

Abstract
The venom from spiders, scorpions, and sea anemone contain a rich diversity of protein toxins that interact with ion channel voltage sensors. Although atomic structures have been solved for many of these toxins, the surfaces that are critical for interacting with voltage sensors are poorly defined. Hanatoxin and SGTx are tarantula toxins that inhibit activation of Kv channels by interacting with each of the four voltage sensors. In this study we set out to identify the active surface of these toxins by alanine-scanning SGTx and characterizing the interaction of each mutant with the Kv2.1 channel. Examination of the concentration dependence for inhibition identified 15 mutants with little effect on the concentration dependence for toxin inhibition of the Kv2.1 channel, and 11 mutants that display moderate to dramatic perturbations. Mapping of these results onto the structure of SGTx identifies one face of the toxin where mutations with pronounced perturbations cluster together, and a backside of the toxin where mutations are well tolerated. The active surface of SGTx contains a ring-like assembly of highly polar residues, with two basic residues that are particularly critical, concentrically arranged around a hydrophobic protrusion containing critical aliphatic and aromatic residues. These results identify the active surface of the toxin and reveal the types of side chains that are important for interacting with voltage sensors.
Keywords: spider venom, scanning mutagenesis, gating modifier, voltage-activated channels
INTRODUCTION
The venom from poisonous animals contains a fascinating array of protein toxins that interact with ion channels embedded in the plasma membrane of excitable cells. For example, venom from arachnids (spiders and scorpions), anthozoans (sea anemone), mollusks (cone snails), and reptiles (snakes) contain toxins that target voltage-gated potassium (Kv), sodium (Nav) or calcium (Cav) channels (Possani et al., 2000; Rash and Hodgson, 2002; Srinivasan et al., 2002). All of these voltage-gated channels are thought to be comprised of two types of domains—a pore-forming domain constructed from the tetrameric arrangement of S5-S6 segments and four voltage-sensing domains, each constructed from the S1-S4 segments (Kubo et al., 1993; Doyle et al., 1998; Li-Smerin and Swartz, 1998; Li-Smerin et al., 2000a; Lu et al., 2001; Jiang et al., 2003a). Interestingly, the toxins that target voltage-gated ion channels segregate into two classes based on the domain that they interact with and their mechanisms of action. The first class of toxins interact with the external vestibule of the ion conduction pore and work like a plug to block the flow of ions (Miller, 1995). Examples of pore-blocking toxins are abundant for Kv channels and other types of K+ channels (Miller, 1995; Rodriguez de la Vega et al., 2003), but also exist for Nav (Cruz et al., 1985; Moczydlowski et al., 1986) and probably also for Cav channels (Boland et al., 1994; Ellinor et al., 1994; Feng et al., 2003). Pore-blocking toxins have been exceptionally useful tools for investigations of channel structure and function, helping to elucidate the pore-forming parts of the channel (MacKinnon and Miller, 1989), to determine subunit stoichiometry (MacKinnon, 1991) and arrangement (Dudley et al., 2000), and to establish structural similarities between prokaryotic channels of known structure and eukaryotic channels of unknown structure (MacKinnon et al., 1998; Legros et al., 2000). The interaction surfaces between toxins and the ion conduction pores of K+ channels have been studied in some detail, and are largely dominated by interactions between polar residues (Park and Miller, 1992b; Stampe et al., 1994; Hidalgo and MacKinnon, 1995; Naranjo and Miller, 1996; Ranganathan et al., 1996). In the case of charybdotoxin, one of the better studied potassium channel pore-blocking toxins, a Lys residue on the active surface of the toxin projects into the pore of the channel and interacts with potassium ions bound within the selectivity filter (Anderson et al., 1988; MacKinnon and Miller, 1988; Park and Miller, 1992a). The second class of toxins interact with the voltage-sensing domains of voltage-gated channels and modify the gating behavior of the channel through allosteric mechanisms. These include the α-scorpion toxins and sea anemone toxins that bind to the voltage-sensors in Nav channels and slow inactivation (Hanck and Sheets, 1995; Rogers et al., 1996; Cestele and Catterall, 2000), thereby facilitating channel activity, and numerous spider toxins that bind to the voltage-sensors in Kv, Cav and Nav channels and inhibit channel activity (McDonough et al., 1997a,b; Swartz and MacKinnon, 1997a,b; Li-Smerin and Swartz, 1998, 2000, 2001; Winterfield and Swartz, 2000; Middleton et al., 2002). In contrast to pore-blocking toxins, the interface between gating modifier toxins and voltage-sensors is poorly understood.
The structure of a Kv channel from aeropyrum pernix (KvAP) was recently solved to atomic resolution (Jiang et al., 2003a). The pore-forming part of KvAP is structurally quite similar to the corresponding regions in other types of potassium channels (e.g., KcsA, KirBac, and MthK channels) (Doyle et al., 1998; Jiang et al., 2002a,b; Kuo et al., 2003). The structure of the voltage-sensing domain (S1–S4) in both the intact KvAP channel and the isolated S1–S4 domain shows that each of the S1–S4 segments adopt α-helical secondary structures, with S3 having two separate helices interrupted by a short nonhelical region (Jiang et al., 2003a), all of which are consistent with helix-scanning studies on eukaryotic Kv channels (Monks et al., 1999; Hong and Miller, 2000; Li-Smerin et al., 2000a; Li-Smerin and Swartz, 2001). An interesting feature of the KvAP structures is the presence of a helix-turn-helix motif termed the voltage-sensor paddle. This paddle motif is comprised of both S3b and S4 helices and is positioned toward the intracellular membrane–water interface in the periphery of the tetrameric complex. From experiments probing the accessibility of biotinylated residues to avidin (Jiang et al., 2003b), the paddles in KvAP were proposed to translocate through the hydrophobic membrane core during activation, moving from a position near the intracellular side when in a resting conformation at negative voltages, to an extracellular position when activated by membrane depolarization.
The model for activation of the KvAP channel is intriguing because several gating modifier toxins interact with residues in the voltage-sensor paddles when applied to the extracellular side of the membrane. Hanatoxin, for example, is a protein toxin from tarantula venom that inhibits the Kv 2.1 channel by interacting with the S3b helix within the voltage-sensor paddle (Swartz and MacKinnon, 1997a,b; Li-Smerin and Swartz, 1998, 2000, 2001). In the KvAP model, the paddle motif would be accessible to the extracellular solution when adopting an activated conformation, but located near the intracellular solution when adopting a resting conformation. However, measurements of gating charge movement suggest that hanatoxin stabilizes a resting state of the voltage sensors when it binds from the extracellular solution (Lee et al., 2003). How could a water soluble toxin access a resting voltage-sensor paddle that is embedded in the lipid membrane? The NMR solution structure of hanatoxin (Takahashi et al., 2000) reveals that one face of the toxin contains a protruding cluster of hydrophobic residues, primarily comprised of W30, L19, L5, F6, Y4, and Y27 (Fig. 1 B). If the interaction between toxin and paddle is dominated by hydrophobic interactions, then perhaps this nonpolar region of the toxin can intercalate into the membrane to reach the resting voltage-sensor paddle (Lee et al., 2003). The perimeter of this face of hanatoxin is comprised of highly polar residues (Fig. 1 B), including six basic residues (R3, K10, K26, R24, K22, K17) and four acidic residues (E1, D25, D31, D14). Although the presence of this large number of polar residues should prevent this region of the toxin from penetrating into the hydrophobic core of the membrane, the alternating arrangement of basic and acidic residues raises the possibility that they might interact to form neutral salt-bridges, thereby reducing the energetic cost of entering the membrane. In this study we set out to identify the molecular surface of the toxin that is important for interacting with voltage sensors. Our attempts to produce hanatoxin using recombinant expression systems or solid phase peptide synthesis met with limited success because of extremely low yields in the folding reaction. Recently, however, a related toxin named SGTx was isolated from the venom of an African baboon spider, Scodra griseipes (Fig. 1 A) (Marvin et al., 1999). SGTx and hanatoxin have 80% sequence identity (Fig. 1 A), similar three-dimensional solution structures (Fig. 1, B and C), and inhibit the Kv2.1 channel by a similar mechanism, yet SGTx folds with high efficiency in vitro (Takahashi et al., 2000; Lee et al., 2004). In the present study we use solid phase peptide synthesis to mutate most residues on the toxin to Ala and electrophysiological techniques to study the interaction of toxin mutants with the Kv2.1 channel.
Figure 1.
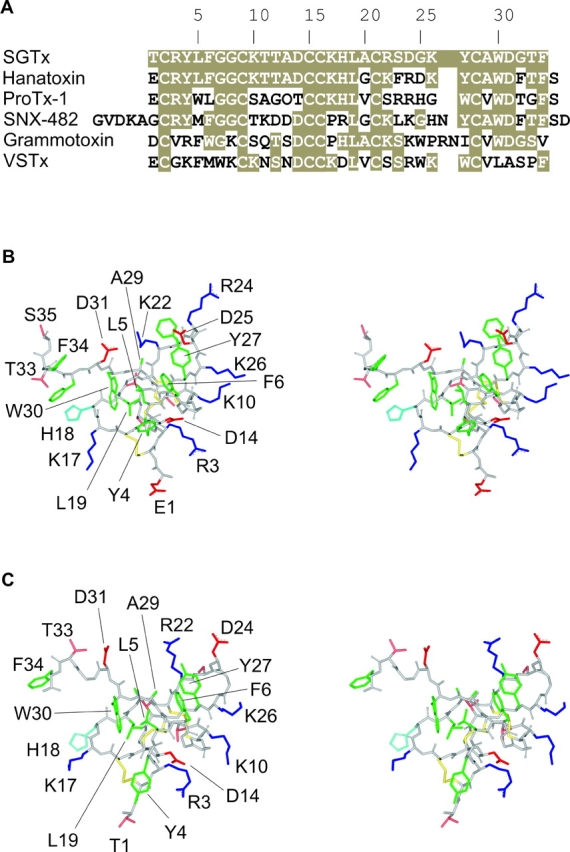
Structures of toxins interacting with the voltage-sensors. (A) Sequence alignment for toxins interacting with voltage sensors. Tan shading indicates similarity to SGTx. (B) Stereo views of the hanatoxin NMR solution structure (PDB accession code 1D1H) shown as a stick model. Side chain coloring as follows: hydrophobic (green), basic (blue), His (light blue), acidic (red), both Ser and Thr (pink), and disulfide bonds in yellow. Backbone atoms are colored light gray. (C) Stereo views of the SGTx NMR solution structure (PDB accession code 1LA4) shown as a stick model. Same color scheme as in B. Structures here and in all subsequent figures were generated using DS Viewer Pro (Accelrys).
MATERIALS AND METHODS
Solid Phase Peptide Synthesis of SGTx
Peptide synthesis was conducted on an Applied Biosystems model 433A peptide synthesizer. The linear precursor of SGTx was synthesized using solid-phase methodology with Fmoc chemistry, starting from Fmoc-Phe-Alko resin using a variety of blocking groups for the protection of the amino acids. After trifluoroacetic acid cleavage, a crude linear peptide was extracted with 2 M acetic acid and diluted to a final concentration of 25 μM. A solution containing 0.1 M ammonium acetate, 2 M urea, and 2.5 mM reduced/0.25 mM oxidized glutathione was adjusted to pH 7.8 with aqueous NH4OH and stirred slowly at 4°C for 3 d. The folding reaction was monitored with RP-HPLC, and the crude oxidized product was purified by successive chromatography steps with CM-cellulose CM-52 and preparative RP-HPLC with a C18 silica column. The purity of the synthetic SGTx was confirmed by analytical RP-HPLC and MALDI-TOF-MS measurements.
The concentration of synthetic SGTx was determined from dry weight of the protein. To confirm the accuracy of toxin concentration we also measured absorbance at 280 nm and calculated concentration using a calculated extinction coefficient of 8.6 × 103 M−1 cm−1 (Gill and von Hippel, 1989). With the exception of W30A, a mutation that removes the only Trp, concentrations determined from dry weight were within 5% of those determined from absorbance.
Circular Dichroism Measurements of SGTx and its Analogs
CD spectra were obtained for wild-type SGTx and 26 mutants using a JASCO J-750 spectropolarimeter (0.01 M sodium phosphate solution in water, pH 7.0) at 20°C with a quartz cell of path length 1 mm. The spectra were expressed as molecular ellipticity [θ] in deg·cm2·dmol−1.
Functional Characterization of SGTx Mutants
The inhibitory activity of synthetic SGTx was examined against the Kv2.1 (Frech et al., 1989) Δ7 channel expressed in Xenopus oocytes. Kv2.1Δ7 contains seven mutations in the S5-S6 linker (Aggarwal and MacKinnon, 1996; Li-Smerin and Swartz, 1998) that render the channel sensitive to the pore-blocking toxin agitoxin-2, enabling the toxin to be used to subtract background conductances. Kv2.1 cDNA in the pBlu-SK- vector was linearized with NotI and transcribed using T7 RNA polymerase. Xenopus laevis oocytes were removed surgically and incubated with agitation for 1–1.5 h in a solution containing (in mM): 82.5 NaCl, 2.5 KCl, 1 MgCl2, 5 HEPES, and 2 mg/ml collagenase (Worthington Biochemical Corp.), pH 7.6 with NaOH. Defolliculated oocytes were injected with cRNA and incubated at 17°C in a solution containing (in mM): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, and 50 μg/ml gentamicin (Invitrogen/GIBCO BRL), pH 7.6 with NaOH.
Macroscopic ionic currents from oocytes expressing the Kv2.1 Δ7 channel were recorded using two-electrode voltage-clamp recording techniques between 1 and 5 d after cRNA injection using an OC-725C oocyte clamp (Warner Instruments). Oocytes were studied in a 200 μl recording chamber that was perfused with a solution containing (in mM): RbCl (50), NaCl (50), MgCl2 (1), CaCl2 (0.3), and HEPES (20), pH 7.6 with NaOH. Data were filtered at 2 kHz (8-pole Bessel) and digitized at 10 kHz. Microelectrode resistances were between 0.2–1.2 MΩ when filled with 3 M KCl. All experiments were performed at room temperature (∼22°C). Linear capacity, leak, and endogenous currents were subtracted after blocking the Kv2.1 channel with 1 μM agitoxin-2 (Garcia et al., 1994).
RESULTS
Characterization of Wild-type SGTx
The objective of the present study was to define the surface of SGTx that interacts with the voltage sensors in the Kv2.1 channel. As a first step we synthesized the wild-type toxin using solid-phase peptide synthesis, folded the toxin in vitro and purified a dominant and correctly folded species using HPLC (Lee et al., 2004). To characterize the activity of SGTx we studied its interaction with the Kv2.1 channel using two-electrode voltage-clamp recording techniques after expressing the channel in Xenopus oocytes. To initially evaluate the effects of SGTx, the Kv2.1 channel was activated repeatedly by weak depolarizations of the membrane while adding the toxin to the recording chamber. At a concentration of 2.5 μM, the toxin rapidly (τ ∼10 s) produces nearly complete inhibition (Fig. 2, A and B) . The voltage-activated currents recover to control levels after removal of the toxin from the recording chamber (Fig. 2 B), indicating that the effects of the toxin are readily reversible. An important observation suggesting that SGTx inhibits the Kv2.1 channel by modifying gating is that the toxin shifts activation of the channel to more depolarized voltages (Fig. 2 C), similar to previously observed for hanatoxin (Swartz and MacKinnon, 1997a). Although the toxin completely inhibits currents activated by weak depolarization to −15 mV, toxin-bound channels can be opened if the applied voltage is sufficiently depolarized (Fig. 2 C).
Figure 2.
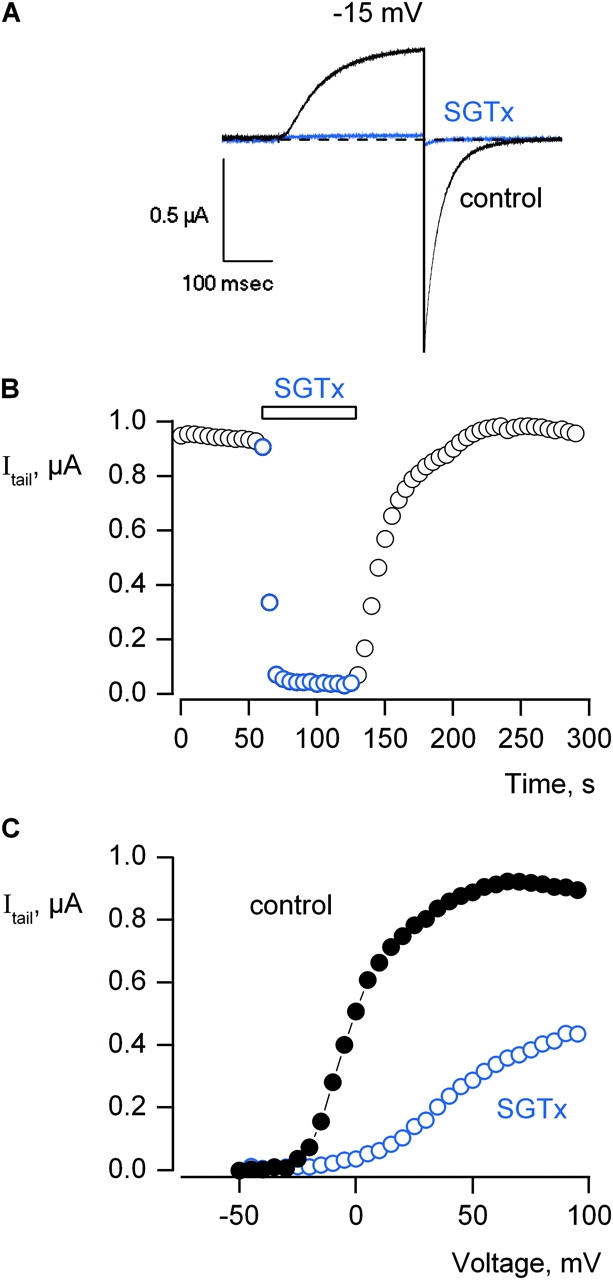
Inhibition of the Kv2.1 channel by SGTx. (A) Current records were elicited by depolarization to −15 mV in the absence (black trace) and presence (blue trace) of 2.5 μM SGTx. Holding voltage was −90 mV and tail voltage was −50 mV. Dashed line indicates the level of zero current. (B) Time course for inhibition of the Kv2.1 channel by 2.5 μM SGTx. Current records were elicited by depolarizations to −15 mV every 5 s. Holding voltage was −90 mV and tail voltage was −50 mV. Same cell as in A. (C) Voltage-activation relations in the absence and presence of 10 μM SGTx. Tail currents obtained following various strength depolarizations were averaged for 0.4 ms beginning 3.4 ms after repolarization to−50 mV. Holding voltage was −90 mV and tail voltage was −50 mV. Same subtraction protocol as in A. In all cases, leak, background, and capacitive currents were subtracted after blocking the channel with agitoxin-2.
To quantify the interaction between SGTx and the resting state of the Kv2.1 channel we examined the concentration dependence for SGTx inhibition of the channel using negative holding voltages (−90 mV) and relatively short (200–300 ms) depolarizing pulses to examine the extent of inhibition. Initially, a range of toxin concentrations (39 nM to 10 μM) were applied as illustrated in Fig. 2 B, and the extent of inhibition monitored until it reached constant values, suggesting that equilibrium had been achieved. The onset of inhibition was slower at lower toxin concentrations (e.g., τ ∼70 s at 156 nM), and thus required more time (up to 200 s) to reach equilibrium. The fractional occupancy of the channel by toxin was estimated, as previously described for hanatoxin (Swartz and MacKinnon, 1997a), by measuring the fractional inhibition of currents activated by depolarizing to voltages between −20 and +40 mV (Fig. 3 A). Since the toxin shifts activation of the channel to more depolarized voltages, the open probability for toxin-bound channels will be low for relatively weak depolarizations, but quite significant for strong depolarizations. Thus, the fraction of uninhibited current (I/I0) will most faithfully approximate the fraction of unbound channels (F) when channel opening is elicited using weak depolarizations where the probability of toxin-bound channels opening is low. If F is estimated from I/I0 in the negative voltage range (−20 to −10 mV) and plotted as a function of SGTx concentration, the data can be well described by a model (smooth curve) with four independent binding sites per channel where occupancy of any single site is sufficient to prevent opening (Swartz and MacKinnon, 1997a; Lee et al., 2003). This analysis yields an equilibrium dissociation constant (Kd) of 2.7 μM for SGTx binding to each of the four voltage-sensor, a value that is ∼20 times higher than what is observed for hanatoxin (Swartz and MacKinnon, 1997a; Lee et al., 2003).
Figure 3.
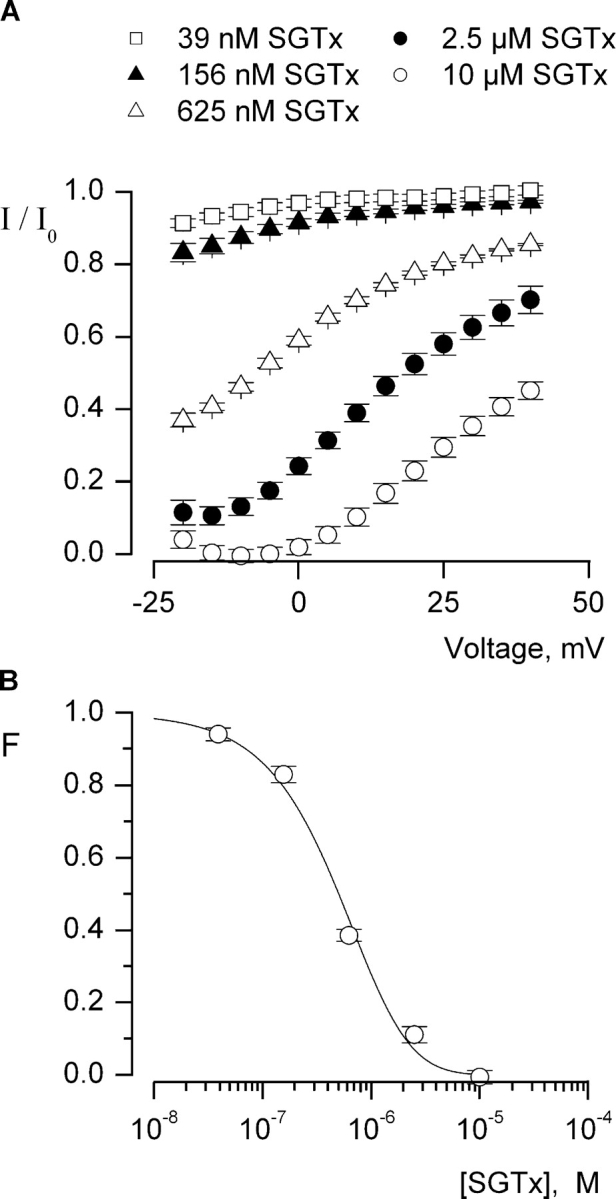
Concentration dependence for fractional occupancy of the Kv2.1 channel by SGTx. (A) Fraction of uninhibited tail currents in the presence of various concentrations of SGTx. Currents were elicited by depolarization to voltages between−25 and +40 mV (5-mV increments) from a holding potential of −90 mV and a tail voltage of −50 mV. I and I0 represent tail currents in the presence and absence of SGTx, respectively, measured 2.3 ms after repolarization to −50 mV and averaged over 0.3 ms. Symbols represent mean ± SEM for 5–10 cells at each concentration. (B) Concentration dependence for fractional occupancy of the Kv 2.1 channel by SGTx. Plot of fraction unbound (F) for varying concentrations of SGTx. Fraction unbound is the fraction of uninhibited tail current for weak depolarizations (taken from −20 to −10 mV). Data points are mean ± SEM for 7 to 11 cells at each concentration of toxin. Solid line is a fit of F = (1 − p)4 to the data, where F is the probability of having a channel with no SGTx bound (with each channel having four equivalent and independent binding sites) and p = [SGTx]/[SGTx] + Kd with a Kd of 2.7 μM.
Characterization of SGTx Mutants
Having characterized the interaction of wild-type SGTx with the Kv2.1 channel, we used solid-phase peptide synthesis to introduce point mutations at all positions except Cys, since mutation of the six Cys residues would disrupt the stabilizing disulfide bond network at the core of the molecule (yellow atoms in Fig. 1 C). 25 mutants were synthesized where Ala was substituted for the wild-type residue, and three mutants were synthesized where Ala residues present in the wild-type toxin were mutated to Ser. We attempted to fold all 28 mutants in vitro and in 26 instances succeeded in obtaining a dominant correctly folded species that could be purified using HPLC. In the cases of L19A and Y27A, the mutations lowered folding efficiency enough so as to prevent identification and isolation of the correctly folded species from the folding reaction. The interaction of each of the 26 correctly folded SGTx mutants with the Kv2.1 channel was then investigated in the same manner as described for the wild-type toxin. For each mutant we determined the Kd from an examination of the concentration dependence for toxin inhibition of the channel, as illustrated for five mutants in Fig. 4 . Table I gives both Kd and perturbation energy (ΔΔG) values for each mutant. 15 mutants exhibit nearly wild-type activity, with Kd values within fourfold of the wild-type toxin and |ΔΔG| values <1 kcal mol−1. Four mutants display moderately weakened interactions with the Kv2.1 channel, with Kd values between 7- and 12-fold higher than the wild-type toxin, corresponding to |ΔΔG| values of 1–1.5 kcal mol−1. Five mutants exhibit greatly weakened interactions, with Kd values >150-fold higher than the wild-type toxin and |ΔΔG| values >3 kcal mol−1. Although the Kd values for this latter group cannot be determined accurately because even the highest concentrations of toxin give relatively high F values (Fig. 4), the mutants in this category clearly exhibit profound perturbations in their interaction with the channel. Interestingly, there are two mutants that exhibit tighter interactions with the Kv2.1 channel, in both cases where an Asp is changed to Ala (Fig. 4; Table I). The Kd for D14A is about fivefold lower and the Kd for D24A is ∼20-fold lower, when compared with the wild-type toxin.
Figure 4.

Concentration dependence for fractional occupancy of the Kv2.1 channel by SGTx mutants. Fraction unbound (F) plotted as a function of SGTx concentration for the wild-type toxin and five Ala mutants. Determinations of F and fitting of a four site model to the data are as described in text and Fig 3. Data points are mean ± SEM for 3 to 11 cells at each toxin concentration. K10A (light gray) is an example of a mutant that does not significantly alter Kd (|ΔΔG| < 1 kcal mol−1). H18A (pink) is an example of a mutant that displays a moderately higher Kd (|ΔΔG| = 1–1.5 kcal mol−1), and both R22A and W30A (red) are examples of mutants with dramatically higher Kd values (|ΔΔG| > 1.5 kcal mol−1). D24A (purple) is an example of a mutant that displays a lower Kd. See Table I for Kd and ΔΔG values for all mutants.
TABLE I.
Perturbations in the Interaction between SGTx and Kv2.1 Channels Resulting from Mutations in the Toxin
| Kd (M) | Kd mut/Kd wt | |ΔΔG| | |
|---|---|---|---|
| kcal mol−1 | |||
| Wild-type | 2.7 × 10−6 | — | — |
| T1A | 2.6 × 10−6 | 1.0 | 0.0 |
| R3A | >5 × 10−4 | >150 | >3.0 |
| Y4A | 2.0 × 10−5 | 7.5 | 1.2 |
| L5A | >5 × 10−4 | >150 | >3.0 |
| F6A | >1 × 10−3 | >300 | >3.4 |
| G7A | 1.0 × 10−5 | 3.8 | 0.8 |
| G8A | 1.9 × 10−6 | 0.7 | 0.2 |
| K10A | 5.2 × 10−6 | 2.0 | 0.4 |
| T11A | 6.3 × 10−6 | 2.4 | 0.5 |
| T12A | 4.0 × 10−6 | 1.5 | 0.2 |
| A13S | 2.4 × 10−6 | 0.9 | 0.1 |
| D14A | 4.5 × 10−7 | 0.2 | 1.1* |
| K17A | 1.6 × 10−6 | 0.6 | 0.3 |
| H18A | 2.0 × 10−5 | 7.5 | 1.2 |
| A20S | 8.3 × 10−6 | 3.1 | 0.7 |
| R22A | >5 × 10−4 | >150 | >3.0 |
| S23A | 1.4 × 10−6 | 0.5 | 0.4 |
| D24A | 1.4 × 10−7 | 0.05 | 1.8* |
| G25A | 1.5 × 10−6 | 0.5 | 0.4 |
| K26A | 6.7 × 10−6 | 2.5 | 0.5 |
| A29S | 2.0 × 10−5 | 7.5 | 1.2 |
| W30A | >1 × 10−3 | >300 | >3.4 |
| D31A | 3.0 × 10−5 | 11.3 | 1.4 |
| G32A | 9.6 × 10−6 | 3.6 | 0.8 |
| T33A | 1.0 × 10−5 | 3.8 | 0.8 |
| F34A | 9.4 × 10−6 | 3.5 | 0.7 |
F values (see Figs. 3 and 4) were determined for 3–6 different toxin concentrations in 3–11 cells at each concentration. Kd values were obtained by fitting a four site model to the F vs. [SGTx] relation, as in Fig. 4. The change in the free energy of toxin binding was calculated as: ΔΔG = −RTln(Kd mut/Kd WT). Two mutants displaying stronger interactions with the channel (lower Kd values) are indicated with asterisks.
In principle, mutations in SGTx could perturb the concentration dependence for inhibition of the Kv2.1 channel by disrupting the structure of the toxin. With the exception of L19A and Y27A, the folding reactions for each of the mutants yield a dominant soluble product. Because SGTx has three disulfide bonds that stabilize the structure, and the toxin contains only 34 residues, success in obtaining a dominant product in the folding reaction argues against gross disruptions of the toxin structure. To further evaluate the structure of each mutant we obtained circular dichroism (CD) spectra and compared them to the wild-type toxin. Fig. 5 shows CD spectra for the wild-type toxin along with all 26 mutants (gray lines). In each case, the shape of the spectra are similar to the wild-type toxin, and any modest deviations are similar for the mutants exhibiting wild-type activity (Fig. 5 A) when compared with the mutants with pronounced perturbations (Fig. 5 B). Aromatic residues can significantly contribute to the CD spectrum of a toxin like SGTx that contains little helical character, however, even in instances where individual aromatic residues are mutated (e.g., F6A, W30A, F34A) the CD spectra are relatively unaltered. These results suggest that all of the mutants studied here do not exhibit substantial changes in structure.
Figure 5.
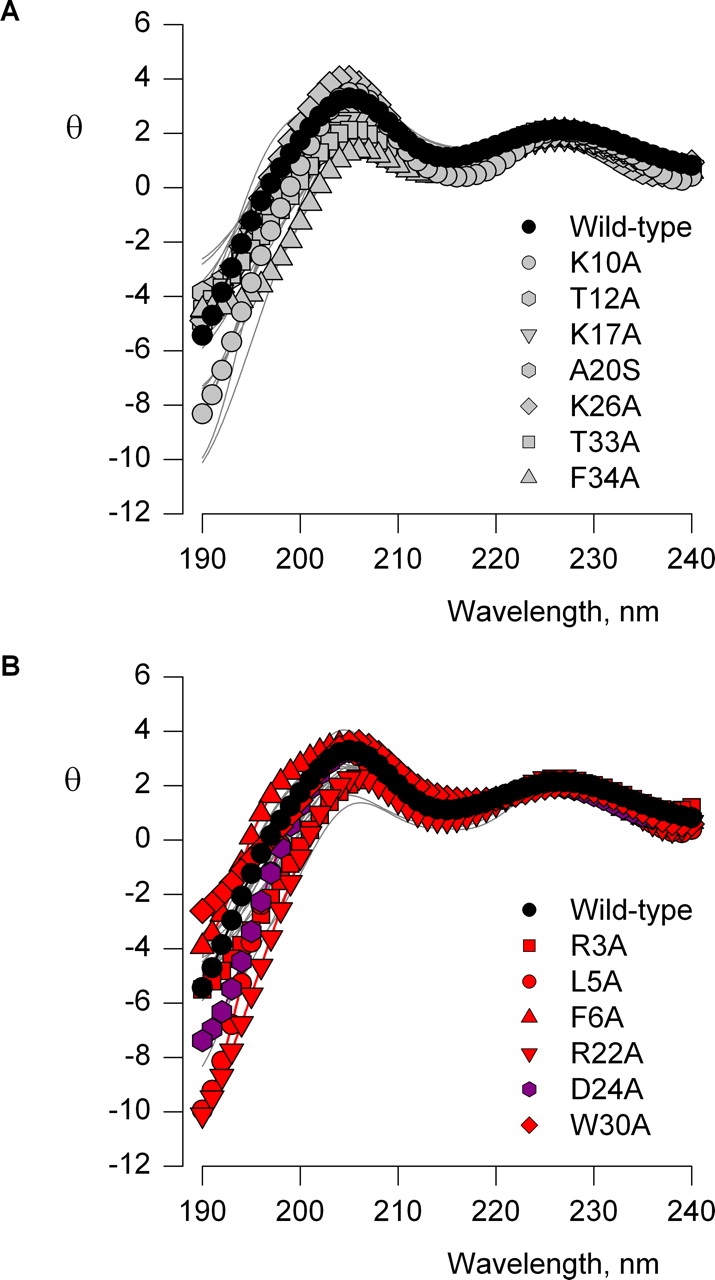
Circular dichroism spectra for SGTx mutants. Ellipticity (θ) plotted against wavelength for wild-type SGTx (filled black circles) and 26 SGTx mutants (light gray lines). θ (× 103) has units of deg cm2 dmol−1. In A, seven mutants are shown in gray symbols where |ΔΔG| < 1 kcal mol−1 and in B, six mutants are shown in red or purple where |ΔΔG| > 1.5 kcal mol−1. The red residues have increased Kd values while the purple residue has a decreased Kd value.
Structural Features of the Active Surface of the Toxin
To define the important regions of SGTx we mapped the mutagenesis results onto the SGTx NMR structure (Fig. 6) (Lee et al., 2004). Stereo views of the structure are shown as a stick representations or surface renderings with side chains colored according to ΔΔG. Mutants displaying only minor perturbations (|ΔΔG| < 1 kcal mol−1) are colored light gray, and mutants displaying increased Kd values are colored pink if the perturbations are moderate (|ΔΔG| = 1–1.5 kcal mol−1) or red if the perturbations are dramatic (|ΔΔG| > 1.5 kcal mol−1). Both unstudied residues (L19, Y27, and the 6 Cys) and backbone atoms are colored dark gray. The two mutants (D24A and D14A) that shift the concentration dependence for inhibition to lower toxin concentrations are colored purple. Inspection of the resulting maps (Fig. 6, A and B) reveals that one face of SGTx stands out as containing a large number of residues (e.g., W30, L5, F6, R3, R22, and D24) where mutations to Ala display dramatic perturbations. This same face of the toxin contains residues (e.g., Y4, H18, A29, and D31) where mutations produce moderate perturbations, but no residues that are tolerant to mutation. The absence of minor perturbations from this face of the toxin is quite dramatic because it stands in stark contrast to the opposite face of the toxin where many mutations having minor perturbations cluster together, producing a tolerant face of the toxin (Fig. 6 C).
Figure 6.
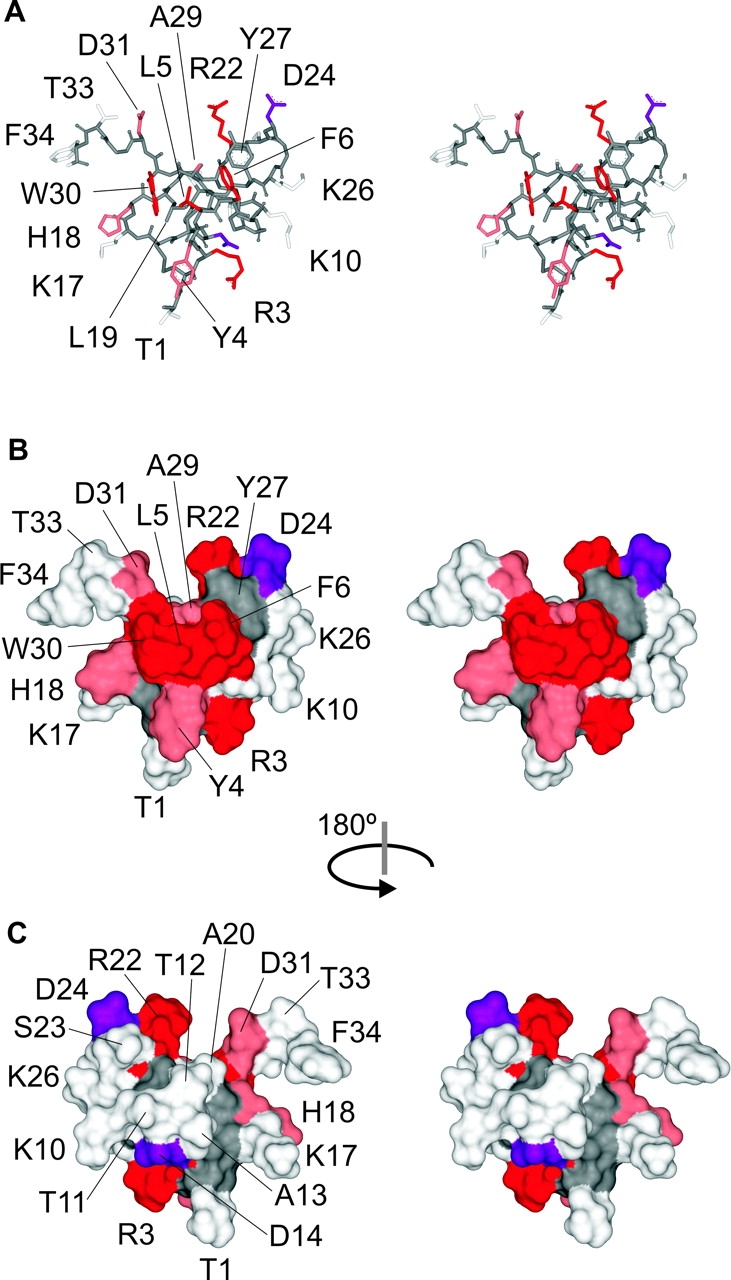
Mapping of perturbations onto the SGTx structure. (A) Stereo pair of the SGTx NMR solution structure shown as a stick representation. Backbone atoms and unstudied residues (L19, Y27, and 6 Cys) are colored dark gray. Mutants that do not significantly alter Kd (|ΔΔG| < 1 kcal mol−1) are colored light gray. Mutants that display weaker binding affinity are colored pink if |ΔΔG| = 1–1.5 kcal mol−1 and red if |ΔΔG| > 1.5 kcal mol−1. Two mutants displaying stronger binding affinity are colored purple. See Table I for Kd and ΔΔG values for all mutants. (B) Stereo pair of the SGTx structure shown as surface renderings with a probe radius of 1 Å. Same coloring scheme as in A. (C) Stereo surface renderings as in B except that the structure was rotated 180° about the indicated axis.
The critical residues on the active surface of the toxin (Fig. 6, A and B) have a rather interesting arrangement. First, the three most critical hydrophobic residues (L5, F6, and W30; ΔΔG values > 3 kcal mol−1) form a protrusion that projects from the active surface, with L5 sandwiched between the two aromatic residues. The aliphatic side chain of L19 is largely buried below the aromatic sandwich, so it is understandable why the L19A mutation is so poorly tolerated, resulting in misfolded protein. Y4 and Y27 are located adjacent to the aromatic sandwich, with the Y4A mutation causing a moderate perturbation (|ΔΔG| = 1.2 kcal mol−1) and the Y27A mutation resulting in misfolded protein. In SGTx, the aromatic ring of Y4 lies flat relative to the surface from which W30, L5, and F6 project, whereas in hanatoxin the ring of Y4 contributes to the hydrophobic protrusion, further encasing L5 in a three-way aromatic sandwich (Fig. 1, B and C). From inspection of the 20 converged NMR structures for both SGTx (Fig. 7) and hanatoxin, the position of Y4 is less well defined than for W30, L5, F6, and Y27 (which exhibit high angular order parameters; Lee et al., 2004), raising the possibility that Y4 may adopt a similar position when the toxins are bound to the voltage sensor. A second prominent feature of the active surface is that the hydrophobic protrusion is surrounded by a concentric ring–like arrangement of polar residues, including T1, R3, K10, K26, D24, R22, D31, and H18. Of these hydrophilic residues, mutations at five positions exhibit either moderate or major perturbations. Mutations at R22 and R3 dramatically increase toxin Kd (|ΔΔG| > 3 kcal mol−1), and mutation of H18 and D31 moderately increase toxin Kd (|ΔΔG| values = 1–1.5 kcal mol−1), consistent with intimate interactions between these residues and residues in the voltage-sensor paddle. In contrast, mutation of D24 to Ala decreases the Kd of the toxin, with a |ΔΔG| value of 1.8 kcal mol−1. We conclude that the face of SGTx containing the hydrophobic protrusion and surrounding polar residues is the surface of the toxin that makes intimate contact with residues within the voltage-sensor in Kv2.1 channels.
Figure 7.

Position of polar residues in SGTx. (A) Stereo pairs of the 20 final converged structures of SGTx shown as a stick representation. Side chain colors are as follows: green for hydrophobic, dark blue for basic, light blue for His, red for acidic, and pink for both Ser and Thr. Backbone atoms are colored light gray. (B) Stereo pair of the lowest XPLOR-energy structure of SGTx.
DISCUSSION
In the present study we investigated the interaction of toxins with the voltage-sensing domains of voltage-gated channels. Although the molecular mechanisms underlying the interaction between pore-blocking toxins and the ion conduction pore of voltage-gated channels has been investigated extensively, most notably for Kv channels (Miller, 1995), the interaction of gating modifiers is not well understood. Indeed, evidence for toxins interacting with voltage sensors is available for only a small number of toxins (Rogers et al., 1996; Swartz and MacKinnon, 1997b; Li-Smerin and Swartz, 1998, 2000; Winterfield and Swartz, 2000; Jiang et al., 2003a,b; Ruta et al., 2003).
The Active Surface of SGTx
The data presented here provides an essentially complete map of the functionally important surface of SGTx. The toxin contains 34 residues and we succeeded in obtaining data for 26 of the 28 mutants that we synthesized. Residues where mutations have similar effects on the concentration dependence for toxin inhibition cluster together to unambiguously identify the active surface of the toxin (Fig. 6, B and C). These mutagenesis results for the active surface of SGTx indicate that several hydrophobic residues on the toxin are energetically influential. In particular, W30, L5, F6, and Y4 form a clear patch of important hydrophobic residues on the active surface. The nearby Y27 may also be important, although this will need to be examined in other ways since the Ala mutant dramatically lowered the yield of the folding reaction. Of the 14 polar residues mutated, the R3A and R22A mutations cause the largest perturbations (|ΔΔG| values > 3 kcal mol−1), suggesting that these two basic residues are the most likely to be involved in intimate polar interactions with residues in the voltage sensors. The H18A and D31A mutants also perturb the concentration dependence for toxin inhibition, with ΔΔG values of 1.2 and 1.4 kcal mol−1, respectively. Mutation of two additional polar residues (D14 and D24) have a pronounced effect on the interaction between toxin and channel, but in these two instances the Kd decreases, suggesting that the Ala substitution removes an unfavorable interaction.
Interaction of SGTx with the Voltage-sensor Paddle
How might toxins like hanatoxin and SGTx interact with the voltage sensor? Previous mutagenesis studies in the Kv2.1 channel suggest that hanatoxin interacts intimately with residues in the S3b and S4 helices of the voltage-sensor paddle, and that both strong hydrophobic and ionic interactions make important contributions to toxin binding (Li-Smerin and Swartz, 2000, 2001). Although the effects of these channel mutations on SGTx binding have not yet been examined, there are several reasons to believe that hanatoxin and SGTx interact in similar ways with the Kv2.1 channel. First, both toxins inhibit the Kv2.1 channel by shifting activation to more depolarized voltages. Second, although there are differences at seven positions in the sequence of the two toxins (Fig. 1 A), most of these positions are tolerant to mutation in SGTx. In contrast, of the nine residues in SGTx where mutations display either moderate or dramatic perturbations, eight are identical between the two toxins and one is a conservative substitution (R22 in SGTx to K22 in hanatoxin). In the Kv2.1 channel F274 is a particularly sensitive hydrophobic residue within the S3b helix where mutations increase the hanatoxin Kd by as much as 500-fold (|ΔΔG| ∼3.7 kcal mol−1) (Li-Smerin and Swartz, 2000). It seems likely that hydrophobic interactions exist between F274 and the toxin because substitution of large hydrophobic residues at 274 are the least disruptive (Li-Smerin and Swartz, 2000). From the results presented here, residues in the hydrophobic protrusion (e.g., W30, L5, F6) are the most likely candidates for making hydrophobic interactions with F274. Another important residue in the S3b helix is E277, a position where the substitution of basic residues are the most disruptive, increasing the hanatoxin Kd by as much as 125-fold (|ΔΔG| ∼2.8 kcal mol−1) (Li-Smerin and Swartz, 2000). One possibility is that E277 forms a salt-bridge with a basic residue on the toxin. Of the five basic residues on SGTx, only mutations at R3 or R22 dramatically weaken the interaction between toxin and channel. Since the guanido groups of these two basic residues are ∼20 Å apart on opposite sides of the toxin (Fig. 6 A), E277 could only form a salt-bridge with one of them. An intimate interaction between F274 in Kv2.1 and residues in the toxin's hydrophobic protrusion is compatible with an interaction between E277 in Kv2.1 and either SGTx R3 or R22, since both of these basic residues are located an equal distance (∼10–12 Å) from the hydrophobic protrusion. Although it should be possible to use mutant cycle analysis (Hidalgo and MacKinnon, 1995) to distinguish whether R3 or R22 interacts with E277, the interaction between these toxin mutants and the wild-type channel is already weaker than can be determined accurately (e.g., Fig. 4; Table I), and thus it is not feasible to study their interaction with channels that contain a mutation at E277. In the future it will be interesting to address this question, perhaps using the D24A mutation to increase the affinity of the toxin–channel interaction, or other toxin homologues that bind more tightly.
Comparison of Homologous Toxins
The sequence alignment in Fig. 1 A lists five spider toxins, including SGTx (Marvin et al., 1999), hanatoxin (Swartz and MacKinnon, 1995), grammtoxin (Lampe et al., 1993), ProTx (Middleton et al., 2002), and SNX-482 (Newcomb et al., 1998), that have been shown to inhibit activation of voltage-gated channels by shifting activation to more positive voltages (McDonough et al., 1997a; Swartz and MacKinnon, 1997a; Bourinet et al., 2001; Middleton et al., 2002; Lee et al., 2004). This group includes inhibitors of all major types of voltage-gated channels, including Kv, Cav, and Nav channels. Several other types of protein toxins (e.g., ω-AgaIVA) have very different sequences (Mintz et al., 1992), yet inhibit activation of Cav channels in a similar fashion by interacting with the S3b helix in voltage-sensor paddles (McDonough et al., 1997b; Winterfield and Swartz, 2000). A recently identified toxin named VSTx was isolated from tarantula venom and shown to inhibit the KvAP channel (Ruta et al., 2003). Although the sequence of VSTx is ∼50% similar when compared with hanatoxin, there are three observations that raise the possibility of distinct inhibitory mechanisms for these toxins. First, the inhibitory effects of VSTx cannot be overcome by membrane depolarizations to voltages as positive as +100 mV. This is unprecedented for toxins that bind to voltage-sensors, as the opening of toxin-bound channels has been demonstrated in all other instances. Second, it has been suggested that binding of VSTx requires depolarization of the membrane (Jiang et al., 2003b), whereas in the case of hanatoxin and SGTx binding occurs when the membrane is constantly held at −100 mV (Lee et al., 2003). A third distinction can be seen from a sequence comparison between VSTx and SGTx when considering the active surface of SGTx identified in the present study. Of the most influential residues in SGTx, W30 is a Leu in VSTx, L5 is a Phe, F6 is a Met, R3 is a Gly, R22 is a Ser, D24 is an Arg, Y4 is a Lys, H18 is an Asp, and D31 is an Ala. This comparison indicates that almost none of the most critical residues in SGTx are conserved in VSTx. It will be interesting to further explore the inhibitory mechanism of VSTx and to examine whether VSTx interacts with different regions of the channel compared with SGTx and hanatoxin.
Significance of the Polar Ring
An important question that we set out to address was how SGTx might access the voltage-sensor paddle from the extracellular solution if the paddle is positioned toward the intracellular side of the membrane in the resting conformation, a state to which the toxin appears to bind tightly (Lee et al., 2003). Although SGTx contains a large number of basic and acidic residues, they are arranged in an alternating fashion around the perimeter of the toxin's active surface, raising the possibility that they form neutral salt bridges that facilitate partitioning into the hydrophobic core of the membrane. Fig. 7 shows the 20 converged NMR structures (A) and the lowest XPLOR-energy structure (B) for SGTx. In the low energy structure the distances between basic and acidic residues range from 5 to 9 Å, considerably further apart than would be compatible with salt bridges. However, inspection of the 20 converged structures shows that the precise positions of the polar residues in question are not well constrained, leaving open the possibility of salt-bridge formation. If this type of mechanism operates to facilitate entry of the toxin into the membrane we would anticipate that charge neutralizing mutations would increase the apparent Kd. Of the five basic and three acidic residues at the perimeter of the active surface, mutations at only two positions result in an increase in toxin Kd. For both R3 and R22, the observed increases are so large that they most likely reflect the loss of important contacts with the voltage sensor. Mutations of the remaining three basic and all three acidic residues result in either no change or a decrease in toxin Kd, counter to what would be predicted for an important salt-bridge network. We conclude that interactions between acidic and basic residues on SGTx are not necessary for the toxin to access the voltage-sensor paddle motif. In this case the large number of residues in the polar ring would suggest that this region of the toxin does not penetrate into the hydrophobic phase of the membrane. The interactions between polar residues on the toxin and the voltage-sensor paddle would likely occur near the aqueous solution (Lee et al., 2003).
Dimpling of the Hydrophobic Protrusion into the Membrane
The hydrophobic protrusion is a prominent feature on the active face of SGTx and hanatoxin. In both toxins the hydrophobic cluster protrudes ∼8–10 Å from the ring of polar residues, and one might imagine that these protrusions might enable these regions of SGTx and hanatoxin to dip into the hydrophobic core of the membrane. There is related toxin named GsMTx-4 that has been shown to inhibit cationic stretch-activated channels (Suchyna et al., 2000). Experiments using gramicidin as a probe for protein interactions with lipid membranes have uncovered an interaction between GsMTx-4 and lipid membranes (Andersen, O.S., personal communication), strengthening the possibility that toxins like hanatoxin and SGTx might dimple into the hydrophobic phase of the membrane. If dimpling is energetically important, some of the mutations studied here may perturb the energetics of inhibition by SGTx because they influence the interaction between the toxin and membranes. The sensitivity of the relevant hydrophobic residues that we observed in this study (Fig. 6; Table I) is consistent with this possibility. It will be interesting to investigate this possibility and to determine the relative energetic contribution of residues to protein-lipid and protein–protein interactions. Interestingly, the hydrophobic protrusion seems to exhibit significant variation among toxins that have been established to interact with voltage-sensors. SGTx (Lee et al., 2004), hanatoxin (Takahashi et al., 2000), and grammotoxin (Takeuchi et al., 2002) have a high degree of sequence similarity and so it is not surprising that their NMR structures show many conserved features (Fig. 8) . However, grammotoxin displays an interesting adaptation on the hydrophobic protrusion, whereby two basic residues are located at the tip of the protrusion. Like SGTx and hanatoxin, Grammotoxin interacts with the S3b helix within the voltage sensor in Kv and Cav channels (Li-Smerin and Swartz, 1998), so this modification of the hydrophobic protrusion doesn't prevent the toxin from accessing the voltage-sensor paddle motif. Anthropleurin-B (Monks et al., 1995), a sea anemone toxin, and BmK M1 (He et al., 1999), an α-scorpion toxin, interact with the S3b helix region of the voltage-sensor in domain four of Nav channels and produce a profound slowing of inactivation (Hanck and Sheets, 1995; Rogers et al., 1996; Sun et al., 2003). Although the interacting surfaces for these toxins have not yet been systematically defined, mutation of the residues labeled in Fig. 8 have been shown to influence toxin binding (Khera et al., 1995; Dias-Kadambi et al., 1996a,b; Zilberberg et al., 1997; Sun et al., 2003; Wang et al., 2003), most likely identifying important interacting faces for these toxins. Although there are clusters of hydrophobic residues on the putative active faces for anthropleurin-B and BmK M1, they do not form a protrusion and there are many polar residues intercalating with hydrophobic residues. This comparison suggests that toxin interactions with the membrane may vary considerably between different toxins that interact with voltage sensors.
Figure 8.

Structures of toxins that bind to voltage-sensors. Surface renderings of five toxins that bind to voltage-sensors in voltage-gated ion channels. Structures to the right were obtained by rotating structures on the left by 90° about the axis shown. Probe radius is 1 Å and coloring as in Fig 7. Structures of hanatoxin, SGTx, grammotoxin (1KOZ), and anthropleurin-B (1APF) were determined using NMR while the structure of BmK M1 (1SN1) was determined using X-ray diffraction. Mutation of residues labeled in anthropleurin-B and BmK M1 cause decreases in toxin binding to Nav channels.
Conclusion
In this study we provide evidence that (a) a defined surface of SGTx is presented to the voltage-sensing domain; (b) the active face of the toxin contains important polar and hydrophobic residues; (c) homologous toxins like SGTx and VSTx may present unique interaction surfaces to their targets and inhibit Kv channels through distinct mechanisms; (d) salt-bridges between basic and acidic residues in the toxin's polar ring do not play a significant role in enabling the toxin to access the voltage-sensor paddle; and (e) there are considerable structural variations in the arrangement of hydrophobic residues in toxins that bind to voltage-sensors.
Acknowledgments
We thank Miguel Holmgren, Mark Mayer, Joe Mindell, Shai Silberberg, and members of both the Swartz and Kim Labs, for helpful discussions and critique of the manuscript.
This study was supported by the Intramural Research Program within the National Institute of Neurological Disorders and Stroke, and by grants from the Ministry of Science and Technology, Korea, the Korea Science and Engineering Foundation through the Research Center for Proteinaceous Materials, and from the Brain Korea 21 program.
Olaf S. Andersen served as editor.
Julia M. Wang and Soung Hun Roh contributed equally to this paper.
References
- Aggarwal, S.K., and R. MacKinnon. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177. [DOI] [PubMed] [Google Scholar]
- Anderson, C.S., R. MacKinnon, C. Smith, and C. Miller. 1988. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J. Gen. Physiol. 91:317–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland, L.M., J.A. Morrill, and B.P. Bean. 1994. ω-Conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J. Neurosci. 14:5011–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet, E., S.C. Stotz, R.L. Spaetgens, G. Dayanithi, J. Lemos, J. Nargeot, and G.W. Zamponi. 2001. Interaction of SNX482 with domains III and IV inhibits activation gating of α(1E) (Ca(V)2.3) calcium channels. Biophys. J. 81:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestele, S., and W.A. Catterall. 2000. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 82:883–892. [DOI] [PubMed] [Google Scholar]
- Cruz, L.J., W.R. Gray, B.M. Olivera, R.D. Zeikus, L. Kerr, D. Yoshikami, and E. Moczydlowski. 1985. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem. 260:9280–9288. [PubMed] [Google Scholar]
- Dias-Kadambi, B.L., K.A. Combs, C.L. Drum, D.A. Hanck, and K.M. Blumenthal. 1996. a. The role of exposed tryptophan residues in the activity of the cardiotonic polypeptide anthopleurin B. J. Biol. Chem. 271:23828–23835. [DOI] [PubMed] [Google Scholar]
- Dias-Kadambi, B.L., C.L. Drum, D.A. Hanck, and K.M. Blumenthal. 1996. b. Leucine 18, a hydrophobic residue essential for high affinity binding of anthopleurin B to the voltage-sensitive sodium channel. J. Biol. Chem. 271:9422–9428. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J.M. Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Dudley, S.C., Jr., N. Chang, J. Hall, G. Lipkind, H.A. Fozzard, and R.J. French. 2000. mu-conotoxin GIIIA interactions with the voltage-gated Na+ channel predict a clockwise arrangement of the domains. J. Gen. Physiol. 116:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinor, P.T., J.F. Zhang, W.A. Horne, and R.W. Tsien. 1994. Structural determinants of the blockade of N-type calcium channels by a peptide neurotoxin. Nature. 372:272–275. [DOI] [PubMed] [Google Scholar]
- Feng, Z.P., C.J. Doering, R.J. Winkfein, A.M. Beedle, J.D. Spafford, and G.W. Zamponi. 2003. Determinants of inhibition of transiently expressed voltage-gated calcium channels by omega-conotoxins GVIA and MVIIA. J. Biol. Chem. 278:20171–20178. [DOI] [PubMed] [Google Scholar]
- Frech, G.C., A.M. VanDongen, G. Schuster, A.M. Brown, and R.H. Joho. 1989. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 340:642–645. [DOI] [PubMed] [Google Scholar]
- Garcia, M.L., M. Garcia-Calvo, P. Hidalgo, A. Lee, and R. MacKinnon. 1994. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 33:6834–6839. [DOI] [PubMed] [Google Scholar]
- Gill, S.C., and P.H. von Hippel. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182:319–326. [DOI] [PubMed] [Google Scholar]
- Hanck, D.A., and M.F. Sheets. 1995. Modification of inactivation in cardiac sodium channels: ionic current studies with Anthopleurin-A toxin. J. Gen. Physiol. 106:601–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X.L., H.M. Li, Z.H. Zeng, X.Q. Liu, M. Wang, and D.C. Wang. 1999. Crystal structures of two alpha-like scorpion toxins: non-proline cis peptide bonds and implications for new binding site selectivity on the sodium channel. J. Mol. Biol. 292:125–135. [DOI] [PubMed] [Google Scholar]
- Hidalgo, P., and R. MacKinnon. 1995. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 268:307–310. [DOI] [PubMed] [Google Scholar]
- Hong, K.H., and C. Miller. 2000. The lipid-protein interface of a Shaker K+ channel. J. Gen. Physiol. 115:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. a. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. b. The open pore conformation of potassium channels. Nature. 417:523–526. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. a. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., V. Ruta, J. Chen, A. Lee, and R. MacKinnon. 2003. b. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 423:42–48. [DOI] [PubMed] [Google Scholar]
- Khera, P.K., G.R. Benzinger, G. Lipkind, C.L. Drum, D.A. Hanck, and K.M. Blumenthal. 1995. Multiple cationic residues of anthopleurin B that determine high affinity and channel isoform discrimination. Biochemistry. 34:8533–8541. [DOI] [PubMed] [Google Scholar]
- Kubo, Y., T.J. Baldwin, Y.N. Jan, and L.Y. Jan. 1993. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 362:127–133. [DOI] [PubMed] [Google Scholar]
- Kuo, A., J.M. Gulbis, J.F. Antcliff, T. Rahman, E.D. Lowe, J. Zimmer, J. Cuthbertson, F.M. Ashcroft, T. Ezaki, and D.A. Doyle. 2003. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 300:1922–1926. [DOI] [PubMed] [Google Scholar]
- Lampe, R.A., P.A. Defeo, M.D. Davison, J. Young, J.L. Herman, R.C. Spreen, M.B. Horn, T.J. Mangano, and R.A. Keith. 1993. Isolation and pharmacological characterization of omega-grammotoxin SIA, a novel peptide inhibitor of neuronal voltage-sensitive calcium channel responses. Mol. Pharmacol. 44:451–460. [PubMed] [Google Scholar]
- Lee, C.W., S. Kim, S.H. Roh, H. Endoh, Y. Kodera, T. Maeda, T. Kohno, J.M. Wang, K.J. Swartz, and J.I. Kim. 2004. Solution structure and functional characterization of SGTx1, a modifier of Kv2.1 channel gating. Biochemistry. 43:890–897. [DOI] [PubMed] [Google Scholar]
- Lee, H.C., J.M. Wang, and K.J. Swartz. 2003. Interaction between extracellular Hanatoxin and the resting conformation of the voltage-sensor paddle in Kv channels. Neuron. 40:527–536. [DOI] [PubMed] [Google Scholar]
- Legros, C., V. Pollmann, H.G. Knaus, A.M. Farrell, H. Darbon, P.E. Bougis, M.F. Martin-Eauclaire, and O. Pongs. 2000. Generating a high affinity scorpion toxin receptor in KcsA-Kv1.3 chimeric potassium channels. J. Biol. Chem. 275:16918–16924. [DOI] [PubMed] [Google Scholar]
- Li-Smerin, Y., D.H. Hackos, and K.J. Swartz. 2000. a. Alpha-helical structural elements within the voltage-sensing domains of a K+ channel. J. Gen. Physiol. 115:33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin, Y., and K.J. Swartz. 1998. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc. Natl. Acad. Sci. USA. 95:8585–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin, Y., and K.J. Swartz. 2000. Localization and molecular determinants of the hanatoxin receptors on the voltage-sensing domain of a K+ channel. J. Gen. Physiol. 115:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin, Y., and K.J. Swartz. 2001. Helical structure of the COOH terminus of S3 and its contribution to the gating modifier toxin receptor in voltage-gated ion channels. J. Gen. Physiol. 117:205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Z., A.M. Klem, and Y. Ramu. 2001. Ion conduction pore is conserved among potassium channels. Nature. 413:809–813. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R. 1991. Determination of the subunit stoichiometry of a voltage-activated potassium channel. Nature. 350:232–235. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., S.L. Cohen, A. Kuo, A. Lee, and B.T. Chait. 1998. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 280:106–109. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., and C. Miller. 1988. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J. Gen. Physiol. 91:335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon, R., and C. Miller. 1989. Mutant potassium channels with altered binding of charybdotoxin, a pore-blocking peptide inhibitor. Science. 245:1382–1385. [DOI] [PubMed] [Google Scholar]
- Marvin, L., E. De, P. Cosette, J. Gagnon, G. Molle, and C. Lange. 1999. Isolation, amino acid sequence and functional assays of SGTx1. The first toxin purified from the venom of the spider scodra griseipes. Eur. J. Biochem. 265:572–579. [DOI] [PubMed] [Google Scholar]
- McDonough, S.I., R.A. Lampe, R.A. Keith, and B.P. Bean. 1997. a. Voltage-dependent inhibition of N- and P-type calcium channels by the peptide toxin omega-grammotoxin-SIA. Mol. Pharmacol. 52:1095–1104. [DOI] [PubMed] [Google Scholar]
- McDonough, S.I., I.M. Mintz, and B.P. Bean. 1997. b. Alteration of P-type calcium channel gating by the spider toxin omega- Aga-IVA. Biophys. J. 72:2117–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton, R.E., V.A. Warren, R.L. Kraus, J.C. Hwang, C.J. Liu, G. Dai, R.M. Brochu, M.G. Kohler, Y.D. Gao, V.M. Garsky, et al. 2002. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry. 41:14734–14747. [DOI] [PubMed] [Google Scholar]
- Miller, C. 1995. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 15:5–10. [DOI] [PubMed] [Google Scholar]
- Mintz, I.M., V.J. Venema, K.M. Swiderek, T.D. Lee, B.P. Bean, and M.E. Adams. 1992. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 355:827–829. [DOI] [PubMed] [Google Scholar]
- Moczydlowski, E., B.M. Olivera, W.R. Gray, and G.R. Strichartz. 1986. Discrimination of muscle and neuronal Na-channel subtypes by binding competition between [3H]-saxitoxin and mu-conotoxins. Proc. Natl. Acad. Sci. USA. 83:5321–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks, S.A., D.J. Needleman, and C. Miller. 1999. Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 113:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks, S.A., P.K. Pallaghy, M.J. Scanlon, and R.S. Norton. 1995. Solution structure of the cardiostimulant polypeptide anthopleurin-B and comparison with anthopleurin-A. Structure. 3:791–803. [DOI] [PubMed] [Google Scholar]
- Naranjo, D., and C. Miller. 1996. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+ channel. Neuron. 16:123–130. [DOI] [PubMed] [Google Scholar]
- Newcomb, R., B. Szoke, A. Palma, G. Wang, X. Chen, W. Hopkins, R. Cong, J. Miller, L. Urge, K. Tarczy-Hornoch, et al. 1998. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 37:15353–15362. [DOI] [PubMed] [Google Scholar]
- Park, C.S., and C. Miller. 1992. a. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel. Neuron. 9:307–313. [DOI] [PubMed] [Google Scholar]
- Park, C.S., and C. Miller. 1992. b. Mapping function to structure in a channel-blocking peptide: electrostatic mutants of charybdotoxin. Biochemistry. 31:7749–7755. [DOI] [PubMed] [Google Scholar]
- Possani, L.D., E. Merino, M. Corona, F. Bolivar, and B. Becerril. 2000. Peptides and genes coding for scorpion toxins that affect ion-channels. Biochimie. 82:861–868. [DOI] [PubMed] [Google Scholar]
- Ranganathan, R., J.H. Lewis, and R. MacKinnon. 1996. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 16:131–139. [DOI] [PubMed] [Google Scholar]
- Rash, L.D., and W.C. Hodgson. 2002. Pharmacology and biochemistry of spider venoms. Toxicon. 40:225–254. [DOI] [PubMed] [Google Scholar]
- Rodriguez de la Vega, R.C., E. Merino, B. Becerril, and L.D. Possani. 2003. Novel interactions between K+ channels and scorpion toxins. Trends Pharmacol Sci. 24:222–227. [DOI] [PubMed] [Google Scholar]
- Rogers, J.C., Y. Qu, T.N. Tanada, T. Scheuer, and W.A. Catterall. 1996. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 271:15950–15962. [DOI] [PubMed] [Google Scholar]
- Ruta, V., Y. Jiang, A. Lee, J. Chen, and R. MacKinnon. 2003. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature. 422:180–185. [DOI] [PubMed] [Google Scholar]
- Srinivasan, K.N., P. Gopalakrishnakone, P.T. Tan, K.C. Chew, B. Cheng, R.M. Kini, J.L. Koh, S.H. Seah, and V. Brusic. 2002. SCORPION, a molecular database of scorpion toxins. Toxicon. 40:23–31. [DOI] [PubMed] [Google Scholar]
- Stampe, P., L. Kolmakova-Partensky, and C. Miller. 1994. Intimations of K+ channel structure from a complete functional map of the molecular surface of charybdotoxin. Biochemistry. 33:443–450. [DOI] [PubMed] [Google Scholar]
- Suchyna, T.M., J.H. Johnson, K. Hamer, J.F. Leykam, D.A. Gage, H.F. Clemo, C.M. Baumgarten, and F. Sachs. 2000. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 115:583–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y.M., F. Bosmans, R.H. Zhu, C. Goudet, Y.M. Xiong, J. Tytgat, and D.C. Wang. 2003. Importance of the conserved aromatic residues in the scorpion α-like toxin BmK M1: the hydrophobic surface region revisited. J. Biol. Chem. 278:24125–24131. [DOI] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1995. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 15:941–949. [DOI] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1997. a. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 18:665–673. [DOI] [PubMed] [Google Scholar]
- Swartz, K.J., and R. MacKinnon. 1997. b. Mapping the receptor site for hanatoxin, a gating modifier of voltage- dependent K+ channels. Neuron. 18:675–682. [DOI] [PubMed] [Google Scholar]
- Takahashi, H., J.I. Kim, H.J. Min, K. Sato, K.J. Swartz, and I. Shimada. 2000. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K+ channels: common surface features of gating modifier toxins. J. Mol. Biol. 297:771–780. [DOI] [PubMed] [Google Scholar]
- Takeuchi, K., J.I. Kim, H. Takahashi, K. Swartz, and I. Shimada. 2002. Solution structure of w-grammotoxin SIA, a gating modifier of P/Q and N type calcium channels. J. Mol. Biol. 321:517–526. [DOI] [PubMed] [Google Scholar]
- Wang, C.G., N. Gilles, A. Hamon, F. Le Gall, M. Stankiewicz, M. Pelhate, Y.M. Xiong, D.C. Wang, and C.W. Chi. 2003. Exploration of the functional site of a scorpion alpha-like toxin by site-directed mutagenesis. Biochemistry. 42:4699–4708. [DOI] [PubMed] [Google Scholar]
- Winterfield, J.R., and K.J. Swartz. 2000. A hot spot for the interaction of gating modifier toxins with voltage-dependent ion channels. J. Gen. Physiol. 116:637–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberberg, N., O. Froy, E. Loret, S. Cestele, D. Arad, D. Gordon, and M. Gurevitz. 1997. Identification of structural elements of a scorpion α-neurotoxin important for receptor site recognition. J. Biol. Chem. 272:14810–14816. [DOI] [PubMed] [Google Scholar]