

Abstract
The positively charged S4 transmembrane segment of voltage-gated channels is thought to function as the voltage sensor by moving charge through the membrane electric field in response to depolarization. Here we studied S4 movements in the mammalian HCN pacemaker channels. Unlike most voltage-gated channel family members that are activated by depolarization, HCN channels are activated by hyperpolarization. We determined the reactivity of the charged sulfhydryl-modifying reagent, MTSET, with substituted cysteine (Cys) residues along the HCN1 S4 segment. Using an HCN1 channel engineered to be MTS resistant except for the chosen S4 Cys substitution, we determined the reactivity of 12 S4 residues to external or internal MTSET application in either the closed or open state of the channel. Cys substitutions in the NH2-terminal half of S4 only reacted with external MTSET; the rates of reactivity were rapid, regardless of whether the channel was open or closed. In contrast, Cys substitutions in the COOH-terminal half of S4 selectively reacted with internal MTSET when the channel was open. In the open state, the boundary between externally and internally accessible residues was remarkably narrow (∼3 residues). This suggests that S4 lies in a water-filled gating canal with a very narrow barrier between the external and internal solutions, similar to depolarization-gated channels. However, the pattern of reactivity is incompatible with either classical gating models, which postulate a large translational or rotational movement of S4 within a gating canal, or with a recent model in which S4 forms a peripheral voltage-sensing paddle (with S3b) that moves within the lipid bilayer (the KvAP model). Rather, we suggest that voltage sensing is due to a rearrangement in transmembrane segments surrounding S4, leading to a collapse of an internal gating canal upon channel closure that alters the shape of the membrane field around a relatively static S4 segment.
Keywords: voltage-gated K+ channels, HCN1 channel, cysteine, sulfhydryl reagents, ion channel gating
INTRODUCTION
A key question in understanding the function of voltage-gated channels is how such channels sense and respond to changes in membrane voltage. Nearly two decades have passed since the initial cloning of the voltage-gated channels revealed highly conserved, regular repeats of positively charged amino acids at every third position along the fourth transmembrane segment (S4) (Noda et al., 1984; Catterall, 1986). This led to the suggestion that S4 forms the voltage sensor. Subsequent mutagenesis experiments confirmed the importance of S4 in voltage gating since neutralization or charge reversal of its basic residues markedly altered activation gating (Stuhmer et al., 1989; Liman et al., 1991; Papazian et al., 1991; Logothetis et al., 1992).
If S4 forms the voltage sensor, its charge must move relative to the membrane electric field in response to changes in voltage, coupling electrical energy to mechanical or chemical energy required to gate the channel. This charge movement could be accomplished either by an outward movement of S4 relative to the membrane electric field or, alternatively, by a change in the shape of the electric field around a static S4 (Yang et al., 1996). Some of the clearest evidence for S4 movements comes from studies on depolarization-activated channels using sulfhydryl-modifying methanethiosulfonate (MTS) (Akabas et al., 1992). By using permanently charged, membrane-impermeant compounds, such as the cationic [2-(trimethylammonium) ethyl] methane thiosulfonate (MTSET), it is possible to determine whether a given residue is accessible to either the extracellular or intracellular aqueous environment and how that accessibility varies when the channel is in the open or closed state.
Several studies on depolarization-activated channels using MTS modification of substituted cysteines (Cys) have identified a series of positions along S4 that show a clear pattern of state-dependent reactivity consistent with S4 movement (Yang and Horn, 1995; Larsson et al., 1996; Yang et al., 1996; Yusaf et al., 1996; Baker et al., 1998). Residues in the external, NH2-terminal half of S4 tend to be accessible to MTS reagents in the open state (when the membrane is depolarized) but not in the closed state (when the membrane is hyperpolarized). Conversely, residues in the internal, COOH-terminal half of S4 tend to be accessible in the closed state (when the membrane is hyperpolarized), but not in the open state (when the membrane is depolarized). Remarkably, some residues in the middle of S4 completely reverse their sidedness of accessibility during activation, switching from internally accessible in the closed state to externally accessible in the open state (Yang et al., 1996; Baker et al., 1998). Such findings are consistent with the idea that S4 undergoes an outward translational movement upon depolarization, due to an electrostatic effect of the membrane field on the positive charges on S4.
Surprisingly, residues that were reactive with external MTS reagents were separated from residues that reacted with internal MTS by a very small distance in a given state. This suggests that S4 lies in a water-filled pore, the gating canal, with a constriction that forms a narrow boundary between the internal and external environment (Goldstein, 1996; Yang et al., 1996; Baker et al., 1998). Subsequent fluorescence studies (Cha et al., 1999; Glauner et al., 1999) have led to a modified model in which S4 undergoes a rotational movement, causing positive charge exposed to the internal mouth of the gating canal in the closed state to become exposed to the external mouth of the gating canal in the open state. We refer to this voltage-dependent translational or rotational movement of S4 through a water-filled crevice (or gating canal) as the classic model (Fig. 1 A).
Figure 1.

Models of voltage sensor movement during changes in membrane voltage. (A) A model for depolarization-activated channels (classic model). For clarity, a single S4 α-helix is shown which sits in a water-filled crevice with a narrow constriction, separating the internal from external environment. In response to membrane depolarization, S4 moves outward, causing translocation of a series of positively charged residues either by translation or rotation. (B) In the KvAP model, a voltage sensor paddle (formed of S3b-S4 helixes; one of four in the channel complex) moves through the lipid bilayer.
A second model for S4 movements comes from the recent X-ray crystal structure and functional analysis of KvAP, a depolarization-activated K+ channel from archaebacteria (Jiang et al., 2003a,b). These studies indicate that S4 forms a mobile helix-loop-helix structure with the external, COOH-terminal half of S3 (S3b), termed the “voltage-sensor paddle.” Surprisingly, this paddle lies on the outer radius of the channel protein buried in the lipid bilayer, nearly perpendicular to the pore axis (the KvAP model, see Fig. 1 B). During activation the paddle is thought to move into a more upright position, displacing charge through the membrane. Despite its ancient nature, the KvAP primary sequence is highly conserved with eukaryotic voltage-gated K+ channels (Ruta et al., 2003), suggesting that voltage-sensor paddle movement may be a common mechanism.
In this study we investigated S4 voltage sensor movement during gating of the mammalian HCN channels, which encode hyperpolarization-activated cation currents (termed Ih or If) that contribute to pacemaker activity in the heart and brain (Robinson and Siegelbaum, 2003). Although the HCN channels are activated by hyperpolarization and not depolarization, they are members of the voltage-gated channel family and contain six transmembrane (TM) segments with a positively charged S4. Thus, these channels provide an interesting experimental system to investigate whether S4 movements are a conserved feature of all six TM segment voltage-gated channels. Indeed, previous studies of MTS reactivity in the sea urchin HCN channel (spHCN; Männikkö et al., 2002), and in hyperpolarization-activated K+ channels from archaebacteria (MVP; Sesti et al., 2003) or plants (KAT1; Latorre et al., 2003) reported that S4 undergoes an inward movement in response to hyperpolarization and resides in a water-filled canal (for spHCN and MVP), similar to depolarization-activated channels. This suggests that the difference in gating between depolarization- and hyperpolarization-activated channels lies in the mechanism that couples S4 movement to channel opening, and not in the movement of S4 itself.
Here we further explore the nature of hyperpolarization-activated gating for the mouse HCN1 channel isoform (mHCN1; Santoro et al., 1997), using MTSET modification of Cys-substituted S4 residues. Although both mammalian HCN channels and spHCN are nonselective cation channels that share the property of hyperpolarization activation, spHCN channels show pronounced inactivation (Gauss et al., 1998), whereas mammalian HCN channels do not (Ludwig et al., 1998; Santoro et al., 1998). We engineered an MTS-resistant HCN1 channel (HCN1-R) in which 11 out of 12 endogenous Cys were either deleted or mutated to non-Cys residues, reducing the likelihood of MTS reaction with native cysteines. The reactivity of substituted Cys residues to MTSET was defined along the length of the HCN1-R S4 voltage sensor, using an irreversible change in Ih as an index of reactivity.
Our results provide evidence that S4 movements during hyperpolarization activation in mHCN1 differ from those in other hyperpolarization-activated (spHCN, MVP, and KAT1) and depolarization-activated channels. We found that the COOH-terminal, internal half of the mHCN1 S4 is accessible to the internal aqueous environment in the open (hyperpolarized) state but becomes buried in the closed (depolarized) state. In contrast, the NH2-terminal half of S4 is highly reactive to external MTSET in both the open and closed state. These results cannot be explained by the classic translation or rotation models for S4 movements, nor are they readily explained by the KvAP voltage-sensing paddle model. Rather, they are most consistent with the view that S4 is relatively immobile and that inward charge displacement during hyperpolarization gating results from a dilation of the gating canal surrounding the COOH-terminal half of S4 that increases exposure of COOH-terminal S4 residues to the internal aqueous environment and alters exposure to the membrane electric field.
MATERIALS AND METHODS
Molecular Biology—Creation of MTS-resistant HCN1-R and Cys Substitution Mutant Channels
To create an MTS-resistant channel, we used an HCN1 truncation mutant, HCN1-ΔCterm (HCN1-S391stop mutation), that lacked nearly the entire COOH terminus, which removed 6 out of 12 endogenous Cys (Wainger et al., 2001). We then removed five of the six remaining Cys in HCN1-ΔCterm using the following substitution mutations: C55S, C298I, C318S, C347S, and C374T. Four of those substitutions were based on a conservative Ser or Thr substitution. In the case of C298I, we substituted the homologous residue present in cyclic nucleotide–gated channels. The resultant construct, HCN1-R (see Fig. 2 A), generated a channel that showed normal hyperpolarization-activated gating and was resistant to modification by MTS reagents (see results). Mutation of the one remaining Cys in HCN1-R, C303, which occurs in the S5 segment, to any one of the other 19 amino acids failed to yield functional channels. Individual Cys substitutions were made along the length of the S4 region of HCN1-R using PCR mutagenesis, yielding 18 individual S4 Cys–substituted mutant channels (R244C, V246C, R247C, T249C, K250C, I251C, L252C, S253C, L254C, L255C, L257C, L258C, R259C, L260C, S261C, I264C, R265C, and H268C; see Fig. 3 B). Mutant channels were subcloned into the pGHEM-HE vector through 5′ EcoRI and 3′ XbaI restriction sites (Liman et al., 1992). All mutant channels were sequenced and verified using dideoxy chain termination sequencing.
Figure 2.

MTSET effects on wild-type mHCN1 and HNC1-R. (A) mHCN1 (wt) channel schematic (left) depicting the six transmembrane segment topology and COOH-terminal cyclic nucleotide–binding domain. S3 is divided into two regions (S3a and S3b) by a conserved proline break (Li-Smerin et al., 2000; Jiang et al., 2003a). 12 endogenous Cys are shown (filled diamonds). The right panel shows HCN1-R, with a COOH-terminal deletion and conservative mutations that removed five of six remaining Cys (open diamonds). Mutations at C303 were not possible without loss of functional expression (filled diamond). (B) The effect of external application of MTSET (5 mM for 2 min) on mHCN1 wt (left) and HCN1-R (right) channels. Currents were recorded using two-electrode voltage clamp (TEVC) by stepping from a holding voltage of −30 mV to a test voltage of −80 mV for 4 s, once every 15 s (voltage pulse protocol above current traces). Current traces are shown before MTSET application (I0, black line) and after washout of MTSET (IMTSET, open circles). (C) Effect of internal MTSET application (5 mM applied for 2 min) on mHCN1 wt (left) and HCN1-R (right) currents in cell-free inside-out patches. Currents were elicited by hyperpolarizing voltage steps (3-s duration) from a holding voltage of −30 mV to a test voltage of −105 mV. Current symbols as in B.
Figure 3.

S4 voltage sensor Cys substitutions. (A) Mean, normalized tail current activation curves for mHCN1 wt (red, filled diamonds), HCN1-R (green, open diamonds), and the 12 Cys substitution mutant channels (see legend inset). Tail currents were fitted by a Boltzmann equation (see materials and methods) to obtain V1/2 and slope factor (mean ± SEM, both in mV, see legend inset); italicized figures in brackets are the number of experiments (n) for each channel. Boltzmann fits to mHCN1 and HCN1-R currents are shown as red and green lines, respectively; fits to Cys substitution mutant channels shown as thin, black lines. (B) Sequence alignment of S4 regions for selected hyperpolarization- and depolarization-activated K+ channels. The first four sequences are of hyperpolarization-activated channels: mouse HCN1 (used in this study, Santoro et al., 1997), sea urchin spHCN (Gauss et al., 1998), plant KAT1 (Anderson et al., 1992; Sentenac et al., 1992), and archaebacterial MVP (Sesti et al., 2003). The final two sequences are from depolarization-activated K+ channels: Drosophila Shaker (Kamb et al., 1987) and archaebacterial KvAP (Ruta et al., 2003). Residue numbering is at the NH2- and COOH-terminal end of each sequence; blue highlights positively charged residues (R or K); red highlights negatively charged residues (D or E); underlined residues form S4 transmembrane segment; arrows above mHCN1 indicate Cys substitution residues studied here. Alignments were adjusted so that residues responsible for gating charge movements of R362, R365, R368, and R371 for Shaker (Baker et al., 1998), and R117, R120, R123, and R126 for KvAP (Jiang et al., 2003b), were aligned with residues in mHCN1 we propose to be important for charge movement.
cRNA Injection and Electrophysiological Recordings in Xenopus Oocytes
RNA was transcribed from NheI-linearized DNA using a T7 RNA polymerase (Message Machine) and injected into Xenopus laevis oocytes as described previously (Goulding et al., 1992; Santoro et al., 1998). For inside-out patch clamp experiments 50 ng of channel cRNA was injected; for two-electrode voltage clamp (TEVC) 5–50 ng cRNA was injected. RNA concentration was measured from the optical density ratio at 260- and 280-nM wavelengths (Sambrook et al., 1989) using a DU640 spectrophotometer (Beckman).
TEVC recordings were routinely performed using an OC-725C voltage-clamp amplifier (Warner Instrument Corp.). The extracellular solution contained (in mM): 112 KCl, 2 MgCl2, 10 HEPES, pH 7.4. Microelectrodes were filled with 3 M KCl and had resistances of 0.5–3 MΩ. Recordings were usually obtained 18–24 h after oocyte injection. Because S253C channels showed poor expression, they were studied 3–5 d post injection. L255C, L258C, R259C, and L260C showed very poor expression and were not studied further. K250C and R265C did not yield detectable current.
Inside-out (IO) patch clamp recordings were made using an EPC-7 patch clamp amplifier (HEKA Elektronik) 5–9 d after cRNA injection. S253C channels were not studied in these conditions due to their very small patch currents. The extracellular (pipette) solution contained (in mM): 112 KCl, 2 MgCl2, 10 HEPES, pH 7.4. The intracellular (bath perfusate) solution contained (in mM): 112 KCl, 1 MgCl2, 1 EGTA, 10 HEPES, pH 7.4.
All recordings were filtered at 2 kHz using an 8-pole low pass Bessel filter (Frequency Devices) and sampled at 1 kHz using an ITC interface (InstruTech Corp.) and Pulse software (HEKA Elektronik). Voltage pulse protocols, written and applied using Pulse, are described below. Ag-AgCl grounding wires were isolated from bath solutions (often containing oxidizing MTSET) by 3M KCl/2% agar bridge electrodes. Analyses were performed using PulseFit (v8.5, HEKA Elektronik) and Microcal Origin software (v6.0, Microcal Software, Inc.).
A 1M MTSET (Toronto Research Chemicals) stock was prepared in double-distilled water and subsequently diluted in the perfusing solution to the desired final concentration. MTSET solutions were made fresh during a recording, resulting in the MTSET being in solution for <90 s before application. No linear leak subtraction was applied to current recordings. All recordings were performed at room temperature (21–24°C).
MTSET Modification Protocols and Modification Rate Calculation
The following voltage-step protocols were used to define accessibility of MTSET in the open and closed state (see Fig. 4 A). A holding voltage of −30 mV was routinely used, at which nearly all channels were completely closed. The following more positive holding potentials were used in four mutant channels that displayed a more positive activation range: −20 mV, for L254C and L257C; −10 mV, for S253C and I264C. Prior to MTSET exposure, the membrane was stepped for 3–4 s to an initial test voltage (V0) that lay between the V1/2 (midpoint voltage of activation) and a voltage 10 mV negative to the V1/2, which was specific to each channel (see Fig. 3 A for V1/2 values). For open state exposures in either TEVC or IO patch clamp, MTSET was applied with the membrane held at an exposure voltage (VEX) that ranged from −75 to −110 mV. For closed state exposures, MTSET was applied with the membrane held at a VEX of +40 mV for TEVC and 0 mV for IO patch clamp (all channels were ∼100% closed at these voltages).
Figure 4.

Effects of MTSET on three representative Cys substitution mutant channels: V246C, T249C, and L257C. The α-helical diagram representing mHCN1 S4 from K238 to H268 (far left) shows positions of the α-carbons (dark gray spheres) of the three residues described in this figure. (A) The voltage pulse protocol used for measuring effects of MTSET application to the channel in either the open or closed state (see materials and methods for details). (B and C) Effect of external application of MTSET on V246C (B1, B2) or T249C (C1, C2) currents measured using TEVC. (B1 and C1) Plot of normalized steady-state Ih current amplitude, ITEST/I0, as function of MTSET exposure time multiplied by MTSET concentration (units of mM·s) for applications to either the open state (open squares) or closed state (filled squares) of the channel. Note the 10,000-fold difference in cumulative exposure scale between B1 and C1. (B2 and C2) Current traces from individual experiments showing steady-state effects of external MTSET application to closed state of channel. Currents before (I0, black line) and after MTSET (IMTSET, open circles), show effect of external MTSET to inhibit Ih. (D and E) Effect on L257C channels in inside-out patches of cumulative exposure to internal MTSET applied to either open (D1) or closed (E1) states. Note the 1,000-fold difference in the cumulative exposure between D1 and E1. (D2) Open state MTSET modification caused channels to open constitutively (IMTSET, open circles), generating a large instantaneous “leakage” current that was blocked by ZD 7288 (50 μM, IZD 7288, open triangles). (E2) Closed state modification of L257C (IMTSET, open circles) had the opposite action, inhibiting channel opening.
MTSET exposure was limited to the open or closed state by restricting the period of application to 3–10 s (black bar above VEX, Fig. 4 A) within the VEX voltage step (9–20 s). The start of the MTSET application occurred ≥3 s after the onset of VEX to ensure complete channel activation/deactivation before MTSET exposure. The end of the MTSET application occurred ≥3 s before the termination of VEX, to ensure complete washout of the compound (achieved in ∼2 s) before channel deactivation/activation. After an exposure in a designated channel state, the membrane voltage was returned to the holding voltage for 3–5 s. The channel was subsequently reopened by hyperpolarizing the membrane to the test voltage (VTEST, equal to the initial preMTSET voltage step, V0). Successive exposures to the desired channel state were performed until MTSET modification reached completion.
Modification rates were determined as follows. The magnitude of the time-dependent Ih was obtained by measuring the difference between steady-state current at the end of the 3–4-s long hyperpolarizing test pulse and the instantaneous current after the decay of the capacitive transient at the beginning of the hyperpolarization (see Fig. 4, B2–E2). Ih values were measured before MTSET exposure (I0) and at various points after repeated MTSET exposures (ITEST). ITEST values were normalized to I0 and the resulting fractional current (ITEST/I0) was plotted versus the cumulative MTSET exposure (MTSET concentration × total exposure time, in mM·s). A single exponential function was fitted to these data to determine a decay constant (in mM·s) for each of the modifications (Fig. 4, B1–E1, solid black lines). Decay rates were converted to second order modification rate constants (in s−1M−1). Modification rates for each experimental determination were averaged to give mean modification rates (n = 3–6 experiments).
Based on the half-life of MTSET (11.2 min, at pH 7.0, 20°C, Karlin and Akabas, 1998) and the upper limit of MTSET concentration used in this study (5 mM), we estimate that any modification rate ≤0.5 s−1M−1 will be too slow to accurately measure. Thus, Cys residues that showed no significant reactivity were arbitrarily assigned a modification rate of 0.5 s−1M−1 for purposes of display (Fig. 5, A and B).
Figure 5.

A summary of external (A) and internal (B) MTSET second order reaction rates for HCN1-R and each of the Cys substitution mutant channels for both closed state (filled symbols) and open state (open symbols) exposure. Mean ± SEM reaction rates (3–6 experiments) were plotted on a Log10 scale for each Cys substitution mutant. The fastest modification rate on each graph (2.12 × 105 s−1M−1) defines the reaction rate of MTSET with free sulfhydryls in aqueous solution (Pascual and Karlin, 1998). The hatched area on each plot represents modification rates that are ≤20 s−1M−1, a conservative guide-line based on the range of reactivity for Cys substitutions buried in the interior of globular proteins (∼50 s−1M−1, A. Karlin, personal communication). Colored bars at the top of each plot highlight this accessibility/buried definition: gray corresponds to buried Cys substitutions; blue corresponds to externally accessible Cys residues; red corresponds to internally accessible Cys substitutions. We arbitrarily assigned a modification rate of 0.5 s−1M−1, our lower limit of detection, to residues that failed to show significant reactivity (see materials and methods). (C) α-Helical diagrams summarizing the reactivity of Cys substitutions. Each of the α-carbons of the Cys substitutions are color-coded according to their accessibility as above. The gray, semiopaque areas delineate the region of the S4 α-helix that is protein and/or lipid buried. Positively charged residues are highlighted by plus symbols: black and white plus symbols indicate buried or accessible residues, respectively.
Boltzmann Fits to Tail Current Activation Curves
Tail current activation curves were obtained by plotting tail current amplitudes measured upon return to a fixed holding voltage from a series of 10-s hyperpolarizing test pulses to different potentials. Tail current values were plotted as a function of test voltage, and the resulting I-V relationship was fitted by the Boltzmann equation:
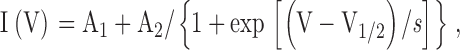 |
where A1 is an offset due to steady holding current, A2 is the maximal tail current amplitude, V1/2 is the midpoint activation voltage, and s is the slope factor of the relation. Mean tail current activation curves were obtained by subtracting the offset (A1) from individual current-voltage curves and then normalizing by the maximal amplitude (A2). The normalized data were then averaged among different experiments and the mean normalized tail current-voltage relation was then fitted with a Boltzmann curve.
Calculation of Predicted State-dependent Changes in External Accessibility of MTSET
We used the following procedure to estimate the predicted magnitude of the state-dependent change in MTSET reactivity for an S4 residue during an external MTSET application at a hyperpolarized exposure voltage that opens a fraction, f, of channels, assuming that a given residue goes from being externally exposed in the closed state to fully buried in the open state. In the simplest model, we assumed that the rate of reaction is directly related to the fraction of channels that remain closed at the exposure voltage. In this case the expected ratio of the rate of reaction at the hyperpolarized voltage to the rate of reaction at a depolarized voltage, where all channels are closed, is given by (1 − f). In typical experiments using steps to −100 mV, the fraction of open channels was ∼0.8, leading to a predicted fivefold slowing of reactivity.
A more realistic model takes into account the presence of four S4 segments per tetrameric channel. We further assume that modification of a single S4 Cys residue is sufficient to alter gating, and that channel activation follows a Hodgkin-Huxley scheme in which all four S4 residues must undergo an independent activation reaction before a channel opens. In this case, the fraction of S4 gates in the active conformation, n, is given by: n = f 1/4. For a channel in the fully closed state in which all four S4 segments are deactivated, and hence MTSET accessible, the rate of MTSET modification will be given by 4 · q, where q is the rate of reaction with a single S4. During hyperpolarization to a voltage that opens a fraction, f, of channels and activates a corresponding fraction, n, of S4 gates, the predicted rate of current modification, q′, is given by:
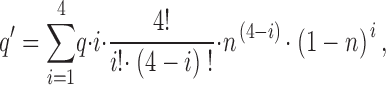 |
where i is the number of S4 segments in the closed (deactivated) position per channel and the other terms are as defined above. This equation states that the rate of modification at a given voltage is equal to the rate of modification of an individual S4 segment (q), multiplied by the number of S4 segments in the closed position (i), multiplied by the probability of observing i closed S4 segments per channel. These normalized rates are then summed over the four possible states of a binomial distribution in which one or more S4 segments are in the closed position. The predicted factor by which the hyperpolarization slows the rate of MTSET reactivity is then given by the ratio (4 · q)/q′, where 4 · q is the modification rate when the channel is completely closed (four S4 closed). Thus, a hyperpolarization that opens ∼0.8 of the channels leads to a predicted ∼60-fold decrease in the rate of reaction with external MTSET, relative to the closed state rate, for a Cys residue that undergoes a change in position from an externally accessible position in the closed state to an externally inaccessible position in the open state.
RESULTS
Methane Thiosulfonate–resistant HCN1 Channel (HCN1-R) and Cys Substitutions
The mouse wild-type HCN1 subunit (wt), with 12 endogenous Cys residues (see Fig. 2 A), showed significant reactivity to either external or internal application of MTSET. Thus a 2-min exposure to 5 mM external MTSET caused a 51.4 ± 4.5% (n = 4) inhibition of mHCN1 current measured under two-microelectrode voltage clamp (Fig. 2 B). A previous study also showed significant inhibition of mHCN1 current after external application of other MTS reagents (MTSEA and MTSES, Xue and Li, 2002). A 2-min application of 5 mM MTSET to the internal surface of the membrane in the inside-out patch configuration caused a 30.9 ± 9.9% (n = 7) current reduction (Fig. 2 C). A methane thiosulfonate–resistant HCN1 channel (HCN1-R, see Fig. 2 A) was therefore created.
Construction of HCN1-R was based on an HCN1 deletion mutant, HCN1-ΔCterm (Wainger et al., 2001), in which the entire COOH terminus was removed by insertion of a stop codon three amino acids COOH-terminal to the end of S6 (HCN1-S391stop). This mutant lacks 6 of the 12 endogenous Cys residues and shows normal hyperpolarization activation, except for a 5–7-mV positive shift (Wainger et al., 2001). We next used a series of point mutations to substitute alternate amino acids for five of the six remaining Cys (C55S, C298I, C318S, C347S, and C374T; see materials and methods), yielding HCN1-R. Mutation of the one remaining Cys, C303 within the S5 segment, to any of the 19 remaining residues resulted in a complete loss of functional expression. Thus, all experiments were performed in the background of HCN1-R.
HCN1-R showed robust functional expression with relatively normal voltage gating and, importantly, a complete resistance to internal or external MTSET. Tail current activation curves for HCN1-R showed a typical S-shaped dependence on voltage that, when fitted by a Boltzmann relation, yielded a midpoint voltage of activation (V1/2) of −76.1 ± 2.1 mV and a slope of 11.6 ± 1.0 mV (n = 7), similar to the values for wt mHCN1 (V1/2 = −79.0 mV; slope = 11.9 mV; see Fig. 3 A). A 2-min application of 5 mM MTSET had no effect on channel current when applied to either the external side of the membrane (peak current equal to 97.8 ± 1.2% of control, n = 5; see Fig. 2 B) or internal side of the membrane (peak current equal to 101 ± 2.3% of control, n = 3; see Fig. 2 C). Thus, HCN1-R channels provide a suitable background for studying MTSET reactivity of substituted S4 Cys residues.
12 out of 18 separate Cys substitutions within the S4 segment of HCN1-R yielded functional channels with good expression levels (see materials and methods). All 12 Cys mutants generated hyperpolarization-activated currents with normal voltage-dependent activation curves that were well-fitted by a Boltzmann relation, similar to the behavior of wt mHCN1 and HCN1-R. However, the midpoint position of the activation curves for some mutants was shifted along the voltage axis relative to the HCN1-R or mHCN1 activation curves (Fig. 3 A). At one extreme, the S253C channel V1/2 was shifted to −45.2 mV, 30.9 mV more positive than HCN1-R, indicating a significant stabilization of the open state. At the other extreme, the 261C channel V1/2 was shifted to −97.4 mV, −21.3 mV negative to HCN1-R, indicating a significant stabilization of the closed state. These shifts in V1/2 are similar to those seen in previous studies that examined point mutants for a subset of these residues, either in the background of wild-type HCN2 (Chen et al., 2000; Vaca et al., 2000) or wild-type spHCN (Männikkö et al., 2002), indicating that the background mutations used to construct HCN1-R have not significantly altered the channel's structure.
An alignment of S4 segments from a number of hyperpolarization- and depolarization-activated channels demonstrates the conservation of the basic residues and allows us to compare results in mHCN1 with previous results from other channels. All six channels shown in Fig. 3 B are members of the voltage-gated K+ channel superfamily, although they differ in the polarity of voltage gating. The S4 of hyperpolarization-activated channels (the first four sequences in Fig. 3 B) are distinguished by a particularly large number of basic residues, with an unusual break in the pattern, in which a neutral serine (or valine, in MVP) is present in place of the usual basic residue in the middle of S4 (S253 in mHCN1, see Fig. 3 B). The Cys substitutions that we have made span the length of the mHCN1 S4 segment (Fig. 3 B).
Studies of MTSET Reactivity of S4 Cys–substituted Residues
Channels containing substituted Cys residues were exposed to MTSET in either the open or closed state from either the internal or external side of the membrane (as described in the materials and methods). We observed two general patterns of reactivity, depending on the position of the residue along the length of S4. Residues in the NH2-terminal portion of S4 proximal to T249C, including R244C, V246C, and R247C, showed a rapid, state-independent reactivity with external but not internal MTSET. MTSET modification of these residues led to irreversible inhibition of channel opening. Conversely, residues in the COOH-terminal portion of S4 between I251C and H268C, including L252C, L254C, L257C, S261C, and I264C showed a rapid reactivity with internal, but not external, MTSET. Moreover, this reactivity was state dependent, proceeding ∼2–3 orders of magnitude faster in the open state compared with the closed state. Finally, modification of these internal-facing residues resulted in an irreversible activation of the channel; that is, the channels were locked in the open state.
These effects are illustrated in Fig. 4 for three representative residues in different regions of S4: V246C, T249C, and L257C. In our protocol, the membrane was typically held at −30 mV and a 3–4-s hyperpolarizing potential step (V0) was applied to record the initial magnitude of Ih (Fig. 4 A). MTSET was then selectively applied to channels in either the closed state, using a prolonged voltage clamp step to a depolarized potential (VEX = 0 to +40 mV, Fig. 4 A), or in the open state, using a prolong step to a hyperpolarized voltage (VEX = −75 to −110 mV, Fig. 4 A; see materials and methods). After various durations of exposure (3–10 s), MTSET was washed out of the bath and the membrane returned to its initial holding potential. To assay reactivity, we measured Ih with a second short (3–4 s) test pulse (VTEST), identical to the initial test pulse. MTSET reactivity is defined by the presence of a persistent irreversible change in Ih amplitude or kinetics of activation.
Residue V246C, located in the NH2-terminal, presumably external, half of S4, showed a very rapid modification during exposure to 3 μM external MTSET under two microelectrode voltage-clamp conditions. There was nearly complete and irreversible inhibition of hyperpolarization-activated current after a 15–18-s cumulative exposure (Fig. 4, B1 and B2). Moreover, this inhibition showed little state dependence. The mean rate of modification of V246C at +40 mV, when the channels are nearly completely closed, was 9.5 × 104 M−1s−1 (n = 3). This rate was 2.2-fold greater than the mean rate of modification at −100 mV, 4.3 × 104 M−1s−1 (n = 4), when the channels are mostly open (fractional activation is 0.81). These rates are close to the rate of reaction of MTSET with free sulfhydryls in aqueous solution (2.12 × 105 s−1M−1; Pascual and Karlin, 1998), indicating that V246C is fully exposed to the external solution in either the open state or closed state of the channel. We observed no effect of internal MTSET on V246C channels, either in the open or closed state (up to 5 mM for 2 min, n = 3).
The 2.2-fold decrease in the rate of reactivity of V246C at hyperpolarized versus depolarized voltages is much smaller than the predicted fivefold decrease if V246C becomes completely buried in the membrane (and therefore inaccessible to MTSET) in the open state (based on the measured fractional opening of 0.8 for V246C at −100 mV; see materials and methods). The discrepancy is even larger for the fraction of S4 segments that will have undergone a Hodgkin-Huxley activation gating reaction at this voltage, which predicts a 60-fold slowing in rate of reactivity at −100 mV (see materials and methods). Thus, the small state-dependent MTSET reactivity of V246C may reflect a local change in the environment of this residue, rather than a translational movement of S4 that buries this residue in the membrane. This conclusion is further supported by results on neighboring S4 residues (see below).
Residue T249C, approximately one helical turn below V246C, also reacted with MTSET, producing a complete and irreversible blockade of hyperpolarization-activated current (Fig. 4, C1 and C2). The reactivity of T249C also showed only a small state dependence, although the absolute rate of modification in either the open or closed state was ∼3 orders of magnitude slower than that of V246C. Thus, the rate of MTSET modification at 0 mV (closed state) was only 26.3 M−1s−1 (n = 4); the rate at −100 mV with channels mostly in the open state was ∼2.5-fold lower, 11 M−1s−1 (n = 5). T249C showed no reactivity with internal MTSET, even at 5 mM (n = 3). Because modification rates of ∼50 s−1M−1 have been found for reaction of MTSET with native Cys residues that lie buried within the interior of the X-ray crystal structure of the globular proteins carbonic anhydrase II and enolase (A. Karlin, personal communication), the slow rate of reactivity of T249C with MTSET suggests that this residue may lie buried within the channel protein or at the external membrane-water interface. Furthermore, ∼0.8 of T249C channels have activated at the exposure voltage of −100 mV, which predicts up to a 60-fold decrease in reactivity rate if T249C becomes completely buried in the activated state (see above). Thus, the 2.5-fold state-dependent change in rate of reactivity may reflect a local change in environment, rather than a complete disappearance of T249C into the membrane.
Residue L257C, approximately two helical turns below T249C, selectively reacted with internal MTSET (Fig. 4, D1 and E1). External application of 5 mM MTSET had no effect on channel current (n = 3). The internal modification of L257C was highly state dependent, occurring ∼1,000-fold faster when MTSET was applied at −90 mV (2.56 × 104 s−1M−1), when channels were mostly open, than when the reagent was applied at 0 mV (17.9 s−1M−1), when channels were fully closed. In contrast to the effects of external MTSET to inhibit channel current, open-state modification of L257C by internal MTSET caused HCN channels to become permanently activated (lock-open). After modification of L257C, there was an increase in holding current at −30 mV and the hyperpolarizing step now elicited a large time-independent instantaneous current (see Fig. 4 D2). The amplitude of the time-independent current was identical to the steady-state current amplitude of Ih activated by the hyperpolarization before MTSET application (Fig. 4 D2), indicating that the current was carried by modified HCN channels. Indeed, the increase in instantaneous current was completely blocked by a specific HCN channel blocker, ZD 7288 (50 μM) (Fig. 4 D2). Surprisingly, internal application of MTSET to L257C channels at 0 mV, when the channels were closed, resulted in an inhibition of opening, even though open-state modification produced an enhancement of opening (Fig. 4 E2).
Summary of HCN1-R S4 MTSET Reactivity
We observed a clear pattern of MTSET modification for all 12 Cys substituted S4 residues, in which the characteristic effect was determined by the linear position of the reactive Cys residue along S4 (Fig. 5) . Thus R244C, V246C, and R247C, which lie in the NH2-terminal portion of S4, proximal to T249C, all showed a rapid, largely state-independent reaction with external MTSET (see Fig. 5 A) that inhibited channel opening. Importantly, R247C showed virtually no state-dependent change in reactivity, with only an ∼1.3-fold difference in reaction rate between positive and negative voltages. The fraction of R247C channels opened during the exposure voltage of −100 mV was ∼0.87, leading to a predicted ∼8-fold decrease in reaction rate at this potential if R247C becomes buried during opening and a >90-fold reduction if R247C accessibility tracks individual independent S4 movements. The fact that R247C, which lies internal to V246, showed almost no state dependence strongly argues that the small state-dependent changes for V246C and R244C do not reflect a global inward translational movement of S4, which should result in burial of R247C, but may reflect changes in local environment.
Residues between I251C and H268C, including L252C, L254C, L257C, S261C, and I264C all showed rapid reactivity with internal MTSET in the open state, and a very large 100–1,000-fold decrease in rate of reactivity in the closed state (see Fig. 5 B). In addition, open state modification by internal MTSET enhanced channel activation for all five internally accessible residues. Reaction with internal MTSET in the closed state also led to enhanced activation of L252C, L254C, S261C, and I264C, although the rate of modification was slow. Indeed, given the very rapid rate of modification in the open state, it is possible that the closed state reactivity results from modification of the small fraction of S4 segments that are in the activated state. For L257C, however, this cannot be the case as its modification in the closed state produced a different functional effect, channel inhibition, compared with the facilitatory effect of open-state modification.
I264C showed a relatively high rate of closed-state reactivity (179 ± 83.3 s−1M−1, n = 3), although even this rate was ∼170-fold slower than the reaction rate in the open state (30,522 ± 17,645 s−1M−1, n = 4). This large state dependence indicates the presence of a significant change in the environment around this residue during activation gating. However, the relatively high rate of closed state reactivity indicates that I264C may be only partially buried in the closed state, suggesting that this residue may lie close to the boundary between the membrane and the aqueous environment.
Three Cys-substituted residues failed to conform to the above linear pattern. I251C, which lies near the center of S4, showed no effect of exposure to MTSET from either side of the membrane, in either the closed or open state. This indicates that either I25IC is permanently buried in the protein or membrane, or that its modification caused no functional effect. S253C showed anomalous behavior in that it reacted with external MTSET (in a state-independent manner), even though it lies at a position in S4 below that of the internally reactive L252C. However, the extremely slow rate of reaction of S253C (∼5 M−1s−1) suggests that this residue is likely to be buried in the protein or membrane and that the reaction with MTSET may occur through a tortuous path from the external solution. S253C was also anomalous in that modification by external MTSET enhanced channel opening, as evidenced by an approximatel +15-mV shift in the activation curve, rather than the usual inhibitory effect observed with all other externally accessible residues. The low levels of expression of S253C prevented us from measuring its internal reactivity in inside-out patches. Finally, H268C, at the very COOH-terminal end of S4, did not show any state dependence of reactivity, unlike other internally accessible residues (see Fig. 5 B). In addition MTSET modification at H268C resulted in an inhibition of channel opening (due to a −30-mV shift in the activation curve), rather than the usual enhancement of channel opening observed for other internally accessible residues.
These results are summarized in Fig. 5 C, which maps the pattern of reactivity of S4 onto an α-helix representation. This map highlights the relatively static nature of the aqueous environment around the NH2-terminal, externally facing portion of S4, where Cys modification rates only differ by 1.3- to 2.8-fold between closed and open states (Fig. 5 A). In contrast, marked state-dependent changes in environment occur in the COOH-terminal internal end of S4, between residues I251 and H268C, where modification rates are ∼170–3,200-fold slower in the closed than open states (Fig. 5 B). The map also highlights the very short stretch of residues that are buried in the closed state (T249 to I264). These buried residues correspond to residues that were deduced to participate in activation gating from experiments that studied effects of charge neutralization on channel gating and assembly (Chen et al., 2000). We also see that the region of buried residues narrows to a remarkably short three-residue barrier in the open state (T249 to I251, Fig. 5 C).
DISCUSSION
Our results provide new insights into the nature of S4 movements in the HCN channels by defining the pattern of MTSET reactivity with a series of Cys-substituted S4 residues. Residues in the NH2-terminal half of S4 (R244 to R247) showed a high rate of reaction with external but not internal MTSET; this reactivity was largely state independent. Residues in the COOH-terminal half of S4 (L252 to I264) showed a high rate of reactivity with internal MTSET, but not external MTSET. These COOH-terminal S4 residues showed a pronounced state dependence; upon channel closure the rate of reactivity decreased by 2–3 orders of magnitude. Moreover, for NH2-terminal residues, MTSET modification inhibited channel opening. For COOH-terminal residues, open-state modification enhanced channel opening. This pattern of accessibility is difficult to reconcile with the idea that S4 moves to translocate charge across the membrane and is incompatible with two leading models for S4 movements, as discussed below.
Previous Models of S4 Voltage Sensing
The nature and movement of the voltage-sensing mechanism in voltage-gated channels has been extensively investigated (for reviews see Bezanilla, 2000, 2002; Gandhi and Isacoff, 2002; Horn, 2002; Larsson, 2002; Cohen et al., 2003). There is a general consensus, based largely on functional and mutagenesis studies of the depolarization-activated Shaker K+ channels, that the S4 segment is an α-helix that traverses the lipid membrane more-or-less perpendicular to the membrane. Voltage sensing is thought to occur through the displacement of a series of positively charged S4 residues through the membrane electric field due to a translocation of S4 relative to the membrane. The outward movement of S4 gives rise to an outward gating current and is coupled to the opening of the channel. One surprising and consistent finding from previous studies is that the barrier between the cytosolic and extracellular environment is very narrow; only a short stretch of ∼10 residues appears to be buried in a given state. Because this is much shorter than the length of an α-helix necessary to span the lipid bilayer, S4 has been proposed to reside in water-filled crevices that form a gating canal through which it moves (Larsson et al., 1996; Mannuzzu et al., 1996; Yang et al., 1996; Baker et al., 1998; Cha et al., 1999; Glauner et al., 1999).
These observations have led to the classic model of S4 voltage sensing (see Fig. 1 A, introduction). The discovery that some members of the voltage-gated channel family are activated by hyperpolarization, and not depolarization, raises a number of questions about the generality of the voltage-sensing mechanism. Does S4 move in response to voltage in a similar manner in both depolarization- and hyperpolarization-activated channels? Recent studies on other hyperpolarization-activated channels, namely the sea urchin spHCN channel (Männikkö et al., 2002), the archaebacterial MVP channel (Sesti et al., 2003), and the plant KAT1 channel (Latorre et al., 2003), suggest that this indeed is the case. Using MTS modification, such studies suggest that S4 residues move from an externally accessible position to an internally accessible position upon hyperpolarization, the same type of movement that occurs in depolarization-activated channels. Such studies further imply that the difference between depolarization- and hyperpolarization-activated channel gating must arise from differences downstream of the voltage-sensing mechanism that couple S4 movement to channel opening.
Recent studies in the archaebacterial, depolarization-activated K+-selective channel, KvAP (Ruta et al., 2003), suggest a radically different protein topography (Jiang et al., 2003a) and voltage-sensing mechanism (Jiang et al., 2003b). The X-ray crystal structure of a KvAP–antibody complex showed that the S4 and S3b segments form a helix-loop-helix structure, the voltage-sensing paddle, which lies on the outer radius of the channel within the lipid bilayer, rather than within a water-filled protein crevice. Furthermore, in this KvAP model S4-S3b lies roughly horizontal to the membrane (perpendicular to the pore axis) in the closed state at negative potentials, and is suggested to move into a more upright position during activation gating in response to depolarization (see Fig. 1 B, introduction).
Comparison with other Results on Hyperpolarization-activated and Depolarization-activated Voltage-gated Channels
Our results on S4 of the mammalian mHCN1 suggest that the mechanism of gating differs from either the classic or KvAP models. One of our most striking results is the presence of a minimal barrier between the external and cytosolic environment that is ∼17 residues long (extending from position 249 to 265, see Fig. 5 C) in the closed state and that narrows to only ∼3-residues long in the open state (extending from position 249 to 251, see Fig. 5 C). Such a short barrier is incompatible with the idea that S4 is buried within the lipid bilayer, as in the KvAP model. The results are, however, consistent with the notion that S4 is surrounded by water-filled crevices that form an hour-glass–shaped gating canal, similar to conclusions reached for depolarization-activated channels. However, our finding that only the internal half of S4 of HCN1 experiences a large change in its surrounding environment whereas the external half of S4 exhibits little state-dependent structural change is inconsistent with either a classic S4 translation or KvAP paddle motion.
The difference between our results on mammalian HCN1 and previous results on the hyperpolarization-activated sea urchin spHCN (Männikkö et al., 2002), archaebacterial MVP (Sesti et al., 2003), and plant KAT1 (Latorre et al., 2003) channels, orthologues of the mammalian HCN channels, might be attributable to the functional differences among the channels. However, our conclusions also differ from those of Vemana et al. (2004)(in this issue) on mHCN1, who used MTS reagents to scan accessibility of substituted Cys residues on the S4 of a truncated mHCN1, in which 7 out of 12 Cys residues were removed. Most of our experimental results are in essential agreement with those of Vemana et al. (2004), who examined 6 of the 12 Cys-substituted residues that we have analyzed. Thus, that study found little state dependence for external MTSET reactivity with R247C but a large open-state–dependent increase in reactivity with internal MTSET for L254C and S261C. However, Vemana et al. (2004) report a larger open-state–dependent decrease in external MTS reactivity for T249C and S253C than we found. Based on these results, the authors conclude that S4 of mHCN1 is likely to move inward upon hyperpolarization, which shields T249C and S253C from the external environment and exposes L254C and S261C to the internal environment. Thus, Vemana et al. (2004) suggest that S4 movements in mHCN1 are similar to those of depolarization-activated K+ channels and the hyperpolarization-activated spHCN (Männikkö et al., 2002), MVP (Sesti et al., 2003), and KAT1 (Latorre et al., 2003) channels.
However, even if T249C does undergo a significant state-dependent change in reactivity, it is difficult to reconcile our results with the idea that S4 moves inward upon hyperpolarization. In particular, a rigid body inward motion of S4 during activation cannot explain how the externally facing R247C, which lies just two residues above T249C, can retain its high rate of reactivity with external MTSET in the open state, whereas a stretch of 13 COOH-terminal S4 residues (L252C-I264C) show a 100 to 1,000-fold increase in internal reactivity. Thus, rather than representing an inward movement of S4, we conclude that the state-dependent reactivity of T249C is more likely to reflect a more local conformational change.
New Models for Mammalian HCN Channel Voltage Sensing
Three models that are consistent with the pattern of substituted Cys accessibility along the mHCN1 S4 segment that we and Vemana et al. (2004) report are shown in Fig. 6 : a swinging-tail model (Fig. 6 A), a collapsing gating canal model (Fig. 6 B), and a rotating helix model (Fig. 6 C).
Figure 6.

Three models for conformational changes in HCN1-R S4 region during channel gating based on MTSET-reactivity experiments. (A) Swinging-tail model, the COOH-terminal tail of S4 is buried within the lipid or protein interface (gray, semi-opaque area) in the closed state and swings into an aqueous environment in the open state. (B) Collapsing gating canal model, an extensive aqueous crevice collapses around and buries the COOH-terminal tail of S4 in the closed state. In the open state the crevice is open. (C) Rotation model, channel opening is associated with an ∼180° rotation of the COOH terminal tail of S4 that exposes one face of the α-helix that is buried within the membrane in the closed state.
According to the swinging tail model, the COOH-terminal tail of S4 lies buried in the membrane when the channel is closed at positive potentials and swings down into an aqueous-accessible environment (or gating canal) when the channel opens in response to hyperpolarization. This movement is somewhat similar to that proposed for KvAP (Jiang et al., 2003b), except in the archaebacterial channel the entire S3b-S4 voltage sensor paddle moves as a unit. Our data showing a stationary external end of S4 argue against a concerted S3b-S4 motion. Moreover, the lack of any obvious structural break in the mHCN1 S4 α-helix, such as the conserved glycine-gating hinge observed in S6 (del Camino et al., 2000; Jiang et al., 2002) or proline interruption in S3 (Li-Smerin et al., 2000; e.g., see Fig. 2 A), makes this model less attractive.
In the rotation model, the COOH-terminal portion of S4 is proposed to lie on the internal surface of the gating canal in the open state. Depolarization causes a rotation of S4, burying charge within the canal (Fig. 6 C). According to this model, the similar state dependence of Cys substitutions at L254, L257, S261, and I264 results from the fact that these residues all lie on one side of the S4 helix. Thus, a rotation of ∼180° can potentially move these Cys substitutions from all-accessible to all-buried. However, this model cannot explain the selective open state accessibility of L252C, which lies on the opposite face to L254C, L257, S261, and I264 (see Figs. 5 C and 6 C).
In the collapsing gating canal, there is a minimal movement of S4 associated with channel gating. Rather, a change in the environment around the COOH-terminal portion of S4 during channel closure is proposed to close the access of this region to the internal aqueous environment and to alter the membrane electric field around S4. The net result is a displacement of charge relative to the field that underlies voltage sensing and gating. The concept of a static S4 in which voltage sensing results from the movement of the environment and membrane electric field was previously suggested as an alternative mechanism for the voltage-gated sodium channel (Yang et al., 1996).
Previous studies on depolarization-activated channels are consistent with certain aspects of the collapsing gating canal model. Movements and crevice formation around S3 have been observed in response to voltage changes in the human skeletal muscle voltage-gated Na+ channel (Nguyen and Horn, 2002). In Shaker potassium channels, the buried region of S4 was found to expand during depolarization (Larsson et al., 1996), similar to what we observe here for mHCN1. Perhaps the most relevant results come from the study of a hyperpolarization-induced switch to a slow mode of opening in depolarization-activated EAG channels (Schonherr et al., 2002). MTS reactivity changes indicate that extreme hyperpolarization narrows the crevice around S4 due to motions of neighboring S2/S3 transmembrane segments. The collapse of the S2/S3 crevice also forms a Mg2+ binding site, coordinated by pairs of acidic residues in S2 and S3 (Silverman et al., 2000). Interestingly, although such negative residues are not present in the homologous positions of other depolarization-activated channels, they are conserved in all HCN family members (D172 and D222 in mHCN1 align with D278 and D327 in EAG). However, hyperpolarization exerts an opposite effect on gating canal architecture in HCN and EAG channels. In EAG, hyperpolarization collapses the external portion of the canal and inhibits opening, whereas in HCN channels hyperpolarization opens the internal portion of the canal and enhances opening.
If S4 is static, how do voltage changes trigger the rearrangement of HCN1 to cause the collapse or expansion of the gating canal? HCN channels contain a total of six highly conserved negative charges in S1, S2, S3, and S5. In Shaker channels, movement of negative charge in S2 is thought to contribute to gating currents (Seoh et al., 1996). Thus, one possibility is that the non-S4 membrane segments that form the gating canal act as voltage sensors, moving in response to an interaction of their negative charge with the membrane electric field. However, the conformation change associated with the gating canal collapse does not require a direct action of voltage on charged non-S4 residues. Rather, the effect of voltage could be purely allosteric. When the membrane is depolarized, the collapse of the gating canal would lower the free energy of the channel (i.e., produce a more favorable energetic state) by enveloping the positively charged S4 residues in a low dielectric environment, partially insulating them from the positive internal voltage.
Total Charge Movement Predicted by Mammalian HCN S4 Voltage-sensing Models
Any successful model must account for the total charge movement relative to the electric field associated with gating. In inside-out patches, the HCN tail current activation curve shows a steep voltage dependence, with activation increasing e -fold for a 4-mV hyperpolarization. This corresponds to a movement of six elementary charges per channel, similar to that observed for depolarization-activated K+ and Na+ channels. Because macroscopic activation curves only provide a lower limit on total charge movement, the actual charge moved in HCN channels could be as high as 13, based on gating current measurements from K+ channels (Schoppa et al., 1992; Aggarwal and MacKinnon, 1996; Seoh et al., 1996). Thus, for each S4 segment, slightly more than three charges would need to traverse the entire electric field.
Assuming a linear drop in the electric field from the external side of the membrane to the internal side, the fractional charge contribution of each positive residue can be approximated by its fractional linear position within the field, whose boundaries are delineated by the boundaries of MTSET accessibility. Our studies indicate that the internally facing charged residues R256, R259, R262, and R265 undergo a transition from a buried position in the closed state to an internally accessible position in the open state. In the closed state, the buried region comprises around 17 residues, from T249 to R265. R256 lies at position 10 out of 17 residues from the internal membrane surface in the closed state (see Fig. 5 C), and thus would contribute 10/18 of an elementary charge as it becomes fully exposed to the internal environment in the open state. Following this logic the contribution for each of the remaining residues (in elementary charges) is: R259, 7/18; R262, 4/18; and R265, 1/18. K250 lies in position 2 out of 3 buried residues (T249 to I251) in the open state and at position 16 out of 17 buried residues in the closed state (see Fig. 5 C). This change in relative position within the electric field will contribute 7/18 of a charge. Thus, each S4 will contribute ∼1.6 elementary charges, totaling ∼6.4 elementary charges for the tetrameric complex. Additional gating charge could be contributed by the movement of some of the 24 negative charges per channel or by a nonlinearity in the electric field around the internal half of S4.
In contrast to the likely contribution of the internal half of S4 to charge displacement through the membrane field, the four positively charged residues (K238, R241, R244, and R247, see Fig. 3 B) in the NH2-terminal half of S4 in mHCN1 are unlikely to contribute to charge displacement, due to their minimal state-dependent change in MTSET accessibility (see Fig. 5 C). Nonetheless, this region of S4 is likely to play an important role in channel function since modification of R247C and R244C by MTSET causes an inhibition of hyperpolarization-activated current. The importance of these positively charged residues in HCN channel gating was previously highlighted by mutagenesis studies, in which charge neutralization caused ∼20-mV negative shifts in activation curves (Chen et al., 2000; Vaca et al., 2000). These results suggest that the NH2-terminal half of S4 may play a more static, structural role, stabilizing channel states by interacting with negatively charged residues in neighboring segments. Indeed, R174 in the plant hyperpolarization-activated channel KAT1 (equivalent to R247 in mHCN1, see Fig. 3 B) was recently suggested to participate in salt-bridge formation with negative charges in S2 or S3, but not to contribute to gating charge movement (Sato et al., 2003). Thus, at these NH2-terminal S4 residues, the effects of charge neutralization shown previously and the effects of MTSET modification observed in this study may be explained by the disruption of structural interactions that alter the stability of the open or closed state of the channel.
In conclusion, the MTSET reactivity of Cys substitutions along the S4 voltage sensor in mammalian HCN channels shows that only the COOH-terminal, internal end of the S4 voltage sensor undergoes a marked and global change in environment during channel gating. Our data are consistent with a model in which the S4 COOH-terminal tail lies in an extensive water-filled canal in the open state. Upon channel closure, this crevice collapses, presumably due to the movement of neighboring transmembrane α-helixes. This gating canal motion is proposed to be coupled to the movement of the activation gate of the channel. A similar gating canal collapse is also thought to underlie a hyperpolarization-dependent slowing in the opening of the depolarization-activated EAG channel. However, the effects of hyperpolarization on gating and gating canal movements is reversed in EAG and HCN channels, implying a fundamental difference in the way voltage is coupled to transmembrane region rearrangements. Specification of the nature of these rearrangements in membrane helixes surrounding S4 during hyperpolarization gating is clearly an important goal for future studies.
Acknowledgments
We thank Nelson B. Olivier for technical assistance with molecular modeling and Edgar Young, Lei Zhou, and Chris Ulens for their helpful discussions and comments on the manuscript.
D.C. Bell was supported by the Wellcome Trust, UK. This work was partially supported by grant NS36658 from the National Institutes of Health.
Olaf S. Andersen served as editor.
Abbreviations used in this paper: HCN1-R, methane thiosulfonate–resistant mHCN1 channel; IO, inside-out; mHCN1, mouse hyperpolarization–activated, cyclic nucleotide–modulated, cation-nonselective; MTS, methane thiosulfonate; MTSET, [2-(trimethylammonium) ethyl] methane thiosulfonate; TEVC, two-electrode voltage clamp.
References
- Aggarwal, S.K., and R. MacKinnon. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177. [DOI] [PubMed] [Google Scholar]
- Akabas, M.H., D.A Stauffer, M. Xu, and A. Karlin. 1992. Acetylcholine receptor channel structure probed in cysteine-substitution mutants. Science. 258:307–310. [DOI] [PubMed] [Google Scholar]
- Anderson, J.A., S.S. Huprikar, L.V. Kochian, W.J. Lucas, and R.F. Gaber. 1992. Functional expression of a probable Arabidopsis thaliana potassium channel in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA. 89:3736–3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker, O.S., H.P. Larsson, L.M. Mannuzzu, and E.Y. Isacoff. 1998. Three transmembrane conformations and sequence-dependent displacement of the S4 domain in shaker K+ channel gating. Neuron. 20:1283–1294. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F. 2000. The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80:555–592. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F. 2002. Voltage sensor movements. J. Gen. Physiol. 120:465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall, W.A. 1986. Molecular properties of voltage-sensitive sodium channels. Annu. Rev. Biochem. 55:953–985. [DOI] [PubMed] [Google Scholar]
- Cha, A., G.E. Snyder, P.R. Selvin, and F. Bezanilla. 1999. Atomic scale movement of the voltage-sensing region in a potassium channel measured via spectroscopy. Nature. 402:809–813. [DOI] [PubMed] [Google Scholar]
- Chen, J., J.S. Mitcheson, M. Lin, and M.C. Sanguinetti. 2000. Functional roles of charged residues in the putative voltage sensor of the HCN2 pacemaker channel. J. Biol. Chem. 275:36465–36471. [DOI] [PubMed] [Google Scholar]
- Cohen, B.E., M. Grabe, and L.Y. Jan. 2003. Answers and questions from the KvAP structures. Neuron. 39:395–400. [DOI] [PubMed] [Google Scholar]
- del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- Gandhi, C.S., and E.Y. Isacoff. 2002. Molecular models of voltage sensing. J. Gen. Physiol. 120:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauss, R., R. Seifert, and U.B. Kaupp. 1998. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature. 393:583–587. [DOI] [PubMed] [Google Scholar]
- Glauner, K.S., L.M. Mannuzzu, C.S. Gandhi, and E.Y. Isacoff. 1999. Spectroscopic mapping of voltage sensor movement in the Shaker potassium channel. Nature. 402:813–817. [DOI] [PubMed] [Google Scholar]
- Goldstein, S.A. 1996. A structural vignette common to voltage sensors and conduction pores: canaliculi. Neuron. 16:717–722. [DOI] [PubMed] [Google Scholar]
- Goulding, E.H., J. Ngai, R.H. Kramer, S. Colicos, R. Axel, S.A. Siegelbaum, and A. Chess. 1992. Molecular cloning and single-channel properties of the cyclic nucleotide-gated channel from catfish olfactory neurons. Neuron. 8:45–58. [DOI] [PubMed] [Google Scholar]
- Horn, R. 2002. Coupled movements in voltage-gated ion channels. J. Gen. Physiol. 120:449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. a. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., V. Ruta, J. Chen, A. Lee, and R. MacKinnon. 2003. b. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 423:42–48. [DOI] [PubMed] [Google Scholar]
- Kamb, A., L.E. Iverson, and M.A. Tanouye. 1987. Molecular characterization of Shaker, a Drosophila gene that encodes a potassium channel. Cell. 50:405–413. [DOI] [PubMed] [Google Scholar]
- Karlin, A., and M.H. Akabas. 1998. Substituted-cysteine accessibility method. Methods Enzymol. 293:123–145. [DOI] [PubMed] [Google Scholar]
- Larsson, H.P. 2002. The search is on for the voltage sensor-to-gate coupling. J. Gen. Physiol. 120:475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, H.P., O.S. Baker, D.S. Dhillon, and E.Y. Isacoff. 1996. Transmembrane movement of the shaker K+ channel S4. Neuron. 16:387–397. [DOI] [PubMed] [Google Scholar]
- Latorre, R., R. Olcese, C. Basso, C. Gonzalez, F. Munoz, D. Cosmelli, and O. Alvarez. 2003. Molecular coupling between voltage sensor and pore opening in the Arabidopsis inward rectifier K+ channel KAT1. J. Gen. Physiol. 122:459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liman, E.R., P. Hess, F. Weaver, and G. Koren. 1991. Voltage-sensing residues in the S4 region of a mammalian K+ channel. Nature. 353:752–756. [DOI] [PubMed] [Google Scholar]
- Liman, E.R., J. Tytgat, and P. Hess. 1992. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 9:861–871. [DOI] [PubMed] [Google Scholar]
- Li-Smerin, Y., D.H. Hackos, and K.J. Swartz. 2000. α-Helical structural elements within the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 115:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logothetis, D.E., S. Movahedi, C. Satler, K. Lindpaintner, and B. Nadal-Ginard. 1992. Incremental reductions of positive charge within the S4 region of a voltage-gated K+ channel result in corresponding decreases in gating charge. Neuron. 8:531–540. [DOI] [PubMed] [Google Scholar]
- Ludwig, A., X. Zong, M. Jeglitsch, F. Hofmann, and M. Biel. 1998. A family of hyperpolarization-activated mammalian cation channels. Nature. 393:587–591. [DOI] [PubMed] [Google Scholar]
- Männikkö, R., F. Elinder, and H.P. Larsson. 2002. Voltage-sensing mechanism is conserved among ion channels gated by opposite voltages. Nature. 419:837–841. [DOI] [PubMed] [Google Scholar]
- Mannuzzu, L.M., M.M. Moronne, and E.Y. Isacoff. 1996. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 271:213–216. [DOI] [PubMed] [Google Scholar]
- Nguyen, T.P., and R. Horn. 2002. Movement and crevices around a sodium channel S3 segment. J. Gen. Physiol. 120:419–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda, M., S. Shimizu, T. Tanabe, T. Takai, T. Kayano, T. Ikeda, H. Takahashi, H. Nakayama, Y. Kanaoka, N. Minamino, et al. 1984. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 312:121–127. [DOI] [PubMed] [Google Scholar]
- Papazian, D.M., L.C. Timpe, Y.N. Jan, and L.Y. Jan. 1991. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 349:305–310. [DOI] [PubMed] [Google Scholar]
- Pascual, J.M., and A. Karlin. 1998. State-dependent accessibility and electrostatic potential in the channel of the acetylcholine receptor. Inferences from rates of reaction of thiosulfonates with substituted cysteines in the M2 segment of the α-subunit. J. Gen. Physiol. 111:717–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, R.B., and S.A. Siegelbaum. 2003. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu. Rev. Physiol. 65:453–480. [DOI] [PubMed] [Google Scholar]
- Ruta, V., Y. Jiang, A. Lee, J. Chen, and R. MacKinnon. 2003. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature. 422:180–185. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E.F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. 2nd edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Santoro, B., S.G. Grant, D. Bartsch, and E.R. Kandel. 1997. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. USA. 94:14815–14820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, B., D.T. Liu, H. Yao, D. Bartsch, E.R. Kandel, S.A. Siegelbaum, and G.R. Tibbs. 1998. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 93:717–729. [DOI] [PubMed] [Google Scholar]
- Sato, Y., M. Sakaguchi, S. Goshima, T. Nakamura, and N. Uozumi. 2003. Molecular dissection of the contribution of negatively and positively charged residues in S2, S3, and S4 to the final membrane topology of the voltage sensor in the K+ channel, KAT1. J. Biol. Chem. 278:13227–13234. [DOI] [PubMed] [Google Scholar]
- Schonherr, R., L.M. Mannuzzu, E.Y. Isacoff, and S.H. Heinemann. 2002. Conformational switch between slow and fast gating modes: allosteric regulation of voltage sensor mobility in the EAG K+ channel. Neuron. 35:935–949. [DOI] [PubMed] [Google Scholar]
- Schoppa, N.E., K. McCormack, M.A. Tanouye, and F.J. Sigworth. 1992. The size of gating charge in wild-type and mutant Shaker potassium channels. Science. 255:1712–1715. [DOI] [PubMed] [Google Scholar]
- Sentenac, H., N. Bonneaud, M. Minet, F. Lacroute, J.M. Salmon, F. Gaymard, and C. Grignon. 1992. Cloning and expression in yeast of a plant potassium ion transport system. Science. 256:663–665. [DOI] [PubMed] [Google Scholar]
- Seoh, S.A., D. Sigg, D.M. Papazian, and F. Bezanilla. 1996. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 16:1159–1167. [DOI] [PubMed] [Google Scholar]
- Sesti, F., S. Rajan, R. Gonzalez-Colaso, N. Nikolaeva, and S.A. Goldstein. 2003. Hyperpolarization moves S4 sensors inward to open MVP, a methanococcal voltage-gated potassium channel. Nat. Neurosci. 6:353–361. [DOI] [PubMed] [Google Scholar]
- Silverman, W.R., C.Y. Tang, A.F. Mock, K.B. Huh, and D.M. Papazian. 2000. Mg2+ modulates voltage-dependent activation in ether-a-go-go potassium channels by binding between transmembrane segments S2 and S3. J. Gen. Physiol. 116:663–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer, W., F. Conti, H. Suzuki, X.D. Wang, M. Noda, N. Yahagi, H. Kubo, and S. Numa. 1989. Structural parts involved in activation and inactivation of the sodium channel. Nature. 339:597–603. [DOI] [PubMed] [Google Scholar]
- Vaca, L., J. Stieber, X. Zong, A. Ludwig, F. Hofmann, and M. Biel. 2000. Mutations in the S4 domain of a pacemaker channel alter its voltage dependence. FEBS Lett. 479:35–40. [DOI] [PubMed] [Google Scholar]
- Vemana, S., S. Pandey, H.P. Larsson. 2004. S4 movement in mammalian HCN. J. Gen. Physiol. 123:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger, B.J., M. DeGennaro, B. Santoro, S.A. Siegelbaum, and G.R. Tibbs. 2001. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 411:805–810. [DOI] [PubMed] [Google Scholar]
- Xue, T., and R.A. Li. 2002. An external determinant in the S5-P linker of the pacemaker (HCN) channel identified by sulfhydryl modification. J. Biol. Chem. 277:46233–46242. [DOI] [PubMed] [Google Scholar]
- Yang, N., A.L. George, Jr., and R. Horn. 1996. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 16:113–122. [DOI] [PubMed] [Google Scholar]
- Yang, N., and R. Horn. 1995. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 15:213–218. [DOI] [PubMed] [Google Scholar]
- Yusaf, S.P., D. Wray, and A. Sivaprasadarao. 1996. Measurement of the movement of the S4 segment during the activation of a voltage-gated potassium channel. Pflugers Arch. 433:91–97. [DOI] [PubMed] [Google Scholar]