

Abstract
The polymerization of RecA on individual double-stranded DNA molecules is studied. A linear DNA (λ DNA, 48.5 Kb), anchored at one end to a cover glass and at the other end to an optically trapped 3-μm diameter polystyrene bead, serves as a template. The elongation caused by RecA assembly is measured in the presence of ATP and ATP[γS]. By using force extension and hydrodynamic recoil, a value of the persistence length of the RecA–DNA complex is obtained. In the presence of ATP, the polymer length is unstable, first growing to saturation and then decreasing. This suggests a transient dynamics of association and dissociation for RecA on a double-stranded DNA, the process being controlled by ATP hydrolysis. Part of this dynamics is suppressed in the presence of ATP[γS], leading to a stabilized RecA–DNA complex. A one-dimensional nucleation and growth model is presented that may account for the protein assembly.
Keywords: genetic recombination, optical tweezers, DNA–protein interactions, nucleation and growth
RecA protein plays an essential role in bacterial recombination and DNA repair and is ubiquitous in nature (1–14, 29). RecA or a homolog of RecA is found in all biological cells so far examined. RecA is an example of a protein having a strong structural effect on DNA. During genetic processes, the structural modification of DNA is a key step in sequence recognition and specificity. From early electron microscopy studies, RecA is found to cooperatively bind to DNA; the resulting complex is observed to be stretched by a factor of 1.5 with respect to the naked form and has a twist of 20° per bp instead of 35° in double-stranded DNA (dsDNA) (4–8). An important function of the protein is the DNA strand exchange reaction, whereby a single-stranded DNA (ssDNA) replaces the homologous strand on a dsDNA. Biochemical and electron microscopy studies have suggested that, during strand exchange reactions, RecA polymerizes both on ssDNA and dsDNA in the presence of ATP. In in vitro experiments, RecA monomers are also found to polymerize in the absence of DNA, and its self assembly is reminiscent of the assembly of actin into F-actin and tubulin into microtubules. The depolymerization of RecA is thought to occur via the hydrolysis of ATP (1–3, 9–14, 29). The wide range of roles played by the small RecA protein (molecular mass 37.8 kDa) makes its study interesting. Although an extensive documentation exists, a complete understanding of the molecular mechanisms of its function remains elusive.
In this paper, one directly measures the kinetics of polymerization of RecA on a single dsDNA molecule and the resulting changes in the entropic elasticity of DNA. Single molecule measurements of the role of ATP hydrolysis in RecA polymerization are performed. The physical parameters of the experiment are T = 37°C, pH 6.8 ± 0.2. For this study, λ-phage DNA is used. Single DNA molecules are attached at one end to a glass cover slide and at the other to a bead of 3-μm diameter. By using an optical tweezer to trap the bead, one can extend the length of DNA away from its equilibrium configuration (15–18). To measure the ensuing tension in the DNA, the location of the bead within the trap is obtained by collecting the backscattered light onto a quadrant detector (18). First, force-extension measurements are performed, as a function of time, during the various stages of the RecA polymerization along DNA (19, 20, 30). This allows one to deduce the changes in the persistence length, A, of the dsDNA molecule. We find that for complete polymerization of RecA on DNA, A is about four times larger than for the naked DNA molecule. Second, one studies the recoil dynamics of a DNA molecule from the fully stretched state back to equilibrium by turning off the optical trap (21). Finally, the length of the DNA–RecA complex is measured at small time intervals both for the case of RecA–ATP and RecA—adenosine 5′-[γ-thio]triphosphate (ATP[γS]) reactions. One thus monitors the detailed kinetics of the reaction showing that it can be described by a nucleation and growth model. For RecA–ATP, nucleation is relatively slow, and the growth is fast; the opposite is observed for RecA—ATP[γS]; namely, nucleation is faster than growth. This reaction is also followed, in parallel, at the macroscopic level via a 32P isotope ATPase assay from which the [ATP]/[ADP] ratio can be monitored throughout the experiment. In the ATP reaction, the length of the polymer increases first to 1.5 its original contour length and, after some time, reverses slowly to its original size, presumably by RecA depolymerization. This reversal starts at an [ATP]/[ADP] ratio of about 5 and ends at a value of about 0.25 and thus is clearly associated with ATP hydrolysis. On the other hand, the dynamics of the ATP[γS] reaction lacks the depolymerization phase.
METHODS
Force-Extension Measurement by Using Backscattering and Optical Tweezers.
A schematic of the experiment is shown in Fig. 1. Light from a near infrared laser diode (power 150 mW, wavelength 830 nm) is focused by using an infinity-corrected Zeiss Neofluar objective (×100, 1.3 numerical aperture, oil immersion) to construct the laser trap. Collinear with the infrared laser beam, light from a red laser (8 mW laser, 633 nm) is used to scatter light from a DNA tethered dielectric polystyrene bead (3 μm in diameter) confined in the trap. The backscattered light from the particle is collected by using the same objective and is then focused onto a quadrant detector. Force extension measurements are recorded by moving the piezo-stage [Physik Instrumente (Auburn, MA); dynamic range 30 μm and 10-nm minimum step size] and simultaneously recording the displacement of the tethered bead. The trapping stiffness is ≈0.05 ± 0.01 pN/nm for a laser power of 100 mW. The details of the measurement setup and calibration of the optical trap have been described elsewhere (18).
Figure 1.

Schematic of the experimental setup.
DNA Attachment by Using a Low pH Method.
Tethered beads are prepared by attaching one end of λ-DNA (Promega) to a cover slide and the other end to a 3-μm latex bead (Polysciences) by using a low-pH method (22). Five μl (≈0.5 μgm/μl) of λ DNA, a 48.5-Kb molecule 16.5 μm in length, is first incubated together with 1 μl (≈106 beads/μl) of beads and 400 μl of PBS buffer (pH 6) for 15 min at room temperature (rt). This way the bead–DNA link is obtained. To link the other end of the DNA to the glass, 40 μl of the solution is pipetted into a cell (6-mm radius, 2-mm height) and incubated for 24 h (rt). After this preparation, beads can be either stuck to the glass, freely floating, or tethered. Because the last two possibilities are indistinguishable when viewed under the microscope (both undergo Brownian motion), it is desirable to reduce the fraction of free beads. This can be achieved either by washing a few times or by inverting the sample. In the latter case, the free beads fall because of gravitation (ρlatex = 1.05 g/ml and so it takes 2 h to cross the 2-mm height of the cell). Typically one in 20 beads found on the sample cell are tethered with a single DNA molecule. The tethered attachment is irreversible, and therefore the pH of the sample cell can be changed. By pulling the bead with the laser trap, one checks whether: (i) the bead is tethered; (ii) the length of the tether is in the right range; and (iii) the length of the tether is the same in all directions. This way one selects a bead with one DNA attached excluding the possibility of multiple tethers (asymmetry) or of several λ-DNA being attached head to tail (different length). For the experiment, we choose only a bead connected to a single DNA attached at its ends.
RecA/DNA Reactions and ATP Assay.
The sample-cell final volume is about 100 μl. All the experiments are carried out at pH 6.8 ± 0.2, 150 mM PBS and temperature T = 37°C. RecA protein comes from an Escherichia coli strain MM294 that carries an overexpressed RecA gene from E. coli (New England Biolabs). The final sample concentrations, in addition to PBS, are RecA = 10 μM, ATP (or ATP[γS]) = 1 mM, MgCl2 = 20 mM, DTT = 20 mM, Tris⋅HCl = 60 mM. For the ATP assay (Fig. 6a), in parallel to the single molecule measurement, small volumes (1 μl step) are removed from the sample reaction containing [γ-32p]ATP at 10-min intervals. Each 1-μl reaction volume is added to 5 μl of stop solution aliquots (20 mM EDTA and 1% SDS) to quench the reaction. Hydrolysis of ATP was assayed by thin layer chromatography by using polyethyleneimine-cellulose plates. Reading off the corresponding radioactivity levels from the plate (analyzed by PhosphorImager from Molecular Dynamics) one obtains the [ATP]/[ADP] ratio throughout the reaction.
Figure 6.

(a) The disassembly of RecA from DNA in the presence of ATP (bullets). The curve represents the best fit of Eq. 10. (b) [ATP]/[ADP] ratio as obtained from the 32P radioactive ATPase assay (bullets). The line represents the best exponential fit.
Model for Nucleation and Growth in One Dimension.
We treat this problem as a one-dimensional analog of three-dimensional crystal growth (23, 24). The two relevant parameters are the rate of nucleation n (per unit time per unit length) and the rate of growth of an individual nucleus v. The following two cases have to be considered: (i) the case of low nucleation rate, 1/(nL0) > L0/v, and (ii) that of high nucleation rate, 1/(nL0) < L0/v, where L0 is the length of the DNA.
(i) Single Nucleus Case.
In the case of low nucleation rate, the process of binding is limited by the initiation of a single nucleus. The expectation time for the formation of a nucleus is tn = 1/(nL0). Once formed, the nucleus grows at constant speed v and covers the entire DNA filament within a time tg = L0/v. Thus, typically the fraction φ of DNA covered with RecA remains zero during a time interval of about tn and then grows linearly between tn and tn + tg (Fig. 5a) until it reaches maximal extension.
Figure 5.

Comparison of the relative change in the length of DNA during the assembly of RecA on DNA in the presence of (a) ATP (bullets) and (b) ATP[γS] (bullets). In a, the line corresponds to a linear fit to the growth phase of the ATP assembly. In b, the curve represents the best fit of Eq. 7 to the data. A schematic illustration of the corresponding mechanisms is shown in the insets of both a and b.
(ii) Multiple Nuclei Case.
In the case of large n (or low v), there are multiple nuclei growing simultaneously. To understand the way in which the fraction φ(t) of DNA covered with RecA changes with time, one needs to calculate the time dependence of the number N(t) of growing nuclei. New nuclei are formed in the parts of DNA free from RecA with the rate nL0(1 − φ(t)). Moreover, nuclei stop growing when their ends collide, each collision corresponding to the merging of two nuclei such that N(t) decreases by one. The rate of collisions can be estimated as follows: the “density” of growth fronts in the free part of DNA is 2N/(L0(1 − φ)). Therefore, the probability per unit time for each growing nucleus to undergo a collision is 2Nv/(L0(1 − φ)) that leads to an overall collision rate of N2v/(L0(1 − φ)). In other words, N(t) is changing according to
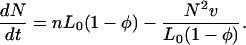 |
1 |
The growth rate of the part of DNA covered with RecA is proportional to N(t)
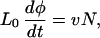 |
2 |
and together with the initial conditions N(0) = 0, φ(0) = 0, the above equations yield
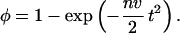 |
3 |
Thus, the solution depends only on a single parameter nv. The characteristic feature of Eq. 3 is a slowing down of the growth at high RecA coverage because of collisions of growth fronts. We verified Eq. 3 by simulating the process of nucleation and growth for different conditions. The details of the simulation are beyond the scope of this paper. We note, however, that our mean-field calculation agrees very well with the averaged growth curve. The spread of the individual growth curves around the average allows, in principle, to separate the nucleation rate and the growth rate. Further experiments will allow to deduce this distribution.
RESULTS AND DISCUSSION
Force-Extension Curve.
The schematic of the experiment is shown in Fig. 1. The force required to extend the coil by a certain amount z is measured, where z = r − r0 , r is the position of the DNA end attached to the bead and r0 corresponds to the end attached to the cover glass. This force, FDNA(z), is obtained by measuring the displacement of the bead from the center of the optical trap, d. The relation between FDNA and d is determined by the shape of the effective potential that describes the effect of the trap, V(d). Moreover, near the center of the trap, d = 0, this potential can be approximated by a harmonic well, that is,
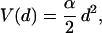 |
and the corresponding stiffness, α, is measured from the range of thermal fluctuations of a bead that is not tethered. In turn, one obtains the value of d by measuring the backscattered light from the trapped bead, by using a second laser and a quadrant detector. The difference in the light intensity that reaches the quadrants is proportional to d. The reading from the detector is calibrated by using a bead stuck to the coverslip and displacing it in steps of 10 nm, by using the piezo-driven sample stage (see Methods). The piezo stage is also used to vary the value of the extension. It takes 2 min to obtain a full FDNA(z) curve.
During the RecA polymerization, the DNA molecule is kept under the pull of a constant 6-pN force exerted by the optical trap, which is enough to reduce the entropic conformations of DNA (15). The activation barrier for RecA binding to DNA is lowered when the molecule is stretched, leading to an acceleration of the polymerization process (19, 20, 30). In the unstretched state, one finds that, aside from the change in the polymerization time scales, the static properties are the same as those of the stretched state. We therefore will present only FDNA(z) curves obtained when polymerization takes place in the stretched state of DNA.
Two studies of the FDNA(z) curve were done; namely, one where the polymerization of RecA on DNA takes place in the presence of ATP (Fig. 2a) and a second where ATP[γS] is used instead (Fig. 2b).
Figure 2.

The force exerted by DNA as a function of the extension at the various stages of RecA assembly. (a) ATP, (b) ATP[γS]. Curves for naked DNA (circles) and DNA fully covered with RecA (diamonds) are shown for both polymerization conditions. In a, the force-extension behavior for partially covered DNA at t2 = 15 min (squares) and the one obtained after depolymerization at t4 = 300 min (triangles) are also shown.
For ATP we present four force-extension curves at times t1 to t4. Time t1 corresponds to the start of polymerization, t2 (15 min) to the middle of the growth phase, t3 (30 min) to full RecA coverage, and t4 (300 min) to complete depolymerization where DNA has returned to its naked form. The most striking feature of Fig. 2a is the variation in the contour length, L0, such that L1 = 15 μm, L2 = 18 μm, L3 = 23 μm, and L4 = L1. In addition, one observes a decrease in the slope of the FDNA(z) curve for z ≪ L0 between t1 and t3, which corresponds to an increase in the persistence length, A (see Eq. 4). We shall refer to this slope as the entropic stiffness of DNA. The FDNA(z) curves corresponding to t1 and t4 are identical within experimental error.
A second experiment was performed in which the polymerization of RecA on DNA occurs in the presence of ATP[γS] rather than ATP. The resulting force-extension curves for the naked DNA (t1 = 0) and for the fully covered DNA (t2 = 30 min) are shown in Fig. 2b. Because ATP[γS] is nonhydrolyzable, at t > t2 we observe a stabilized RecA–DNA complex and correspondingly an unchanging FDNA(z) curve. Within experimental error, there is no difference between the fully covered force-extension curves resulting from the ATP and ATP[γS] reactions.
To model the force extension-curves for naked DNA, Marko and Siggia (25) used a worm-like chain model. A good approximation to their result is
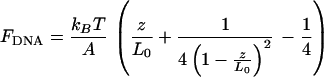 |
4 |
where kB is the Boltzmann constant, T is the temperature, z the extension, L0 the contour length, and A the persistence length. Whereas the first linear term in Eq. 4 dominates in the z ≪ L0 regime, the second term describes the sharp rise in the force that occurs where z approaches L0. Eq. 4 fails when z > 0.97 L0. Eq. 4 provides a qualitative description for both the naked and fully covered DNAs. Accordingly, the change in the persistence length is determined by extracting the slope from the range z < 10 μm. The values of Ar/An, (where Ar is the persistence length of DNA–RecA complex and An, the naked DNA one) for fully covered DNA in the presence of ATP or ATP[γS] has a lower bound value equal to 4 (Ar = 0.21 μm). This value is smaller than the one measured from electron microscopy experiments (7). A quantitative comparison with electron microscopy studies will require fitting the force extension data to an elastic model of the RecA–DNA complex.
Recoil Experiments.
The relaxation dynamics of the DNA–bead system is monitored via image processing of the video recording at 10 frames/sec. The DNA is first stretched to its full length, L0, by pulling the optically trapped bead. Next, the trapping laser is turned off, allowing the DNA to relax to its equilibrium configuration, pulling along the 3-μm bead that must overcome fluid drag. The time scale for the motion of the bead, τb ≈ 3s, is much larger than the equilibration time for the DNA (the Zimm time), τz ≈ 0.1s, and therefore the DNA relaxation is quasistatic. That is, during relaxation DNA is passing only through equilibrium stretched states (21). Because inertia is negligible, the static force of the DNA in each such state, FDNA, is balanced by the Stokes drag, FStokes,
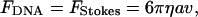 |
5 |
where η is the viscosity, a is the radius of the bead, and v is the velocity of the bead. One observes that the dynamics of fully covered DNA (in the presence of ATP[γS]) is significantly slower than that for the naked DNA (see Fig. 3), indicating that the former has a larger persistence length. One can use Eq. 4 for FDNA to show that a simple rescaling of the variables makes the recoil dynamic dependent only on A. Replacing v by dz/dt in Eq. 5 and rescaling both the extension, z′ = z/L0 and the time, t′ = t/L0, the differential equation for z′(t′) becomes dependent only on the persistence length A,
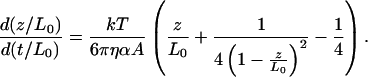 |
6 |
Moreover, the value of t′ during which the DNA relaxes to some fraction of its length, z′, t′z′, is proportional to the persistence length, t′z′ = c(z′) A. In Fig. 3, the lower curve corresponds to three different naked DNA molecules with different L0 values, and within experimental error they coincide. On the other hand, the upper curve is that of a DNA covered with RecA following an ATP[γS] reaction. From the ratio of the measured recoil time scales for both the naked DNA and the fully covered case the relative change in A is obtained. We find that the value of the ratio Ar/An is 2.5 ± 0.3. This value is even smaller than the one obtained by force extension measurement. This might be caused by imperfect coverage, as this dynamical experiment is particularly sensitive to it. The resolution in the length measurements is ±0.2 μm (which amounts to 588.2 bp of DNA ≈ 1.2% of λ-DNA). The imperfect coverage we are referring to is of this order.
Figure 3.

The dynamics of recoil of DNA-bead initially stretched by a 6-pN force. Both extension and time are normalized by the contour length (L) of the DNA. Even though the naked DNA curves (bullets, triangles and squares) correspond to slightly different lengths, in the normalized variables they coincide within experimental error. The recoil is much slower when DNA is fully covered with RecA-ATP[γS] (diamonds).
Kinetics of RecA Assembly and Disassembly.
The rate at which RecA polymerizes can be obtained by measuring the variation in the length of the tether, L(t), that is expected to vary between L0 and Lmax ≈ 1.5L0, as observed in electron microscopy (4). We locate a bead that is tethered to the cover slide via a single λ-DNA molecule, and at t = 0 we add RecA with either ATP or ATP[γS]. The value of L(t) is measured by pulling the bead, by using the optical trap, to the point of escape along the horizontal axis on both sides of the equilibrium point. Accordingly, we define L(t) as the half distance between two opposite escape points at a trapping power that corresponds to a force of 6 pN. The measurement is performed at time intervals that vary between 30 sec and 5 min, depending on the rate of variation of L(t), and in between the tether is stretched to the brink of escape. The experiments are ended after several hours when L(t) has clearly stopped changing, reaching Lf .
In the dynamics of L(t) in the presence of ATP, one can distinguish four qualitatively different regimes (Fig. 4). First, we observe a lag phase during which L(t) = L0, and which typically extends for about 22 min, and this value fluctuates with experiment. Next, between 22 min and about 33 min, there is a growth phase during which L(t) steeply rises to Lmax. A third regime follows in the time range of (33 min, 70 min), during which L(t) stays, unchanged, aside from small fluctuations. We refer to this as the stationary phase. Finally, for t > 70 min, L(t) decreases such that at the end of the run it returns to the original length, Lf = L0. Because this decrease is a consequence of the gradual loss of RecA, we coin this regime the dissociation phase. The detachment of RecA from DNA is associated with ATP hydrolysis. A control experiment was performed, with ATP replaced by ATP[γS]. As expected, the dissociation phase was absent. Furthermore, the nucleation lag time was reduced, and the growth kinetic was found to be different.
Figure 4.

The relative change in the length of DNA during the assembly and disassembly of RecA on DNA in the presence of ATP (bullets).
Clearly, ATP plays a central role in the mechanism of polymerization and depolymerization of RecA on dsDNA. Indeed, RecA requires an ATP molecule to bind to the DNA, and in the first step a RecA-ATP complex is formed. Next, either these complexes or polymeric filaments of those bind to the DNA at several locations that play the role of nucleation sites. From each of these sites, growth fronts emerge where the RecA–ATP complex is significantly more likely to bind than elsewhere. A competing reaction is the DNA-dependent hydrolysis of ATP, whereby a RecA–ATP complex becomes a RecA–ADP complex that may dissociate from DNA if ATP does not quickly replace the ADP. Whereas in the growth phase the DNA binding is the dominating process, in the dissociation phase it is the hydrolysis that dominates. This is shown as we perform in parallel a 32P isotope ATPase assay where for a fraction of the ATP the P atom that is released during hydrolysis to form ADP is replaced with the radioactive 32P isotope (26). One obtains the [ATP]/[ADP] ratio, RTD, throughout the assembly process. Because the rate of DNA-independent hydrolysis is negligible, this ratio can be viewed as an indicator for the extent of RecA polymerization on DNA throughout the sample. For the DNA molecule of Fig. 4, we find that the beginning of depolymerization corresponds to an [ATP]/[ADP] ratio of about 5, while the end occurs around 0.25.
A Model of Nucleation and Growth.
The single DNA molecule study allows us to probe the real time kinetics of RecA polymerization. We describe this process as occurring through the nucleation of RecA on DNA followed by the growth of RecA filaments. The derivation of this model is presented in Methods.
The shape of the growth phase of RecA polymerization in the presence of ATP (Fig. 5a) shows features that are characteristic of the regime of growth from a single nucleus, namely long lag phase, constant growth rate, and abrupt transition to the stationary phase. Because the growth velocity appears to be constant (since there is no inflection point, Fig. 5a), it suggests either, (i) the nucleation starts exactly at the center, or (ii) it starts at one or both of the ends of the DNA; it can be easily argued that ii is more likely than i. Nucleation from a random site would imply that the growth velocity reduces by half when one of the growing fronts reaches the end of DNA. From earlier studies, we can safely assume that the nucleation starts from both ends of λ-DNA, which contain 12 bases single-stranded cohesive ends (27). Our results allow to estimate the nucleation rate n ≈ 10−6/min per bp (from the lag time) and the growth rate v = 12 monomers/sec (from the linear regime). This number corresponds to ≈2.15 kb/min.
On the other hand, polymerization of RecA in the presence of ATP[γS] (Fig. 5b) shows features characteristic of growth from multiple nuclei, that is, relatively short lag time and sigmoidal type growth curve (23, 24). The experimental data is well described (see Methods) as an exponential
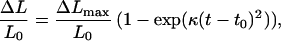 |
7 |
where ΔL = change in length, L0 = original length, Lmax = maximum extended length, ΔLmax = Lmax − L0, and t = time. Eq. 7 is essentially the same as Eq. 3. The prefactor is added to convert from coverage, φ, to length, and the lag time t0 is related to the homogenization of the added protein. This equation has only two adjustable parameters, namely, t0 and κ = nv/2, where n = rate of nucleation and v = rate of growth of an individual nucleus. The best fit is given by κ = 0.022 ± 0.002 min−2. One can obtain a rough estimate for the number of nuclei, M, by assuming that the growth rate is comparable to the ATP one. This gives M ≈ 6. However, judging from the smoothness of the growth curve, this may be underestimating the value of M. These results are consistent with the observation that the use of ATP[γs] in place of ATP can overcome the initial kinetic barrier for RecA to bind to DNA. In the case of ATP, the nucleation events at the end of the molecule are always the same with similar growth curve between different single molecule experiments except that the stochastic nature of nucleation events alters the lag time. For ATP[γs], the multiple nucleation events lead to both variation in the lag time as well the shape of the sigmoidal growth curves. We have also verified this by numerical simulation.
The kinetics of dissociation can be described by using a phenomenological argument, then compared with the experimental data of Fig. 6a. The growth of a filament results from the competition between rates of binding and unbinding of RecA monomers and depends on the [ATP]/[ADP] ratio c (28). At c ≫ 1, the rate of binding is much greater than the rate of dissociation, and the net growth of the filament is favored. However, the growth rate is a decaying function of c, and for a value of c = c0, the binding rate becomes equal to the dissociation rate. Thus, at c < c0, the net disassembly of the filament is preferred. To quantitatively relate c to the dynamics of RecA polymerization, we expand the net growth rate v around c0,
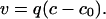 |
8 |
Moreover, we use the observation that in the stationary phase of the length dynamics, the [ATP]/[ADP] ratio decays exponentially as seen in Fig. 6b,
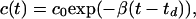 |
9 |
where β = decay exponent, t = time, td is the time when c = c0, and the disassembly of the filament begins. Combining Eqs. 8 and 9 and integrating v(t), we obtain for the change in length:
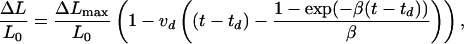 |
10 |
where vd = qc0 is the maximal dissociation rate. The depolymerization phase is well described by Eq. 10 by using the experimental value of β = 0.02 min−1 obtained from Fig. 6b and having vd and td as free parameters. The dissociation rate we obtain, vd = 2.4 monomers/sec, as well as the association rate, is in good agreement with the results of biochemical studies (3). The onset of dissociation (Fig. 6 a and b) occurs at td = 70 min, which corresponds to c(td) ≈ 5.
CONCLUSIONS
The main conclusions of this paper are:
(i) The kinetics of RecA protein polymerizing on an individual dsDNA molecule are measured. We find that, in the presence of ATP, the rate of polymerization is ≈12 monomers/sec and the rate of depolymerization is ≈2 monomers/sec under the conditions used.
(ii) We present direct evidence that RecA polymerization in the presence of ATP leads to end nucleation and linear growth process, while in the presence of ATP[γs], growth emerges from multiple nuclei and a sigmoidal growth. An effect of ATP hydrolysis is the observation of a longer lag time for the formation of a critical nucleus. This may suggest the formation of the nucleus near a mismatched region more favorable and further the depolymerization stage allowing strand exchange reactions to be completed for proofreading and editing.
(iii) A direct measure of the change in relative persistence length between naked DNA and a RecA-decorated DNA is obtained. In the presence ATP or ATP[γs], the lower bound value of this ratio Ar/An ≈ 4 (Ar = 0.21 μm). The possibility to directly observe changes in the elastic state of individual DNA molecule, in real time, may enable the study of RecA mediated recombination events.
(iv) A phenomenological description is presented that provides a good characterization of our data. A schematic model of nucleation and growth, in analogy with three-dimensional crystal growth, is used to describe RecA assembly.
Acknowledgments
We thank K. Adzuma, D. Chatenay, J. F. Marko, P. Model, E. Siggia, D.Thaler, and M. Turner for useful discussions. This work was supported by the Mathers Foundation. M. F. acknowledges partial support from the Sloan Foundation.
ABBREVIATIONS
- dsDNA
double-stranded DNA
- ssDNA
single-stranded DNA
- ATP[γS]
adenosine 5′-[γ-thio]triphosphate
References
- 1.Kowalczykowski S C, Eggleston A K. Annu Rev Biochem. 1994;63:991–1043. doi: 10.1146/annurev.bi.63.070194.005015. [DOI] [PubMed] [Google Scholar]
- 2.Roca A I, Cox M M. Crit Rev Biochem Mol Biol. 1990;25:415–456. doi: 10.3109/10409239009090617. [DOI] [PubMed] [Google Scholar]
- 3.Cox M M. Trends Biochem Sci. 1994;19:217–222. doi: 10.1016/0968-0004(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 4.Stasiak A, Di Capua E, Koller T. J Mol Biol. 1981;151:557–564. doi: 10.1016/0022-2836(81)90010-3. [DOI] [PubMed] [Google Scholar]
- 5.Stasiak A, Di Capua E. Nature (London) 1982;299:185–186. doi: 10.1038/299185a0. [DOI] [PubMed] [Google Scholar]
- 6.Howard-Flanders P, West S C, Stasiak A. Nature (London) 1984;309:215–220. doi: 10.1038/309215a0. [DOI] [PubMed] [Google Scholar]
- 7.Egelman E H, Stasiak A. J Mol Biol. 1986;191:677–697. doi: 10.1016/0022-2836(86)90453-5. [DOI] [PubMed] [Google Scholar]
- 8.Cassuto E, Howard-Flanders P. Nucleic Acids Res. 1986;14:1149–1157. doi: 10.1093/nar/14.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kowalczykowski S C, Clow J, Krupp R A. Proc Natl Acad Sci USA. 1987;84:3127–3131. doi: 10.1073/pnas.84.10.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Honigberg S M, Radding C M. Cell. 1988;54:525–532. doi: 10.1016/0092-8674(88)90074-8. [DOI] [PubMed] [Google Scholar]
- 11.Brenner S L, Zlotnick A, Griffith J D. J Mol Biol. 1988;204:959–972. doi: 10.1016/0022-2836(88)90055-1. [DOI] [PubMed] [Google Scholar]
- 12.Pugh B F, Cox M M. J Mol Biol. 1988;203:479–493. doi: 10.1016/0022-2836(88)90014-9. [DOI] [PubMed] [Google Scholar]
- 13.Lindsley J F, Cox M M. J Mol Biol. 1989;205:695–711. doi: 10.1016/0022-2836(89)90315-x. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi M, Norden B. Adv Biophys. 1994;30:1–35. doi: 10.1016/0065-227x(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 15.Smith S B, Finzi L, Bustamante C. Science. 1992;258:1122–1126. doi: 10.1126/science.1439819. [DOI] [PubMed] [Google Scholar]
- 16.Simmons R M, Finer J T, Chu S, Spudich J. Biophys J. 1996;70:1813–1822. doi: 10.1016/S0006-3495(96)79746-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang M D, Yin H, Landick R, Gelles J, Block S M. Biophys J. 1997;72:1335–1346. doi: 10.1016/S0006-3495(97)78780-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shivashankar G V, Stolovitzky G, Libchaber A. Appl Phys Lett. 1998;73:291–293. [Google Scholar]
- 19.Shivashankar G V, Libchaber A. Biophys J. 1998;74:A242. [Google Scholar]
- 20.Leger J F, Robert J, Bourdieu L, Chatenay D, Marko J F. Proc Natl Acad Sci USA. 1998;95:12295–12299. doi: 10.1073/pnas.95.21.12295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perkins T T, Quake S R, Smith D E, Chu S. Science. 1994;264:822–826. doi: 10.1126/science.8171336. [DOI] [PubMed] [Google Scholar]
- 22.Allemand J-F, Bensimon D, Jullien L, Bensimon A, Croquette V. Biophys J. 1997;73:2064–2070. doi: 10.1016/S0006-3495(97)78236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolmogorov A N. Izv Akad Nauk SSSR, Ser Khem. 1937;3:355–359. [Google Scholar]
- 24.Avrami M. J Chem Phys. 1939;7:1103–1112. [Google Scholar]
- 25.Marko J F, Siggia E D. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 26.Weinstock G M, McEntee K, Lehman I R. J Biol Chem. 1981;256:8829–8834. [PubMed] [Google Scholar]
- 27.West S C, Cassuto E, Mursalim J, Howard-Flanders P. Proc Natl Acad Sci USA. 1980;77:2569–2573. doi: 10.1073/pnas.77.5.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee J W, Cox M M. Biochemistry. 1990;29:7666–7676. doi: 10.1021/bi00485a016. [DOI] [PubMed] [Google Scholar]
- 29.Brenner S L, Zlotnick A, Griffith J D. J Mol Biol. 1990;216:949–964. doi: 10.1016/S0022-2836(99)80013-8. [DOI] [PubMed] [Google Scholar]
- 30.Hegner M, Bustamante C. Biophys J. 1998;74:A150. [Google Scholar]