

Abstract
Symbiotic nitrogen-fixing rhizobia are able to trigger root deformation in their Fabaceae host plants, allowing their intracellular accommodation. They do so by delivering molecules called Nod factors. We analyzed the patterns of nucleotide polymorphism of five genes controlling early Nod factor perception and signaling in the Fabaceae Medicago truncatula to understand the selective forces shaping the evolution of these genes. We used 30 M. truncatula genotypes sampled in a genetically homogeneous region of the species distribution range. We first sequenced 24 independent loci and detected a genomewide departure from the hypothesis of neutrality and demographic equilibrium that suggests a population expansion. These data were used to estimate parameters of a simple demographic model incorporating population expansion. The selective neutrality of genes controlling Nod factor perception was then examined using a combination of two complementary neutrality tests, Tajima's D and Fay and Wu's standardized H. The joint distribution of D and H expected under neutrality was obtained under the fitted population expansion model. Only the gene DMI1, which is expected to regulate the downstream signal, shows a pattern consistent with a putative selective event. In contrast, the receptor-encoding genes NFP and NORK show no significant signatures of selection. Among the genes that we analyzed, only DMI1 should be viewed as a candidate for adaptation in the recent history of M. truncatula.
THE barrel medic (Medicago truncatula) is an annual, highly self-fertilizing diploid species with a natural geographical distribution covering the Mediterranean region. It is used as a model for plant–microbe interactions and for various topics in Fabaceae genetics (Cook 1999; http://www.noble.org/MedicagoHandbook/). Most Fabaceae form nitrogen-fixing root nodules containing symbiotic bacteria known as rhizobia. Rhizobial infection is controlled by molecular recognition of Nod factors, which are molecules released by rhizobia (Cullimore et al. 2001). Recently, the understanding of the genetics of this recognition has progressed substantially through the identification of genes specifically involved in Nod factor perception and signaling (Geurts et al. 2005). Five genes controlling the earliest stages of this signaling pathway (Figure 1) are thought to be the main components of the recognition pathway. NFP encodes a LysM domain-containing receptor kinase and is the Nod factor receptor candidate (Arrighi et al. 2006). NORK encodes a leucine-rich repeat receptor kinase involved in rhizobial and mycorrhizal perception (Endre et al. 2002). DMI1 encodes an ion channel and seems to mediate responses triggered by symbiotic (Nod factor and mycorrhizal) signals (Ané et al. 2004). DMI3 encodes a Ca2+/calmodulin-dependent kinase that can control gene expression (Lévy et al. 2004). Finally, NIN shows homologies to transcription factors (Schauser et al. 1999).
Figure 1.—

Genes involved in early responses to Nod factor inoculation in M. truncatula. Genes are ordered after phenotypic alterations of responses observed in gene mutants. The phenotypic responses observed in the wild type are displayed. No responses are observed in NFP mutants, but the earliest responses are observed in all other mutants. No infection/nodulation occurs in mutants of any of these genes (Schauser et al. 1999; Catoira et al. 2000; Ben Amor et al. 2003).
Resistance to pathogenic micro-organisms in plants relies on signaling systems triggered by resistance proteins interacting with the pathogens' avirulence proteins (Van Der Biezen and Jones 1998). In response to evolutionary constraints generated by pathogens, resistance genes undergo strong selective pressures (De Meaux and Mitchell-Olds 2003; Tiffin and Moeller 2006). Together, genes involved in resistance to pathogens and pathogen virulence represent the most frequently described instances of positive selection, i.e., fast evolution at the protein level driven by natural selection (Nielsen 2005; Yang 2006). Whether mutually beneficial interactions such as the Fabaceae–rhizobia symbiosis generate this type of selection is currently unclear. One could view mutualism as a situation where no species should be selected for evading a beneficial symbiotic partner. Then mutualism should promote the stability of a specific recognition system, thereby inducing strong purifying selection on both genes encoding the symbiotic signal and genes controlling perception of that signal. But, on the other hand, mutualism is best viewed as a continuum that encompasses features such as conflicts of interest among partners and parasitism of the mutualism (Thompson 2006). In such a context, the selection for avoiding cheaters (nonbeneficial genotypes counterfeiting a symbiotic signal like Nod factors) would induce periodic shifting in symbiotic signals and their receptors. Such episodes would affect both genes encoding the symbiotic signals and their perception and are able to generate patterns of selection essentially similar to those generated by host–pathogen interactions. To our knowledge, the evolutionary dynamics of symbiotic signaling between two different species has not been addressed theoretically. Thus, it is still unclear whether mutualism can be a driving force for fast evolution at the molecular level.
Empirical studies are rare and have focused mostly on Wolbachia models, which include parasitic and mutualistic lineages (Fenn and Blaxter 2006). These studies have uncovered evidence of positive selection in parasitic lineages only (Jiggins 2006). However, a recent study suggests that mutualistic Wolbachia lineages may undergo positive evolutionary constraints, although at a reduced level compared to parasitic lineages (Brownlie et al. 2007). This is consistent with a previous study of the Fabaceae–rhizobia model symbiosis (De Mita et al. 2006) where we detected evidence for positive selection in the gene NORK. Note that footprints of positive selection were detected among various species of the Fabaceae but no recent episode of positive selection was found when examining patterns of nucleotide polymorphism within M. truncatula.
The pattern of molecular diversity of genes records the signatures of selective pressures undergone by the products of these genes. These selective pressures can be specifically detected by appropriate statistical analysis methods (Bamshad and Wooding 2003). If the function of the gene examined (in our case the perception of mutualistic partners) is under strong selection to remain identical through time, one would expect purifying selection to constrain amino acid sequence evolution and patterns of within-species polymorphism to be mostly neutral (Kimura 1983). Conversely, if the function induces episodes of positive selection, evolution is comparatively faster and the polymorphism of sequences near the site of selection is episodically reduced and distorted by selective sweeps (Maynard Smith and Haigh 1974). Therefore, searching for signatures of natural selection in genes involved in the partner recognition in the Fabaceae–rhizobia symbiosis is a way of understanding the mode of evolution of the symbiosis itself.
Statistical methods for testing whether naturally occurring nucleotide variability fits the predictions of the standard neutral model (SNM) are based on the frequency spectrum of mutations, e.g., Tajima's D (Tajima 1989). The SNM assumes that samples were collected in a nonstructured, constant-sized population. Tests of selective neutrality can therefore reject the null hypothesis in the absence of selection if any other assumptions of the SNM are violated (Depaulis et al. 2003). Genomewide patterns of polymorphism in Arabidopsis thaliana exhibit substantial deviation from the hypothesis of a nonstructured, constant-sized population (Nordborg et al. 2005; Ostrowski et al. 2006). The case of A. thaliana is more likely the rule than the exception, and recent surveys of patterns of polymorphism in plants illustrate the need to account for genomewide deviation from the SNM due to either population structure or demographic history (Wright and Gaut 2004).
The SNM is the simplest of a family of demographic models allowing for population structure and population growth or decline, among others. Thus, in a given study and depending on the model species, there must be a (potentially unknown) scenario best describing the multi-locus pattern of polymorphism. Although searching through all possible scenarios is impossible, one can identify a scenario that accounts for most features of the data without invoking selection. A popular way to do so is to compare a set of statistics summarizing the polymorphism at many loci to those calculated in data sets of comparable size simulated under a given scenario (Weiss and Von Haeseler 1998). The chosen scenario can then be substituted for the standard neutral model as a null hypothesis, incorporating nonstandard demography but no selection, to explicitly test the hypothesis of selective neutrality. This approach allowed the detection of candidate loci that have experienced recent episodes of selection in a number of studies (Tenaillon et al. 2004; Haddrill et al. 2005; Schmid et al. 2005; Williamson et al. 2005; Wright et al. 2005; Hamblin et al. 2006). In each of these studies, simplified models were used to account for the specific demographic history of each species (e.g., bottleneck associated with domestication in maize and population expansion in humans and Drosophila melanogaster).
Here, we study patterns of nucleotide diversity in 5 focal loci located in genes involved in Nod factor perception (NFP, NORK, DMI1, DMI3, and NIN) and 24 control loci located in gene fragments chosen without considering their function a priori. We find that patterns of polymorphism of control loci in our sample are not compatible with the SNM, with lower-than-expected values for both Tajima's D and Fay and Wu's H tests. Among major demographic models (population subdivision, demographic growth, and demographic decline), only demographic growth is expected to result in negative D. H is very sensitive to errors in orienting polymorphisms (misorientations), which result in negative values. We use data from the 24 control loci to fit a model featuring a single population but allowing for any form of population expansion (population expansion model, or PEM). We do so by using coalescent simulations and a rejection-sampling method to obtain an approximate posterior distribution for the scaled mutation rate, the scaled intragenic recombination rate, and the scaled population growth parameter. In addition, we incorporate an estimation of the rate of error when orienting polymorphisms. We find that this simple model substantially improves the fit of various statistics describing our data. Then, under the PEM, we generate expectations for the joint distribution of D and H, allowing testing of the selective neutrality of the polymorphism in the focal genes involved in early Nod factor perception and signaling (NFP, NORK, DMI1, DMI3, and NIN) while taking into account deviation from the equilibrium standard model.
MATERIALS AND METHODS
Plant material:
We used 30 genotypes from a reference collection of M. truncatula accessions (Table 1). These accessions were obtained after two consecutive generations of selfing of plants grown from seeds collected in the wild. Given that M. truncatula exhibits high selfing rates in the wild (estimated as 0.97; http://www.noble.org/MedicagoHandbook/), all 30 genotypes of M. truncatula are a priori expected to be highly homozygous. This was confirmed by genotyping the 30 genotypes at 15 microsatellite loci (Ronfort et al. 2006). All accessions that we use originate from sampling a set of natural populations located in a well-defined geographic region, the southeastern limit of the species range (the Morocco–Spain area), and belong to one of four groups of related individuals previously detected when analyzing the structure of the diversity in this species using microsatellite markers (Ronfort et al. 2006). Outgroup genotypes were chosen from M. tornata whenever possible and from M. ciliaris and M. rigidula when not.
TABLE 1.
Accessions used in this study
| Accession no. | Accession label | Country of origin | Species |
|---|---|---|---|
| L0166B | SA025654 | Morocco | M. truncatula |
| L0216B | SA027961 | Morocco | M. truncatula |
| L0233C | SA024576 | Morocco | M. truncatula |
| L0239B | SA026063 | Morocco | M. truncatula |
| L0306C | SA008623 | Morocco | M. truncatula |
| L0330B | SA002806 | Morocco | M. truncatula |
| L0357B | DZA202-4 | Algeria | M. truncatula |
| L0369B | PRT180-A | Portugal | M. truncatula |
| L0372B | PRT177-C | Portugal | M. truncatula |
| L0400C | DZA323-3 | Algeria | M. truncatula |
| L0401C | ESP031-A | Spain | M. truncatula |
| L0404B | ESP039-A | Spain | M. truncatula |
| L0410B | ESP043-B | Spain | M. truncatula |
| L0414C | ESP050-B | Spain | M. truncatula |
| L0421B | ESP098A-C | Spain | M. truncatula |
| L0425C | ESP100-G | Spain | M. truncatula |
| L0430B | ESP104-A | Spain | M. truncatula |
| L0448B | ESP175-D | Spain | M. truncatula |
| L0482C | ESP095-C | Spain | M. truncatula |
| L0514C | ESP163-C | Spain | M. truncatula |
| L0526C | PRT179-F | Portugal | M. truncatula |
| L0543B | DZA327-7 | Algeria | M. truncatula |
| L0544A | ESP105-L | Spain | M. truncatula |
| L0545C | ESP158-A | Spain | M. truncatula |
| L0546B | ESP159-11 | Spain | M. truncatula |
| L0547B | ESP165-D | Spain | M. truncatula |
| L0640C | DZA213-K | Algeria | M. truncatula |
| L0648D | Salses 42B | France | M. truncatula |
| L0673C | DZA319 | Algeria | M. truncatula |
| L0679C | F66017 | France | M. truncatula |
| Outgroup genotypes | |||
| L0750D | ESP050 clock | Spain | M. tornata |
| L0897E | DZA204 | Algeria | M. ciliaris |
| L0901E | ES024 | Spain | M. rigidula |
Accession numbers correspond to the Institut National de la Recherche Agronomique (INRA) collection maintained at Station de Génétique et Amélioration des Plantes, INRA, Montpellier, France.
Choice of sequenced loci:
Overall, 29 loci were used for this study (Table 2). Five loci are fragments located in the genes DMI1, DMI3, NFP, NIN, and NORK that are involved in various stages of the signaling pathway (called Nod factor signaling) controlling the entry of symbiotic Sinorhizobium sp. This set of fragments is hereafter referred to as focal loci. A set of 24 control loci chosen regardless of their putative function was also sequenced. These control loci are gene fragments located on various linkage groups of the M. truncatula genome (see supplemental Table S1 at http://www.genetics.org/supplemental/).
TABLE 2.
Loci sequenced in this study
| Locus name | LG | Clone | Outgroup |
|---|---|---|---|
| Control loci | |||
| AAT | 5 | AC126783 | M. tornata |
| AGT | 7 | AC173291 | M. tornata |
| ASPP | 7 | AC157894 | M. tornata |
| ERF (F) | 5 | CR955012 | M. tornata |
| EXRN (F) | 2 | AC167330 | M. tornata |
| FT160 (F) | 7 | AC123593 | M. tornata |
| GLUT (F) | 8 | AC171266 | M. tornata |
| HAPc (R) | M. tornata | ||
| JUNBP | 3 | AC140026 | M. ciliaris |
| LEG195 (R) | 1 | AC146755 | M. tornata |
| LEG196 (F) | 5 | AC122165 | M. tornata |
| LEG219 (F) | 3 | CT030174 | M. tornata |
| LEG202 (F) | 5 | CR936948 | M. rigidula |
| LEG295 (F) | 3 | AC140849 | M. tornata |
| LEG391 (F) | 5 | CR931738 | M. tornata |
| LEG722 | Ca | AC093544 | M. tornata |
| LEG725 (F) | 4 | AC135605 | M. tornata |
| MAAP (F) | 8 | AC140032 | M. tornata |
| MADS (F) | 5 | CR931728 | M. tornata |
| MADS27 | 5 | AC126010 | No outgroup |
| MSL83 | AC157984 | M. tornata | |
| MTU | M. tornata | ||
| SULF (R) | 6 | AC139842 | M. tornata |
| UNK29 | M. tornata | ||
| Focal loci | |||
| DMI1 | 2 | AC140550 | M. tornata |
| DMI3 | 8 | AY508219 | M. tornata |
| NFP | 5 | AC126779 | M. tornata |
| NIN | 5 | CR931808 | M. tornata |
| NORK | 5 | CT030174 | M. tornata |
“Clone” is the BAC clone accession number (when applicable) yielding the highest BLAST score when the sequenced fragment was searched in the BAC contig assembly at http://www.medicago.org/genome. “LG” refers to the linkage group where the fragment is located (when applicable). F/R, loci for which only one strand was sequenced.
The best hit of homology search for LEG722 is located in chloroplast sequences.
DNA sequencing:
Sequences of the primers used for PCR and sequencing are available in supplemental Table S1. PCR reactions were performed as described in Fourmann et al. (2002). The PCR products were purified using P100 resin or using AMPure magnetic beads (Agencourt). Fragments were sequenced using the BigDye sequencing kit (ABI, Courtaboeuf, France) according to the manufacturer's instructions. Sequencing reactions were purified using G50 resin and loaded onto ABI3730 (with 36-cm capillaries) or onto ABI3130XL (with 50-cm capillaries) 96-capillary sequencers.
Analysis of nucleotide polymorphism:
For each locus, all available sequence reads (both forward and reverse) were assembled and aligned using the Staden package (Staden 1996) and the Genalys software (Takahashi et al. 2002). Sites with more than two nucleotide variants within M. truncatula and sites with >50% unexploitable data (alignment gaps and ambiguous nucleotides) were excluded from the analysis. In some instances, the locus was sequenced on one strand only (F or R, depending on the fragment; see Table 2). The number of polymorphic sites (S), nucleotide diversity (π), Watterson's estimate of the scaled mutation rate (θw), the number of distinct haplotypes (K), and the minimum number of recombination events (Rm) were computed at each locus. When applicable (i.e., S > 0), Tajima's D (Tajima 1989) and the standardized Fay and Wu's H (Zeng et al. 2006) statistics were computed at each locus. For each control locus, transition and transversion rates were estimated taking into account all diallelic polymorphic and fixed mutations, relative to an outgroup sequence. The probability of an observable mutation in the outgroup branch (not causing homoplasy) is estimated as the proportion of polymorphic sites with a third state in the outgroup. The proportion of unobservable mutations in the outgroup branch (causing homoplasy and the misorientation of a polymorphic site) is computed as in Baudry and Depaulis (2003). This allowed us to estimate the probability, PM, of incorrectly inferring the ancestral/derived status of polymorphisms detected at each locus.
Fit of a PEM:
We used a model featuring a single population undergoing constant demographic growth. The population size at time t in the past is given by 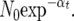 Time t is measured in units of 4N0 generations, where N0 is the present population size. An infinite-site model of mutation is assumed, with a scaled mutation rate (per site) of θ = 4N0μ, where μ is the mutation rate. Recombination occurs within each locus at a rate ρL, where ρ is the scaled recombination rate 4N0c, c the (per site) recombination rate, and L the number of sites in the locus. It is assumed that θ and ρ are identical among loci. Within the coalescent framework, parameters left to be estimated are α, θ, and ρ. We used an approximate Bayesian method using the rejection-sampling method of Haddrill et al. (2005) and Thornton and Andolfatto (2006) to estimate the parameters of the PEM. First, a uniform prior distribution of θ, ρ, and α is assumed with wide bounds: θ from 0 to 0.01, ρ from 0 to 0.02, and α from 0 to 40. A set of parameters is randomly sampled in this prior distribution, and coalescent simulations are performed to generate a simulated data set. A simulated data set consists of 24 independent fragments, each matching the number of sequences and alignment length of the 24 control fragments that we sequenced. The simulated data set is accepted only if the average values of S, π, and K in simulated data are all within a range defined as x ± ex, where x is the average value of S, π, or K in the observed data and e a tolerance factor. e is empirically chosen to maintain an acceptance rate close to 0.001. If the simulated data set is accepted, the values of the parameters θ, ρ, and α used to seed the coalescent simulation are recorded. The rejection sampling is stopped as soon as 1000 simulated data sets are accepted. The 1st and 99th percentiles of each marginal posterior distribution are used as bounds for new prior distributions on the parameters. In a second round of rejection sampling, the same procedure is followed using this new prior, using a smaller e (chosen for maintaining an acceptance rate close to 0.001) and stopping when 10,000 values are retained. The distribution of accepted values of θ, ρ, and α in this second run is used to approximate the joint posterior distribution of (θ, ρ, α), given the empirical data. Coalescent simulations are performed using ms (Hudson 2002). Since the coalescent implementation of ms generates sequence data with oriented polymorphism, the ancestral vs. derived status of alleles at polymorphic sites is switched around at a rate PM before processing simulated data, with the purpose of incorporating the level of misorientation observed in the data. The rejection-sampling procedure and the calculation of all summary and test statistics on simulated and empirical data sets has been written in C++ and Python (available from S. De Mita). The visualization of the marginal posterior distributions of the parameters has been done in R (R Development Core Team 2007).
Time t is measured in units of 4N0 generations, where N0 is the present population size. An infinite-site model of mutation is assumed, with a scaled mutation rate (per site) of θ = 4N0μ, where μ is the mutation rate. Recombination occurs within each locus at a rate ρL, where ρ is the scaled recombination rate 4N0c, c the (per site) recombination rate, and L the number of sites in the locus. It is assumed that θ and ρ are identical among loci. Within the coalescent framework, parameters left to be estimated are α, θ, and ρ. We used an approximate Bayesian method using the rejection-sampling method of Haddrill et al. (2005) and Thornton and Andolfatto (2006) to estimate the parameters of the PEM. First, a uniform prior distribution of θ, ρ, and α is assumed with wide bounds: θ from 0 to 0.01, ρ from 0 to 0.02, and α from 0 to 40. A set of parameters is randomly sampled in this prior distribution, and coalescent simulations are performed to generate a simulated data set. A simulated data set consists of 24 independent fragments, each matching the number of sequences and alignment length of the 24 control fragments that we sequenced. The simulated data set is accepted only if the average values of S, π, and K in simulated data are all within a range defined as x ± ex, where x is the average value of S, π, or K in the observed data and e a tolerance factor. e is empirically chosen to maintain an acceptance rate close to 0.001. If the simulated data set is accepted, the values of the parameters θ, ρ, and α used to seed the coalescent simulation are recorded. The rejection sampling is stopped as soon as 1000 simulated data sets are accepted. The 1st and 99th percentiles of each marginal posterior distribution are used as bounds for new prior distributions on the parameters. In a second round of rejection sampling, the same procedure is followed using this new prior, using a smaller e (chosen for maintaining an acceptance rate close to 0.001) and stopping when 10,000 values are retained. The distribution of accepted values of θ, ρ, and α in this second run is used to approximate the joint posterior distribution of (θ, ρ, α), given the empirical data. Coalescent simulations are performed using ms (Hudson 2002). Since the coalescent implementation of ms generates sequence data with oriented polymorphism, the ancestral vs. derived status of alleles at polymorphic sites is switched around at a rate PM before processing simulated data, with the purpose of incorporating the level of misorientation observed in the data. The rejection-sampling procedure and the calculation of all summary and test statistics on simulated and empirical data sets has been written in C++ and Python (available from S. De Mita). The visualization of the marginal posterior distributions of the parameters has been done in R (R Development Core Team 2007).
Test of neutrality:
We use a test of selective neutrality employing the joint observed values of D and H as test statistics, expecting an increased power due to the complementary information contained by these two tests (Zeng et al. 2006). Significance levels are obtained for each locus using 105 coalescent simulations under the PEM fitted above to generate the null joint distribution of D and H. This joint distribution is binned in 1024 evenly sized classes. The P-value of a joint observation (Dobs, Hobs) is computed as the sum of frequencies of all classes with a frequency equal to or lower than the class comprising (Dobs, Hobs). For generating a simulated data set under the fitted PEM, random values for (θ, ρ, α) are sampled from the posterior distribution of (θ, ρ, α), given the data at the 24 control loci. The three-dimensional space (θ, ρ, α) is binned in 1000 classes. For each simulation, one parameter set is determined first by drawing one class (classes being weighted by their frequency) and then by drawing values assuming a uniform distribution within the class. This procedure aims to take into account the uncertainty over parameter estimates.
RESULTS
Data on sequence polymorphism in M. truncatula were obtained by sequencing 24 control loci (on average 618 bp/fragment), representing a total of 14,830 bp sequenced and aligned, on average, in 27 genotypes and 5 focal loci (12,393 bp) sequenced in 30 genotypes (Tables 3 and 4). Below we describe patterns of polymorphism in control loci and how the SNM fits these observed data. We then present the results obtained when fitting a PEM to the control data. Finally, patterns of diversity found in the 5 focal loci are presented using the fitted PEM as a null model for selective neutrality.
TABLE 3.
Polymorphism at control loci
| Locus | n | Sites | S | θw | D | H | P(D, H) |
|---|---|---|---|---|---|---|---|
| AAT | 30 | 878 | 5 | 0.00144 | −0.56 | 0.42 | 0.890 |
| AGT | 29 | 755 | 9 | 0.00304 | 0.67 | 0.56 | 0.410 |
| ASPP | 30 | 734 | 5 | 0.00172 | 0.38 | 0.78 | 0.600 |
| ERF | 6 | 777 | 3 | 0.00169 | −0.45 | 0.00 | 0.634 |
| EXRN | 30 | 640 | 1 | 0.00039 | −1.13 | 0.15 | 0.773 |
| FT160 | 30 | 425 | 2 | 0.00119 | −0.43 | −1.45 | 0.293 |
| GLUT | 28 | 647 | 3 | 0.00119 | 0.50 | −0.25 | 0.230 |
| HAPc | 27 | 397 | 2 | 0.00131 | 0.58 | 0.26 | 0.435 |
| JUNBP | 28 | 753 | 6 | 0.00205 | −1.35 | 0.50 | 0.928 |
| LEG195 | 30 | 367 | 0 | 0 | NC | NC | NC |
| LEG196 | 30 | 284 | 3 | 0.00267 | −1.36 | 0.36 | 0.640 |
| LEG219 | 30 | 610 | 5 | 0.00207 | −1.29 | −0.68 | 0.355 |
| LEG202 | 13 | 360 | 5 | 0.00448 | −0.11 | −1.51 | 0.032* |
| LEG295 | 24 | 550 | 11 | 0.00536 | −0.70 | 0.14 | 0.609 |
| LEG391 | 29 | 648 | 11 | 0.00432 | −0.73 | −0.15 | 0.458 |
| LEG722 | 24 | 500 | 0 | 0 | NC | NC | NC |
| LEG725 | 30 | 633 | 5 | 0.00199 | 0.52 | 0.51 | 0.552 |
| MAAP | 29 | 572 | 5 | 0.00223 | −2.01 | −1.66 | 0.007* |
| MADS | 26 | 590 | 9 | 0.00400 | −0.16 | −2.63 | 0.025* |
| MADS27 | 13 | 495 | 4 | 0.00260 | 0.52 | NC | NC |
| MSL83 | 30 | 985 | 15 | 0.00384 | −0.95 | −0.04 | 0.676 |
| MTU | 30 | 1113 | 21 | 0.00476 | −1.75 | −1.64 | 0.081** |
| SULF | 30 | 378 | 8 | 0.00534 | 1.17 | −1.26 | 0.038* |
| UNK29 | 30 | 739 | 3 | 0.00102 | −0.83 | 0.50 | 0.950 |
n, number of accessions sequenced; sites, number of sites analyzed (excluding sites discarded from the analysis; see materials and methods); P(D, H), P-value associated with the joint D and H values under the PEM; NC, not computable. *P < 0.05; **P < 0.1.
TABLE 4.
Polymorphism at focal loci
| Locus | n | Sites | S | θw | D | H | P(D, H) |
|---|---|---|---|---|---|---|---|
| DMI1 | 30 | 2716 | 22 | 0.00204 | −1.86 | −1.49 | 0.022* |
| DMI3 | 30 | 3772 | 155 | 0.01037 | −0.47 | −0.23 | 0.908 |
| NFP | 30 | 1467 | 7 | 0.00120 | −0.03 | −1.29 | 0.127 |
| NIN | 30 | 2999 | 9 | 0.00076 | −0.14 | 0.01 | 0.745 |
| NORK | 29 | 1439 | 15 | 0.00068 | −1.23 | −0.68 | 0.410 |
n, number of accessions sequenced; sites, number of sites analyzed (excluding sites discarded from the analysis; see materials and methods); P(D, H), P-value associated with the joint D and H values under the PEM. *P < 0.05.
Patterns of nucleotide diversity in the sample are not compatible with the SNM:
Patterns of diversity averaged over the control loci are summarized in Table 5. We found an average of 5.9 polymorphic sites per locus, yielding a fairly low estimate of θ (θw = 0.00245/bp). This value was used as a fixed parameter for coalescent simulations to generate expectations for other parameters under the SNM. The average Tajima's D observed in our set of control loci is more negative than predicted by the SNM (average D = −0.43; P < 0.05). The negative value of D indicates that polymorphic sites tend to exhibit more skewed allele frequencies than expected under the SNM. The average value of H in the data is also more negative than expected (average H = −0.34; P < 0.05), indicating that the derived alleles tend to be at higher frequency than predicted by the SNM. The Rm per locus is 0.38 instead of 0 as expected under the SNM with an infinite-site model of mutation and without intragenic recombination. Similarly, K is higher than expected in simulations of the SNM (comparatively smaller values of K are expected in the absence of recombination). The average transition rate, transversion rate, and proportion of polymorphic sites with a third state in the outgroup were, respectively, 0.65, 0.18, and 0.07 in 22 control loci for which computations were possible. We estimated the rate of undetected back mutations (and therefore misorientations in our polymorphic positions) in the outgroup branch as PM = 0.07. As expected, incorporating this rate of misorientation in the SNM coalescent simulations does not affect any statistic other than H. The expected value of H under the SNM is decreased and closer to the observed value in the data (Table 5).
TABLE 5.
Observed and simulated multi-locus summary statistics
| Statistic | S | θw | π | D | H | K | Rm |
|---|---|---|---|---|---|---|---|
| Observed | 5.88 | 0.00245 | 0.00211 | −0.43 | −0.34 | 5.17 | 1 |
| SNM | |||||||
| PM = 0 | 5.74 | 0.00245NS | 0.00245NS | −0.05* | 0.04* | 4.80NS | 0*** |
| PM = 0.07 | 5.74 | 0.00245NS | 0.00245NS | −0.05* | −0.26NS | 4.80NS | 0*** |
| PEM | |||||||
| PM = 0 | 5.73 | 0.00244NS | 0.00209NS | −0.41NS | 0.24*** | 5.63NS | 0.10*** |
| PM = 0.07 | 5.72 | 0.00244NS | 0.00208NS | −0.41NS | −0.13NS | 5.63NS | 0.10*** |
“Observed” refers to the average summary statistics in 24 control loci. SNM, simulations under the standard neutral model with θ = 0.00245/site and no recombination; PEM, simulations under the population expansion model with parameters following the posterior distribution; PM, proportion of site misorientation. For simulations under SNM and PEM, 10,000 random data sets have been generated. For each data set, the averages of statistics are computed. The means of these 10,000 averages and results of unilateral tests are given (NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001). The tests are defined as the proportion of simulated 24-locus data sets with the average of D, H, etc., more extreme than the observed average.
Fit of a model incorporating demographic growth and recombination using control loci:
The PEM that we considered has three free parameters: θ (the scaled mutation rate), ρ (the scaled intragenic recombination rate), and α (the exponential growth rate). The initial uniform prior distributions used for each free parameter were θ from 0 to 0.01, ρ from 0 to 0.02, and α from 0 to 40. A first round of rejection sampling was conducted. The tolerance rate was first set to e = 0.1, and we ran coalescent simulations until 1000 simulated data sets were accepted (with an acceptance rate of 0.00155). The 1st and 99th percentiles of the resulting marginal distributions were, respectively: θ, 0.00237 and 0.00806; ρ, 0.00004 and 0.01621; and α, 0.116 and 19.36. These values were used as bounds for a new set of uniform priors for θ, ρ, and α. A second round of rejection sampling was then conducted using these new priors and setting e to 0.07. The run was stopped when 10,000 simulated data sets were accepted (acceptance rate: 0.00106; total simulations performed: 9,404,213). The 1st and 99th percentiles in the 10,000-point posterior distribution were, respectively: θ, 0.00252 and 0.00685; ρ, 0.00007 and 0.01016; and α, 0.273 and 12.28 (thus showing an increase in precision of the estimation procedure). The marginal posterior distributions obtained in the second round of rejection sampling are displayed in Figure 2. θ and α are strongly and positively correlated in the posterior distributions (Figure 2E). However, there are obvious modes in the marginal posterior distributions of individual parameters (Figure 2, A–C), suggesting that there is enough information in the data to estimate the model parameters. ρ seems to be less well estimated than θ. An obvious explanation would be that only K contains information about the recombination rate while S, π, and K yield complementary information regarding the scaled mutation rate. However, the use of more powerful estimators of ρ (Hudson 2001; McVean et al. 2002) would require inordinate computing effort, given that even an approximate estimate of ρ is sufficient for our purpose in this study. Maximum a posteriori (MAP) estimates of each parameter were obtained by binning the posterior distribution in 8000 classes (1019 nonempty classes). The MAP estimates were then taken as the mid-values of the most frequent class: θ* = 0.00337, ρ* = 0.00044, and α* = 1.57. We then assessed the goodness of fit of the PEM to the data in control loci. This was done by comparing expected vs. observed values for (1) the summary statistics used in the fitting procedure and (2) the summary statistics of the site frequency spectrum (D and H), which were not used to fit the PEM. As expected, the PEM tends to produce data sets with skewed allele frequencies, as indicated by a negative average D (Table 5). The statistics summarizing the level of diversity (θw and π) are very well fitted (P = 0.48 and 0.45, respectively). K is also well fitted, although marginally less so (P = 0.11), with a tendency for the PEM to generate more haplotypes than actually observed. Simulated data sets accepted under the PEM also exhibited, on average, positive H values (H = 0.04 under the SNM, 0.24 under the PEM), which is far off from the observed value (H = −0.34). Incorporating some level of misorientation of polymorphic sites in the PEM reduces this discrepancy (PEM simulations with PM = 0.07: average H = −0.13; P = 0.14).
Figure 2.—

Approximate posterior distributions of the PEM. Marginal distributions of the individual parameters θ (A), ρ (B), and α (C). (D–F) Joint bivariate distributions for pairs of parameters.
Test of selective neutrality:
D and H could not be computed for a few control loci: LEG195 and LEG722 display no polymorphic sites, and no outgroup sequence could be obtained for MADS27. The P-value associated with the joint (D, H) was computed for 24 control loci and for the 5 focal loci using the PEM as a null model (Tables 3 and 4). The results of all individual D and H and joint (D, H) tests under the different models are shown in supplemental Table S2 (at http://www.genetics.org/supplemental/). We find one focal locus, DMI1, with a significant deviation from the PEM: DMI1 exhibits both D and H values smaller than expected (P = 0.033; Figure 3). Among control loci, we find four loci significant at the 5% level: LEG202, MAAP, MADS, and MTU. For LEG202, D is larger and H more negative than expected; for MAAP, MADS, and MTU, both D and H are more negative than expected. In addition, SULF is significant at the 10% level and displays a larger D and a smaller H. DMI3 exhibits an outstanding level of polymorphism (θw = 0.01037, all other loci lying in the range of 0–0.00536). However, the test for neutrality, given that D and H are not affected by the raw level of polymorphism, shows no departure from the PEM (P > 0.5; Table 4).
Figure 3.—

Distribution of D and H under the PEM. Shaded areas indicate 10,000 simulations performed assuming a locus comprising 3013 nucleotides (the average length of focal loci) sequenced in 30 genotypes. Solid areas indicate observed values for each of the five focal loci.
Nonsynonymous variation:
A comparison of the amount of nonsynonymous and synonymous variation was performed (supplemental Table S3 at http://www.genetics.org/supplemental/). In all cases, the amount of nonsynonymous variation is lower than the synonymous (whatever the estimate), consistent with the hypothesis that all genes are currently evolving under purifying selection. Nonsynonymous variation is moderate in DMI1 and NFP (∼30% of synonymous) and low in DMI3 and NIN (<5%), and no nonsynonymous variation in NORK was detected in this study (a single nonsynonymous polymorphic site was detected using a different sample of genotypes in our earlier study). For NORK and all control loci, the number of exon sites sequenced is always <500, making the estimates of the nonsynonymous-to-synonymous ratio of variation somewhat unreliable. The nonsynonymous-to-synonymous ratio of divergence is of the same order of magnitude as the nonsynonymous-to-synonymous polymorphism ratio. The exceptions are NIN and SULF for which there is an excess of nonsynonymous divergence. McDonald–Kreitman tests were performed at each locus (control and focal) using M. tornata as an outgroup for estimating divergence (McDonald and Kreitman 1991). No test was significant (supplemental Table S3).
DISCUSSION
We analyzed five genes involved in the Nod factor perception pathway. These genes are the probable Nod factor receptor NFP, the symbiotic receptor NORK, the ion channel DMI1, the regulator gene DMI3, and the transcription factor NIN. The null hypothesis tested in this article is that of the selective neutrality of segregating polymorphism, allowing for any rate of population growth and accounting for possible misorientations of segregating sites. The expected joint distribution of D and H tests was obtained by simulations under a PEM with parameters fitted to 24 control loci chosen regardless of their function. Within the Nod factor perception pathway, only DMI1 displays a departure from neutrality (P < 0.05; Table 4). The four other genes, including the receptor-encoding genes NFP and NORK, display patterns of polymorphism consistent with selective neutrality under our PEM. We discuss below which selective constraints are most likely shaping the evolution of genes in the Nod factor perception pathway.
Selective constraints on Nod factor signaling genes:
No intense diversifying or balancing selection in the Nod factor perception pathway:
Overall, patterns of nucleotide variation at NORK, DMI3, and NIN reveal strong constraints by purifying selection, as indicated by low amounts of nonsynonymous variation as compared to synonymous variation (supplemental Table S3 at http://www.genetics.org/supplemental/). NFP and DMI1 seem less constrained in that regard. The McDonald–Kreitman test using M. tornata as the outgroup supports the neutrality hypothesis for these genes. In the case of NORK, this result shows that the events of positive selection previously described in De Mita et al. (2006) occurred before the divergence between M. truncatula and M. tornata. The gene DMI3 exhibits strikingly large amounts of variability. Presumably, the local rate of mutation is larger than in the other loci that we examined, producing a higher level of neutral variation. Very little of this variability appears to be nonsynonymous, indicating a strong level of purifying selection.
At the intraspecific level, genes involved in pathogen recognition often present signatures of balancing selection. Such footprints of selection are usually detected through high levels of nonsynonymous variation and positive values of Tajima's D (Tiffin and Moeller 2006). None of the five genes involved in the response to symbiotic bacteria appears to correspond to this model when examining within-species polymorphism.
The effects of most forms of selection on polymorphism are transient. The exception is balancing selection, which is able to generate specific signatures (including a positive D) over a long time (Charlesworth 2006). On average and under a neutral model, genes sampled within a population find a common ancestor <4N generations ago (N is the effective number of individuals in the population; Hein et al. 2005). In this article, we estimated θ = 4Nμ as 0.00337 (maximum a posteriori estimate). Assuming a mutation rate μ of 10−8 (per site) and considering that M. truncatula has a generation time of 1 year, the time when this common ancestor lived is ∼300,000 years ago. This estimate assumes a neutral model with constant population size, which we showed to be unlikely for our sample. Demographic growth tends to produce shorter gene genealogies and therefore 300,000 years are an overestimate. Therefore, our results do not exclude that some events of molecular adaptation in genes other than DMI1 have occurred earlier in the history of the Medicago genus and Fabaceae family (as demonstrated in the case of NORK).
A putative selective sweep in DMI1:
DMI1 is the only focal locus that can be rejected from a neutral model. Its pattern of polymorphism consists of negative D and H. The negative means of D and H across other loci has been explained by, respectively, population growth and site misorientation due to mutations in the branch leading to the outgroup. Even accounting for both processes, DMI1 still displays an unexpected deviation that can be explained by selection. Positive selection can produce a negative D through a fast increase of the positively selected mutation (a so-called selective sweep; Tajima 1989). It produces a negative H only if the polymorphism has not been completely eliminated by directional selection, e.g., because of recombination between the selected site and the rest of the sequence (Fay and Wu 2000). Supporting this hypothesis, five possible points of recombination are found in the DMI1 sequence data (using the four-gamete test; data not shown). Another explanation is that a few individuals have escaped the selective sweep, either because it occurred only in a limited range of populations or because the favored allele has not yet reached fixation. This event cannot have involved more than a few amino acid changes because the divergence between M. tornata includes few amino acid substitutions.
Functional interpretation of a potential event of selection in DMI1:
The current working model of Nod factor signaling (Stacey et al. 2006) assumes that DMI1 plays a role in initiating Ca2+ spiking and that the spiking mediates the gene activation (through DMI3) required for the formation of the infection thread (the first step mediating the entry of the symbiotic bacteria into the roots of Fabaceae). It is not known whether DMI1 is activated by NORK directly or indirectly, or if it is activated by independent means. The localization of the protein on the nuclear envelope (Riely et al. 2006) suggests that a direct activation is unlikely. Therefore, DMI1 is probably an intermediary step during the Nod factor signaling pathway. We hypothesize that modifications of DMI1 could alter the sensitivity of the signaling pathway and allow plants to be more sensitive to alteration of the Nod factor structure. A range of alterations in Nod factor structure lead to failure of infections or abnormal infections (Ardourel et al. 1994; Limpens et al. 2003). These observations suggest that an imperfect binding of altered Nod factors to their receptor still triggers residual responses. For example, if a DMI1 mutant has an increased affinity to either NORK or Nod factor receptors, then responses to altered Nod factors may be amplified because even marginal binding would be sufficient to initiate responses. Conversely, a more stringent interaction may abolish any responses to altered Nod factors. Therefore, positive selection of DMI1 mutations could reflect the selection for a new level of tolerance to alternative Nod factor structures.
However, more complicated models with several receptors with different structures cannot yet be ruled out and DMI1 could intervene in one of them, as suggested by biochemical observations (Hogg et al. 2006). DMI1 might therefore participate directly in Nod factor recognition. In this case, the fixation of new mutations can even more straightforwardly be interpreted by the selection for a new specificity. Lotus japonicus has two homologs to DMI1 (Imaizumi-Anraku et al. 2005). Therefore, it seems that DMI1 has been duplicated in the Lotus lineage or that one of the two copies of Lotus has been deleted in the Medicago lineage. These observations and our results suggest that the mode of evolution of DMI1 involves significant changes at both large and small evolutionary timescales. This gene may therefore have a more important role in controlling the specificity of Nod factor recognition than expected a priori.
Testing selective neutrality using the PEM:
Underestimation of the rate of homoplasy:
The estimated recombination rate is not sufficient for explaining the observed values of Rm. An explanation is that Rm can be inflated by multiple mutations at the same site within M. truncatula. We modeled only multiple hits in the interspecific divergence. The obvious outcome of multiple hits is a site with more than two alleles. We detected such sites in DMI3 only, which was an order of magnitude more polymorphic than for the other loci and was not used in fitting the PEM. However, since all mutations are not equally likely, due to the bias toward transitions, multiple mutations are statistically more likely to be back mutations and therefore undetectable (Baudry and Depaulis 2003). We repeated the test (D, H) using PM = 0.15 instead of PM = 0.07 (supplemental Table S2 at http://www.genetics.org/supplemental/). This change makes our test far more conservative regarding H but not D. Only one control locus with a strong departure of D (MAAP) remains significant, but note that patterns of variation at DMI1 are still suggestive (P = 0.06).
Simplicity of the model:
Even if some simplified demography and intragenic recombination are incorporated into the PEM, our model cannot account for all features of demography as well as sequence evolution. This probably explains why some summary statistics observed in the data are not perfectly fitted. However, the fit with the PEM is dramatically improved relative to the SNM. Additional features may have to be included for an accurate modeling of the data, like substructuration, migration from other geographical regions, as well as more complicated forms of population growth (recovery after a bottleneck, for example). However, modeling more complex models would require accurate knowledge of the present and past structure of populations of M. truncatula and enough data to simultaneously fit more parameters. Neither are available at the moment. This prospect is relevant for issues of accurate modeling of past demographic history and less relevant for detecting natural selection per se. The model that we used here provides a relatively simple extension of the SNM, appropriate for detecting loci exhibiting atypical patterns of polymorphism suggestive of selection while controlling for the genomewide effect of demography.
Selection at control loci:
A final potential caveat to our approach is the possible occurrence of selection in control loci. The method for designing the null hypothesis of the neutrality test assumes that all control loci are neutral when fitting the PEM. In this case, we still expect 1 locus of 20 with P < 0.05. We observed 4. For one (LEG202), the orientation of polymorphisms was performed using M. rigidula as an outgroup, which is less closely related to M. truncatula than M. tornata is. The true misorientation rate is therefore theoretically higher for this locus than for the others that were oriented using M. tornata. Since misorientation negatively biases H, the P-value associated with this test for LEG202 is likely to be too liberal. Note that, if selection occurred at 1 of the 3 other significant control loci (MAAP, MADS, and MTU, the best candidate being MAAP), then it would have made our test too conservative regarding the same kind of deviation (negative D and H values). Therefore, our test would not have generated a false positive for DMI1. SULF displays an unusual pattern with a positive D and a negative H and a marginally significant test of neutrality (P < 0.10). This pattern could be the consequence of balancing selection. The test for SULF may be less powerful because the region sequenced is rather short. Finally, we note that the distribution of P-values associated with the (D, H) test at the 24 control loci is fairly evenly distributed between 0 and 1 (Table 3). This finding is quite compelling, given that under a proper null model we would expect the distribution of P-values of control loci to be uniform in the interval [0,1].
Conclusion:
The selective neutrality of five genes involved in early responses of symbiotic microbes in M. truncatula was tested using data from anonymous control loci. These loci were used to fit a model featuring an exponentially growing population. We found that this simple model substantially improved the fit of several statistics describing our data. DMI1 showed a significant departure from this model, and it is possible that directional selection on DMI1 is operating in the population studied. Under this hypothesis, DMI1 may play a significant part in controlling the specificity of symbiotic infections and further research focusing on this gene would be of particular interest.
Acknowledgments
We thank S. Santoni and A. Weber for help with sequencing; P. Ratet, J. Burstin, A. Niebel, L. Godiard, B. Julier, C. Gough, and F. Debellé for sharing unpublished information for the sequencing of gene fragments located in loci FT160, NIN, NFP, SULF, DMI3, and HAPc; L. Madsen and J. Fredslund for information on the LEG markers and primer aliquots; P. Ratet and R. Geurts for discussion; and A. Alexander Smith, M. Lascoux, J. de Meaux, D. Brunel, and two anonymous reviewers for reading through this manuscript. Sequencing was funded by Institut National de la Recherche Agronomique. The European Science Foundation supported part of this research through an Exchange Grant (ConGen 2006-EX/1135) awarded to S.D.M.
References
- Ané, J.-M., G. B. Kiss, B. K. Riely, R. V. Penmetsa, G. E. D. Oldroyd et al., 2004. Medicago truncatula DMI1 required for bacterial and fungal symbioses in legumes. Science 303: 1364–1367. [DOI] [PubMed] [Google Scholar]
- Ardourel, M., N. Demont, F. Debellé, F. Maillet, F. de Billy et al., 1994. Rhizobium meliloti lipooligosaccharide nodulation factors: different structural requirements for bacterial entry into target root hair cells and induction of plant symbiotic developmental responses. Plant Cell 6: 1357–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrighi, J.-F., A. Barre, B. Ben Amor, A. Bersoult, L. Campos Soriano et al., 2006. The Medicago truncatula LysM-receptor kinase gene family includes NFP and new nodule-expressed genes. Plant Physiol. 142: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamshad, M., and S. P. Wooding, 2003. Signatures of natural selection in the human genome. Nat. Rev. Genet. 4: 99–111. [DOI] [PubMed] [Google Scholar]
- Baudry, E., and F. Depaulis, 2003. Effect of misoriented sites on neutrality tests with outgroup. Genetics 165: 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Amor, B., S. L. Shaw, G. E. D. Oldroyd, F. Maillet, R. V. Penmetsa et al., 2003. The NFP locus of Medicago truncatula controls an early step of Nod factor signal transduction upstream of a rapid calcium flux and root hair deformation. Plant J. 34: 495–506. [DOI] [PubMed] [Google Scholar]
- Brownlie, J. C., M. Adamski, B. Slatko and E. A. McGraw, 2007. Diversifying selection and host adaptation in two endosymbiont genomes. BMC Evol. Biol. 7: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catoira, R., C. Galera, F. de Billy, R. V. Penmetsa, E.-P. Journet et al., 2000. Four genes of Medicago truncatula controlling components of a Nod factor transduction pathway. Plant Cell 12: 1647–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, D., 2006. Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet. 2: e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook, D. R., 1999. Medicago truncatula: a model in the making! Curr. Opin. Plant Biol. 2: 301–304. [DOI] [PubMed] [Google Scholar]
- Cullimore, J., R. Ranjeva and J.-J. Bono, 2001. Perception of lipo-chitooligosaccharidic Nod factors in legumes. Trends Plant Sci. 6: 24–30. [DOI] [PubMed] [Google Scholar]
- de Meaux, J., and T. Mitchell-Olds, 2003. Evolution of plant resistance at the molecular level: ecological context of species interactions. Heredity 91: 345–352. [DOI] [PubMed] [Google Scholar]
- De Mita, S., S. Santoni, I. Hochu, J. Ronfort and T. Bataillon, 2006. Molecular evolution and positive selection of the symbiotic gene NORK in Medicago truncatula. J. Mol. Evol. 62: 234–244. [DOI] [PubMed] [Google Scholar]
- Depaulis, F., S. Mousset and M. Veuille, 2003. Power of neutrality tests to detect bottlenecks and hitchhiking. J. Mol. Evol. 57: S190–S200. [DOI] [PubMed] [Google Scholar]
- Endre, G., A. Kereszt, Z. Kevei, S. Mihacea, P. Kalò et al., 2002. A receptor kinase gene regulating symbiotic nodule development. Nature 417: 962–966. [DOI] [PubMed] [Google Scholar]
- Fay, J. C., and C.-I. Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155: 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenn, K., and M. Blaxter, 2006. Wolbachia genomes: revealing the biology of parasitism and mutualism. Trends Parasitol. 22: 60–65. [DOI] [PubMed] [Google Scholar]
- Fourmann, M., P. Barret, N. Froger, C. Baron, F. Charlot et al., 2002. From Arabidopsis thaliana to Brassica napus: development of amplified consensus genetic markers (ACGM) for construction of a gene map. Theor. Appl. Genet. 105: 1196–1206. [DOI] [PubMed] [Google Scholar]
- Geurts, R., E. Fedorova and T. Bisseling, 2005. Nod factor signaling genes and their function in the early stages of Rhizobium infection. Curr. Opin. Plant Biol. 8: 346–352. [DOI] [PubMed] [Google Scholar]
- Haddrill, P. R., K. R. Thornton, B. Charlesworth and P. Andolfatto, 2005. Multilocus patterns of nucleotide variability and the demographic and selection history of Drosophila melanogaster populations. Genome Res. 15: 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin, M. T., A. M. Casa, H. Sun, S. C. Murray, A. H. Paterson et al., 2006. Challenges of detecting directional selection after a bottleneck: lessons from Sorghum bicolor. Genetics 173: 953–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein, J., M. H. Schierup and C. Wiuf, 2005. Gene Genealogies, Variation and Evolution: A Primer in Coalescent Theory. Oxford University Press, Oxford.
- Hogg, B. V., J. V. Cullimore, R. Ranjeva and J. J. Bono, 2006. The DMI1 and DMI2 early symbiotic genes of Medicago truncatula are required for a high-affinity nodulation factor-binding site associated to a particulate fraction of roots. Plant Physiol. 140: 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their application. Genetics 159: 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Imaizumi-Anraku, H., N. Takeda, M. Charpentier, J. Perry, H. Miwa et al., 2005. Plastid proteins crucial for symbiotic fungal and bacterial entry into plant roots. Nature 433: 527–531. [DOI] [PubMed] [Google Scholar]
- Jiggins, F. M., 2006. Adaptive evolution and recombination of Rickettsia antigens. J. Mol. Evol. 62: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Lévy, J., C. Bres, R. Geurts, B. Chalhoub, O. Kulikova et al., 2004. A putative Ca2+ and calmodulin-dependent protein kinase required for bacterial and fungal symbioses. Science 303: 1361–1364. [DOI] [PubMed] [Google Scholar]
- Limpens, E., C. Franken, P. Smit, J. Willemse, T. Bisseling et al., 2003. LysM domain receptor kinases regulating rhizobial Nod factor-induced infection. Science 302: 630–633. [DOI] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23: 23–55. [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. [DOI] [PubMed] [Google Scholar]
- McVean, G., P. Awadalla and P. Fearnhead, 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160: 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., 2005. Molecular signatures of natural selection. Annu. Rev. Genet. 39: 197–218. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., T. T. Hu, Y. Ishino, J. Jhaveri, C. Toomajian et al., 2005. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 3: e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowski, M. F., J. David, S. Santoni, H. McKhann, X. Reboud et al., 2006. Evidence for a large-scale population structure among accessions of Arabidopsis thaliana: possible causes and consequences for the distribution of linkage disequilibrium. Mol. Ecol. 15: 1507–1517. [DOI] [PubMed] [Google Scholar]
- R Development Core Team, 2007. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna.
- Riely, B. K., G. Lougnon, J.-M. Ané and D. R. Cook, 2006. The symbiotic ion channel homolog DMI1 functions in the nuclear membrane of Medicago truncatula roots. Plant J. 49: 208–216. [DOI] [PubMed] [Google Scholar]
- Ronfort, J., T. Bataillon, S. Santoni, M. Delalande, J. David et al., 2006. Microsatellite diversity and broad scale geographic structure in a model legume: building a set of nested core collections for studying naturally occurring variation in Medicago truncatula. BMC Plant Biol. 6: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauser, L., A. Roussis, J. Stiller and J. Stougaard, 1999. A plant regulator controlling development of symbiotic root nodules. Nature 402: 191–195. [DOI] [PubMed] [Google Scholar]
- Schmid, K. J., S. Ramos-Onsins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey, G., M. Libault, L. Brechenmacher, J. Wan and G. D. May, 2006. Genetics and functional genomics of legume nodulation. Curr. Opin. Plant Biol. 9: 110–121. [DOI] [PubMed] [Google Scholar]
- Staden, R., 1996. The Staden sequence analysis package. Mol. Biotechnol. 5: 233–241. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, M., F. Matsuda, N. Margetic and M. Lathrop, 2002. Automated identification of single nucleotide polymorphisms from sequencing data. Proceedings of the IEEE Computer Society Bioinformatics Conference, Stanford, CA, pp. 87–93. [PubMed]
- Tenaillon, M. I., J. U'Ren, O. Tenaillon and B. S. Gaut, 2004. Selection versus demography: a multilocus investigation of the domestication process in maize. Mol. Biol. Evol. 21: 1214–1225. [DOI] [PubMed] [Google Scholar]
- Thompson, J. N., 2006. The Geographic Mosaic of Coevolution. University of Chicago Press, Chicago.
- Thornton, K., and P. Andolfatto, 2006. Approximate Bayesian inference reveals evidence for a recent, severe bottleneck in a Netherlands population of Drosophila melanogaster. Genetics 172: 1607–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiffin, P., and D. A. Moeller, 2006. Molecular evolution of plant immune system genes. Trends Genet. 22: 662–670. [DOI] [PubMed] [Google Scholar]
- Van der Biezen, E. A., and J. D. G. Jones, 1998. Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem. Sci. 23: 454–456. [DOI] [PubMed] [Google Scholar]
- Weiss, G., and A. von Haeseler, 1998. Inference of population history using a likelihood approach. Genetics 149: 1539–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson, S. H., R. Hernandez, A. Fledel-Alon, L. Zhu, R. Nielsen et al., 2005. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102: 7882–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S. I., and B. S. Gaut, 2004. Molecular population genetics and the search for adaptive evolution in plants. Mol. Biol. Evol. 22: 506–519. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., I. V. Bi, S. G. Schroeder, M. Yamasaki, J. F. Doebley et al., 2005. The effects of artificial selection on the maize genome. Science 308: 1310–1314. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 2006. Computational Molecular Evolution. Oxford University Press, Oxford.
- Zeng, K., Y. X. Fu, S. Shi and C. I. Wu, 2006. Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics 174: 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]