androgen-dependent cells to an androgen-independent state.
Androgen ablation induced multiple deletions of mtDNA and depletion of mtDNA in prostate cancer cell lines; however, the mechanisms involved have not yet been understood. It is suggested that point mutation can be the cause for multiple large-deletion mutant mtDNA and depletion of mtDNA (Nishigaki et al., 2004). In 1989, Zeviani et al. (1989) described autosomal dominant progressive external ophthalmoplegia (adPEO) with multiple mtDNA deletion, the first of several diseases attributed to defects of nuclear DNA leading to the disorder of mtDNA. It is conceivable that androgen ablation may generate similar changes, possibly inhibition of certain enzymes coded in nuclear DNA, leading to multiple deletion and depletion of mtDNA. Mutations in the mitochondrial proteins adenine nucleotide translocator 1 (ANT1) (Kaukonen et al., 2000), Twinkle (Spelbrink et al., 2001) and polymerase γ (Van Goethem et al., 2001) have been found to cause autosomal dominant progressive external ophthalmoplegia with multiple deletion of mtDNA. Mitochondrial Neurogastrointestinal Encephalomyopathy (MINGIE) is an autosomal recessive disorder due to loss-of-function mutations in the gene encoding thymidine phosphorylase, associated with multiple deletions, depletion and site-specific point mutations of mtDNA (Hirano et al., 1994; Papadimitriou et al., 1998; Nishigaki et al., 2003). ANT1 forms a homodimeric inner mitochondrial membrane channel that translocates ADP into ATP out of the mitochondrial matrix. Therefore, this protein regulates concentration of adenine nucleotides in the cytoplasm and mitochondria. Twinkle is a mitochondrial protein with homology to phage T7prim ase/helicase, and the mutation to Twinkle enhances dNTPase activity (Washington et al., 1996). The identification of mutations of genes encoding ANT1 and Twinkle in patients with adPEO and thymidine phosphorylase in patients with MINGIE indicate that imbalance of mitochondrial nucleotide pools may cause multiple deletion of mtDNA and depletion of mtDNA. In contrast, mutations of polymerase γ and Twinkle leading to the change of mtDNA may be caused by the defects in the mtDNA repair and replication machinery. Similar mechanisms might be induced by androgen ablation and lead to the changes in mtDNA.
Another possible pathway to induce mtDNA mutation is the ROS-associated pathway. The mitochondrial genome is extremely susceptible to damages from constant exposure to ROS produced endogenously from MRC. Mitochondrial DNA has been shown to accumulate high levels of 8-hydroxy-2′-deoxyguanosine (8-oxo-G), the product of hydroxylation of guanine at carbon 8, which is a mutagenic lesion. The base excision repair pathway repairs most of these small-base modifications. The 8-oxoguanine-DNA glycosylase 1 (OGG1) protein is the major DNA glycosylase for the repair of 8-oxo-G lesions in the DNA. Inactivation of OGG1 leads to the accumulation of point mutations and deletion mutations in mtDNA (Singh et al., 2001). Polymerase γ is another key enzyme in the repair of 8-oxo-G lesions in the DNA induced by ROS, and transgenic mice expressing a proofreading-deficient polymerase γ exhibit accumulation of point and deletion mutations in mtDNA (Zhang et al., 2000). To protect against the effects of ROS, mitochondria metabolize superoxide and hydrogen peroxide with MnSOD and Se-containing glutathione peroxidase, respectively. ROS have been thought to be involved in the increase in the proportion of both point mutant and deletion mutant mtDNA (Ozawa, 1997). It has also been reported that androgen can regulate ROS generation (Ripple et al., 1997). Our preliminary results indicate that androgen inhibited superoxide generation (data not shown). Given these observations, it is likely that the inhibition of the repair system for ROS-mediated damage to mtDNA, detoxification of ROS, or increase in ROS generation might be possible causes for point mutations of mtDNA, accumulation of large-deletion mutant mtDNA, and the depletion of mtDNA induced by androgen ablation.
Another possible explanation is that androgen ablation selected a small subpopulation of androgen-independent cells with reduced normal mtDNA and accumulated deleted mtDNA that already existed in LNCaP. We are now investigating the exact mechanisms of how androgen ablation reduces normal mtDNA and accumulates deletion mtDNA in prostate cancer cells.
The mechanisms by which reduction of normal mtDNA and accumulation of large-deletion mutant mtDNA contributes to androgen independence during prostate cancer progression are not clearly understood. However, induction of antiapoptotic and/or growth-stimulating signals such as constitutive activation of NF-κB (Palayoor et al., 1999; Catz and Johnson, 2001; Huang et al., 2001), Bcl-2 overexpression (McDonnell et al., 1992; Catz and Johnson, 2001), and constitutive phosphorylation of Akt (Graff et al., 2000) can contribute to the androgen independence of prostate cancers. Depletions of mtDNA induce activation of CREB (Arnould et al., 2002), NF-kB (Higuchi et al., 2002), and other stress-related pathways (Biswas et al., 1999). Thus, it is conceivable that inhibition of respiratory function followed by depletion of normal mtDNA and accumulation of large-deletion mutant mtDNA might induce resistance to androgen ablation through activation of survival and/or antiapoptotic pathways. Further, blocking the loss of mtDNA and accumulation of large-deletion mutant mtDNA or the pathways induced by this process could provide novel effective treatment strategies. We are also investigating whether depletion of mtDNA can be induced after androgen ablation in patients’ tissue samples.
The concept of a mutator phenotype in cancer was formulated to account for the disparity between the rarity of mutations in normal cells and the large number of mutations present in a variety of human malignancies (Loeb, 2001). Rasmussen et al. (2003) reported that deletion or depletion of mtDNA generates a mutator phenotype. Therefore, depletion or deletion of mtDNA by androgen ablation can be responsible for the generation of mutator phenotype leading to further progression of the cancer. A progressive phenotype showing androgen independence was reversed by the transfer of normal mtDNA, suggesting that generation of mutator phenotype is not the direct cause for the shift of androgen dependence in C4-2 cells. However, it is very likely that androgen-independent prostate cancer with reduced normal mtDNA might acquire a more progressive phenotype by inducing mutations to nuclear DNA. Understanding the mechanisms involved in the generation of deletion and depletion of mtDNA induced by androgen ablation and the induction of antiapoptotic pathways induced by mtDNA changes could provide novel insights into the development of androgen independence in cancer cells, cancer heterogeneity and cancer progression, and could thus lead to novel and effective therapies for prostate cancer.
Materials and methods
Cell lines
LNCaP and C4-2 were purchased from UROCOR (Oklahoma City, OK, USA). LNCaP, C4-2, LNρ0 and LNρ0-8 and cybrid LNCyb4 were incubated in DMEM supplemented with 10% FCS and gentamicin (50 μg/ml). The media for LNρ0 and LNρ0-8 was additionally supplemented with 50 μg/ml uridine and 100 μg/ml sodium pyruvate.
Extraction of DNA
Total DNA from approximately 5 × 106 cells was extracted using the Qiagen Blood and Cell Culture DNA Mini kit (Qiagen Inc.) as recommended by the manufacturer. Mitochondrial DNA from approximately 3 × 106 cells was extracted using the mtDNA Extraction CT kit (Wako Pure Chemical Industries Ltd) as recommended by the manufacturer.
MtDNA amplification
The long range PCR employed the primers MITOF (Forward-TGAGGCCAAATATCATTCTGAGGGGC) and MITOR (Reverse-TTTCATCATGCGGAGATGTTGGATGG) and AccuPrime Taq DNA Polymerase High Fidelity (Invitrogen Co.) to generate approximately 16 k bp molecule from normal mtDNA. The amplification reaction mixtures contained 5 μl of 10 × AccuPrime PCR Buffer II, 0.5 μM of each of the primers, 2.5U of enzyme, and 500 ng of the DNA samples. PCR amplifications were performed in thin-wall PCR tubes (USA Scientific Inc.) using a P × E Thermal Cycler (Thermo Electron Co.). The PCR profiles consisted of initial denaturation at 94°C for 2 min, followed by 10 cycles of denaturation at 94°C for 10 s, annealing at 65°C for 30 s, and extension at 68°C for 14 min, and then followed by 25 cycles of denaturation at 94°C for 10 s, annealing at 65°C for 30 s, and extension at 68°C for 14 min plus 10 additional seconds per cycle, and a final extension at 68°C for 7min.
Southern blot analysis
In order to quantify mtDNA levels, 10 μg of total DNA extracted from each cell line was digested with BamH1 (Roche Applied Science, Indianapolis, IN, USA), which cut the mitochondrial genome at a single site, electrophoresed through a 0.8% agarose gel, and transferred to Zeta-Probe membranes (Bio-Rad, Hercules, CA, USA). PCR was used to generate a 2.5-kb fragment of mtDNA (forward primer: 5′-ccactccaccttactaccagac- 3′, reverse primer: 5′-gtaatgctagggtgagtggtagg-3′) and probe was prepared by DIG high prime labeling kit (Roche Applied Science). Filter was hybridized with approximately 25 ng/ml of mtDNA probe. Hybridization and washings were performed at 68°C. Filter was scanned with a Gel Doc System.
Assay for respiratory activity
Oxygen consumption was measured with a Clark oxygen electrode (model 5300; Yellow Spring Instrument Co.). First, 0.8 ml of cells (1 × 107 cells/ml) suspended in respiration medium (0.25M sucrose; 20mM Hepes, pH 7.2; 2mM KH2PO4; 1mM EGTA) were injected into the respiration chamber (37°C) and permeabilized with 0.025% digitonin. ADP and succinate were then added to give a final concentration of 1 and 5mM to initiate State 3 respiration, respectively. Oxygen consumption was calculated as the rate of change in the O2 concentration following the addition of a substrate, assuming an initial O2 concentration of 217 μM.
Establishment of LNρ0
LNCaP cells were incubated in DMEM supplemented with 5% FCS, 50 μg/ml uridine, and 100 μg/ml sodium pyruvate in the presence of 200 ng/ml ethidium bromide for 8 weeks to select mtDNA-deficient cells as described previously (King and Attardi, 1989). Subcloning was performed by limiting dilution and we obtained 20 clones (LNρ0-1 to 20).
Detection of cell viability by crystal violet
Cells (5 × 103 cells/well) suspended in DMEM medium with 1% FCS and 5% charcoal stripped bovine serum were added to the wells of 96-well flat-bottomed micro titer plate. For the experiment specific to Figure 3, cells were incubated for overnight. Cells were then incubated with or without a test sample, and then the plates were incubated at 37°C for the indicated times. Subsequently, the supernatants were discarded and the remaining viable adherent cells were stained with 0.2% crystal violet in 20% ethanol for 5 min. The micro titer plate was then rinsed with water and 0.1 ml of solution (H2O:methanol:ethanol = 5:1:4) was added to each well to solubilize the stained cells. The absorbance of each well was read at 590nm with a micro plate spectrophotometer. All culture was performed in triplicate and results were expressed by mean ± s.e.
Fusion of LNρ0-8 with platelets
Heparinized whole blood was centrifuged for 15 min at 150 g at 4°C, and platelet-rich plasma was recovered. PBS was added and the mixture was centrifuged for 15 min at 150 g at 4°C. The platelet fraction, which contained no other cell types, was centrifuged for 30 min at 2000 g at 4°C. Platelets (1 × 107) and LNρ0-8 (5 × 105) cells were mixed in PBS and centrifuged at 160 g for 10 min. The supernatant was aspirated and a polyethylene glycol solution (45% of polyethylene glycol 2000 in Hanks balanced salt solution) was added. After 1 min incubation at 37°C, 9ml of RPMI 1640 medium was added slowly. The cells were then centrifuged for 5 min at 150 g, and suspended in DMEM supplemented with 10% FCS. In these conditions only cybrid cells could grow.
Karyotyping analysis
Chromosome studies were performed by standard cytogenetic methods. Briefly, cells in culture were exposed to colcemid for 1 h. The cells were washed with alkaline Pucks saline solution and treated with trypsin to remove the cells from the flasks. Cells were treated in hypotonic solution (0.05M KCl) and fixed in Carnoy’s fixative (3:1 methanol:acetic acid). Cell suspensions were dropped onto slides and trypsin G-banded. A total of 20 G-banded metaphase cells were counted and three cells karyotyped for each cell line.
Assessment of in vivo tumor growth
Athymic nude mice, 6–8-week-old, (20–25 g) (BALB/c strain; Charles River Laboratory) were used for all in vivo experiments. Cells (1 × 106) were resuspended in 50 μl of medium and mixed into 50 μl of Matrigel™ for injection per site. Two xenografts were placed on the back of each mouse via a 27G needle. Tumors were measured weekly and their volumes calculated by the formula L × W × H × 0.5236. The mice were divided into three groups: uncastrated mice, mice castrated before tumor inoculation (castrated mice) and mice castrated 28 days after tumor inoculation (postcastrated). Each group had five mice (10 tumors in each group). After 6 weeks, tissues from tumors and organs were harvested and frozen immediately in liquid nitrogen and stored at −80°C for histological examination.
Detection of apoptotic cells
To determine the induction of cell apoptosis, the tumor tissues were immunostained with a polyclonal antibody against single-stranded DNA (DAKO Japan Co., Ltd) to detect the DNA fragmentation (Kawarada et al., 1998).
Acknowledgments
We thank Drs Tetsuo Ashizawa and Gordon B Mills for careful review of the manuscript. We thank Dr Yasuhisa Nakayama for his help in establishing long range PCR. We thank Dr Yutaka Nishigaki for his help in establishing Southern blotting. This work was supported by Charlotte Geyer Foundation, Taiho Pharmaceutical Co., Ltd, Tobacco Settlement at State of Arkansas and NIH grant RO1 CA100846 to M Higuchi.
References
- Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, Avadhani NG. EMBO J. 2001;20:1910–1920. doi: 10.1093/emboj/20.8.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Arnould T, Vankoningsloo S, Renard P, Houbion A, Ninane N, Demazy C, et al. EMBO J. 2002;21:53–63. doi: 10.1093/emboj/21.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas G, Adebanjo OA, Freedman BD, Anandatheerthavarada HK, Vijayasarathy C, Zaidi M, et al. EMBO J. 1999;18:522–533. doi: 10.1093/emboj/18.3.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgart LJ, Zheng J, Shu Q, Strickler JG, Shibata D. Am J Pathol. 1995;147:1105–1111. [PMC free article] [PubMed] [Google Scholar]
- Catz SD, Johnson JL. Oncogene. 2001;20:7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- Chen JZ, Gokden N, Greene GF, Mukunyadzi P, Kadlubar FF. Cancer Res. 2002;62:6470–6474. [PubMed] [Google Scholar]
- Culig Z, Klocker H, Eberle J, Kaspar F, Hobisch A, Cronauer MV, et al. Prostate. 1993;22:11–22. doi: 10.1002/pros.2990220103. [DOI] [PubMed] [Google Scholar]
- Diamond DA, Barrack ER. J Urol. 1984;132:821–827. doi: 10.1016/s0022-5347(17)49881-8. [DOI] [PubMed] [Google Scholar]
- Graff JR, Konicek BW, McNulty AM, Wang Z, Houck K, Allen S, et al. J Biol Chem. 2000;275:24500–24505. doi: 10.1074/jbc.M003145200. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Aggarwal BB, Yeh ET. J Clin Invest. 1997;99:1751–1758. doi: 10.1172/JCI119339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Manna KS, Sasaki R, Aggarwal BB. Antioxidants Redox signal. 2002;4:945–955. doi: 10.1089/152308602762197489. [DOI] [PubMed] [Google Scholar]
- Hirano M, Silvestri G, Blake DM, Lombes A, Minetti C, Bonilla E, et al. Neurology. 1994;44:721–727. doi: 10.1212/wnl.44.4.721. [DOI] [PubMed] [Google Scholar]
- Horton TM, Petros JA, Heddi A, Shoffner J, Kaufman AE, Graham SD, Jr, et al. Genes Chromosomes Cancer. 1996;15:95–101. doi: 10.1002/(SICI)1098-2264(199602)15:2<95::AID-GCC3>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Oncogene. 2001;20:4188–4197. doi: 10.1038/sj.onc.1204535. [DOI] [PubMed] [Google Scholar]
- Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, et al. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- Jeronimo C, Nomoto S, Caballero OL, Usadel H, Henrique R, Varzim G, et al. Oncogene. 2001;20:5195–5198. doi: 10.1038/sj.onc.1204646. [DOI] [PubMed] [Google Scholar]
- Jessie BC, Sun CQ, Irons HR, Marshall FF, Wallace DC, Petros JA. Exp Gerontol. 2001;37:169–174. doi: 10.1016/s0531-5565(01)00153-x. [DOI] [PubMed] [Google Scholar]
- Kaukonen J, Juselius JK, Tiranti V, Kyttala A, Zeviani M, Comi GP, et al. Science. 2000;289:782–785. doi: 10.1126/science.289.5480.782. [DOI] [PubMed] [Google Scholar]
- Kawarada Y, Miura N, Sugiyama T. Jbiochem. 1998;123:492–498. doi: 10.1093/oxfordjournals.jbchem.a021963. [DOI] [PubMed] [Google Scholar]
- Kimura K, Markowski M, Bowen C, Gelmann EP. Cancer Res. 2001;61:5611–5618. [PubMed] [Google Scholar]
- King MP, Attardi G. Cell. 1988;52:811–819. doi: 10.1016/0092-8674(88)90423-0. [DOI] [PubMed] [Google Scholar]
- King MP, Attardi G. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Loeb LA. Cancer Res. 2001;61:3230–3239. [PubMed] [Google Scholar]
- McDonnell TJ, Troncoso P, Brisbay SM, Logothetis C, Chung LW, Hsieh JT, et al. Cancer Res. 1992;52:6940–6944. [PubMed] [Google Scholar]
- Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Science. 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- Nishigaki Y, Marti R, Hirano M. Hum Mol Genet. 2004;13:91–101. doi: 10.1093/hmg/ddh010. [DOI] [PubMed] [Google Scholar]
- Nishigaki Y, Marti R, Copeland WC, Hirano M. J Clin Invest. 2003;111:1913–1921. doi: 10.1172/JCI17828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T. Physiol Rev. 1997;77:425–464. doi: 10.1152/physrev.1997.77.2.425. [DOI] [PubMed] [Google Scholar]
- Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD. Oncogene. 1999;18:7389–7394. doi: 10.1038/sj.onc.1203160. [DOI] [PubMed] [Google Scholar]
- Papadimitriou A, Comi GP, Hadjigeorgiou GM, Bordoni A, Sciacco M, Napoli L, et al. Neurology. 1998;51:1086–1092. doi: 10.1212/wnl.51.4.1086. [DOI] [PubMed] [Google Scholar]
- Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. Proc Natl Acad Sci USA. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen AK, Chatterjee A, Rasmussen LJ, Singh KK. Nucleic Acids Res. 2003;31:3909–3917. doi: 10.1093/nar/gkg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripple MO, Henry WF, Rago RP, Wilding G. J Natl Cancer Inst. 1997;89:40–48. doi: 10.1093/jnci/89.1.40. [DOI] [PubMed] [Google Scholar]
- Simonnet H, Alazard N, Pfeiffer K, Gallou C, Beroud C, Demont J, et al. Carcinogenesis. 2002;23:759–768. doi: 10.1093/carcin/23.5.759. [DOI] [PubMed] [Google Scholar]
- Singh KK, Sigala B, Sikder HA, Schwimmer C. Nucleic Acids Res. 2001;29:1381–1388. doi: 10.1093/nar/29.6.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer A, Haendler B. Current Opinion in Drug Discovery & Development. 2003;6:702–711. [PubMed] [Google Scholar]
- Spelbrink JN, Li FY, Tiranti V, Nikali K, Yuan QP, Tariq M, et al. Nature Genetics. 2001;28:223–231. doi: 10.1038/90058. [DOI] [PubMed] [Google Scholar]
- Van Goethem G, Dermaut B, Lofgren A, Martin JJ, Van Broeckhoven C. Nat Genet. 2001;28:211–212. doi: 10.1038/90034. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Annu Rev Biochem. 1992;61:1175–1212. doi: 10.1146/annurev.bi.61.070192.005523. [DOI] [PubMed] [Google Scholar]
- Washington MT, Rosenberg AH, Griffin K, Studier FW, Patel SS. J Biol Chem. 1996;271:26825–26834. doi: 10.1074/jbc.271.43.26825. [DOI] [PubMed] [Google Scholar]
- Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Int J Cancer. 1994;57:406–412. doi: 10.1002/ijc.2910570319. [DOI] [PubMed] [Google Scholar]
- Zeviani M, Servidei S, Gellera C, Bertini E, DiMauro S, DiDonato S. Nature. 1989;339:309–311. doi: 10.1038/339309a0. [DOI] [PubMed] [Google Scholar]
- Zhang D, Mott JL, Chang SW, Denniger G, Feng Z, Zassenhaus HP. Genomics. 2000;69:151–161. doi: 10.1006/geno.2000.6333. [DOI] [PubMed] [Google Scholar]