

Abstract
To study the role of human papillomavirus (HPV) infection in the development of genetic instability, we transduced normal human airway and anogenital epithelial cells with various combinations of HPV-16 E6, E7, and the reverse transcriptase component of telomerase (hTERT). Cell lines generated by co-expression of E7 with E6 and/or hTERT (i.e., E6/E7, E7/hTERT, and E6/E7/hTERT) exhibited extra copies of chromosome 20 and specific amplification of the 20q12-ter region, whereas those generated without E7 (i.e., hTERT alone or E6/hTERT) did not. Co-expression of hTERT and a dominant-negative version of cdk4 that has been shown to inactivate the retinoblastoma (pRb) pathway also resulted in 20q amplification. Interestingly, extra copies of chromosome 20 were observed in early passage keratinocytes that expressed E7 alone, and microarray expression analysis revealed that genes in the 20q region and on chromosome 5 were specifically upregulated in these cells. Our results indicate that chromosome 20q amplification is an early event that may be specifically caused by expression of E7 through inactivation of the pRb pathway in human epithelial cells.
Keywords: Papillomavirus, HPV, Chromosome 20, Instability, E7, Airway epithelial cells, Immortalization, Telomerase, Keratinocytes
Introduction
Human papillomaviruses (HPVs) are involved in the development of several different epithelial malignancies (Franceschi et al., 1996; Howley, 1991). The association of HPV with cervical cancer and other anogenital cancers is well known, with up to 99% of cervical cancers containing high-risk HPV sequences (Walboomers et al., 1999). There is now considerable evidence that high-risk HPV types can also play a role in the development of certain head and neck cancers, including tonsillar, laryngeal, and some respiratory carcinomas (Gillison et al., 2000; van Houten et al., 2001). As with cervical cancer, the most common HPV type found in these malignancies is HPV-16. The main transforming genes of HPV are E6, which, among other functions, degrades the p53 tumor suppressor protein and activates telomerase, and E7, which is known to inactivate the retinoblastoma (pRb) protein (Dyson et al., 1989; Klingelhutz et al., 1996; Scheffner et al., 1990). Head and neck cancers that are HPV positive and express E6 and E7 generally do not have mutations in genes involved in the p53 and pRb pathways, while HPV-negative cancers have a high incidence of p53 mutation and often lack expression of p16INK4a, an inhibitor of the cdk4/cyclin D complex that phosphorylates pRb (van Houten et al., 2001; Wiest et al., 2002). A recent study examining head and neck cancers found that HPV-positive and HPV-negative cancers exhibit different chromosome aberrations (Braakhuis et al., 2004). These results indicate that HPV-positive and HPV-negative cancers are distinctly different entities.
Both HPV E6 and E7 have been shown to be involved in the development of genetic instability. For example, it has been demonstrated that HPV-16 E7 expression causes centrosome abnormalities that can lead to aneuploidy (Duensing and Munger, 2003; Duensing et al., 2001; Southern et al., 2004). HPV-16 E6 has been implicated in inducing premature mitotic chromosome segregation as well as anaphase bridge formation (Duensing and Munger, 2002; Plug-Demaggio and McDougall, 2002). Thus, expression of HPV E6 and E7 not only initiates the process of transformation but also creates an environment that can lead to genetic instability and malignant progression.
Chromosome 20 gain has been observed to be associated with HPV-mediated immortalization of uroepithelial cells, prostate epithelial cells, and ovarian epithelial cells (Cuthill et al., 1999; Jin et al., 2004; Macoska et al., 2000; Savelieva et al., 1997). It has been proposed that chromosome 20q amplification, in the context of E7 expression, represents a dominant genetic alteration that is necessary for immortalization (Cuthill et al., 1999). Amplification of chromosome 20 has also been observed in breast cancer, bladder cancer, and head and neck cancers, and several different genes on chromosome 20q have been found to be upregulated in cell lines and cancers that exhibit chromosome 20 amplification, although the role of these genes in transformation remains controversial (Collins et al., 1998; Gollin, 2001; Guo et al., 2004; Hodgson et al., 2003; Reshmi et al., 2004). In this study, we show that expression of HPV-16 E7 is associated with amplification of the 20q region in human airway epithelial cells and human foreskin keratinocytes. Our data indicate that chromosome 20 abnormalities and amplification of genes on chromosome 20 are an early event in the process of transformation by E7 and may be a specific consequence of E7 expression associated with pRb inactivation.
Results
Our previous work demonstrated that adult human airway epithelial (HAE) cells could be efficiently immortalized by co-expressing HPV-16 E6/E7 and hTERT (Zabner et al., 2003). Using this strategy, independent immortal HAE cell lines derived from six different donors were generated. Examination of these cell lines revealed specific cytogenetic abnormalities. In particular, extra copies of chromosomes 5 and 20 were observed in all the cell lines (Zabner et al., 2003). Of particular interest was that none of the E6/E7/hTERT HAE lines went through a crisis (slow down in growth) during the immortalization process that would indicate a requirement for other alterations aside from those conferred by expression of E6, E7, and hTERT. Thus, the acquisition of chromosomes 5 and 20 abnormalities in these cells did not appear to be a rare rate-limiting event.
To determine what factors were specifically involved in the development of chromosome aberrations during the immortalization process, we transformed early passage HAE by using various combinations of retroviruses containing E6, E7, and hTERT. After introduction of the different genes by retroviral transduction and selection in appropriate antibiotics, the cells were passaged to determine whether they had an extended lifespan. Cells that were transduced with vector alone, E6 alone, or E7 alone senesced, whereas transduction of E6 and E7 together resulted in cell lines with a significantly extended lifespan of greater than 100 population doublings (pd) (Fig. 1A). Transduction of hTERT alone also resulted in a significantly extended lifespan of greater than 100 pd but only after a crisis period at approximately 65 pd (Fig. 1B). Co-expression of hTERT with E6, E7, or E6/E7 resulted in efficient extension of lifespan without an apparent crisis, although E7/hTERT cells had a slower growth rate overall (Fig. 1B). All transduced cells that reached 100 pd or more continue to proliferate in culture and are apparently immortal. A more detailed characterization of the immortal HAE cells will be reported elsewhere (unpublished data). Karyotype analysis revealed that all extended lifespan cells that expressed E7 had extra copies of chromosome 20, whereas extended lifespan cells that were transduced with hTERT alone or a combination of E6 and hTERT did not (Table 1). This was apparent in two independently derived E7/hTERT and hTERT-transduced cell lines. To determine whether the chromosome 20 defect was associated with inactivation of the pRb pathway, we also transduced HAE using a combination of hTERT with a dominant-negative version of cdk4, R24C, which has been shown to be constitutively active in phosphorylating and inactivating pRb (Wei et al., 2003). HAE cells generated using R24C and hTERT also exhibited an extra copy of chromosome 20, suggesting that R24C and E7 may be similar with regard to their association with the chromosome 20 abnormality (Table 1). All of the cell lines except E6/hTERT also had extra copies of chromosome 5 (Table 1).
Fig. 1.
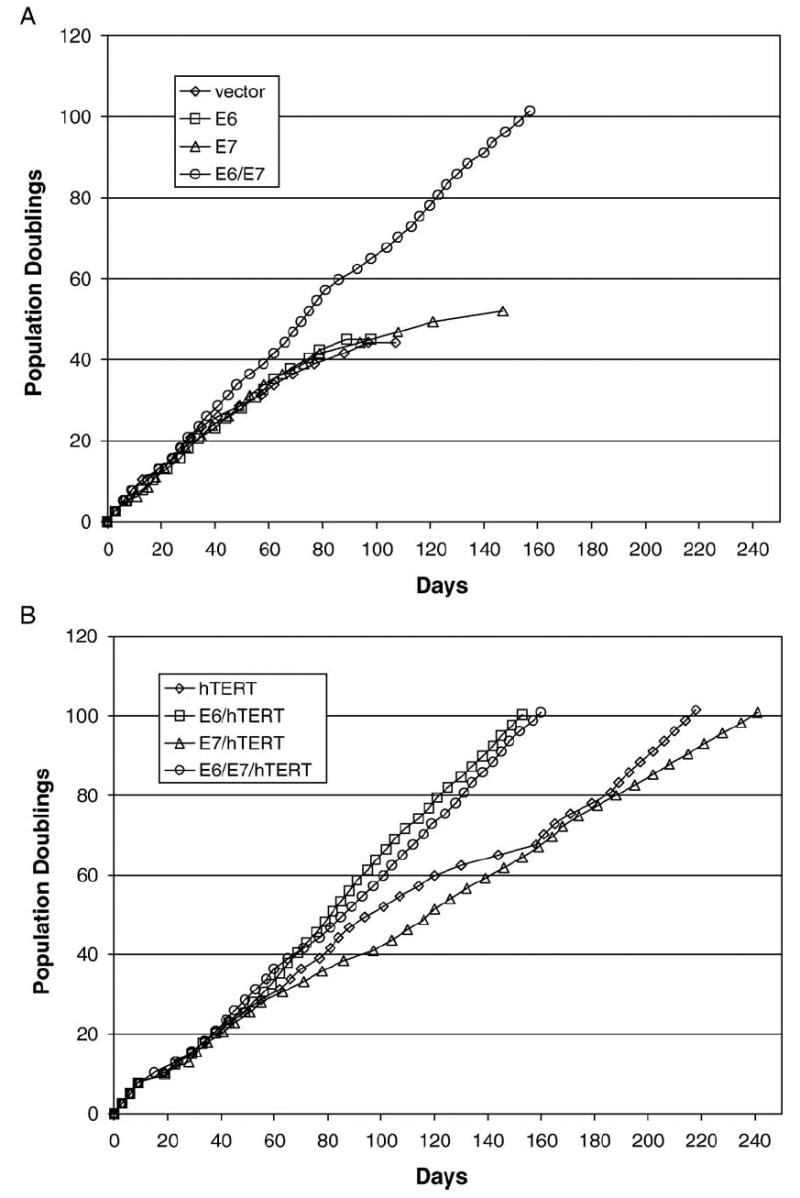
Effects of HPVoncogenes and hTERT on growth of airway epithelial (HAE) cells. (A) Growth of HAE transduced with vector alone, HPV-16 E6, HPV-16 E7, or HPV-16 E6/E7. Only those cells that expressed both E6 and E7 became immortal. (B) Growth of HAE transduced with hTERT and E6, E7, or E6/E7. Those cells expressing hTERT alone exhibited a crisis before reaching 100 pd, whereas those cells that co-expressed hTERT with any of the viral oncogenes reached 100 pd without any significant slow down in growth after the initial transduction and selection period between pd10 and pd15.
Table 1.
Karyotype analysis of HAE cells transduced with combinations of E6, E7, TERT, and R24C
| Cell typea | Karyotypeb |
|---|---|
| Vectorc | 46, XX[20] |
| E6/E7 | 48, XX, +5, +20[15] |
| E7/hTERT-1 | 95–105, XXXX, +5, +6, +7, +11, −16, +18, +20, +20[cp10]d |
| E7/hTERT-2 | 48, XX, +7, +20, add(22)(q13)[10] |
| E6/hTERT | 46, XX, der(22)t(1;22)(q21;q14)[5]/46, XX[11] |
| E6/E7/hTERT | 47, XX, +20[8]/48, XX, +5, +20[4]/48, XX, +20, +20[3] |
| hTERT-1 | 47, XX, +5[10] |
| hTERT-2 | 46, XX[10] |
| R24C/hTERT | 49, XX, +5, +7, der(7)t(7;8)(q22;q13), +20[7]/48, XX, +5, +20, del(20)(p11.2)[3] |
The different cells in this experiment were derived from the same normal HAE cell strain. E7/hTERT-1 and E7/hTERT-2 as well as hTERT-1 and hTERT-2 represent derivatives that were generated in independent retroviral transductions. Cells were karyotyped between 80 and 90 pd.
At least 10 metaphase spreads were karyotyped in detail. Number in brackets represents the number of metaphases with that particular karyotype.
Vector alone did not have an extended lifespan and was karyotyped at approximately 8 pd post-transduction.
This is a composite (cp) karyotype. The range of 95–105 is due to heterogeneity in the population with the modal number of chromosomes being 98. The range of chromosome 20 copies was 6–7 with a modal number of 6.
FISH analysis was used to characterize the chromosome defects in the cells in more detail. Using a 20q12 probe (20q) and a chromosome 20 centromeric probe (20cen), it was found that a large number of interphase nuclei in E7 expressing cells exhibited specific amplification of the 20q region beyond that observed by karyotype analysis (Fig. 2A). This was particularly evident in E7/hTERT cells where some nuclei had 4- or 5-fold increases in 20q signals as compared to 20cen signals (Fig. 2B). Overall, amplification of the 20q region was specifically observed in E7/hTERT, E6/E7, E6/E7/hTERT, and R24C/hTERT cells, whereas extra copies were not generally observed in cells expressing E6/hTERT or hTERT alone. Identical results were obtained with a 20qter probe (data not shown). Thus, amplification of the 20q12-qter region was specifically associated with expression of E7 or R24C. Extra copies of chromosome 5, on the other hand, were evident in all of the cell lines except E6/hTERT, and chromosome 5 copy numbers, as ascertained by FISH, were the same as what was observed by karyotype analysis (data not shown).
Fig. 2.
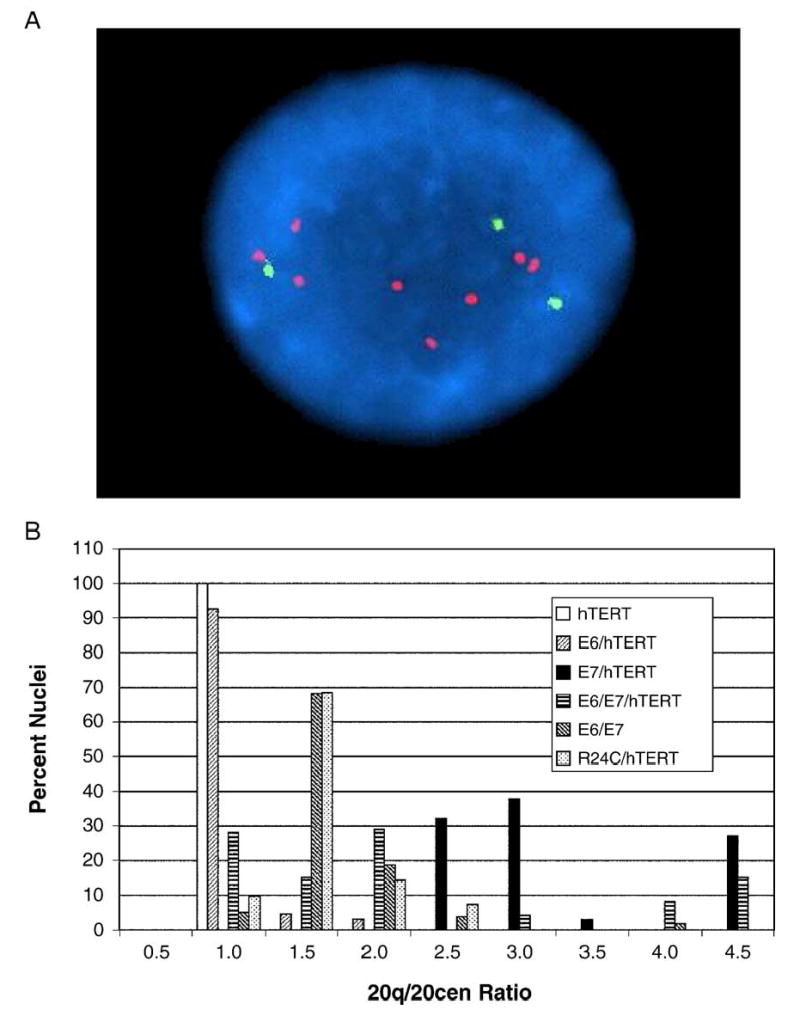
Amplification of 20q in E7 expressing human airway epithelial (HAE) cells. (A) Interphase FISH of E6/E7/TERT airway epithelial cells using dual-color 20q and 20cen probes. This particular cell shows three copies of 20cen signal (green) and multiple copies of 20q signals (red). (B) Composite graph showing ratios of signal from a 20q probe over signal from a 20cen probe in the various HAE lines. Two hundred or more nuclei were examined for each cell type. Note that any cell line that expressed E7 exhibited amplification of 20q signals as compared to 20cen signals.
To determine whether chromosome 20q amplification was associated with E7 expression in another cell type, we also examined human foreskin keratinocytes (HFK) that were transduced by E6/E7, E7/hTERT, R24C/hTERT, or hTERT alone. As with HAE, the E6/E7 E7/hTERT, and R24C/hTERT cell lines exhibited extra copies of chromosome 20, whereas cells transfected with vector alone or immortalized by hTERT alone did not (Table 2).
Table 2.
Karyotype analysis of HFK cells
| Cell type | Karyotypea |
|---|---|
| Normalb | 46, XY[10] |
| E6/E7 | 47–49, XY, +5, +6, +11, add(19)(p13.3), +20, −22[cp15]c |
| E7/hTERT | 91–92, XXYY, −15, +20, +20, −22, +mar[cp10]c |
| hTERTb | 47, XY, iso(9)(q)[10] |
| R24C/hTERT | 49, XY, +1, add(1)(q22), del(7)(q22q32), +20, +mar[10] |
| Vectord | 46, XY[10] |
| E7d | 48, XY, +9, +20[10]/48, XY, +5, +20[5] |
At least 10 metaphase spreads were karyotyped in detail. Number in brackets represents the number of metaphases with that particular karyotype.
Reported previously (Farwell et al., 2000). Cells were derived from the same individual.
These are composite (cp) karyotypes. The ranges of 47–49 for E6/E7 and 91–92 for E7/hTERT are due to heterogeneity in the population with modal numbers being 47 and 91, respectively. Listed aberrations were found in the majority of metaphase spreads. All E6/E7 cells contained 3 copies of chromosome 20, whereas the range of chromosome 20 copies in E7/hTERT was 4–7 with a modal number of 6.
Karyotypes were performed on vector and E7 cells within 8 populations doublings post-transduction and selection.
In a previous study, we reported the results of a microarray expression analysis of HFK transduced with various combinations of HPV oncogenes (Duffy et al., 2003). In that study, data analysis with Affymetrix software indicated that a large number of cellular genes were affected by E6 and/or E7 expression within 3–4 passages after introduction of the genes into the cells by retroviral transduction. To determine whether genes from specific chromosome regions or functional groups were upregulated in early passage E7 expressing cells, we reanalyzed the Affymetrix expression data by using a software program called D-Chip, which was recently developed at Harvard (Li and Wong, 2001). This program allows an assessment of statistically significant gene clusters based on function and genome map location (see Materials and methods). We focused on those genes that were upregulated by 3-fold or more in E7 expressing cells. This resulted in assignment of several functionally significant clusters, mostly related to DNA metabolism and replication, as might be expected of cells in which pRb has been inactivated. Strikingly, the only two genome location clusters that were statistically significant contained genes on chromosome 20q and chromosome 5 (Table 3). Karyotype analysis of the early passage E7 expressing HFK (i.e., within 3 passages after retroviral transduction) revealed that these cells contained extra copies of chromosome 20 and chromosome 5 in a significant fraction of the metaphase spreads (Table 2), whereas vector control cells were normal. Thus, induction of extra copies of chromosomes 20q and 5 occurred at a relatively early stage in E7 expressing HFKs and was not necessarily associated with extension of lifespan or immortalization, as HFKs that expressed E7 alone eventually senesced.
Table 3.
Genes upregulated 3 or more fold in E7 keratinocytes that map to chromosome 20q or chromosome 5
| Upregulated chromosome 20q genes |
| TAF4A RNA polymerase II, TBP-associated factor |
| Protein kinase C binding protein 1 |
| Nuclear receptor coactivator 6 |
| Splicing factor (CC1.3) |
| Tumor differentially expressed 1 |
| RAE1 RNA export 1 homolog |
| Upregulated chromosome 5 genes |
| Chondroitan sulfate proteoglycan 2 (Versican) |
| Phosphoinositol-3-kinase, regulatory subunit |
| KIAA0073 protein |
| Transcription factor IIF |
| X-ray repair gene |
| Fer tyrosine kinase NCP94 |
| Thyroid hormone receptor coactivating protein |
| Cyclophilin C |
| 3-Oxoacid CoA transferase |
| Cyclin H |
| Forkhead Box D1 |
| IK cytokine downregulator of HLA II |
| Beta-2 adrenergic receptor |
| MAP kinase 9 |
| Death-associated protein |
| TAF7 RNA polymerase (II), TBP-associated factor |
| Purine-rich element binding protein A |
| Aldehyde dehydrogenase 7 |
| Hydroxysteroid dehydrogenase 4 |
| LNS-CAI/LNS-CAII |
| Calcium modulating ligand |
| KIAA0202 protein |
| ADP-ribosylation factor domain protein 1 |
| Ras p21 protein activator 1 |
| Histidyl–tRNA synthetase |
Discussion
Amplification of the 20q11–13 region has been implicated as a critical event in the transformation of certain epithelial cell types, including uroepithelial and mammary epithelial cells (Cuthill et al., 1999; Hodgson et al., 2003). Our current study indicates that amplification of 20q is also associated with HPV-mediated extension of lifespan and immortalization of airway and foreskin epithelial cells. Several genes which map to the 20q11–13 region have been identified as potential oncogenes that are amplified during bladder and breast carcinogenesis, but the role of these genes in the development of cancer is not completely clear (Collins et al., 1998; Guo et al., 2004). Our studies suggest that 20q amplification and upregulation of genes on chromosome 20q may be a specific consequence of HPV E7 expression and inactivation of pRb.
The mechanism of 20q amplification in E7 expressing cells remains unknown. In the generation of many of the cell lines in our study, there was never an apparent crisis, suggesting that chromosome 20 aberrations occur at high frequency. E7’s role in causing centrosome defects and inducing genetic instability certainly could play a role in this process (Duensing and Munger, 2002; Duensing et al., 2001). It is interesting to note that both E7/hTERT-1 HAE and E7/hTERT HFK were tetraploid, as might be expected in cells with abnormal centrosome regulation. Since one of the main functions of E7 is the inactivation of pRb, it can be speculated that inactivation of the pRb pathway is somehow involved in the development of 20q abnormalities. Our studies using the dominant-negative cdk4 construct support this hypothesis. Interestingly, extra copies of chromosome 20 were also observed in a recent study in which bronchial epithelial cells were immortalized by co-expression of hTERT and wild-type cdk4, which would also be expected to inactivate the pRb pathway (Ramirez et al., 2004).
Recently, a model has been proposed to explain how defects in the pRb pathway promote genetic instability (Hernando et al., 2004). In this model, pRb inactivation leads to deregulation of E2F transcription factors, which results in upregulation of Mad2, a mitotic checkpoint protein. Aberrant Mad2 expression can promote mitotic defects that lead to aneuploidy. Thus, inactivation of pRb by E7 may be involved in generating chromosome defects, but how this relates to the development of chromosome 20q abnormalities specifically is unknown. It is interesting to note that, in our experiments, E7/hTERT cells had a slower growth rate overall than E6/hTERT, E6/E7/hTERT, or E6/E7 cells. One possibility is that abrogation of the pRb pathway may also lead to a condition that leads to a growth block that must be compensated for by amplification of a gene or genes on chromosome 20. Expression of E6 may partially overcome this block and allow faster growth. In our study, cells that expressed hTERT alone did not exhibit 20q amplification, even though they exhibited a significant crisis before becoming apparently immortal. No apparent defects in the pRb pathway, including loss of p16INK4a, were detected in the hTERT HAE cells (data not shown). However, loss of p16INK4a was observed in hTERT immortalized HFK, but these cells did not exhibit chromosome 20 abnormalities (Farwell et al., 2000; Kiyono et al., 1998). Thus, p16INK4a loss, unlike constitutive expression of cdk4, may not be associated with induction of chromosome 20 abnormalities. Further work, using other factors that inactivate the pRb pathways, and the identification and characterization of genes on 20q that are specifically upregulated during immortalization will be useful in understanding the mechanism and function of 20q amplification.
Clearly, other genetic abnormalities besides chromosome 20q amplification are important for the malignant progression of HPV-infected cells. In addition to chromosome 20 abnormalities, we also consistently observed extra copies of chromosome 5 in our HAE and HFK cell lines. Whether amplification of genes on chromosome 5 is important for immortalization of HAE or HFK is unknown. In a recent study, chromosome 5 abnormalities were observed in airway epithelial cells that were immortalized by expression of hTERT alone (Piao et al., 2005). We also observed extra copies of chromosome 5 in our hTERT-transduced HAE, but this was not apparent in E6/hTERT HAE or hTERT HFK, indicating that hTERT expression and chromosome 5 alterations are unrelated. Interestingly, chromosome 5 alterations have also been observed in a number of different cancers, including head and neck cancers (Gollin, 2001). Performing wide-scale genomic analyses on HPV-positive and -negative cancers, such as head and neck cancers, will be useful for determining which genetic alterations are specifically associated with HPV infection in vivo and which ones are not. These studies could lead to the discovery of novel genes that may be involved in the development of HPV-associated cancer. They may also provide valuable information that could be used for better diagnosis and treatment of HPV-positive and -negative malignancies.
Materials and methods
Generation of cell lines
Human airway epithelial cells (HAE) used in this study were isolated from bronchial or tracheal tissue of normal adult cadaver specimens and human foreskin keratinocytes (HFK) were obtained from foreskin tissue of neonates in accordance with protocols approved by the University of Iowa Institutional Review Board and HIPAA guidelines. HAE cells were isolated and grown as previously described in bronchial epithelial growth medium (BEGM, Cambrex) on placental collagen (type VI)-coated plates (Karp et al., 2002). HFK were grown in keratinocyte serum-free media (KSFM, Invitrogen) as described (Klingelhutz et al., 1994).
For transduction of exogenous genes, primary cells at passages 2–3 post-isolation were infected as described (Farwell et al., 2000; Kiyono et al., 1998; Zabner et al., 2003) with various retrovirus constructs in combinations that allowed for selection in hygromycin (10 μg/ml) or G418 (50 μg/ml). The LXSN, LXSH, 16 E7 LXSH, and 16 E6 LXSN, and 16 E6/E7 LXSN replication-incompetent MLV retroviral constructs were obtained from Denise Galloway (Fred Hutchinson Cancer Research Center). The hTERT neo pBABE and hTERT hygro-pBABE replication-incompetent retroviral constructs were obtained from Robert Weinberg (Whitehead Institute), and the retroviral construct expressing a dominant-negative cdk4 (R24C) was obtained from William Hahn (Harvard). Cells infected with single vectors were selected for 8 days after infection in the appropriate antibiotic. Cells infected with two vectors were sequentially selected. All cell lines were checked for expression of various genes by RT-PCR or Western blot and for telomerase activity as previously described (Sprague et al., 2002; Zabner et al., 2003). Culture splits were 1:6 for HAE and 1:5 for HFK. Cells were passaged until senescence or until they had exceeded 100 populations doublings or more, which is approximately 2.5- to 3.0-fold their normal lifespan in these culture conditions.
Cytogenetic and FISH analyses
Chromosomal analyses were performed on HAE cells from in situ cultures harvested on coverslips by TeCan robotic harvester using standard protocols. Briefly, cells were arrested in metaphase by adding ethidium bromide (final concentration12.5 μg/ml) for 40 min followed by colcemid (final concentration 6 μg/ml). After 1–2 h, the cells were incubated for 25 min at room temperature with hypotonic solution (3:1 mixture of 0.8% sodium citrate and 0.075 molar potassium chloride). Cells were then fixed three times with a 3:1 methanol/acetic acid. Chromosome spreading on coverslips was done on Thermotron and mounted on glass slides after the drying process. Ten to twenty G-banded metaphases were analyzed for each cell type. Karyotypes were performed for most cells (except normal, vector, and early passage E7 cells) after they had reached 80–90 pd or approximately 2- to 2.5-fold beyond their normal lifespan. The images were captured and karyotyped in CytoVision computerized imaging system (Applied Imaging, USA).
For FISH analysis, probes for the 20q subtelomeric (mixture #15), 20q12, and a chromosome 20 centromeric region probe, 20cen (VYSIS, Downer’s Grove, IL, USA), were used in this study. The 20q12 probe (D20S108) has been previously used to define the region of 20q amplification in breast cancer (Collins et al., 1998). Chromosome 5p and 5cen probes (Vysis) were also used for several of the analyses. Protocols were followed according to manufacturer’s specifications. Briefly, hybridization was performed on Hybrite programmed for melt temperature at 75 °C and time for 1 min. After overnight hybridization at 37 °C, the slides were washed in 0.4× SSC/0.3% NP-40 for 2 min at 72 °C and in 2× SSC/0.1% NP-40 for 1 min at room temperature. The slides were then counterstained with DAPI. For each cell line, five metaphases cells and over 200 interphase nuclei were analyzed. The image acquisition was also through CytoVision. Counts of signals were made for the various probes and amplification of signals in the 20q region was compared to signals from the 20cen region.
Microarrays
Microarray analysis was done as described previously (Duffy et al., 2003) using Affymetrix HuFL arrays. Data were initially analyzed using Affymetrix Microarray Suite software. The data in CEL format (Affymetrix Protocol) were then normalized and analyzed by using dChip version 1.3 software (Harvard) (Li and Wong, 2001). This program allows an assessment of functionally significant gene clusters using GeneOntology terms, as well as an assessment of significant clusters of genes that map to specific chromosome locations (see http://www.biostat.harvard.edu/complab/dchip/clustering.htm for a detailed explanation of how significance is established). P values smaller than 0.001 are considered significant. Only those genes that were upregulated by 3-fold or more in E7-expressing cells were included in the clustering analysis.
Acknowledgments
We are grateful to Kevin Ault, Mike Welsh, and Joseph Zabner for procurement of foreskin and airway tissue and Phil Karp for his extensive help with culturing of airway cells. We thank Kevin Knudtson of The University of Iowa DNA Core for assistance with microarrays and William Hahn, Robert Weinberg, and Denise Galloway for retroviral constructs. We are also grateful to the other members of the Klingelhutz laboratory for helpful discussion. This work was supported by NIA grant AG18265 (AJK). BWD was supported in part through a training grant from the NHLBI, T32 HL07638, and the University of Iowa Medical Scientist Training Program, T32 GM07337.
References
- Braakhuis BJ, Snijders PJ, Keune WJ, Meijer CJ, Ruijter-Schippers HJ, Leemans CR, Brakenhoff RH. Genetic patterns in head and neck cancers that contain or lack transcriptionally active human papillomavirus. J Natl Cancer Inst. 2004;96:998–1006. doi: 10.1093/jnci/djh183. [DOI] [PubMed] [Google Scholar]
- Collins C, Rommens JM, Kowbel D, Godfrey T, Tanner M, Hwang SI, Polikoff D, Nonet G, Cochran J, Myambo K, Jay KE, Froula J, Cloutier T, Kuo WL, Yaswen P, Dairkee S, Giovanola J, Hutchinson GB, Isola J, Kallioniemi OP, Palazzolo M, Martin C, Ericsson C, Pinkel D, Gray JW, et al. Positional cloning of ZNF217 and NABC1: genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc Natl Acad Sci USA. 1998;95:8703–8708. doi: 10.1073/pnas.95.15.8703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthill S, Agarwal P, Sarkar S, Savelieva E, Reznikoff CA. Dominant genetic alterations in immortalization: role for 20q gain. Genes Chromosomes Cancer. 1999;26:304–311. [PubMed] [Google Scholar]
- Duensing S, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075–7082. [PubMed] [Google Scholar]
- Duensing S, Munger K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J Virol. 2003;77:12331–12335. doi: 10.1128/JVI.77.22.12331-12335.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duensing S, Duensing A, Flores ER, Do A, Lambert PF, Munger K. Centrosome abnormalities and genomic instability by episomal expression of human papillomavirus type 16 in raft cultures of human keratinocytes. J Virol. 2001;75:7712–7716. doi: 10.1128/JVI.75.16.7712-7716.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy CL, Phillips SL, Klingelhutz AJ. Microarray analysis identifies differentiation associated genes regulated by HPV-16 E6. Virology. 2003;314:196–205. doi: 10.1016/s0042-6822(03)00390-8. [DOI] [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–937. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- Farwell DG, Shera KA, Koop JI, Bonnet GA, Matthews CP, Reuther GW, Coltrera MD, McDougall JK, Klingelhutz AJ. Genetic and epigenetic changes in human epithelial cells immortalized by telomerase. Am J Pathol. 2000;156:1537–1547. doi: 10.1016/S0002-9440(10)65025-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi S, Munoz N, Bosch XF, Snijders PJ, Walboomers JM. Human papillomavirus and cancers of the upper aerodigestive tract: a review of epidemiological and experimental evidence. Cancer Epidemiol Biomarkers Prev. 1996;5:567–575. [PubMed] [Google Scholar]
- Gillison ML, Koch WM, Capone RB, Spafford M, Westra WH, Wu L, Zahurak ML, Daniel RW, Viglione M, Symer DE, Shah KV, Sidransky D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J Natl Cancer Inst. 2000;92:709–720. doi: 10.1093/jnci/92.9.709. [DOI] [PubMed] [Google Scholar]
- Gollin SM. Chromosomal alterations in squamous cell carcinomas of the head and neck: window to the biology of disease. Head Neck. 2001;23:238–253. doi: 10.1002/1097-0347(200103)23:3<238::aid-hed1025>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Guo Z, Linn JF, Wu G, Anzick SL, Eisenberger CF, Halachmi S, Cohen Y, Fomenkov A, Hoque MO, Okami K, Steiner G, Engles JM, Osada M, Moon C, Ratovitski E, Trent JM, Meltzer PS, Westra WH, Kiemeney LA, Schoenberg MP, Sidransky D, Trink B. CDC91L1 (PIG-U) is a newly discovered oncogene in human bladder cancer. Nat Med. 2004;10:374–381. doi: 10.1038/nm1010. [DOI] [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- Hodgson JG, Chin K, Collins C, Gray JW. Genome amplification of chromosome 20 in breast cancer. Breast Cancer Res Treat. 2003;78:337–345. doi: 10.1023/a:1023085825042. [DOI] [PubMed] [Google Scholar]
- Howley PM. Role of human papillomavirus in human cancer. Cancer Res. 1991;51:5019–5022. [Google Scholar]
- Jin Y, Zhang H, Tsao SW, Jin C, Lv M, Strombeck B, Wiegant J, Wan TS, Yuen PW, Kwong YL. Cytogenetic and molecular genetic characterization of immortalized human ovarian surface epithelial cell lines: consistent loss of chromosome 13 and amplification of chromosome 20. Gynecol Oncol. 2004;92:183–191. doi: 10.1016/j.ygyno.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Karp PH, Moninger TO, Weber GF, Nesselhauf TS, Launsbach J, Zabner J, Welsh M. Developing an in vitro model of differentiated human airway epithelia: methods for establishing primary cultures. In: Wise TM, Totowa NJ, editors. Epithelial Cell Culture Protocols. Humana; 2002. pp. 115–137. [DOI] [PubMed] [Google Scholar]
- Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- Klingelhutz AJ, Barber SA, Smith PP, Dyer K, McDougall JK. Restoration of telomeres in human papillomavirus-immortalized human anogenital epithelial cells. Mol Cell Biol. 1994;14:961–969. doi: 10.1128/mcb.14.2.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingelhutz AJ, Foster SA, McDougall JK. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature. 1996;380:79–82. doi: 10.1038/380079a0. [DOI] [PubMed] [Google Scholar]
- Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci USA. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macoska JA, Beheshti B, Rhim JS, Hukku B, Lehr J, Pienta KJ, Squire JA. Genetic characterization of immortalized human prostate epithelial cell cultures. Evidence for structural rearrangements of chromosome 8 and i(8q) chromosome formation in primary tumor-derived cells. Cancer Genet Cytogenet. 2000;120:50–57. doi: 10.1016/s0165-4608(99)00248-4. [DOI] [PubMed] [Google Scholar]
- Piao CQ, Liu L, Zhao YL, Balajee AS, Suzuki M, Hei TK. Immortalization of human small airway epithelial cells by ectopic expression of telomerase. Carcinogenesis. 2005;26:725–731. doi: 10.1093/carcin/bgi016. [DOI] [PubMed] [Google Scholar]
- Plug-Demaggio AW, McDougall JK. The human papillomavirus type 16 E6 oncogene induces premature mitotic chromosome segregation. Oncogene. 2002;21:7507–7513. doi: 10.1038/sj.onc.1205903. [DOI] [PubMed] [Google Scholar]
- Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- Reshmi SC, Saunders WS, Kudla DM, Ragin CR, Gollin SM. Chromosomal instability and marker chromosome evolution in oral squamous cell carcinoma. Genes Chromosomes Cancer. 2004;41:38–46. doi: 10.1002/gcc.20064. [DOI] [PubMed] [Google Scholar]
- Savelieva E, Belair CD, Newton MA, DeVries S, Gray JW, Waldman F, Reznikoff CA. 20q gain associates with immortalization: 20q13.2 amplification correlates with genome instability in human papillomavirus 16 E7 transformed human uroepithelial cells. Oncogene. 1997;14:551–560. doi: 10.1038/sj.onc.1200868. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Southern SA, Lewis MH, Herrington CS. Induction of tetrasomy by human papillomavirus type 16 E7 protein is independent of pRb binding and disruption of differentiation. Br J Cancer. 2004;90:1949–1954. doi: 10.1038/sj.bjc.6601827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprague D, Phillips S, Mitchell C, Berger K, Lace M, Turek L, Klingelhutz A. Telomerase activation in cervical keratinocytes containing stably replicating human papillomavirus type 16 episomes. Virology. 2002;301:247. doi: 10.1006/viro.2002.1542. [DOI] [PubMed] [Google Scholar]
- van Houten VM, Snijders PJ, van den Brekel MW, Kummer JA, Meijer CJ, van Leeuwen B, Denkers F, Smeele LE, Snow GB, Brakenhoff RH. Biological evidence that human papillomaviruses are etiologically involved in a subgroup of head and neck squamous cell carcinomas. Int J Cancer. 2001;93:232–235. doi: 10.1002/ijc.1313. [DOI] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189:12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Wei W, Jobling WA, Chen W, Hahn WC, Sedivy JM. Abolition of cyclin-dependent kinase inhibitor p16INK4a and p21Cip1/Waf1 functions permits Ras-induced anchorage-independent growth in telomerase-immortalized human fibroblasts. Mol Cell Biol. 2003;23:2859–2870. doi: 10.1128/MCB.23.8.2859-2870.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiest T, Schwarz E, Enders C, Flechtenmacher C, Bosch FX. Involvement of intact HPV16 E6/E7 gene expression in head and neck cancers with unaltered p53 status and perturbed pRb cell cycle control. Oncogene. 2002;21:1510–1517. doi: 10.1038/sj.onc.1205214. [DOI] [PubMed] [Google Scholar]
- Zabner J, Karp P, Seiler M, Phillips SL, Mitchell CJ, Saavedra M, Welsh M, Klingelhutz AJ. Development of cystic fibrosis and noncystic fibrosis airway cell lines. Am J Physiol: Lung Cell Mol Physiol. 2003;284:L844–L854. doi: 10.1152/ajplung.00355.2002. [DOI] [PubMed] [Google Scholar]