

Summary
Protein phosphorylation is a ubiquitous mechanism for cellular signal propagation, and signaling network complexity presents a challenge to protein kinase substrate identification. Few targets of Polo-like kinases are known, despite their significant role in coordinating cell cycle progression. Here, we combine chemical genetic, bioinformatic, and proteomic tools for Polo-like kinase substrate identification. Monospecific pharmacological inhibition of budding yeast Polo-like kinase, Cdc5, resulted in a misaligned pre-anaphase spindle and subsequently delayed anaphase nuclear migration, revealing a novel Cdc5 function. A cellular screen for Cdc5 substrates identified Spc72, a spindle pole body (SPB) component and microtubule anchor required for nuclear positioning. Spc72 bound to the Cdc5 PBD in a mitosis-specific manner, was phosphorylated by Cdc5 in vitro, and demonstrated a loss of mitotic phosphorylation in vivo upon Cdc5 inhibition. Finally, an examination of Cdc5 binding by SPB-localized proteins expanded our knowledge of Cdc5 function at the SPB.
Introduction
Ensuring accurate chromosome segregation is fundamental to survival of a species. Temporally and spatially regulated signals are required to monitor and direct multiple cellular events during cell division. The Polo-like family of serine/threonine protein kinases (Plks) has emerged as an important class of cell cycle regulators that coordinate mitotic progression, with multiple roles from mitotic entry to cytokinesis [1]. Humans have four Plks (Plk1-4), of which Plk1 is most thoroughly characterized [1]. The budding yeast S. cerevisiae has a single Plk, Cdc5, with 49% identity to Plk1 in its kinase domain [2]. Cdc5 regulates multiple cellular functions, including progression through G2/M phase, cohesin cleavage at anaphase entry, and adaptation to the DNA damage checkpoint. Cdc5 also has an essential role in promoting mitotic exit and cytokinesis as part of two signaling networks called FEAR and MEN [2].
Despite the multiple mitotic functions of Cdc5, only a few of its substrates have been conclusively identified [3-6]. Cdc5 substrates are difficult to identify for several reasons. First, cell-cycle regulators are generally low abundance proteins, and both Cdc5 and its known substrates are present at less than 1500 copies per cell [7]. Second, characterized phosphorylation sites in Plk substrates have considerable sequence variation [8] and are of limited utility in identifying potential substrates. Third, Cdc5 is a particularly promiscuous kinase when used in in vitro reactions. Fourth, different cdc5-ts (temperature-sensitive) alleles have given rise to differing terminal phenotypes making the study of Cdc5 function in vivo complex [4].
The variation in phosphorylation site preference of Cdc5 suggests that alternative specificity determinants exist, such as temporal and spatial regulation. Indeed, Cdc5 levels are tightly regulated during with maximal activity in mitosis, due to both cell-cycle transcription and APC-mediated proteolysis [2]. Also, Cdc5 has a distinct localization pattern including the cytoplasmic face of the spindle pole body (SPB, the functional equivalent to the mammalian centrosome), chromosomes, and the bud neck (the future site of cytokinesis) [5, 9-11]. This localization is driven by a C-terminal region of Plks, called the Polo-box domain (PBD), as mutations in it severely disrupt the Cdc5 localization pattern [10]. The PBD is a phospho-serine/threonine binding module that targets Plks to their substrates after prior “priming” phosphorylation of the substrate by an upstream kinase [8, 12].
The importance of cellular localization for Cdc5 substrate specificity motivated us to develop an in vivo screen for identifying bona fide Cdc5 substrates. To accomplish this, we generated a mono-specific Cdc5 inhibitor using a chemical genetic approach, which allowed for the selective and temporal inhibition of Cdc5 in cells. Using this inhibitor, we found a novel role for Cdc5 in pre-anaphase spindle orientation and anaphase spindle migration. Candidate Cdc5 substrates were identified by bioinformatic filtering of the proteome using Cdc5 phosphorylation and PBD binding site preferences, as well as functional criteria. The candidates were screened by chemical inhibition of Cdc5 in vivo. This approach identified novel Cdc5 substrates, including the SPB component Spc72. The SPB projects cytoplasmic microtubules required for positioning of the nucleus, orientation of the mitotic spindle, and subsequent movement of one set of chromosomes across the predetermined cleavage plane at the bud neck [13]. Spc72 anchors cytoplasmic microtubules at the SPB and accordingly functions in nuclear position and spindle orientation, including anaphase spindle migration into the bud [14-17]. We demonstrate that Spc72 (along with other SPB components) binds the Cdc5 PBD. Thus, Cdc5 has a previously unidentified function at the SPB and in spindle positioning.
Results
CMK selectively inhibits an analog sensitive allele of Cdc5
To evaluate cellular roles of budding yeast Cdc5, we engineered Cdc5 to be selectively inhibited by a cell-permeable small molecule using a chemical genetic approach. This method involves introducing a space-creating mutation at the gatekeeper position coupled with a space-filling bulky derivative of the pyrazolopyrimidine (PP1) kinase inhibitor scaffold [18]. We previously found the cdc5-as1 allele (analog-sensitive) encoding the L158G gatekeeper mutation to poorly complement a cdc5-ts strain at restrictive (37°C) temperature [19]. However, Cdc5(L158G) had only a modest 6-fold reduction in kcat/Km [20], and when CDC5 was replaced at its endogenous locus with the cdc5-as1 allele, cdc5-as1 cells were viable with a 47% increase in strain doubling time at 30°C, indicating that Cdc5(L158G) is a functional kinase in the context of this study (data not shown). Since functional Cdc5 is required for cellular proliferation, we used growth inhibition to assess Cdc5 inactivation. Surprisingly, we were unable to obtain significant inhibition of Cdc5(L158G) using a variety of PP1 analogs despite the diverse collection of other protein kinases that have proven amenable to this approach [21] (data not shown). Thus, the ATP binding site of Cdc5 differs enough from other kinases that a new inhibitor strategy was necessary.
Fortunately, the Cdc5 active site contains a nonconserved cysteine (Cys96) homologous to a cysteine in the mammalian p90 RSK family kinases that was recently exploited for the rational design of selective, irreversible inhibitors (Figure 1A). These inhibitors are pyrrolopyrimidines containing either a chloromethylketone (CMK) or a fluoromethylketone (FMK) electrophile at the C-2 position, and like PP1, exploit a threonine (or smaller) gatekeeper residue [22]. We therefore hypothesized that these irreversible inhibitors would inactivate Cdc5(L158G) by virtue of the absence of a bulky gatekeeper residue and the presence of a naturally occurring Cys at position 96 (Figure 1B). Growth of the cdc5-as1 strain, but not the wild type strain (CDC5), was prevented by treatment with CMK, but not its parent pyrrolopyrimidine (scaffold), which does not contain the chloromethyl ketone electrophile (Figure 1C). Similarly, the in vitro IC50 of CMK for Cdc5(L158G) was 36 nM, compared with greater than 30 μM for wild type Cdc5 (Figure S2). Additionally, CMK did not inhibit growth of yeast containing a Cdc5 allele with both L158G and C96V mutations (Figure S1A). This suggests that inactivation of Cdc5(L158G) with CMK is mediated by irreversible covalent bond formation with Cys96. Notably, although a threonine gatekeeper residue is sufficient for inhibition of RSK by CMK [22], Cdc5 required a smaller gatekeeper residue (Figure S1B). Together, the cdc5-as1 strain and CMK provide a means to study cellular roles of Cdc5.
Figure 1. Analog-sensitive Cdc5 is inhibited by CMK.
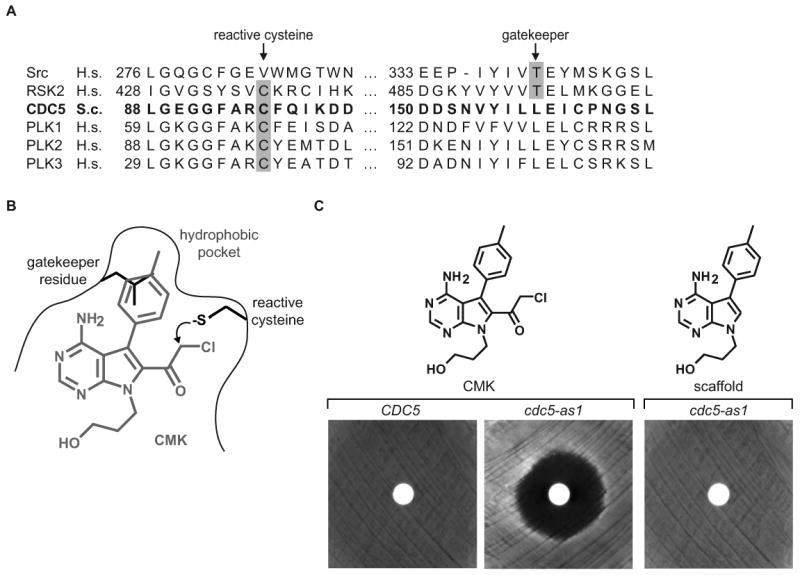
(A) Sequence alignment of kinase domain regions spanning the gatekeeper residue and the reactive cysteine. The Cdc5 sequence is in bold, and the specificity filters critical for RSK2 inhibition by CMK [22] are highlighted in gray.
(B) Chemical structure of CMK. CMK (in gray), with features of the kinase active site depicted, including a cysteine to react with the electrophilic chloromethyl ketone and a gatekeeper residue that controls access to a hydrophobic binding pocket. A predicted steric clash between the Cdc5 leucine gatekeeper residue and CMK is illustrated.
(C) Cell viability halo assay of wild type (CDC5) and cdc5-as1 yeast. Inhibition of cell growth in the region surrounding a disc spotted with 1 nmol CMK or scaffold molecule is observed only upon CMK application to cdc5-as1 (center), indicating a requirement for both the gatekeeper mutation and the electrophilic reactivity of CMK.
Cdc5 is required for proper spindle orientation and migration
We sought to determine the nature of the growth arrest caused by CMK addition to cdc5-as1 yeast. Wild type and cdc5-as1 strains were released from a G1 arrest (unbudded cells) into media containing increasing concentrations of CMK, and cell cycle progression was determined by microscopic observation of budding (Figure 2A). CMK exhibited a concentration-dependent first cell cycle mitotic arrest in the cdc5-as1 strain with an IC50 of 1.1 μM (Figure 2B). By contrast, up to 15 μM CMK had no effect on cell cycle progression in the wild type strain (Figure 2A). We examined in precise detail the arrest phenotype at 5 μM. Notably, arrested cells were large-budded with separated DNA masses and elongated spindles. Pds1 degradation occurred with similar kinetics in both strains, indicating that anaphase onset in budding yeast is unaffected by Cdc5 inhibition (Figure 2C). Strikingly, we also observed a high percentage of anaphase cdc5-as1 cells (23%) in which spindle elongation occurred entirely in the mother cell, rather than through the bud neck (Figures 2E and 2F). This was a rare event in anaphase wild type cells (2%) and implies a failure of the nucleus to migrate into the bud upon anaphase onset. Expression of wild type Cdc5 in the cdc5-as1 cells rescued this defect (4% anaphase spindles in mother), while expression of Cdc5 containing a defective Polo-box domain could not (17% anaphase spindles in mother). Inhibited cdc5-as1 cells were able to elongate their spindles to greater than 5 μm without spindle migration into the bud (Figure 2F). The spindles eventually recovered from this defect, and the cells later arrested in telophase with high Clb2 levels (Figure 2C) and DNA masses segregated properly into mother and daughter (Figure 2D).
Figure 2. CMK inhibition of Cdc5(L158G) leads to a first cell cycle anaphase arrest and delay in anaphase spindle migration.
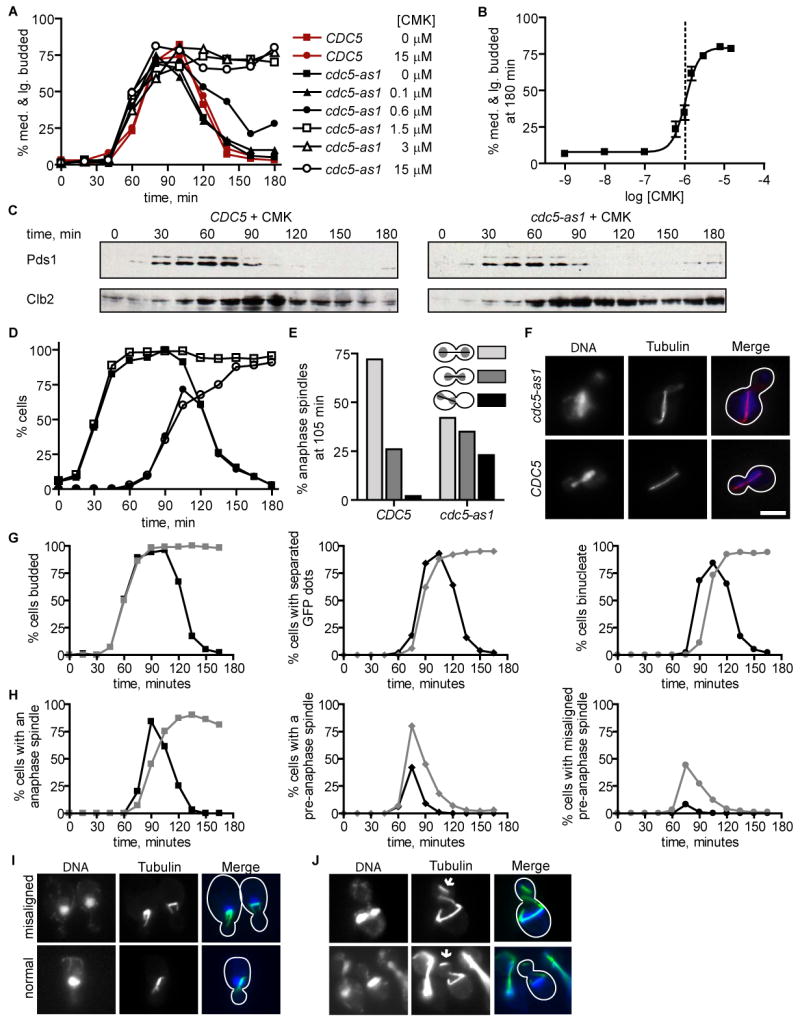
(A) CMK treatment causes of a first cell cycle arrest of cdc5-as1, but not wild type, cells in a dose-dependent manner. Time course of cell cycle synchronized CDC5 and cdc5-as1 cultures released from G1 arrest (unbudded) into the indicated concentrations of CMK (n = 200 cells for each point) is plotted. The y-axis represents the percentage of cells in S/G2 and M phases, as judged by the presence of a medium- to large-sized bud. A second cell cycle was prevented by re-arrest in the subsequent G1.
(B) cdc5-as1 cells arrest with an extracellular 50% inhibitory CMK concentration of 1.1 μM (dotted line, 95% confidence interval from 0.9 to 1.2 μM). The percent cdc5-as1 cells in S/G2 and M cell cycle phases at 180 min is shown for CMK-treated cultures prepared as in (A). Error bars represent standard errors of the mean for three experiments (n = 200 cells for each), and the data were fit to a sigmoidal dose response curve (R2 = 0.97).
(C) CMK-treated cdc5-as1 cells degrade Pds1 with wild type kinetics but maintain stabilized Clb2. Cell extracts from CDC5 and cdc5-as1 strains expressing Pds1-HA3 released from G1 into 5 μM CMK and re-arrested in the subsequent G1 were blotted for HA (Pds1) and Clb2.
(D) CMK-treated cdc5-as1 cells arrest as budded cells with segregated chromosomes. Budded cells (squares) and budded cells with DNA masses separated into mother cell and bud (circles) were quantified in CMK-treated CDC5 (closed symbols) and cdc5-as1 (open symbols) strains for the experiment shown in (C) (n = 100 cells per point).
(E) Mother cell localized anaphase spindles are transiently observed in CMK-treated cdc5-as1 cells. The percentage anaphase spindles with localization depicted (n = 100 cells) in CMK-treated cdc5-as1 and CDC5 cells at 105 min in the experiment shown in (C and D).
(F) Examples of CMK-treated cells with anaphase spindles (tubulin) observed at 90 min in the experiment shown in (C through E). The images show aberrant (cdc5-as1) and wild type (CDC5) anaphase spindle localization, but are not representative of all cells in the population. The scale bar represents 5 μm.
(G) CMK-treated cdc5-as1 cells have a delay in completing chromosome separation. CDC5 (black) and cdc5-as1 (gray) strains expressing tetR-GFP and containing a centromere-proximal tetO array, released from G1 into 5 μM CMK and re-arrested in the subsequent G1 were analyzed for budding (squares), separation of fluorescent GFP chromosomal dots (diamonds), and separation of DNA masses (circles) (n = 100 cells per point).
(H) CMK-treated cdc5-as1 cells have misaligned pre-anaphase spindles. Cells with an anaphase spindle (squares), with a pre-anaphase short spindle (diamonds), and with a misaligned pre-anaphase spindle (circles) in CMK-treated CDC5 (black) and cdc5-as1 (gray) strains were quantified for the experiment shown in (G) (n = 100 cells per point).
(I) Examples of cells with pre-anaphase spindles (tubulin) observed in the experiment shown in (G through H). The images show misaligned and normal pre-anaphase spindles, which are quantified in H.
(J) Detached cytoplasmic microtubules observed in a small number of CMK-treated cdc5-as1 cells. Examples of tubulin-stained cells observed in the experiment shown in (G through I) with detached microtubules indicated (arrows). The images represent only a small number of cells in the cdc5-as1 population. No detached microtubules were observed in CMK-treated CDC5 cells.
To futher clarify the phenotype resulting from Cdc5(l158G) inhibition, we introduced cdc5-as1 into cells in which a centromere-proximal chromosomal array is bound GFP [23], thus allowing visualization of sister chromatid separation. Consistent with the previously described role for Cdc5 in promoting Scc1 cleavage [24], we observe a slight delay in the separation of centromeres in CMK-treated cdc5-as1 cells, as monitored by the separation of centromere-proximal GFP arrays, and a protracted delay in the complete separation of DAPI-stained DNA masses, relative to CMK-treated CDC5 yeast (Figure 2G). Furthermore, a thorough analysis of the mitotic spindle with time revealed that short, pre-anaphase spindles were frequently misaligned in CMK-treated cdc5-as1 cells (55% of pre-anaphase spindles in CMK-treated cdc5-as1 versus 19% in CMK-treated CDC5 at 75 minutes) (Figure 2H). The pre-anaphase nucleus was correctly positioned at the bud neck (Figure 2I and data not shown), indicating that cytoplasmic microtubule guidance was intact. Whereas in cells with properly aligned pre-anaphase spindles only the bud-proximal SPB directs cytoplasmic microtubules into the bud, in cells with misaligned pre-anaphase spindles, cytoplasmic microtubules from both poles were directed into the budneck (Figure 2I). This implies that Cdc5 activity is required to prevent a daughter-directed force guiding cytoplasmic microtubules emanating from the mother bound SPB, as an active mother-directed force is not thought to exist prior to anaphase. As shown in Figure 2J, we also observed a small number of CMK-treated cdc5-as1 cells with detached cytoplasmic microtubules, which were not observed in CMK-treated CDC5 cells. Clearly, chemical Cdc5(L158G) inhibition results in pluripotent defects in cell cycle progression, including both previously described Cdc5 functions (sister chromatid separation and late anaphase progression), and a newly described Cdc5 function in properly positioning the mitotic spindle.
Cdc5 substrates are identified through a candidate-based in vivo screen
A role for Cdc5 in spindle positioning was not previously known and cannot be easily explained by its previously identified substrates. Proper anaphase spindle migration required both Cdc5 catalytic activity and Polo-box domain function, suggesting that the critical substrates would likely contain sequences recognized by both the Polo-box domain and kinase domain (Figure 3A). Therefore, a candidate approach to identifying substrates was designed to incorporate both of these elements (Figure 3B).
Figure 3. A candidate-based in vivo screen identifies Spc72 as a Cdc5 substrate.
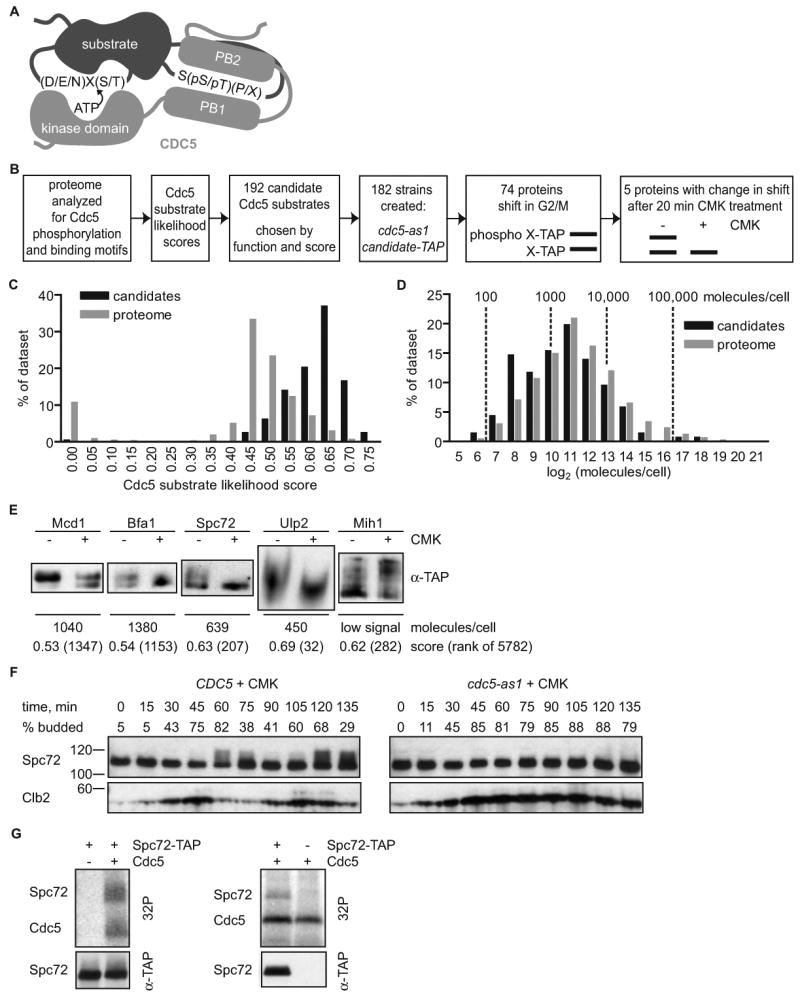
(A) Schematic representation of substrate recognition by Cdc5. The substrate is depicted to contain a Cdc5 phosphorylation motif, (D/E/N)X(S/T) [58], and a binding motif, S(pS/pT)(P/X), which binds the Cdc5 Polo-boxes (PB1 and PB2) [27]. X represents any amino acid, p represents phosphorylation.
(B) Approach to screening for substrates phosphorylated by Cdc5 in vivo.
(C) Bioinformatic mining of the yeast proteome for candidate Cdc5 substrates. The distribution of assigned Cdc5 substrate likelihood scores is shown for 192 Cdc5 candidate substrates compared with the proteome, with low scores reflecting likely candidates.
(D) Cdc5 candidate substrates are enriched in low abundance proteins. Normalized distribution of protein abundance [7] comparing candidate Cdc5 substrates to the entire proteome. The data set means were statistically different (P=0.0003) by unpaired t-test. Proteins without abundance values [7] were excluded from the analysis.
(E) Result of the screen. The gel mobility of five TAP-tagged candidate substrates is altered upon Cdc5 inhibition with 10 μM CMK (+) as compared with a DMSO control treatment (-).
(F) Inhibition of Cdc5 with CMK eliminates the mitotic Spc72 upshift observed in a synchronized cell cycle. Cell cycle progression of CDC5 or cdc5-as1 cells expressing Spc72-TAP released from G1 into 10 μM CMK is indicated by budding index and Clb2 western blot. Spc72 is visualized by anti-TAP western blot (Spc72).
(G) In vitro phosphorylation of Spc72 by Cdc5. Immunopurified TAP-tagged Spc72 was incubated with [γ-32P]ATP, with and without purified Cdc5 (left panels). Phosphorylation (32P) and Spc72 western blot (α-TAP) are observed. No Spc72 phosphorylation is seen when Cdc5 is added to a mock pulldown reaction (untagged Spc72, - Spc72-TAP, right panels).
Briefly, the roughly six thousand predicted yeast proteins were searched with the Scansite profile scanning algorithm [25, 26] for Cdc5 binding and phosphorylation sequence motifs, generating a “Cdc5 substrate likelihood score” for each protein. The Cdc5 binding motif was determined previously by oriented library screening [27]. A predicted Cdc5 phosphorylation motif was generated by combining information on Cdc5, Plk1, and Plk3 substrates [8] with the published kinase phosphorylation motif of Plk1 [8, 28], since both Plk1 and Plk3 can substitute for Cdc5 function in yeast [2]. The distribution of scores is represented in Figure 3C, with high scores representing likely substrates. Notably, the group of best-scoring potential Cdc5 substrates was enriched in low abundance proteins relative to the entire proteome, requiring a sensitive test of candidate phosphorylation (Figure 3D). Cdc5 substrate likelihood scores combined with functional criteria led us to choose 192 total candidate substrates (Table S1). Among the proteins identified by this approach were the known Cdc5 substrates Mcd1, Bfa1, and Swe1 [4, 5, 24]. Swe1 is the only of the three previously shown to directly bind the PBD [29] and was ranked 12th highest overall (Table S1).
To test these candidate substrates for Cdc5-dependent phosphorylation in vivo, we used a library of strains each encoding a candidate substrate, at its endogenous locus, fused to a tandem affinity purification (TAP) tag, which provided an ultra-sensitive handle for immunological detection [7]. The phosphorylation state of the TAP-tagged candidates was monitored by gel shift, a straightforward and well-established method for determining the phosphorylation state of many proteins in vivo [30]. We focused on the candidates that exhibited a gel shift in a mitotic arrest, when Cdc5 is active, but not in a G1 arrest, when Cdc5 is inactive [31] (Figure 3B). Mitotically arrested cells were treated for 20 minutes with 10 μM CMK or DMSO (control). To avoid false positive phosphorylation, cdc5-as1 was the sole source of Cdc5, and CMK was added for only 20 minutes to limit indirect effects of Cdc5 inhibition.
Of the 74 proteins that displayed a gel mobility shift when isolated from mitotic cells, five exhibited reproducible Cdc5-dependent changes in gel mobility (Figure 3E and Table S1). Significantly, of the three known Cdc5 substrates included in our screen, Bfa1 and Mcd1 [4, 24] downshifted upon Cdc5 inhibition. Similarly, Spc72 and Ulp2 also downshifted upon Cdc5 inhibition. Spc72 is a previously described phosphoprotein, but the upstream kinase had not been identified [32]. Intriguingly, Cdc5 inhibition resulted in an upshift of Mih1. The chemical nature of this shift is unclear, since it is unusual for loss of phosphorylation to result is a slower migrating form.
The SPB component Spc72 is a Cdc5 substrate and binding partner in mitosis
Spc72 has several hallmarks of a Cdc5 substrate important for the regulation of spindle positioning. It contains both Cdc5 PBD-binding and phosphorylation motifs, exhibits Cdc5-dependent phosphorylation, and is a SPB component with a previously known function in nuclear positioning [14-17]. Spc72 phosphorylation was examined through the cell cycle to further explore this connection. In wild type cells, Spc72 phosphorylation peaked in late mitosis, just as Clb2 levels begin to decrease (Figure 3F). This Spc72 mobility shift was prevented when Cdc5 was inhibited by treating cdc5-as1 cells with CMK (Figure 3F). Finally, purified 6xHis-tagged Cdc5 directly phosphorylated immunoprecipitated TAP-tagged Spc72 in vitro (Figure 3G), suggesting that Spc72 is a direct substrate of Cdc5.
Spc72 was previously reported to bind to Cdc5 [33, 34], and we investigated the nature of this interaction. To determine whether binding could be mediated by phospho-dependent interactions between Spc72 and the Cdc5 Polo-box domain, we performed pulldown experiments for Spc72 in mitotic extracts using either the PBD region of wild type Cdc5, or a PBD variant (PBD*) in which we introduced mutations corresponding to conserved residues in Plk1 required for phosphospecific binding [27] (Figure 4A). Spc72 efficiently bound to the PBD, relative to a 5% input control, but failed to bind to PBD*, indicating that prior Spc72 phosphorylation by a priming kinase creates a Cdc5 binding site (Figure 4B). Consistent with this, the interaction was also regulated by cell cycle stage, since Spc72 bound the PBD less well in a G1 extract than in a mitotic extract (Figure 4C). Spc72 contains several minimal PBD binding sites and a site at S232 with the 74th best Scansite score out of 6833 matches to the PBD binding site in the yeast proteome. Mutation of S231 and S232 at this site significantly reduced binding to the PBD (Figure 4E), suggesting that Spc72 and Cdc5 bind directly. Residual binding to this Spc72 mutant may reflect contributions from other sites in Spc72 or Cdc5 PBD interactions with Spc72 binding partners.
Figure 4. Spc72 binds to the Cdc5 Polo-box domain in a cell cycle and phosphospecific manner.
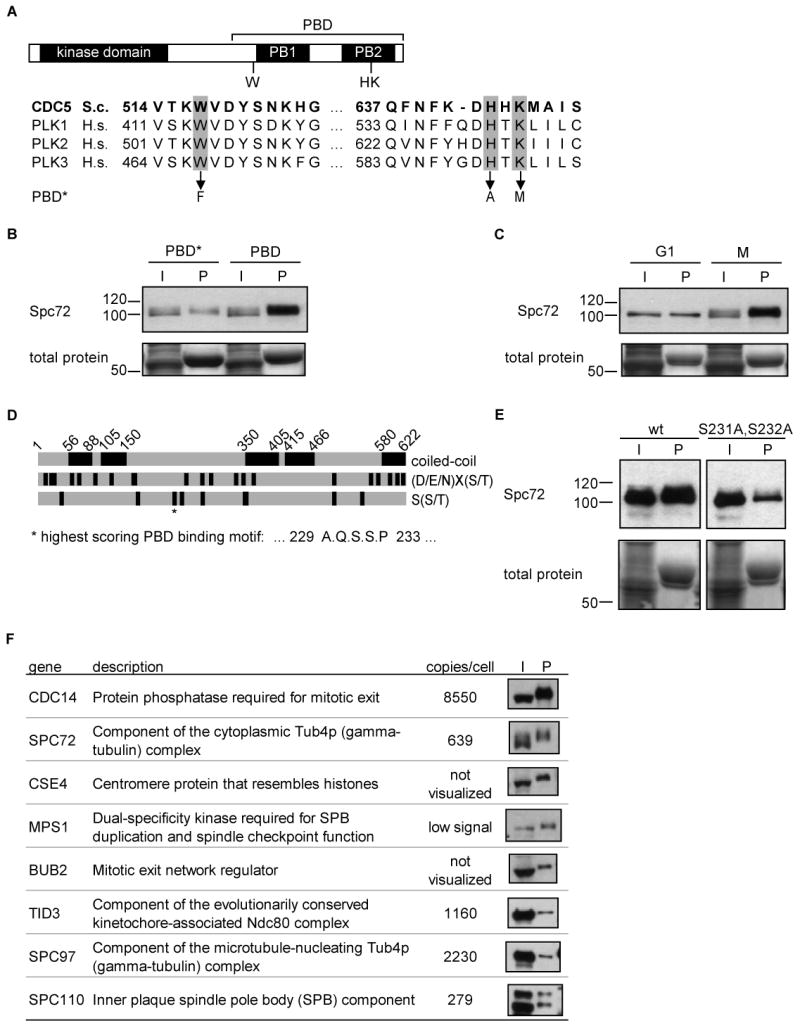
(A) Cdc5 domain structure indicating the kinase domain and Polo-box domain (PBD), and sequence alignment showing three conserved PBD residues required for phosphopeptide binding by Plk1 (highlighted in gray). The PBD* mutant contains mutations of the highlighted Cdc5 residues to the amino acids indicated for elimination of phospho-specific motif binding.
(B) Spc72 is bound by the Cdc5 PBD, and PBD* has reduced Spc72 binding. Anti-TAP (Spc72) western blot indicates Spc72 present in the input mitotic cell extract (I) or pulled down (P) with GST-PBD (PBD) or GST-PBD* (PBD*). Total protein staining indicates the amount of GST-fusion protein in pulldown lanes (P). I = 5% input, P = pulldown.
(C) Cdc5 preferentially binds mitotic Spc72. Wild type PBD pulldowns from mitotic cell extracts (as in (B)) or G1 phase cell extracts were probed for Spc72.
(D) Domain structure of Spc72 including coiled-coils predicted by COILS [59] and sites matching Cdc5 phosphorylation, (D/E/N)X(S/T) [58], and PBD binding, S(S/T) [27], minimal motifs. The best scoring PBD binding motif in Spc72 is indicated (*).
(E) Mutation of consensus Cdc5 binding residues in Spc72 reduces binding to the PBD. PBD binding to Spc72 or Spc72(S231A,S232A) as in (B).
(F) SPB proteins efficiently bound to the Cdc5 PBD. Anti-TAP western blot indicates tagged SPB proteins present in the input mitotic cell extract (5%, I) or pulled down (P) with GST-PBD. Selected SPB proteins efficiently detected in the pulldown are shown, along with functional information and protein abundance [7].
To identify other mediators of Cdc5 binding at the SPB, we tested the ability of the Cdc5 PBD to bind 90 proteins that were either annotated as SPB-localized or identified as SPB-localized in a proteomic study [35]. Notably, PBD binders at the SPB may include Cdc5 substrates involved in anaphase spindle migration, since the PBD is required for both Cdc5 localization to the SPB [10] and proper anaphase spindle migration. SPB-localized proteins were examined for PBD binding in extracts of mitotically arrested cells (Table S2). Components of the SPB cytoplasmic face, Spc72, Cnm67, and Nud1, bound to the PBD, as did known Spc72-binding proteins, Spc97, Stu2, and Kar1 (Table S2). The most efficient PBD binders, determined by the ratio of input and PBD-bound protein levels, are shown in Figure 4F. These included mitotic exit signaling network components, Cdc14 and Bub2, kinetochore proteins, Cse4 and Tid3, and the SPB component, Spc110 (Figure 4F). Significantly, Spc72 was among the most efficient binders, implying both that it directly interacts with the PBD and is a major site of Cdc5 binding at the SPB (Figure 4F).
Discussion
Here we report a chemical genetic and bioinformatic approach to investigate the cellular roles of the yeast Plk, Cdc5. Specific chemical inhibition of Cdc5 by CMK both revealed a new role for Cdc5 in mitotic spindle positioning and facilitated identification of Spc72 as a Cdc5 substrate. Analysis of Spc72 regulation supports a model in which a kinase primes Spc72 for binding to Cdc5 at the SPB in mitosis, resulting in direct Spc72 phosphorylation by Cdc5. Interestingly, the phenotype resulting from loss of Spc72, although more severe than from Cdc5 inhibition, is consistent with Spc72 being a Cdc5 effector in spindle positioning [14-17].
Chemical genetic engineering of Cdc5 posed a significant challenge, as Cdc5 was unexpectedly incompatible with our previously extensively validated approach. Unlike other engineered kinases, including Plk1 [36], Cdc5 was not inhibited by non-electrophilic PP1 analogs. Although a Plk1 kinase domain structure was recently published [37], no Cdc5 structure is available. Our results indicate that access to the hydrophobic pocket in Cdc5 is reduced relative to other protein kinases we have studied. Similar to RSKs, Plks have a nonconserved active site cysteine, which provided a handle for potent irreversible Cdc5 inhibition. Although Cdc5 is the only budding yeast Plk, since the cysteine is conserved across the family, the chemical genetic inhibition strategy should be applicable to studying any Plk family member. Several potent Plk inhibitors have recently been described, and, although some exhibit selectivity for Plk family kinases, they do not discriminate between the closely related mammalian Plk isoforms [38-43]. Chemical genetic methods present a unique pharmacological capability to study the function of a single Plk isoform in the presence of other active family members [36]. One extension of the strategy described here could be in studying mammalian Plk isoforms when irreversible kinase inhibition is preferable, such as for active site labeling [44].
Small molecule addition rapidly inhibits only the catalytic activity of the kinase, both avoiding compensatory mechanisms occurring in the absence of the kinase and allowing for study of temporally resolved processes, and has often revealed new aspects of kinase function missed by genetic approaches [45]. In addition to delaying chromosome segregation and blocking mitotic exit and cytokinesis, inhibition of Cdc5(L158G) by CMK resulted in aberrant pre-anaphase spindle orientation, followed by a delay in anaphase spindle migration into the bud. The defect was corrected eventually, and the spindle moved into the neck, either due to redundant activities or by forces generated by the elongating spindle itself. Since cytoplasmic microtubules from both poles of misaligned pre-anaphase spindles were bud-directed, Cdc5 activity is likely required to prevent a pre-anaphase daughter-directed pulling force. Interestingly, other components of the FEAR pathway act during anaphase to either prevent a daughter-directed force or activate a mother-directed force, a similar but temporally and phenotypically distinct function from Cdc5 [46]. Our chemical genetic approach to Cdc5 inhibition provides a tool to probe the molecular mechanisms coordinating spindle position with the cell cycle. In yeast, movement of the anaphase spindle into the bud is required for proper chromosome segregation due to a predetermined cleavage plane. In higher organisms, spindle position is critical for developmental cell fate decisions [13]. Previous studies with cdc5-ts alleles revealed only the essential Cdc5 functions, as they are more conducive to endpoint assays. Also, elevated temperatures required for inactivating ts alleles accelerate the cell cycle, potentially masking transient effects such as a role in spindle positioning. Thus, this novel Cdc5 function was revealed only by the pharmocologic modulation of Cdc5 activity.
Our substrate screen combined several recent technological advances to meet the stringent requirement of low abundance Cdc5 substrate identification in vivo. Identification of direct Cdc5 targets necessitated selective and rapid Cdc5 inactivation for a brief period, and chemical genetics ideally suited this purpose. Unlike mass spectrometry approaches, which suffer from limited dynamic range [7], utilization of the TAP-tag proteomic library facilitated the visualization of even low abundance phosphoproteins by gel shift. Phosphorylation reactions occurred only in a cellular context, and, crucially, both analog-sensitive Cdc5 and the candidates were at endogenous levels. A candidate approach made this screen feasible, and while the Cdc5 binding and phosphorylation motifs are degenerate, a useful set of candidates was obtained by utilizing the extra information contained in weighting amino acids at each position based on their preference by Cdc5. Functional criteria are a useful filter for substrate sets [47] and were also incorporated into the selection process. However, we were limited by the proportion of phosphoproteins that demonstrated significant phosphorylation shifts. Further advances in mass spectrometric approaches that quantitatively detect differences in low stoichiometry phosphopeptides in a complex mixture are necessary to improve this approach [48].
Although the screen was not limited to these proteins, we were interested in finding substrates consistent with the Cdc5 spindle position phenotype, such as molecular motors required to generate a bud-directed pulling force, cortical determinants that interact with cytoplasmic microtubules penetrating into the bud, or proteins that stabilize or nucleate cytoplasmic microtubules [13]. Of the Cdc5 substrates identified, only Spc72 has a known role in spindle position, which is similar, but not identical, to Cdc5(L158G) inhibition. Loss of Spc72 causes reduced cytoplasmic microtubules, resulting in defects in cytoplasmic microtubule-dependent processes, such as nuclear position and spindle orientation, and a delay in mitotic exit due to anaphase spindles that have not properly migrated into the bud [14-17]. Several lines of evidence indicate that Spc72 is a bona fide Cdc5 substrate. Cdc5 phosphorylated Spc72 in vitro, Cdc5 activity was required for the phosphorylation shift of Spc72 in vivo, and the Cdc5 PBD bound Spc72 in a cellular extract. Further, binding of Spc72 to the Cdc5 PBD was dependent on phosphoselectivity elements, cell cycle stage, and a binding site at S232 in Spc72. These results are consistent with a model in which a kinase primes Spc72 for binding and subsequent phosphorylation by Cdc5. The unidentified kinase would likely localize to the SPB and be active in mitosis but not G1, and the sequence surrounding S232 indicates phosphorylation by a proline-directed kinase, such as cyclin-dependent kinases or mitogen activated protein kinases.
The multitude of potential Cdc5 phosphorylation sites in Spc72 (Figure 4D) and low endogenous levels of these proteins, coupled with residual binding to Spc72(S231A,S232A), presented a technical challenge to studying the downstream effect of Cdc5 regulation of Spc72. Cell cycle regulated Spc72 phosphorylation has been implicated in shuttling of the γ-tubulin complex between distinct substructures of the SPB, and it is tempting to speculate that Cdc5 is the kinase responsible [32, 49]. If so, Cdc5 inhibition will provide a system to further study this event. Additionally, regulation of Spc72 binding to microtubule regulators would be consistent with the Cdc5 inhibition phenotype. The microtubule motor, Kar9, is asymmetrically recruited to a single SPB prior to anaphase, symmetric Kar9 SPB localization results in a similar phenotype to Cdc5(L158G) inhibition [50]. Although Kar9 is not known to bind Spc72, we observed weak binding of Bim1 and Clb4, reported determinants of Kar9 localization, to the Cdc5 PBD (Table S2). Additionally, Stu2, a regulator of microtubule dynamics binds Spc72 [17, 51] and the Cdc5 PBD (Table S2), as well as Kar9 [52], and is genetically placed into the Kar9 pre-anaphase spindle orientation pathway [53]. Conditional depletion of Stu2 results in fewer and less dynamic cytoplasmic microtubules, yielding a misaligned pre-anaphase spindle [53]. When regions of Spc72 that bind Stu2 are deleted, cytoplasmic microtubules are reduced in number and have altered dynamics, with excessively long and stabilized cytoplasmic microtubules observed in anaphase [51]. In G1 they appear normal, but often detach from the SPB prior to anaphase. We observed detached cytoplasmic microtubules in a small number of cdc5-as1 cells treated with CMK, but not in CMK-treated wild type cells. Additionally, we found the Cdc5 PBD to interact with several SPB components, indicating the potential for modulation of Cdc5 function at the SPB by a number of redundant mechanisms.
We also identified Ulp2 and Mih1 as novel Cdc5 substrates, and both are involved in Cdc5-related functions. Mih1 is the yeast homolog of Cdc25, which is phosphorylated by Plks during mitotic entry [2]. Ulp2 is a desumoylase with roles in centromeric cohesion and recovery from checkpoint arrest [54, 55]. Advantageously, many kinetochore proteins were incorrectly annotated as SPB-localized and included in the PBD binding screen. Of these, Cse4 and Tid1 bound the PBD, potentially reflecting a role for Cdc5 at the yeast kinetochore, consistent with other Plks [1]. At the SPB, we find the PBD binds to Spc110, the nuclear equivalent of Spc72 [56], and therefore Cdc5 may also regulate nuclear microtubule dynamics. Likewise, Plk1 spindle functions include centrosome maturation by phosphorylation of Nlp, which (like Spc72 and Spc110) nucleates microtubules by binding to the γ-tubulin ring complex [1].
The low abundance and importance of Cdc5 localization for substrate specificity demanded assays that preserved cellular context and endogenous expression levels. Consequentially, like Cdc5 itself, many of its interactors identified here, including Spc72, are present at less than 1500 molecules per cell. Furthermore, the in vivo method for identifying kinase-dependent phosphoproteins can potentially be extended to additional kinases functioning in this pathway or in other dynamic cell processes.
Significance
Selective small-molecule inhibition of protein kinases is a powerful tool for studying the biological context in which they function. This work presents a novel chemical genetic approach to protein kinase inhibition, applied to the Polo-like kinase Cdc5, thereby expanding the set of kinases amenable to mono-specific pharmacological regulation. We demonstrate the value of the chemical genetic approach for studying Cdc5 function on both the phenotypic and molecular level, revealing a previously unappreciated function for Cdc5 in spindle positioning and identifying the cellular Cdc5 target, Spc72. Contrasting with Polo-like kinases other organisms, it is not likely that Cdc5 is required for bipolar spindle formation in yeast. However, these results provide an indication that Cdc5 may indeed regulate microtubule function, an avenue for further study. This study additionally presents the first stringent, in vivo screen for Polo-like kinase substrates and highlights the utility of chemical genetics for studying rapid signal transduction events in vivo. The importance of designing substrate screens in which low abundance proteins are evaluated is underscored by the identification of a low abundance substrate, Spc72. Together, these results not only illuminate budding yeast Polo-like kinase function, but also form a template for kinase substrate identification.
Experimental Procedures
Plasmids, strains, and yeast methods
A CDC5 (YMR001C) genomic fragment in pRS315 (p315-CDC5) and pRS306 (p306-CDC5) were gifts of J. Charles. The L158G mutation was introduced into p315-CDC5 and p306-CDC5, generating p315-cdc5-as1 and p306-cdc5-as1. The PBD from p315-CDC5 was cloned into pGEX-3X (GE Healthcare) to produce pGST-PBD, and W517F,H641A,K643M mutations were introduced to make pGST-PBD*. The PBD mutations were cloned into p315-CDC5 from pGST-PBD*. CDC5 was cloned into pFastBacHT-A (Invitrogen) to produce pFastBac-CDC5. SPC72 (YAL047C) was amplified from W303 genomic DNA and recombined with a URA3-marked 2 μm plasmid bearing the GAL1 promoter and TAP tag (E.K. O'Shea, Harvard University) to produce pSPC72. The S231A,S232A mutations were introduced into pSPC72 to produce pSPC72-AA. Mutations were introduced by QuikChange site-directed mutagenesis (Stratagene). Open reading frames were fully sequenced after amplification. SPC72 plasmids encoded an I302N difference from published sequence, which was confirmed in a W303 strain.
Standard yeast media and genetic techniques were used [57], except as specifically described. Strains were MATa and W303 (Figures 1 and 2, and Figure S1) or S288C (isogenic with EY1274, all other Figures) strain background. cdc5-as1 was introduced at the CDC5 locus by two-step gene replacement, except as follows. A MATα strain derived from EY1274 carrying a can1Δ::MFA1p-LEU2 selectable marker (E.K. O'Shea, Harvard University) was transformed with a marker fusion PCR product generated by amplification of cdc5-as1 and K.lactis URA3 genes. The entire cdc5-as1 gene was sequenced in the resultant integrated strain, which was then crossed to selected MATa HIS3 marked TAP-tagged library strains [7] (Open Biosystems) using a manual 96-pinning tool (V&P Scientific). After sporulation, TAP-tagged MATa haploids were selected on synthetic media lacking histidine, arginine, leucine, and uracil and containing 50 μg/ml canavanine (Sigma).
Cell cycle synchronization was obtained by G1 arrest with alpha factor for 3 hours, followed by washing to release (0 min time point). Alpha factor was readded at 70-80 minutes when arresting in the following G1. Cell cycle time courses were at 25 °C except for in Figure 3F, which was at 30 °C. Alpha factor was used at 1 μg/ml for bar- cells (Figure 2) and 20 μg/ml for bar+ cells (all other experiments). Mitotic arrests were obtained by nocodazole treatment at 15 μg/ml for 3 hours.
Protein expression and purification
6xHis-Cdc5 was prepared as follows. Bacmid was produced from pFastBac-CDC5 and transfected into Sf9 insect cells using the Bac-to-Bac Baculovirus expression system (Invitrogen). Sf9 cells were harvested after a 2 day infection with Pass 3 baculovirus at 2×106 cells/ml, and cell extract prepared by douncing in Cdc5 lysis buffer (CLB: 2 5 mM HEPES pH 7.4, 300 mM NaCl, 10% glycerol, 50 mM NaF, 50 mM beta-glycerophosphate, 35 nM okadaic acid) with protease inhibitors (1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatin). The cleared extract was applied to a HiTrap Chelating HP column (Amersham) chelated with CoCl2. After washing, peak 6xHis-Cdc5 containing fractions eluted with CLB containing an imidazole gradient were combined.
GST-PBD and GST-PBD* were expressed from pGST-PBD and pGST-PBD* in BL21-CodonPlus(DE3)-RIL cells (Stratagene). Cleared extracts prepared in GST lysis buffer (GLB: 50 mM Tris, pH 8.0, 1 M NaCl, 1% NP40, 1mM PMSF, 1 μg/ml aprotinin, 10 nM DTT) were loaded onto a glutathione agarose (Sigma) column. After washing, GST fusion proteins were eluted in 50 mM Tris, pH 8.0, 250 mM KCl, 5 mM reduced glutathione. Glutathione was subsequently removed using a PD-10 desalting column (GE Healthcare).
Kinase assays
For Cdc5 kinase assays, Spc72-TAP was isolated from cell extract by pulldown with IgG sepharose (GE Healthcare) or rabbit IgG (Sigma) coupled to M-270 epoxy dynabeads (Dynal) and incubated in kinase buffer (25 mM HEPES, pH 8.0, 60 mM KCl, 15 mM MnCl2, 100 μg/ml BSA, 80 nM microcystin, 1mM DTT, 100 μM 200 μCi/ml [γ-32P]ATP) in the absence or presence of 100 ng purified baculovirus expressed 6xHis-Cdc5. 32P incorporation was visualized on a Typhoon PhosphorImager (GE Healthcare), and images were processed using ImageQuant software (GE Healthcare).
Western blotting and pulldowns
Blots were probed with anti-TAP (Open Biosystems), anti-Clb2 (kind gift D. Kellogg), and anti-HA (16B12, Covance) primary antibodies and anti-rabbit (Amersham) and anti-mouse (Pierce) HRP-conjugated secondary antibodies. Total protein was stained using Ponceau S (Sigma).
For phosphorylation gel shifts, cell extracts were prepared in urea lysis buffer as described [30], and 5 μg was loaded on 5%, 7.5%, or 10% Criterion gels (Biorad) for western blotting.
For PBD binding assays, extracts from TAP-tagged library strains were prepared in TAP lysis buffer (TLB: 20 mM HEPES, pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.2% NP40, 50 mM NaF, 50mM beta-glycerophosphate, 100 μM Na3VO4, 20 nM microcystin, Complete EDTA-free protease inhibitors (Roche), 1 μg/ml pepstatin) with 1 mM DTT. Cleared extracts were incubated with purified recombinant GST-PBD or GST-PBD* and glutathione agarose (Sigma) in TLB with 10 mM DTT for 1 hour at 4 degrees. Bead bound complexes were washed and analyzed by western blot for presence of the TAP-tagged protein. For Figure 4E, TAP-tagged Spc72 and Spc72(S231A,S232A) were expressed from pSPC72 and pSPC72-AA in an untagged strain. Expression was induced for 1 hour in mitotically arrested cells with 2% galactose, resulting in tagged protein levels lower than that of endogenously tagged Spc72 (data not shown).
Microscopy and image processing
Cells were fixed with 3.7% formaldehyde overnight at 4 degrees. DNA was stained with 1 μg/ml DAPI. Tubulin was stained with YOL 1/34 (Abcam) and Cy3-conjugated anti-Rat secondary (Jackson Laboratories). Images were acquired at 63x magnification on a Zeiss Axiovert 200 M microscope using Axiovision software. Images were cropped and minimum and maximum pixel values adjusted in Photoshop.
Bioinformatic and statistical analysis
Candidate Cdc5 substrates were initially selected based on information available in September 2003 as follows. First, proteins involved in Cdc5-dependent processes were identified by Gene Ontology (GO) term searches and selected if they contained a minimal phosphorylation or PBD motif. Second, yeast proteins in SWIS-PROT were evaluated with the Scansite algorithm, which assigns final scores (Sf) reflecting how well the query sequence matches a position-specific scoring matrix representing the optimal Cdc5 PBD or kinase motif, normalized to all proteins in the database [25, 26]. These range from 0 (perfect match) to 1 (complete lack of even a minimal binding or phosphorylation motif). The Cdc5 substrate likelihood score was defined as 1-[0.5 (SfPBD+SfKin)], where SfPBD and SfKin were the final Scansite scores for the individual PBD and kinase motifs, respectively. Highest scoring proteins were selected and the remaining proteins considered in turn by score, selecting those with cell cycle function and nuclear or cytoplasmic localization. Table S1 contains updated scoring and functional information from October 2006.
Annotations used for identifying SPB-localized proteins are from SGD (Hong, E.L., et al. “Saccharomyces Genome Database”, http://www.yeastgenome.org, July 2005).
Curve fitting, regression analysis, and statistical tests were performed with Prism 4 (GraphPad software).
Supplementary Material
Figure S1. Additional analysis of Cdc5 sequence requirements for inhibition by CMK
(A) Cell viability of strains grown on 10 μM CMK. Growth of strains requiring the indicated plasmid-borne CDC5 alleles for survival, showing that both the space creating L158A mutation and a cysteine at position 96 are required in combination for inhibition by CMK.
(B) CMK inhibition of Cdc5 alleles. cdc5-1 ts strains bearing a plasmid expressing Cdc5 or the mutant indicated was plated and 10 nmol scaffold molecule (left) or CMK (right) applied to the filter disc. CMK dependent growth inhibition of only the cdc5-L158G and cdc5-L158A cells was apparent after 3 days at restrictive temperature (37 degrees).
Figure S2. IC50 for CMK inhibition of Cdc5-as1. 10 ng purified recombinant His6-Cdc5 (open circles) or His6-Cdc5-as1 (closed circles) was pre-incubated at room temperature for 5 minutes with 10 μg alpha-casein (dephosphorylated) in 25 mM HEPES, pH 8.0, 60 mM KCl, 15 mM MnCl2, and 100 ug/ml BSA in the presence or absence of the indicated concentrations of CMK. After 5 minutes, [γ-32P] ATP (to 10 μM, 100 μCi / ml) and DTT (to 1 mM) were added to begin the kinase reaction. After 30 minutes at room temperature, 10 μl of each reaction solution was spotted on a nitrocellulose disk, washed 4 times (5 min each) in 1 M NaCl, 1% phosphoric acid, dried under a heat lamp 15 min, and counted with a scintillation counter. Data was analyzed using GraphPad Prism software. The IC50 for CMK inhibition of Cdc5-as1 was determined by non-linear regression (R2 = 0.98) to be 36 nM (95% confidence interval, 20 nM to 62 nM).
Acknowledgments
We thank EK O'Shea and J Weissman for sharing of reagents prior to publication and AN Snead for comments on the manuscript.
This work was supported by National Institute of General Medical Sciences grant GM53270 to DOM, NIH grant GM60594 and a Burroughs-Wellcome Career Development Award to MBY, NIH grant GM71434 to JT, NCI training grant 5T32CA09270 and a NSF pre-doctoral fellowship to JLP, a HHMI pre-doctoral fellowship to DML, and NIH grants EB001987 and AI44009 to KMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 2.Lee KS, Park JE, Asano S, Park CJ. Yeast polo-like kinases: functionally conserved multitask mitotic regulators. Oncogene. 2005;24:217–229. doi: 10.1038/sj.onc.1208271. [DOI] [PubMed] [Google Scholar]
- 3.Alexandru G, Zachariae W, Schleiffer A, Nasmyth K. Sister chromatid separation and chromosome re-duplication are regulated by different mechanisms in response to spindle damage. Embo Journal. 1999;18:2707–2721. doi: 10.1093/emboj/18.10.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu F, Wang Y, Liu D, Li Y, Qin J, Elledge SJ. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell. 2001;107:655–665. doi: 10.1016/s0092-8674(01)00580-3. [DOI] [PubMed] [Google Scholar]
- 5.Sakchaisri K, Asano S, Yu LR, Shulewitz MJ, Park CJ, Park JE, Cho YW, Veenstra TD, Thorner J, Lee KS. Coupling morphogenesis to mitotic entry. Proc Natl Acad Sci U S A. 2004;101:4124–4129. doi: 10.1073/pnas.0400641101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoshida S, Kono K, Lowery DM, Bartolini S, Yaffe MB, Ohya Y, Pellman D. Polo-like kinase Cdc5 controls the local activation of Rho1 to promote cytokinesis. Science. 2006;313:108–111. doi: 10.1126/science.1126747. [DOI] [PubMed] [Google Scholar]
- 7.Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 8.Lowery DM, Lim D, Yaffe MB. Structure and function of Polo-like kinases. Oncogene. 2005;24:248–259. doi: 10.1038/sj.onc.1208280. [DOI] [PubMed] [Google Scholar]
- 9.Shirayama M, Zachariae W, Ciosk R, Nasmyth K. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. Embo Journal. 1998;17:1336–1349. doi: 10.1093/emboj/17.5.1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song S, Grenfell TZ, Garfield S, Erikson RL, Lee KS. Essential function of the polo box of Cdc5 in subcellular localization and induction of cytokinetic structures. Molecular and Cellular Biology. 2000;20:286–298. doi: 10.1128/mcb.20.1.286-298.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hornig NC, Uhlmann F. Preferential cleavage of chromatin-bound cohesin after targeted phosphorylation by Polo-like kinase. Embo J. 2004;23:3144–3153. doi: 10.1038/sj.emboj.7600303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elia AE, Cantley LC, Yaffe MB. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science. 2003;299:1228–1231. doi: 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 13.Pearson CG, Bloom K. Dynamic microtubules lead the way for spindle positioning. Nat Rev Mol Cell Biol. 2004;5:481–492. doi: 10.1038/nrm1402. [DOI] [PubMed] [Google Scholar]
- 14.Hoepfner D, Schaerer F, Brachat A, Wach A, Philippsen P. Reorientation of mispositioned spindles in short astral microtubule mutant spc72Delta is dependent on spindle pole body outer plaque and Kar3 motor protein. Mol Biol Cell. 2002;13:1366–1380. doi: 10.1091/mbc.01-07-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knop M, Schiebel E. Receptors determine the cellular localization of a gamma-tubulin complex and thereby the site of microtubule formation. Embo J. 1998;17:3952–3967. doi: 10.1093/emboj/17.14.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Soues S, Adams IR. SPC72: a spindle pole component required for spindle orientation in the yeast Saccharomyces cerevisiae. J Cell Sci. 1998;111(Pt 18):2809–2818. doi: 10.1242/jcs.111.18.2809. [DOI] [PubMed] [Google Scholar]
- 17.Chen XP, Yin H, Huffaker TC. The yeast spindle pole body component Spc72p interacts with Stu2p and is required for proper microtubule assembly. J Cell Biol. 1998;141:1169–1179. doi: 10.1083/jcb.141.5.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 19.Zhang C, Kenski DM, Paulson JL, Bonshtien A, Sessa G, Cross JV, Templeton DJ, Shokat KM. A second-site suppressor strategy for chemical genetic analysis of diverse protein kinases. Nat Methods. 2005;2:435–441. doi: 10.1038/nmeth764. [DOI] [PubMed] [Google Scholar]
- 20.Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hubner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, Katayama CD, Hedrick SM, Shokat KM. A semisynthetic epitope for kinase substrates. Nat Methods. 2007;4:511–516. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bishop AC, Kung CY, Shah K, Witucki L, Shokat KM, Liu Y. Generation of monospecific nanomolar tyrosine kinase inhibitors via a chemical genetic approach. Journal of the American Chemical Society. 1999;121:627–631. [Google Scholar]
- 22.Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka T, Fuchs J, Loidl J, Nasmyth K. Cohesin ensures bipolar attachment of microtubules to sister centromeres and resists their precocious separation. Nat Cell Biol. 2000;2:492–499. doi: 10.1038/35019529. [DOI] [PubMed] [Google Scholar]
- 24.Alexandru G, Uhlmann F, Mechtler K, Poupart MA, Nasmyth K. Phosphorylation of the cohesin subunit Scc1 by Polo/Cdc5 kinase regulates sister chromatid separation in yeast. Cell. 2001;105:459–472. doi: 10.1016/s0092-8674(01)00362-2. [DOI] [PubMed] [Google Scholar]
- 25.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol. 2001;19:348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- 27.Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- 28.Nakajima H, Toyoshima-Morimoto F, Taniguchi E, Nishida E. Identification of a consensus motif for Plk (Polo-like kinase) phosphorylation reveals Myt1 as a Plk1 substrate. J Biol Chem. 2003;278:25277–25280. doi: 10.1074/jbc.C300126200. [DOI] [PubMed] [Google Scholar]
- 29.Asano S, Park JE, Sakchaisri K, Yu LR, Song S, Supavilai P, Veenstra TD, Lee KS. Concerted mechanism of Swe1/Wee1 regulation by multiple kinases in budding yeast. Embo J. 2005;24:2194–2204. doi: 10.1038/sj.emboj.7600683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- 31.Cheng L, Hunke L, Hardy CFJ. Cell cycle regulation of the Saccharomyces cerevisiae polo-like kinase cdc5p. Molecular and Cellular Biology. 1998;18:7360–7370. doi: 10.1128/mcb.18.12.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gruneberg U, Campbell K, Simpson C, Grindlay J, Schiebel E. Nud1p links astral microtubule organization and the control of exit from mitosis. Embo J. 2000;19:6475–6488. doi: 10.1093/emboj/19.23.6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 34.Park JE, Park CJ, Sakchaisri K, Karpova T, Asano S, McNally J, Sunwoo Y, Leem SH, Lee KS. Novel functional dissection of the localization-specific roles of budding yeast polo kinase Cdc5p. Mol Cell Biol. 2004;24:9873–9886. doi: 10.1128/MCB.24.22.9873-9886.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 36.Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, Jallepalli PV. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci U S A. 2007;104:4383–4388. doi: 10.1073/pnas.0701140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kothe M, Kohls D, Low S, Coli R, Cheng AC, Jacques SL, Johnson TL, Lewis C, Loh C, Nonomiya J, Sheils AL, Verdries KA, Wynn TA, Kuhn C, Ding YH. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry. 2007;46:5960–5971. doi: 10.1021/bi602474j. [DOI] [PubMed] [Google Scholar]
- 38.Gumireddy K, Reddy MV, Cosenza SC, Boominathan R, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;7:275–286. doi: 10.1016/j.ccr.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, Noro N, Furuta M, Emmitte KA, Gilmer TM, Mook RA, Jr, Cheung M. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol Cancer Ther. 2007;6:450–459. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Shreder KR, Gai W, Corral S, Ferris DK, Rosenblum JS. Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chem Biol. 2005;12:99–107. doi: 10.1016/j.chembiol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 41.McInnes C, Mazumdar A, Mezna M, Meades C, Midgley C, Scaerou F, Carpenter L, Mackenzie M, Taylor P, Walkinshaw M, Fischer PM, Glover D. Inhibitors of Polo-like kinase reveal roles in spindle-pole maintenance. Nat Chem Biol. 2006;2:608–617. doi: 10.1038/nchembio825. [DOI] [PubMed] [Google Scholar]
- 42.Peters U, Cherian J, Kim JH, Kwok BH, Kapoor TM. Probing cell-division phenotype space and Polo-like kinase function using small molecules. Nat Chem Biol. 2006;2:618–626. doi: 10.1038/nchembio826. [DOI] [PubMed] [Google Scholar]
- 43.Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ. BI 2536, a Potent and Selective Inhibitor of Polo-like Kinase 1, Inhibits Tumor Growth In Vivo. Curr Biol. 2007;17:316–322. doi: 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- 44.Blair JA, Rauh D, Kung C, Yun CH, Fan QW, Rode H, Zhang C, Eck MJ, Weiss WA, Shokat KM. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat Chem Biol. 2007;3:229–238. doi: 10.1038/nchembio866. [DOI] [PubMed] [Google Scholar]
- 45.Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 46.Ross KE, Cohen-Fix O. A role for the FEAR pathway in nuclear positioning during anaphase. Dev Cell. 2004;6:729–735. doi: 10.1016/s1534-5807(04)00128-5. [DOI] [PubMed] [Google Scholar]
- 47.Dephoure N, Howson RW, Blethrow JD, Shokat KM, O'Shea EK. Combining chemical genetics and proteomics to identify protein kinase substrates. Proc Natl Acad Sci U S A. 2005;102:17940–17945. doi: 10.1073/pnas.0509080102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morandell S, Stasyk T, Grosstessner-Hain K, Roitinger E, Mechtler K, Bonn GK, Huber LA. Phosphoproteomics strategies for the functional analysis of signal transduction. Proteomics. 2006;6:4047–4056. doi: 10.1002/pmic.200600058. [DOI] [PubMed] [Google Scholar]
- 49.Pereira G, Grueneberg U, Knop M, Schiebel E. Interaction of the yeast gamma-tubulin complex-binding protein Spc72p with Kar1p is essential for microtubule function during karyogamy. Embo J. 1999;18:4180–4195. doi: 10.1093/emboj/18.15.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liakopoulos D, Kusch J, Grava S, Vogel J, Barral Y. Asymmetric loading of Kar9 onto spindle poles and microtubules ensures proper spindle alignment. Cell. 2003;112:561–574. doi: 10.1016/s0092-8674(03)00119-3. [DOI] [PubMed] [Google Scholar]
- 51.Usui T, Maekawa H, Pereira G, Schiebel E. The XMAP215 homologue Stu2 at yeast spindle pole bodies regulates microtubule dynamics and anchorage. Embo J. 2003;22:4779–4793. doi: 10.1093/emboj/cdg459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller RK, Cheng SC, Rose MD. Bim1p/Yeb1p mediates the Kar9p-dependent cortical attachment of cytoplasmic microtubules. Mol Biol Cell. 2000;11:2949–2959. doi: 10.1091/mbc.11.9.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kosco KA, Pearson CG, Maddox PS, Wang PJ, Adams IR, Salmon ED, Bloom K, Huffaker TC. Control of microtubule dynamics by Stu2p is essential for spindle orientation and metaphase chromosome alignment in yeast. Mol Biol Cell. 2001;12:2870–2880. doi: 10.1091/mbc.12.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bachant J, Alcasabas A, Blat Y, Kleckner N, Elledge SJ. The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol Cell. 2002;9:1169–1182. doi: 10.1016/s1097-2765(02)00543-9. [DOI] [PubMed] [Google Scholar]
- 55.Li SJ, Hochstrasser M. The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol. 2000;20:2367–2377. doi: 10.1128/mcb.20.7.2367-2377.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jaspersen SL, Winey M. The budding yeast spindle pole body: structure, duplication, and function. Annu Rev Cell Dev Biol. 2004;20:1–28. doi: 10.1146/annurev.cellbio.20.022003.114106. [DOI] [PubMed] [Google Scholar]
- 57.Guthrie C, Fink GR. Guide to yeast genetics and molecular and cell biology. San Diego, Calif.: Academic Press; 2002. [Google Scholar]
- 58.Brar GA, Kiburz BM, Zhang Y, Kim JE, White F, Amon A. Rec8 phosphorylation and recombination promote the step-wise loss of cohesins in meiosis. Nature. 2006;441:532–536. doi: 10.1038/nature04794. [DOI] [PubMed] [Google Scholar]
- 59.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Additional analysis of Cdc5 sequence requirements for inhibition by CMK
(A) Cell viability of strains grown on 10 μM CMK. Growth of strains requiring the indicated plasmid-borne CDC5 alleles for survival, showing that both the space creating L158A mutation and a cysteine at position 96 are required in combination for inhibition by CMK.
(B) CMK inhibition of Cdc5 alleles. cdc5-1 ts strains bearing a plasmid expressing Cdc5 or the mutant indicated was plated and 10 nmol scaffold molecule (left) or CMK (right) applied to the filter disc. CMK dependent growth inhibition of only the cdc5-L158G and cdc5-L158A cells was apparent after 3 days at restrictive temperature (37 degrees).
Figure S2. IC50 for CMK inhibition of Cdc5-as1. 10 ng purified recombinant His6-Cdc5 (open circles) or His6-Cdc5-as1 (closed circles) was pre-incubated at room temperature for 5 minutes with 10 μg alpha-casein (dephosphorylated) in 25 mM HEPES, pH 8.0, 60 mM KCl, 15 mM MnCl2, and 100 ug/ml BSA in the presence or absence of the indicated concentrations of CMK. After 5 minutes, [γ-32P] ATP (to 10 μM, 100 μCi / ml) and DTT (to 1 mM) were added to begin the kinase reaction. After 30 minutes at room temperature, 10 μl of each reaction solution was spotted on a nitrocellulose disk, washed 4 times (5 min each) in 1 M NaCl, 1% phosphoric acid, dried under a heat lamp 15 min, and counted with a scintillation counter. Data was analyzed using GraphPad Prism software. The IC50 for CMK inhibition of Cdc5-as1 was determined by non-linear regression (R2 = 0.98) to be 36 nM (95% confidence interval, 20 nM to 62 nM).