

Abstract
The Fab-7 boundary is required to ensure that the iab-6 and iab-7 cis-regulatory domains in the Drosophila Bithorax complex can function autonomously. Though Fab-7 functions as a boundary from early embryogenesis through to the adult stage, this constitutive boundary activity depends on subelements whose activity is developmentally restricted. In the studies reported here, we have identified a factor, called early boundary activity (Elba), that confers Fab-7 boundary activity during early embryogenesis. The Elba factor binds to a recognition sequence within a Fab-7 subelement that has enhancer-blocking activity during early embryogenesis, but not during mid-embryogenesis or in the adult. We found that the Elba factor is present in early embryos but largely disappears during mid-embryogenesis. We show that mutations in the Elba recognition sequence that eliminate Elba binding in nuclear extracts disrupt the early boundary activity of the Fab-7 subelement. Conversely, we find that early boundary activity can be reconstituted by multimerizing the Elba recognition site.
Eukaryotic chromosomes are subdivided into a series of functionally and structurally autonomous domains by special elements called boundaries or insulators (15, 17, 27). These elements define the limits of chromosomal domains and function to establish independent units of gene activity (5, 30). They do so by preventing enhancers or silencers in one domain from influencing the activities of genes or other regulatory elements located in adjacent domains. Elements that have these properties have been found in a diverse array of organisms ranging from yeast to humans.
One of the first insulators identified was the constitutive nuclease-hypersensitive site 5′HS4 at the 5′ end of chicken β-globin locus (3). Like insulators in other organisms, 5′HS4 can block an enhancer from activating a reporter gene when interposed between the enhancer and the reporter gene promoter, but it has no effect on enhancer activity when placed distally to the enhancer. The enhancer-blocking activity of the 5′HS4 element depends on the zinc finger protein CTCF (CCCTC-binding factor) (2). There is a single CTCF site in the 5′HS4 element, and this site is both necessary and sufficient for its insulator activity. CTCF sites have subsequently been found in many other vertebrate boundary elements. Moreover, recent studies on the distribution of CTCF in human fibroblast chromosomes indicate that the ∼13,000 in vivo binding sites for this protein bracket transcription units in a manner predicted by the domain model (16).
While most of the known boundary elements in vertebrates appear to depend solely on CTCF for their insulator activity, this is generally not true for Drosophila melanogaster. One known exception in flies is the su(Hw) (suppressor of Hairy wing) insulator from the gypsy transposon (7). The boundary activity of this insulator depends on ∼12 binding sites for the su(Hw) protein (8). Though several endogenous su(Hw) insulators have now been identified (9, 24, 25), these have only one or a few su(Hw) binding sites, and their boundary activities appear to require the activities of other DNA binding proteins. Thus far, six different DNA binding proteins plus several accessory factors have been shown to be important for the boundary activities of the different Drosophila insulators. The DNA binding proteins include the fly CTCF homolog (22), su(Hw), CP190 (23), BEAF (31), Zw5 (6), and the GAGA factor (28).
Both the fly CTCF protein and the GAGA factor have been implicated in the functioning of boundary elements from the Drosophila bithorax complex (BX-C). Boundary elements in BX-C subdivide the complex into a series of functionally autonomous cis-regulatory domains (19). There are nine BX-C cis-regulatory domains, (abx/bx, bxd/pbx, iab-2, iab-3, iab-4, iab-5, iab-6, iab-7, and iab-8), and each is responsible for driving the expression of one of the three homeotic genes in the complex (Ultrabithorax [Ubx], abdominal-A [abd-A], and Abdominal-B [Abd-B]) in a specific parasegment (1, 18). For example, Abd-B expression in parasegments PS10, PS11, PS12, and PS13 is directed by the iab-5, iab-6, iab-7, and iab-8 cis-regulatory domains, respectively. The activity state of the cis-regulatory domains is set early in embryogenesis by the products of the segmentation genes. The segmentation genes are expressed only transiently in the embryo, and once their gene products disappear, maintaining the on/off state of the cis-regulatory domains depends on the action of the trithorax (trx-G) and Polycomb (Pc-G) group genes (26).
Mutations inactivating one of the BX-C cis-regulatory domains have a simple loss-of-function phenotype, transforming the parasegment specified by that domain into a copy of the parasegment immediately anterior. By contrast, mutations inactivating a boundary element have a complex mixture of both gain-of-function and loss-of-function phenotypes in the affected parasegment. A mixed phenotype is produced because boundary elements function to prevent adventitious interactions between positive and negative regulatory elements in adjacent domains. For example, the Fab-7 boundary separates the iab-6 (PS11) and iab-7 (PS12) cis-regulatory domains, and these two domains are fused into a single domain in Fab-7 mutants (4, 11). In some PS11 cells, positive elements in iab-6 ectopically activate iab-7, and these cells assume a PS12 identity. In other PS11 cells, negative elements in iab-7 block the normal activation of iab-6, and these cells assume a PS10 identity (conferred by iab-5). While the specification of PS11 is disrupted in the Fab-7 mutants, there is no effect on the specification of PS12 by iab-7, because Abd-B is appropriately activated in PS12 cells.
Mutational analysis and transgene experiments have localized the Fab-7 boundary to a ∼1.2-kb DNA segment that spans three nuclease-hypersensitive regions: *, HS1, and HS2 (Fig. 1) (12, 14, 20, 32). Just distal to the boundary is the Polycomb response element for the iab-7 cis-regulatory domain, iab-7 PRE (20, 21). This PRE has been mapped to HS3 (Fig. 1) and is functionally and physically distinct from the Fab-7 boundary. Though the full-length 1.2-kb Fab-7 boundary appears to be functional in all cells throughout the life cycle, this constitutive activity differs from other known fly boundaries in that it depends on subelements that are active only at specific points in development. This was first noticed in mutational studies on the Fab-7 GAGA sites (28). The largest hypersensitive region, HS1, has six binding sites for the GAGA factor, arranged in three pairs, GAGA1-2, GAGA3-4, and GAGA5-6, proximally to distally. Mutations in GAGA1-2 were found to weaken boundary activity in early embryos but to have no effect in mid-embryogenesis or in adults. Conversely, mutations in GAGA3-4 have little effect on embryos but weaken boundary activity in adults.
FIG. 1.

Map of the Fab-7 region and the probes used for EMSA. A schematic structure of the D. melanogaster Fab-7 region is shown at the top. Four DNase I-hypersensitive regions, HS1, HS2, HS3, and the minor region (asterisked), are shown as shaded boxes. The open box represents a 93-bp “high”-homology region that is conserved among Drosophila species. The binding sites for GAGA factor (Trithorax-like) are shown as dark (GAGAG) or light (GAGAA) shaded ovals. The structure of the pHS1 region is shown enlarged below. The name and location of each of the probes from pHS1 used in EMSA are given at the bottom.
Stage-specific effects were also evident when sequences from HS1 were multimerized and tested in enhancer-blocking assays. For example, when 4 copies of a 236-bp fragment (pHS1) extending from the proximal edge of HS1 past the GAGA1-2 pair (Fig. 1) were placed between an hsp70-LacZ reporter and the fushi-tarazu (ftz) UPS (upstream) and NE (neurogenic) enhancers, strong boundary activity was observed in early embryos and there was little or no UPS-dependent stripe expression of β-galactosidase (29). In contrast, blocking of the NE enhancer in the central nervous system (CNS) during mid-embryogenesis was weak and position dependent. In adult flies, the pHS1×4 multimer had no detectable boundary activity, and in transgene assays with a mini-white reporter, it was unable to block the white enhancer from activating white expression in the eye and testes. A different result was obtained with 4 copies of a 198-bp fragment, dHS1A (Fig. 1), from near the middle of HS1. This multimer had little effect on the ftz UPS and NE enhancers in the embryo, while in adults it had even stronger blocking activity than the full-length Fab-7 boundary. Finally, there are two Fab-7 mutations that delete all of HS2 and the distal half of HS1 including GAGA sites 3 to 6. These deletions differ from larger deletions that remove all of HS1 in that they have boundary activity in early embryos. As predicted by the transgene assays, this boundary activity is transient and largely disappears by mid-embryogenesis. Moreover, the early boundary activity of the larger of the two deletion constructs is sensitive to a twofold reduction in the dose of the GAGA gene Trithorax-like (Trl).
While our previous studies indicated that GAGA is required for the boundary activity of the different Fab-7 subelements, it seemed unlikely that this protein was the primary factor responsible for their stage-specific boundary activity. Besides the fact that GAGA has been implicated in the functioning of other types of cis elements including promoters and PREs, the GAGA protein by itself does not appear to have insulator activity (28). Additionally, though the two major GAGA isoforms are expressed differently during development (10), their expression patterns would not be consistent with a stage-specific function in Fab-7. These observations suggest that GAGA most likely functions either in the generation of a nucleosome-free region of chromatin or as a scaffold for the assembly of the stage-specific factors. If this suggestion is correct, then other factors must be responsible for generating the developmentally restricted boundary activity of the different Fab-7 subelements. In the studies reported here, we have searched for sequences and factors that confer Fab-7 boundary activity in the early embryo. We have identified a sequence in pHS1 that is both necessary and sufficient for the early boundary activity of this Fab-7 subelement. We also show that this sequence is specifically recognized by a DNA binding factor that exhibits a developmental activity profile which resembles that observed for pHS1 boundary activity.
MATERIALS AND METHODS
Preparation of nuclear extracts from embryos.
Oregon R embryos were collected from apple juice plates kept for 6 h or 12 h in a population cage. For 6- to 12-h embryos, 0- to 6-h embryos were aged for 6 h on apple juice plates at room temperature. Collected embryos were dechorionated with 50% bleach (2.6% sodium hypochloride) for 3 min, washed first with 0.12 M NaCl-0.04% Triton X-100 and then with 0.12 M NaCl, and stored at −80°C until extraction. All extraction steps were processed at 4°C. About 10 g of frozen embryos was placed in a 40-ml Potter homogenizer (Wheaton Scientific, Millville, NJ) and homogenized with pestle A (loose) for 10 strokes in 30 ml homogenization buffer (HB; 3.75 mM Tris-HCl [pH 7.4], 0.5 mM EDTA-KOH [pH 7.4], 20 mM KCl, 0.05 mM spermine, 0.125 mM spermidine, 0.5% 2,2′-thiodiethanol, 2 μg/ml aprotinin, 0.1 mM phenylmethylsulfonyl fluoride [PMSF], 0.1% digitonin). The lysate was then filtered through two layers of Miracloth (Calbiochem, La Jolla, CA) and centrifuged at 1,900 × g for 5 min using a swing bucket rotor in a Beckman TJ-6 centrifuge at 4°C. The resulting nuclear pellet was washed with 40 ml of HB four times and resuspended in 2 ml of nuclear extraction buffer 20 (NEB 20; 10 mM HEPES-KOH [pH 7.4], 20 mM KCl, 3 mM MgCl2, 0.1 mM EDTA, 10% glycerol, 1 mM dithiothreitol, 0.2 mM PMSF, 2 μg/ml aprotinin). The nuclear suspension was moved to a polyallomer ultracentrifuge tube (Sarstedt 65-90219), and an equal volume of NEB 700 (the same as NEB 20 except that the KCl concentration was 700 mM) was added to the suspension so that the final concentration of KCl would be 360 mM. After a 30-min incubation at 4°C, the sample was centrifuged at 150,000 × g (average) at 4°C for 1 h by using a Beckman SW50.1 swing rotor. The resulting supernatant was dialyzed against dialysis buffer (10 mM HEPES [pH 7.4], 250 mM KCl, 0.1 mM EDTA-KOH, 10% glycerol, 1 mM 2-mercaptoethanol, 0.2 mM PMSF, 2 μg/ml aprotinin) at 4°C for 10 to 12 h. The sample was recovered and divided into aliquots for long-term storage at −80°C. Protein concentrations were measured by the Bradford method (Bio-Rad protein assay) using bovine serum albumin for the standard curve.
Probes.
Probes pHS1A, pHS1B, 1, 2, 5, 6, 1Δ, and 2Δ were obtained from PCRs using a 3.35-kb fragment including Fab-7 inserted into pBluescript as the template. Primers were used in the following combinations: for probe 1, SES23 and SES25; for probe 2, SES26 and SES27; for probe pHS1A, SES23 and SES27; for probe pHS1B, SES28 and SES24; for probe 5, SES28 and SESA02; for probe 6, SESA01 and SES24; for probe 1Δ, SES23 and SESA04; and for probe 2Δ, SESA03 and SES27. The sequences of these primers are as follows: SES23, GTGGCAAAAGCTGGCAAAG; SES25, CTCAAAAGTCCTGCGCAAG; SES26, CTTGCGCAGGACTTTTGAG; SES27, CTTTCCCCCGCCACACAGC; SES28, GCTGTGTGGCGGGGGAAAG; SES24, GCGTTGATATGCCCCAATG; SESA02, CGTGACAGCTGCCATTTG; SESA01, CAAATGGCAGCTGTCACGG; SESA04, TTTCGCTGCTTTGGGGA; SESA03, ATTCTATTAAATTCTAAC. After PCRs, products were purified on a 3% agarose-1× Tris-borate-EDTA (TBE) gel. Probes recovered from the gels were diluted to make stock solutions at a final concentration of 0.5 pmol/μl.
Other probes were created from the synthesized DNAs by annealing two complementary strands. For example, probes 12, 5-1, and 5-5 were derived from the combinations of SES25 with SES26, SES27 with SES28, and SESA01 with SESA02, respectively. Other synthetic probes within region 5 and probe 5-4 derivatives are shown in Fig. 4A and Fig. 7. The precise sequences of the synthesized DNAs for these probes are available upon request. Probe ICD2, an 87-bp fragment from a part of Fab-8, was also a synthetic probe. The top-strand (5′ [proximal]-to-3′ [distal]) sequence of ICD2 is CTCCGACAGTGGACATGTCGCGTAAAAAATGTTCGATAACTTTCAATGGTTCGATTGAACAGACAATAAGTGTATTTAAGACACCAG. The top-strand sequences for the putative Elba consensus probes used in Fig. 8C are as follows: Fab-3 CS1, GGCCACGCCCACTTATTGGCACTGCCGATC; MCP CS1, GAGACAACAGGCTTATTGATGTAGTCTTCC; Fab-7 CS2, ATATCAACGCGCCAAAAAGAAAAACAAAAA; Fab-8 CS1, TGGTTGGTCTGCATATTGGAGGGAATTTTC; Fab-8 CS2, CGATTGAACAGACAATAAGTGTATTTAAGA. (The 8 bases of the putative Elba recognition sequences are underlined.)
FIG. 4.

Localization of the binding sequence for activity b. (A) Probes used for mapping the binding sequence for activity b. The sequences of both strands of probe 5 are shown at the top. Binding site 1 for GAGA factor is boxed. The positions of the overlapping smaller DNAs 5-1 to 5-5 are indicated by lines below the probe 5 sequence. For the 3-base mutations in probe 5-4, the “top”-strand (5′ [proximal] to 3′ [distal]) sequence of DNA 5-4 [5-4(T)] and the corresponding bases of mutations in mutant probes are shown. Each base alteration was introduced so that a purine was converted to a pyrimidine and A/T and C/G were interconverted. (B) Activity b recognizes sequences included in the 5-4 region of probe 5. The EMSA experiment was performed as described for Fig. 2B except that DNA subfragments spanning probe 5 were used as cold competitors. End-labeled probe 5 DNA was incubated with (lanes 2 to 16) or without (lane 1) 15 μg (protein) of nuclear extracts derived from 0- to 12-h embryos in the absence (lanes 1 and 2) or presence (lanes 3 to 18) of cold competitor DNAs (given above the lanes). Either a 50-fold (left lane of each set) or a 100-fold (right lane of each set) excess of cold competitor DNA (lanes 3 to 16) was used for the competition experiments. The competitor used in lanes 17 and 18 was a 100-fold excess of single-strand 5-4 DNA. T and B represent the “top” strand (5′ [proximal] to 3′ [distal]) and the “bottom” strand (5′ [distal] to 3′ [proximal]) of DNA 5-4, respectively. The positions of the band for activity b and the free probe are indicated by a solid arrowhead and the letter F, respectively. (C) Mapping of bases critical for the binding of activity b. The EMSA experiment was performed as described for panel B except that 5-4 DNA fragments containing different 3-base mutations were used as the cold competitors. Labeled probe 5 was incubated with (lanes 2 to 18) or without (lane 1) 15 μg (protein) of nuclear extracts from 0- to 12-h embryos in the absence (lanes 1 and 2) or presence (lanes 3 to 18) of unlabeled competitor probes (given above the lanes). Either a 50-fold (left lane of each set) or a 100-fold (right lane of each set) excess of unlabeled DNA (lanes 3 to 18) was used for each competition. 5-4 wt, wild-type 5-4 probe; mut1, the 5-4 probe with mutation 1, which has the 3-base alteration in the 5-4 sequence shown in panel A. (D) EMSA with mutant probes support the results of the competition experiments. 5-4 wt and mut1 to mut5 were end labeled and used in the EMSA experiments as described above. The labeled probes are shown above the lanes. Nuclear extracts of 15 μg (protein) from 0- to 12-h embryos were used except in lanes 1, 4, 8, 12, 16, and 20. In the competition experiments with wild-type or mutant 5-4 DNAs, a 100-fold excess of unlabeled DNA was used as indicated above the individual lanes.
FIG. 7.

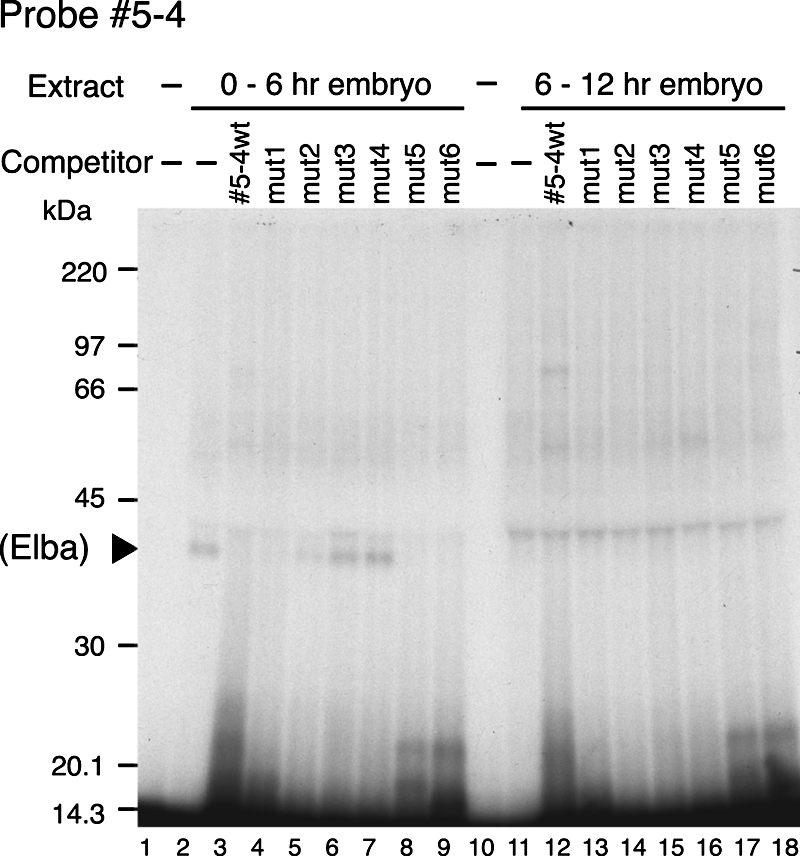
Mapping of the Elba recognition sequence in probe 5-4. Labeled 5-4 DNA was incubated with nuclear extracts from 0- to 12-h embryos in the presence of a 100-fold excess of cold competitor DNA as indicated. DNAs used as cold competitors are diagramed below. The sequence for the top (T) strand (5′ [proximal] to 3′ [distal]) of probe 5-4 is shown. For 3-bp mutations, the changes in the DNA sequence are given below the corresponding bases of 5-4. The rule for changing bases was the same as that described in the legend to Fig. 4. Bold lines delineate the positions of the truncated DNAs. The 8-bp core recognition sequence and the flanking sequences in the top strand of probe 5-4 that contribute to efficient binding in the EMSA experiment are bracketed.
FIG. 8.

The Elba recognition sequence is conserved in other Drosophila species and is present elsewhere in D. melanogaster BX-C. (A) The Elba recognition sequence is conserved in the Fab-7 boundaries of other Drosophila species. The Fab-7 sequences of D. melanogaster and 11 other Drosophila species, available at www.flybase.org, were collected and piled up with the ClustalW program to align the high-homology region conserved among the species. To refine the alignment in the region containing the Elba recognition sequence, a smaller DNA sequence of 120 to 150 bp was piled up again. This procedure aligned the 8-bp Elba recognition sequence (boldfaced and boxed) better in the different species. Only a part of the aligned Fab-7 sequence is shown. The sequence of the putative Elba site in D. mojavensis, which differs in 2 bases (italicized) from that of D. melanogaster, is at the bottom. The bases conserved among the other 11 species are asterisked. The number in parentheses on the left of each sequence represents the distance (in bases) from the “high”-homology region of each sequence. Numbers elsewhere are the numbers of bases omitted in the sequence. Potential binding sites for the GAGA factor (Trithorax-like) are underlined. (B) Distribution of Elba consensus sequences in the BX-C region. About 330 kb of the bithorax complex region is shown as a horizontal line in the center. The cis-regulatory regions of BX-C are indicated on the top. Positions of 13 matches (CS) to the 8-bp core sequence in pHS1 are indicated above the horizontal line. CS10 (Fab-7 CS1) is the Elba recognition sequence in Fab-7. At the bottom, the three homeotic transcription units in BX-C are represented by combinations of thin lines (introns) and thick lines (exons). The arrow indicates the orientation of each transcription. (C) Elba can also bind to sites in Fab-3 and Fab-8. Several of the potential Elba recognition sites (precise matches and sites that differ at a single base) in BX-C were tested in EMSA competition experiments as described for preceding figures. Labeled probe 5-4 was incubated with (lanes 2 to 20) or without (lane 1) 15 μg of 0- to 12-h nuclear extracts in the absence (lanes 1 and 2) or presence (lanes 3 to 20) of the competitor DNAs indicated. The conditions of EMSA experiments were same as those described in Fig. 2. Either a 50-fold (left lane of each set) or a 100-fold (right lane of each set) excess of cold DNA was used in the competition experiments. Fab-3 CS1 (CS8) has a precise match to the 8-bp core recognition sequence in pHS1. Other competitors are probes from known BX-C boundaries that have sequences resembling the Elba 8-bp core recognition sequence in pHS1. Their sequences are as follows: MCP CS1, TCAATAAG; Fab-7 CS2, CCAAAAAG; Fab-8 CS1, CCAATATG; Fab-8 CS2, ACAATAAG. The three mutant probes from Fig. 4C, mut1, mut2, and mut3, were also tested for comparison with the different BX-C probes.
Electrophoretic mobility shift assays (EMSA).
In the labeling reaction, 1 pmol of probe was phosphorylated using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (crude product of 259 TBq/mmol; MP Biomedicals) in 50 μl of 1× T4 polynucleotide kinase buffer. After a 45-min incubation at 37°C, the probe was separated from nonreacted ATP using a Sephadex G-50 fine gel (Amersham Biosciences). The volume of eluted probe was adjusted to 100 μl by adding water so that the final concentration of the probe would be 10 fmol/μl.
For the binding reactions with a volume of 20 μl, 15 μg (as protein) of nuclear extracts or an equal volume of dialysis buffer was mixed with 0.5 μl (5 fmol) of labeled probe in the following buffer: 25 mM Tris-Cl (pH 7.4), 100 mM KCl, 1 mM EDTA, 0.1 mM dithiothreitol, 0.1 mM PMSF, 0.3 mg/ml bovine serum albumin, 10% glycerol, 0.25 mg/ml poly(dI-dC)/poly(dI-dC). In some samples, unlabeled competitor DNA was included so that the final concentration of the competitor would be in either a 50-fold (0.25 pmol) or a 100-fold (0.5 pmol) excess. After a 30-min incubation at room temperature, the samples were loaded onto a 4% acrylamide (mono/bis, 29:1)-0.5× TBE-2.5% glycerol slab gel. Electrophoresis was performed at 4°C and 180 V for 3 to 4 h using 0.5× TBE-2.5% glycerol as a running buffer. Gels were dried and exposed to BioMax MR film (Kodak).
UV cross-linking experiment.
Samples including labeled probe 5-4 were prepared as for the EMSA experiments except that half the final volume of the reaction mixture (10 μl total) was used and the mixture included 20 μg/sample of nuclear extract and 0.025 mg/ml of poly(dI-dC)/poly(dI-dC). After a 30-min incubation at room temperature, the samples were exposed to UV irradiation in a UV Stratalinker 1800 (Stratagene) for 10 min with the lids of the tubes open. Samples were mixed with 5 μl of 3× Laemmli's sample buffer, incubated in boiled water for 3 min, and loaded onto a 10% acrylamide-sodium dodecyl sulfate (SDS) gel. After SDS-polyacrylamide gel electrophoresis, the separating gel was dried and exposed to BioMax MR film.
P-element transgenes.
For the “pHS1mut3×4” construct, mutation 3 was introduced into a single pHS1 fragment cloned into the XhoI-SalI sites of pBluescript (29) using Kunkel's method. (The oligonucleotide DNA sequence used for introducing the mutation is available upon request.) The resulting fragment was cut out with XhoI and SalI and multimerized by ligation in the presence of XhoI and SalI to obtain the tandem repeats of the fragment. The 4-copy repeat fragment was purified from the agarose gel and introduced into pBluescript. The fragment was cut with XhoI and NotI and inserted between the ftz enhancer and the hsp70 promoter of vector pCfhL (12). For the “binding site×8” construct, the 8-copy tandem repeat of the binding site was created from synthesized DNA. A pair of 36-bp oligonucleotide DNAs was designed so that the 30 bp of the binding site would have a SalI site and a XhoI site on either end. The top-strand sequence was TCGACAAGTGCAGCGCCCAATAAGCAAATGGCAGCC, and the bottom-strand sequence was TCGAGGCTGCCATTTGCTTATTGGGCGCTGCACTTG. The oligonucleotide DNAs were annealed and then phosphorylated with T4 polynucleotide kinase. Oligomerization of the fragment and subcloning into pCfhL were performed as described above.
Staining of embryos.
Embryos (0 to 12 h old) from the various transgenic lines were collected and stained in 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) solution as described by Hagstrom et al. (12). Embryos homozygous for the transgene were stained for about 12 h. Lines heterozygous for the transgene (a single copy of the transgene) were stained for 20 to 24 h. Heat shock induction of β-galactosidase was tested for all the “binding site×8” lines to verify that the reduction of expression was not due to silencing of the hsp70-LacZ gene. The embryos were collected in a mesh cup and placed on a prewarmed, water-covered apple juice plate at 37°C for 1 h. The staining was stopped within 2 h in the heat shock experiments.
Genomic sequence analysis.
The Fab-7 sequence of D. melanogaster was downloaded from FlyBase (www.flybase.org). The orthologs of Fab-7 in other Drosophila species were obtained by a reciprocal BLAST search of the genomic sequences starting from the sequence of D. melanogaster. The sequences obtained were aligned with the ClustalW program either by downloading the program from the European Bioinformatics Institute (www.ebi.ac.uk/clustalw) or by using the Web application at GenomeNet (align.genome.jp).
RESULTS
Two major and multiple minor pHS1 DNA-binding activities are observed in EMSA.
In our previous studies, we subdivided the 236-bp pHS1 fragment into two smaller proximal and distal fragments: pHS1A, which contains a ∼90-bp region that is conserved in distant Drosophila species, and pHS1B, which contains the GAGA1-2 pair (Fig. 1) (29). We found that pHS1A had little or no boundary activity in the ftz:hsp70-LacZ enhancer blocking assay. In contrast, pHS1B resembled the larger pHS1 fragment in that it blocked the UPS stripe enhancer in early embryos but showed little blocking of the NE neurogenic enhancer in older embryos. However, unlike that of pHS1, the UPS-blocking activity of pHS1B was quite sensitive to chromosomal position effects, and no blocking was observed in about half of the transgenic lines. Taken together, these findings suggested that pHS1B contains the key cis-acting elements for early blocking activity, while elements in pHS1A play a secondary or supporting role.
To identify potential stage-specific boundary factors, we subdivided pHS1 into four overlapping fragments, probes 1, 2, 5, and 6, and used them in EMSA with nuclear extracts prepared from 0- to 12-h embryos. Probes 1 and 2 cover pHS1A, while probes 5 and 6 cover pHS1B. When labeled probe 1 was used, one major (a) and several minor (a′) shifted bands were observed (Fig. 2A, lane 2). All of these bands were competed by excess unlabeled probe 1 (Fig. 2A, lanes 3 and 4) but were not competed by the unrelated pHS1B (lanes 7 and 8) or by a control fragment, ICD2, that is derived from the Fab-8 boundary (lanes 11 and 12). Surprisingly, unlabeled probe 2 also competed the binding activities to the same extent as probe 1 (Fig. 2A, lanes 5 and 6). Since probes 1 and 2 overlap, we suspected that the binding activities were recognizing motifs in the 19-bp common sequence. This seems to be the case. Probes 1Δ and 2Δ, which lack the 19-bp overlap, did not compete (Fig. 2A, lanes 13 to 16), while an unlabeled 19-bp double-stranded oligonucleotide corresponding to the overlapping sequence competed all the specific bands (lanes 9 and 10). Taken together, these findings map the specific binding activities seen with probe 1 to the overlapping 19-bp sequence. This conclusion was confirmed by using labeled probe 2, which gave exactly the same results as probe 1 (data not shown).
FIG. 2.

Multiple binding activities to the pHS1 region are observed in 0- to 12-h embryo extracts by EMSA. (A) EMSA with labeled probe 1. Probe 1 DNA was 5′ end labeled with 32P and incubated with (lanes 2 to 16) or without (lane 1) 15 μg (protein amount) of nuclear extracts derived from 0- to 12-h embryos in the absence (lanes 1 and 2) or presence (lanes 3 to 16) of unlabeled cold competitor probes as indicated above the lanes. The competitor ICD2 is an 87-bp fragment from Fab-8, used as a heterologous control DNA. Either a 50-fold (left lane of each set) or a 100-fold (right lane of each set) excess of the cold competitor was added to the reaction mixture. After a 30-min incubation at room temperature, the samples were electrophoresed on a 4% acrylamide-0.5× TBE-2.5% glycerol gel. The identity of the shifted band is indicated by a solid arrowhead or half-parentheses on the right. The letter F represents the position of “free”’ (unbound) probe. (B) EMSA with probe 5. End-labeled probe 5 DNA was incubated with (lanes 2 to 12) or without (lane 1) 15 μg (protein amount) of nuclear extracts from 0- to 12-h embryos in the absence (lanes 1 and 2) or presence (lanes 3 to 12) of competitor probes as indicated. Other EMSA experimental conditions were the same as those described for panel A. The free probe and the identities of shifted bands are indicated as described for panel A. (C) EMSA with probe 6. Probe 6 was incubated with (lanes 2 to 7) or without (lane 1) 15 μg (protein amount) of nuclear extracts from 0- to 12-h embryos in the absence (lanes 1 and 2) or presence (lanes 3 to 7) of the competitor probes indicated. A 100-fold excess of unlabeled DNA was used for each competition.
We next tested probes 5 and 6 from the distal half of pHS1. For probe 5, one strong, slowly migrating shift (b) and several more rapidly migrating but weaker shifts (c) were detected (Fig. 2B, lane 2). As expected for a sequence-specific binding factor, the strong shift (b) was competed by unlabeled fragment 5 (Fig. 2B, lanes 3 and 4) and also by unlabeled pHS1B (which includes all of probe 5) (lanes 7 and 8). In contrast, the b shift was not competed by two heterologous fragments, probes 6 (Fig. 2B, lanes 5 and 6) and pHS1A (lanes 9 and 10). On the other hand, we did find that our control fragment, ICD2, from the Fab-8 boundary also competed with probe 5 (Fig. 2B, lanes 11 and 12). This would suggest that this Fab-8 fragment contains a recognition sequence for the factor that generates the b shift. The weaker, more rapidly migrating shifts (c) were also competed by probes 5 and pHS1B but not by probe 6 or pHS1A, showing that these shifts are specific for probe 5 sequences. However, unlike the stronger band b, the c shifts do not seem to be competed by the Fab-8 fragment ICD2. Thus, it is likely that the factors represented by the c shifts have different DNA recognition properties than factor b. Unlike the three other pHS1 probes, sequence-specific DNA binding activities were not detected with probe 6 under our conditions (Fig. 2C). While several bands are evident in the probe 6 EMSA (Fig. 2C, lane 2), they are not efficiently competed by any of the cold competitors, including probe 6 itself (lanes 3 to 7).
Stage-specific factors bind to different pHS1 sequences.
Both our transgene assays and the phenotype of incomplete Fab-7 HS1 deletions suggest that factors conferring early pHS1 boundary activity should be present from some point very early in embryogenesis through germ band extension (20, 29). Since pHS1 boundary activity was greatly reduced later in embryogenesis in germ band-retracted embryos, the factor(s) conferring early boundary activity should be present at only low levels or absent altogether in older embryos. To determine whether any of the factors detected in our EMSA experiments with different pHS1 probes have this activity profile, we prepared nuclear extracts from 0- to 6-h and 6- to 12-h embryos.
Figure 3A shows that the major binding activities, a and a′, detected with probe 1 in 0- to 12-h embryos are present only at very low levels in 0- to 6-h embryos (lanes 2 to 8), while they are much more abundant in 6- to 12-h embryos (lanes 10 to 16). In addition, we detected two new mobility shifts, a″ and a‴, in the 6- to 12-h sample that were not readily apparent in the 0- to 6-h sample. Based on the competition experiments, these two factors appear to have sequence specificities that are different from the factors that generate the a and a′ shifts. The developmental profile of these different binding activities suggests that they are unlikely to correspond to the factor(s) that confers early boundary activity on pHS1. This is not the case for one of the shifts observed with probe 5. As shown in Fig. 3B, the most strongly labeled band seen in 0- to 12-h embryos, b, is present in high yield in 0- to 6-h embryos (lane 2). Though it is also present in the 6- to 12-h nuclear extracts (Fig. 3B, lane 9), the yield of band b is greatly reduced from that for the 0- to 6-h sample. As observed in 0- to 12-h extracts, this band is specifically competed by probe 5 (Fig. 3B, lane 3), pHS1B (lane 5), and the ICD2 control (lane 7). While band b has the expected developmental profile for the early boundary activity, the collection of more rapidly migrating shifts (c) do not. The c shifts are present only at low levels in 0- to 6-h embryos, while they are comparatively enriched in 6- to 12-h embryos.
FIG. 3.

pHS1-binding activities change during embryonic development. Nuclear extracts were prepared from 0- to 6-h embryos or 6- to 12-h embryos and used in EMSA experiments as described in the legend to Fig. 2. (A) DNA binding proteins recognizing probe 1 are enriched in nuclear extracts from 6- to 12-h embryos. Nuclear extracts (15 μg protein) derived from 0- to 6-h embryos (lanes 2 to 8) or 6- to 12-h embryos (lanes 10 to 16), or buffer only (lanes 1 and 9), were incubated with labeled probe 1 in the absence (lanes 1, 2, 9, and 10) or presence (lanes 3 to 8 and 11 to 16) of a 100-fold excess of cold competitor DNA (indicated above each lane). Positions of shifted bands or free probe are shown as described for Fig. 2. (B) High levels of binding activity b are found in nuclear extracts from 0- to 6-h embryos, while only residual amounts of this activity are evident in nuclear extracts from 6- to 12-h embryos. Nuclear extracts (15 μg of protein) from 0- to 6-h embryos (lanes 2 to 7) or 6- to 12-h embryos (lanes 9 to 14), or buffer only (lanes 1 and 8), were incubated with labeled probe 5 in the absence (lanes 1, 2, 8, and 9) or presence (lanes 3 to 7 and 10 to 14) of a 100-fold excess of cold competitor DNA (indicated above the lanes). Positions of shifted bands or free probe are shown as described for Fig. 2.
Localizing the binding site for factor b in probe 5.
Since band b has the developmental profile expected for an early boundary factor, we decided to try to more precisely pinpoint the binding site for this activity within probe 5. For this purpose we used a series of overlapping 19- to 27-bp double-stranded DNAs (DNAs 5-1 to 5-5 in Fig. 4A) as cold competitors in the EMSA with nuclear extracts from 0- to 12-h embryos. As shown in Fig. 4B, four of the five DNAs (DNAs 5-1, 5-2, 5-3, and 5-5) did not compete with probe 5 for binding to factor b (lanes 7 to 12, 15, and 16). By contrast, double-stranded 5-4 DNA competed for factor b binding about as effectively as the full-length DNA 5 (Fig. 4B; compare lanes 13 and 14 with lanes 3 and 4), while it did not appear to compete for the binding of any of the c factors. These findings would map the b binding site to the 5-4 DNA sequence. Since no competition was seen when either the “top” or the “bottom” single-stranded 5-4 DNA was used as the cold competitor (Fig. 4B, lanes 17 and 18, respectively), it would appear that factor b recognizes double-stranded but not single-stranded DNA.
To further narrow down the factor b recognition site, we introduced a series of 3-bp mutations across the 5-4 DNA sequence (Fig. 4A) and used each of the mutant DNAs as a cold competitor in the gel shift assay. Three of six mutant DNAs, mut1, mut5, and mut6 (Fig. 4A), competed with probe 5 for factor b binding about as effectively as the wild type 5-4 DNA or full-length cold probe 5, and as shown in Fig. 4C (lanes 7, 8, and 15 to 18), they almost completely eliminated the early stage-specific b shift. Thus, these mutations do not disrupt the binding site for factor b. In contrast, only partial competition is observed with mut2, while mut3 and mut4 did not appear to compete at all. For these mutant DNAs, the yield of band b is close to that observed in the absence of added competitor. These findings suggest that sequences critical for recognition of the 5-4 DNA by factor b are altered in mut2, mut3, and mut4.
To confirm the results of the competition experiments, we used a gel shift assay to compare the ability of factor b to bind to the various mutant 5-4 DNAs with its binding to wild type 5-4 DNA. As expected from the competition experiments, mut1, mut5 (Fig. 4D), and mut6 (data not shown) gave shifts comparable to that seen for the wild-type 5-4 probe. These shifts were also competed by both cold wild-type and mutant DNA with roughly equal efficiency. Since mut3 and mut4 showed no evidence of binding to factor b in the competition experiments, we did not expect to observe a shift with either of these mutants. Figure 4D, lanes 12 to 19, shows that this is the case. mut2, in contrast, showed evidence of partial competition (Fig. 4C, lanes 9 and 10). Consistent with this finding, we detect a very weakly labeled band b in the mut2 EMSA (Fig. 4D, lanes 8 to 11). Moreover, the shift is more efficiently competed by cold wild-type 5-4 DNA than by mut2 (Fig. 4D, lanes 10 and 11, respectively). Taken together, these findings localize the recognition sequence for the putative early boundary activity to a 27-bp sequence in probe 5, sequence 5-4. Within this 27-bp sequence, the critical bases for binding likely include the 9 bases that are altered in mut2, mut3, and mut4.
A 40-kDa protein is specifically cross-linked to the factor b sequence.
We next used UV cross-linking in combination with competition experiments to further characterize the putative early boundary activity. In the experiment for which results are shown in Fig. 5, 32P-labeled probe 5-4 was incubated with nuclear extracts prepared from 0- to 6-h and 6- to 12-h embryos with or without different cold competitors under the conditions used for EMSA. We then cross-linked proteins bound to the labeled probe with UV irradiation for 10 min and analyzed the 32P-labeled proteins by electrophoresis on SDS-polyacrylamide gels and autoradiography. Based on the results of the EMSA, the putative early boundary factor b should be present in nuclear extracts from 0- to 6-h embryos, while it should be absent or greatly reduced in activity in extracts from 6- to 12-h embryos. As can be seen in Fig. 5, this is true for a band of ∼40 kDa. This protein species is cross-linked to the 5-4 DNA when it is incubated with nuclear extracts from 0- to 6-h embryos (Fig. 5, lane 2), while this band is greatly reduced in yield or absent when nuclear extracts from 6- to 12-h embryos are used for cross-linking (lane 11). There are also a number of additional weakly labeled bands in the 0- to 6-h sample (e.g., the more slowly migrating ∼43-kDa species); however, these bands are present in higher yield in the cross-linked proteins from 6- to 12-h nuclear extracts and would not be good candidates for the putative early boundary activity. The competition experiments also suggest that the ∼40-kDa protein corresponds to the putative early boundary factor b. Cross-linking to the ∼40-kDa protein is strongly suppressed by excess cold 5-4 DNA (Fig. 5, lane 3) and by 5-4 mutations that do not affect factor b binding in the EMSA (lanes 4, 8, and 9). By contrast, mutations 3 and 4, which eliminate factor b binding to 5-4 DNA in the EMSA, have little or no effect on the yield of the cross-linked 40-kDa protein (Fig. 5, lanes 6 and 7), while mut2, which weakens factor b binding in the EMSA, weakly suppresses cross-linking (lane 5).
FIG. 5.
A 40-kDa protein in nuclear extracts from 0- to 6-h embryos is UV cross-linked to probe 5-4. End-labeled probe 5-4 was incubated with nuclear extracts from 0- to 6-h embryos (lanes 2 to 9) or 6- to 12-h embryos (lanes 11 to 18), or with buffer only (lanes 1 and 10), under the same conditions as those used for EMSA except that a lower concentration of poly(dI-dC) was used. After a 30-min incubation at room temperature, the samples were treated with UV light (using a UV cross-linker) for 10 min and then analyzed on a 10% acrylamide-SDS gel. Competition experiments using either wild-type (wt) or mutant 5-4 probes (lanes 3 to 9 and 12 to 18) were performed as described for the EMSA experiments in Fig. 4C. The positions of size markers and the ∼40-kDa band (solid arrowhead) are indicated on the left.
“Elba” is necessary and sufficient for early boundary activity.
The results described above localize the recognition sequence for the putative early boundary activity protein to an approximately 10 bp sequence in pHS1. If this ∼40-kDa DNA binding protein is important for the early blocking activity of pHS1, one would expect that mutations in the recognition sequence that disrupt its binding would compromise the boundary function of pHS1 DNA. To test this possibility, we introduced the mut3 mutations into pHS1, multimerized the mut3-pHS1 DNA, and inserted it in the blocking position in the ftz:hsp70-LlacZ transgene. As shown in Fig. 6, this transgene has two ftz enhancers, the UPS stripe enhancer and the NE neurogenic enhancer. The UPS enhancer drives β-galactosidase expression in seven stripes during early embryogenesis, while the NE enhancer drives β-galactosidase expression in the CNS during mid-embryogenesis (Fig. 6A and B). The activities of these two enhancers are not especially sensitive to distance, and insertion of a “random” DNA fragment between the two enhancers and the hsp70 promoter has little or no effect on β-galactosidase expression. However, when a boundary such as Fab-7 is inserted between the two enhancers and the hsp70 promoter, it blocks enhancer action, and the levels of β-galactosidase in stripes in early embryos (Fig. 6, compare panels A and C) and in the CNS in older embryos (compare panels B and D) are substantially reduced.
FIG. 6.

DNA-binding activity b is necessary and sufficient for the stage-specific boundary activity of pHS1. (A to N) LacZ expression in embryos of representative lines homozygous for transgenes that have different DNA fragments placed between the ftz enhancers and the hsp70 promoter-LacZ reporter. The schematic structure of each transgene is shown on the left. (A, C, E, G, I, K, and M) Images of germ band-extended embryos. Although the UPS enhancer activates stripe expression earlier in development, this is when the highest levels of UPS enhancer-dependent β-galactosidase accumulation are typically observed. (B, D, F, H, J, L, and N) Images of germ band-retracted-stage embryos. Again, though NE-dependent CNS expression comes on earlier, this is when the highest levels of β-galactosidase accumulation from the ftz NE enhancer are observed. (A and B) Nonspecific DNA fragment; (C and D) 1.2-kb (full-length) Fab-7 boundary; (E and F) 4 copies of the pHS1 fragment; (G through J) 4 copies of pHS1 with mutation 3 in line 1 (G and H) and line 2 (I and J); (K through N) 8 copies of the Elba-binding site in line 1 (K and L) and line 2 (M and N). (O to Q) Reporter genes are not silenced in the “Elba binding site ×8” lines. These two lines, as well as a control “spacer DNA”’ line, were treated at 37°C for 1 h to induce LacZ expression from the heat shock promoter. The staining was stopped sooner than for panels A to N. Germ band-extended embryos are shown for each line.
Unlike the full-length Fab-7 boundary, a 4× multimer of the 236-bp pHS1 fragment is fully active only in early embryos (29). As shown in Fig. 6, when the 4× pHS1 multimer is placed between the ftz enhancers and the hsp70 promoter, it blocks the UPS stripe enhancer, and stripe expression is greatly reduced (Fig. 6E) in all transgenic lines. In contrast, the NE-blocking activity of pHS1×4 in older embryos is weak and position dependent. In about half of the pHS1×4 transgenic lines, the NE enhancer drives β-galactosidase expression in the CNS (Fig. 6F) at a level equivalent to that for the random DNA control (Fig. 6B). In the remaining lines, pHS1×4 appears to have some residual blocking activity, and β-galactosidase expression is reduced relative to that for the random DNA control (data not shown). To test for silencing activity, pHS1×4 was also placed upstream of the UPS and NE enhancers. In the upstream position, it had no effect on stripe or CNS β-galactosidase expression.
A quite different result was obtained for the mutant pHS1 multimer pHS1mut3×4. Shown in Fig. 6 is the β-galactosidase expression pattern observed for two representative pHS1mut3×4 lines, 130.34 and 130.249. Unlike the result with pHS1, there is little evidence of boundary activity during early embryogenesis, and the UPS enhancer drives high levels of stripe expression (Fig. 6G and I) that are equivalent to that observed with the random DNA control. As expected, the mutant pHS1 multimer also does not block the NE enhancer (Fig. 6H and J). Similar results were obtained for six out of seven pHS1mut3×4 lines. In the seventh line, the levels of both β-galactosidase stripe and CNS expression were intermediate between those for the random DNA control and Fab-7; however, since the levels were reduced to an equivalent extent at both stages, this appears to be due to a position effect rather than to some residual early blocking activity of the pHS1mut3×4 fragment. From these results we conclude that the DNA sequence recognized by the ∼40-kDa DNA binding protein identified in our EMSA and UV-cross-linking assays is, in fact, required for the early boundary activity of the pHS1 fragment. We have tentatively called this factor the “early boundary activity” protein (Elba).
As described above, previous studies suggested that two GAGA factor binding sites, sites 1 and 2, that flank the Elba recognition sequence are important for early boundary activity; however, we found that multimerized (8×) GAGA binding sites are not by themselves sufficient to reconstitute boundary activity in the ftz:LlacZ transgene assay (28). If anything, the multimerized GAGA sites seemed to stimulate transcription. For this reason we wondered whether a multimer of the Elba recognition sequence would be able to confer boundary activity and, if so, whether this activity would be largely restricted to the early embryo. To address this question, we generated a 5-4×8 multimer (which has 3 extra bases for cloning purposes) and inserted this small fragment between the ftz enhancers and the hsp70 promoter. Figure 6K to N show results for two representative transgenic lines, 131.133D and 131.189. Like pHS1, the 5-4×8 multimer has boundary activity in early embryos and blocks the UPS stripe enhancer (Fig. 6K and M). Similar results were obtained for two other autosomal insertions and for an insertion on the X chromosome. In the case of the NE enhancer, there was no evidence of blocking in two of the autosomal lines and a high level of β-galactosidase expression was observed in the CNS. In the two other autosomal lines, some residual blocking activity was observed and the level of β-galactosidase in the CNS was reduced compared to that for the random DNA control. A similar position-dependent and weak blocking activity was found previously for pHS1×4 and presumably could reflect the residual Elba activity that we see in EMSA using 6- to 12-h embryos (Fig. 3B). To determine if the reiterated 5-4×8 element is silencing the LacZ reporter rather then interfering with enhancer-promoter communications, we tested whether the hsp70 promoter in the transgene is still heat inducible. As illustrated by the examples in Fig. 6P and Q, the 5-4×8 lines showed the same level of β-galactosidase induction as the random DNA control when incubated at 37°C for 1 h. These findings indicated that the 5-4 sequence by itself is sufficient to establish boundary activity in early Drosophila embryos. Based on the effects of mutations in 5-4 DNA sequences that simultaneously abolish the binding of the Elba factor and the early enhancer boundary activity of pHS1, it seems likely that the Elba factor is responsible for the blocking activity of the 5-4 multimer.
Elba recognition sequence in pHS1.
In our EMSA we found that the Elba factor appeared to bind to a “control” DNA fragment from Fab-8. Though Fab-8 contains several sequences that resemble to a greater or lesser extent the Fab-7 ∼10-bp sequence in probe 5-4 that appears to be recognized by the Elba factor, there was no precise match. For this reason we thought it would be of interest to further narrow down the Elba recognition sequence. For this purpose we synthesized some additional mutant versions of the 5-4 probe with 3-base alterations (Fig. 7) and tested these mutants as competitors in EMSA. On the proximal side, mut2 showed partial competition (Fig. 7, lane 7) with probe 5-4, suggesting that this 3-bp mutation weakened but did not eliminate Elba binding. This appears to be due to the fact that of the 3 bases changed in mut2, only the third is important for recognition by the Elba factor. Thus, neither mut1.2 nor mut1.3 seems to affect Elba factor binding (Fig. 7, lanes 5 and 6). On the distal side, mut4.2 and mut4.3 have no apparent affect on Elba binding, while mut4.1 disrupts binding. Taken together, these mutations define an 8-bp core recognition sequence for the Elba protein, CCAATAAG. We also determined the “minimal” DNA sequence that is needed for efficient binding by the Elba protein by using a series of deletions from the proximal and distal ends. Competitors Pd4 and Dd7 only partially competed Elba binding activity (Fig. 7, lanes 14 and 18, respectively), while Pd5 and Dd8 lost the ability to compete (lanes 15 and 19, respectively). From these results, it would appear that 5 additional bases both upstream and downstream of the core recognition sequence are important for full binding activity.
Elba binding sequences in BX-C.
If early boundary activity is important for the functioning of the Fab-7 element in BX-C regulation, then the Elba recognition sequence, CCAATAAG, might be conserved in the Fab-7 boundaries of other Drosophila species. Using a BLAST search, we identified the Fab-7 region from BX-C in 10 other Drosophila species. In these other species, Fab-7 is characterized by multiple conserved sequence blocks extending over a ∼300- to 500-bp DNA segment (including the ∼90-bp high-homology region [Fig. 1]); however, more extensive homology is evident in species closely related to D. melanogaster. As shown in Fig. 8C, we found that 9 of the 10 other Drosophila species examined had the 8-bp Elba binding site. The region of precise homology between these 10 different species is actually slightly larger (13 bp), while in species that are closely related to D. melanogaster, the homology block extends further into the flanking sequences. As observed for D. melanogaster, there are also nearby sites for the GAGA factor. In close relatives of D. melanogaster, (e.g., Drosophila simulans), the Elba recognition sequence is flanked by sites for the GAGA factor. In other species, GAGA sites are only evident to one side or the other. The only species that did not have a precise match to the 8-bp Elba binding site was Drosophila mojavensis. As shown in Fig. 8, two of the A residues (residues 4 and 7) are changed to G. However, the immediately flanking sequences are conserved, and there is also a nearby GAGA site.
We also searched the D. melanogaster genome and the BX-C region for sequences matching the 8-bp Elba recognition sequence. There were more than 3,000 matches in the D. melanogaster genome, while there were 13 matches in the BX-C region. As shown in Fig. 8B, the first two are 17 kb downstream of the Ubx gene (CS1 and CS2) and the last is just upstream of the Abd-B gene (CS13). Several of the sites are located in introns or close to promoters. For example, CS3, CS4, and CS5 are in Ubx introns, while CS13 is located 123 bp upstream of the most distal Abd-B transcription start site. There is also one site, CS8, located 5 kb upstream of the abd-A gene, that is near a recently identified in vivo binding site for CTCF (13). Other than the original Fab-7 binding site, this is the only site that is located close to a known or suspected boundary element (Fab-3). Since our EMSA experiments suggested that the Fab-8 fragment ICD2 might also be recognized by the Elba factor, we looked for sequences in this Fab-8 fragment and in the rest of Fab-8 that resemble but are not identical to the 8-bp recognition sequence in Fab-7. We found two sequences that were similar to the Fab-7 recognition sequence. The sequence we found in ICD2 was ACAATAAG (Fab-8 CS2). It differs from the Fab-7 sequence only in the substitution of an A residue for C at base 1. Interestingly, this results in a sequence identical to that of mut2, which was recognized poorly by the Elba factor both as a competitor and as a probe in EMSA experiments. The other Fab-8 sequence is CCAATATG (Fab-8 CS1), which differs from the Fab-7 recognition sequence in the substitution of a T residue for an A at base 7. Mcp also has the sequence TCAATAAG (MCP CS1), which differs from the recognition sequence in Fab-7 by the substitution of a T residue for a C residue at base 1. Finally, in the distal half of HS1, there is a Fab-7 sequence, CCAAAAAG, that resembles the Elba recognition sequence (Fab-7 CS2).
We synthesized 27-bp double-strand DNAs for each of these sites as well as the “Fab-3” sequence and tested them as competitors in EMSA (Fig. 8C). The “Fab-3” sequence, which fully matched the core recognition sequence in Fab-7, strongly competed Elba binding to the labeled 5-4 probe (Fig. 8C, lanes 5 and 6). The competition was comparable to that of the wild-type probe 5-4 (Fig. 8C, lanes 3 and 4), supporting the idea that this “Fab-3” site is recognized by the Elba protein. The Fab-8 CS2 DNA also competed probe 5-4 binding (Fig. 8C, lanes 13 and 14). The competition was not as strong as that by wild-type 5-4 DNA (Fig. 8C, lanes 3 and 4) but appeared to be stronger than competition by mut2 (compare lanes 13 and 17 or lanes 14 and 18). This result indicates that Elba binding activity in nuclear extracts is influenced by sequences outside of the 8-bp core recognition sequence. In contrast, the other three candidate sites, MCP CS1 (with a core sequence of TCAATAAG), Fab-7 CS2 (CCAAAAAG), and Fab-8 CS1 (CCAATATG), did not show evidence of competition, even though they also had only a 1-base change from the original Elba recognition sequence in Fab-7 (Fig. 8C, lanes 7 to 12). Similar results were obtained when we tested each of these DNAs as labeled probes in EMSA (data not shown).
DISCUSSION
Since Fab-7 is the first example of a constitutively active boundary that depends on subelements with developmentally restricted activities, it clearly would be of interest to learn more about the mechanisms that generate boundary activity at different points in development. In the studies reported here, we have focused on the early boundary activity of the pHS1 subelement. pHS1 contains two closely spaced binding sites for the GAGA factor. While we have shown previously that these two sites are important for early boundary activity (28), the available evidence suggests that GAGA is likely to function as an accessory factor, generating a nucleosome-free region of chromatin and/or providing a scaffold for the assembly of other factors, rather than being itself responsible for boundary activity. Consistent with this idea, we have localized another protein recognition motif in pHS1 that plays a critical and more direct role in early boundary activity. This motif is recognized by a novel DNA binding protein, tentatively called Elba (early boundary activity). The Elba protein exhibits an activity profile that parallels the boundary activity of the pHS1 DNA fragment: it is highly enriched in early, 0- to 6-h embryos but is present at only low levels in older, 6- to 12-h embryos. EMSA and EMSA competition experiments localize the core recognition motif in pHS1 to an 8-bp sequence, CCAATAAG; however, binding efficiency seems to be influenced by sequences on either side of the core motif. UV cross-linking experiments with a small DNA fragment spanning the Elba recognition sequence labels a ∼40-kDa protein species. Two lines of evidence argue that this 40-kDa protein likely corresponds to the Elba protein detected in EMSA experiments. First, like the binding activity detected in the EMSA experiments, the 40-kDa protein is present in nuclear extracts from 0- to 6-h embryos but is absent or greatly reduced in those from 6- to 12-h embryos. Second, competition experiments suggest that the 40-kDa protein has the same sequence preference as the binding activity seen in the EMSA experiments.
The recognition sequence for the Elba factor also seems to be critical for the early boundary activity of pHS1. We found that mutations in the Elba recognition motif that eliminate binding in vitro in EMSA experiments compromise the early boundary function of the multimerized pHS1 subelement in the ftz:LacZ enhancer-blocking assay. Furthermore, we found that multimerizing a short DNA sequence spanning the Elba recognition motif is sufficient to confer early boundary activity in the ftz:LacZ assay. Like pHS1, this Elba multimer (5-4) efficiently blocks the UPS stripe enhancer, while it has only a small residual effect on the NE neurogenic enhancer. In the case of pHS1, we tested for silencing activity both by placing the pHS1×4 multimer upstream of the ftz enhancers and by assaying for the hsp70 promoter for heat induction. As is the case for a classical boundary, the pHS1×4 multimer had no effect when placed distally to the enhancer, nor did it silence the hsp70 promoter when placed next to it. While we did not test the Elba multimer upstream of the ftz enhancers, it did not silence the hsp70 promoter when placed adjacent to it. With the caveat that the multimerized 5-4 DNA has some extra sequences besides the Elba binding site (the 8-bp core plus the 11 nucleotides to either side), these findings argue that the Elba binding site is both necessary for the early boundary activity of pHS1 and, when multimerized, sufficient to confer early boundary activity.
One important question is whether the protein, Elba, detected in our EMSA and UV-cross-linking experiments is actually responsible for the early boundary activity of pHS1 and the 8× 5-4 DNA multimer. Several lines of evidence would argue that this 40-kDa protein is likely to correspond to the early boundary activity. First, it is the only DNA binding protein observed in our gel shift assays that has the appropriate developmental profile. Second, mutations that compromise Elba binding in vitro disrupt early enhancer-blocking activity in vivo. Third, early boundary activity can be reconstituted by multimerizing the Elba binding site. On the other hand, conclusive evidence that this protein is the early boundary factor will require identifying the corresponding gene and demonstrating that mutations in this gene disrupt pHS1 boundary activity.
The fact that multimerizing the Elba binding site is sufficient to generate boundary activity contrasts with the activities of the two pHS1 GAGA binding sites, 1 and 2. While these two GAGA binding sites appear to be necessary for the early boundary activity of Fab-7, an equivalent 8× multimer of GAGA binding sites is not sufficient to generate boundary activity. On the other hand, though the Elba multimer functions as a boundary, it seems unlikely that the single Elba site in pHS1 would be sufficient to generate a functional boundary on its own. In fact, the available evidence suggests that other cis-acting elements are likely to be required. For one, when we subdivided pHS1 into two smaller fragments, pHS1A and pHS1B, the UPS-blocking activity of the fragment containing Elba and the two GAGA binding sites (pHS1B) was position dependent. This would suggest that there are cis elements in pHS1A that are important for full early boundary activity. While we tested fragments spanning pHS1A, the binding activities detected under the conditions used in our EMSA experiments were present at only low levels in 0- to 6-h embryos and thus probably do not correspond to these collaborating factors. Another reason to think that a single Elba site would not be sufficient is the fact that mutations in GAGA sites 1 and 2 compromise the early boundary activity of the full-length Fab-7 element (which has other sequences that contribute to its early boundary activity). This finding would suggest that when there is only a single Elba site, other accessory factors are required for boundary function. Supporting the idea that the GAGA factor plays an important collaborative role is the finding that there is a juxtapositioning of Elba and GAGA binding sites in the Fab-7 boundaries of other Drosophila species.
Though the evolutionary conservation of the Elba binding site in the Fab-7 boundaries of different Drosophila species seems to point to an important role in BX-C regulation, this observation also raises a number of questions. One question is whether the Elba factor is used by any of the other BX-C boundaries. The BX-C region contains 13 sequences that precisely match the 8-bp core recognition motif in pHS1. Only one of these, Fab-3, maps close to a putative BX-C boundary, and as might be expected, this sequence appears to be recognized by the Elba factor in vitro. Whether the Elba sequence/factor is also important for the boundary activity of Fab-3 (assuming that it is a boundary) remains to be determined. The fact that precise matches to the Elba recognition sequence in pHS1 are not present in other known or predicted BX-C boundaries could mean that only Fab-7 (and perhaps also Fab-3) depends on this factor for early boundary activity. On the other hand, there is a related sequence in Fab-8 that appears to be recognized by the Elba factor. Though the Elba factor in nuclear extracts does not seem to bind as strongly to this variant site as it does to the site in pHS1, it is possible that other proteins associated with Fab-8 promote Elba binding in vivo. Presumably this could also be true for some of the other BX-C boundaries. Clearly, this issue will have to be addressed once antibodies against the Elba protein are available for chromatin immunoprecipitations. Other questions of interest include the following. Why would the Fab-7 boundary be composed of subelements that are active only at specific stages of development? Are the boundaries in BX-C (which do not appear to have Elba recognition sites) also composed of subelements that have developmentally restricted activities? More generally, what is the function of the many other Elba binding sites in the fly genome? Are these sites associated with boundaries like Fab-7 that are functional throughout the life cycle but are composed of subelements whose activities are developmentally restricted? Alternatively, are some of these boundaries “developmentally” regulated, being active only early in embryogenesis, when the Elba factor is present? Answers to these questions will require the isolation of the Elba factor and identification of the Elba gene.
Acknowledgments
We thank Greg Shanower, Daryl Gohl, Girish Deshpande, Gretchen Kappes, Jill Penn, and other members of our laboratory for helpful discussions, encouragement, and technical support during the course of these studies. We also thank Gordon Gray for providing fly food.
This work was supported by a National Institutes of Health grants to P.S. T.A. was supported by an Uehara Fellowship from the Uehara Memorial Foundation during 2001.
Footnotes
Published ahead of print on 26 November 2007.
REFERENCES
- 1.Akbari, O. S., A. Bousum, E. Bae, and R. A. Drewell. 2006. Unraveling cis-regulatory mechanisms at the abdominal-A and Abdominal-B genes in the Drosophila bithorax complex. Dev. Biol. 293294-304. [DOI] [PubMed] [Google Scholar]
- 2.Bell, A. C., A. G. West, and G. Felsenfeld. 1999. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 98387-396. [DOI] [PubMed] [Google Scholar]
- 3.Chung, J. H., A. C. Bell, and G. Felsenfeld. 1997. Characterization of the chicken beta-globin insulator. Proc. Natl. Acad. Sci. USA 94575-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galloni, M., H. Gyurkovics, P. Schedl, and F. Karch. 1993. The bluetail transposon: evidence for independent cis-regulatory domains and domain boundaries in the bithorax complex. EMBO J. 121087-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaszner, M., and G. Felsenfeld. 2006. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat. Rev. Genet. 7703-713. [DOI] [PubMed] [Google Scholar]
- 6.Gaszner, M., J. Vazquez, and P. Schedl. 1999. The Zw5 protein, a component of the scs chromatin domain boundary, is able to block enhancer-promoter interaction. Genes Dev. 132098-2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gdula, D. A., T. I. Gerasimova, and V. G. Corces. 1996. Genetic and molecular analysis of the gypsy chromatin insulator of Drosophila. Proc. Natl. Acad. Sci. USA 939378-9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geyer, P. K., and V. G. Corces. 1992. DNA position-specific repression of transcription by a Drosophila zinc finger protein. Genes Dev. 61865-1873. [DOI] [PubMed] [Google Scholar]
- 9.Golovnin, A., I. Birukova, O. Romanova, M. Silicheva, A. Parshikov, E. Savitskaya, V. Pirrotta, and P. Georgiev. 2003. An endogenous Su(Hw) insulator separates the yellow gene from the Achaete-scute gene complex in Drosophila. Development 1303249-3258. [DOI] [PubMed] [Google Scholar]
- 10.Greenberg, A. J., and P. Schedl. 2001. GAGA factor isoforms have distinct but overlapping functions in vivo. Mol. Cell. Biol. 218565-8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gyurkovics, H., J. Gausz, J. Kummer, and F. Karch. 1990. A new homeotic mutation in the Drosophila bithorax complex removes a boundary separating two domains of regulation. EMBO J. 92579-2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagstrom, K., M. Muller, and P. Schedl. 1996. Fab-7 functions as a chromatin domain boundary to ensure proper segment specification by the Drosophila bithorax complex. Genes Dev. 103202-3215. [DOI] [PubMed] [Google Scholar]
- 13.Holohan, E. E., C. Kwong, B. Adryan, M. Bartkuhn, M. Herold, R. Renkawitz, S. Russell, and R. White. 2007. CTCF genomic binding sites in Drosophila and the organisation of the Bithorax complex. PLoS Genet. 3e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karch, F., M. Galloni, L. Sipos, J. Gausz, H. Gyurkovics, and P. Schedl. 1994. Mcp and Fab-7: molecular analysis of putative boundaries of cis-regulatory domains in the bithorax complex of Drosophila melanogaster. Nucleic Acids Res. 223138-3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kellum, R., and S. C. Elgin. 1998. Chromatin boundaries: punctuating the genome. Curr. Biol. 8R521-R524. [DOI] [PubMed] [Google Scholar]
- 16.Kim, T. H., Z. K. Abdullaev, A. D. Smith, K. A. Ching, D. I. Loukinov, R. D. Green, M. Q. Zhang, V. V. Lobanenkov, and B. Ren. 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 1281231-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Labrador, M., and V. G. Corces. 2002. Setting the boundaries of chromatin domains and nuclear organization. Cell 111151-154. [DOI] [PubMed] [Google Scholar]
- 18.Maeda, R. K., and F. Karch. 2006. The ABC of the BX-C: the bithorax complex explained. Development 1331413-1422. [DOI] [PubMed] [Google Scholar]
- 19.Mihaly, J., I. Hogga, S. Barges, M. Galloni, R. K. Mishra, K. Hagstrom, M. Muller, P. Schedl, L. Sipos, J. Gausz, H. Gyurkovics, and F. Karch. 1998. Chromatin domain boundaries in the Bithorax complex. Cell. Mol. Life Sci. 5460-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mihaly, J., I. Hogga, J. Gausz, H. Gyurkovics, and F. Karch. 1997. In situ dissection of the Fab-7 region of the bithorax complex into a chromatin domain boundary and a Polycomb-response element. Development 1241809-1820. [DOI] [PubMed] [Google Scholar]
- 21.Mishra, R. K., J. Mihaly, S. Barges, A. Spierer, F. Karch, K. Hagstrom, S. E. Schweinsberg, and P. Schedl. 2001. The iab-7 polycomb response element maps to a nucleosome-free region of chromatin and requires both GAGA and Pleiohomeotic for silencing activity. Mol. Cell. Biol. 211311-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon, H., G. Filippova, D. Loukinov, E. Pugacheva, Q. Chen, S. T. Smith, A. Munhall, B. Grewe, M. Bartkuhn, R. Arnold, L. J. Burke, R. Renkawitz-Pohl, R. Ohlsson, J. Zhou, R. Renkawitz, and V. Lobanenkov. 2005. CTCF is conserved from Drosophila to humans and confers enhancer blocking of the Fab-8 insulator. EMBO Rep. 6165-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pai, C. Y., E. P. Lei, D. Ghosh, and V. G. Corces. 2004. The centrosomal protein CP190 is a component of the gypsy chromatin insulator. Mol. Cell 16737-748. [DOI] [PubMed] [Google Scholar]
- 24.Parnell, T. J., E. J. Kuhn, B. L. Gilmore, C. Helou, M. S. Wold, and P. K. Geyer. 2006. Identification of genomic sites that bind the Drosophila suppressor of Hairy-wing insulator protein. Mol. Cell. Biol. 265983-5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parnell, T. J., M. M. Viering, A. Skjesol, C. Helou, E. J. Kuhn, and P. K. Geyer. 2003. An endogenous suppressor of hairy-wing insulator separates regulatory domains in Drosophila. Proc. Natl. Acad. Sci. USA 10013436-13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ringrose, L., and R. Paro. 2007. Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134223-232. [DOI] [PubMed] [Google Scholar]
- 27.Schedl, P., and J. R. Broach. 2003. Making good neighbors: the right fence for the right job. Nat. Struct. Biol. 10241-243. [DOI] [PubMed] [Google Scholar]
- 28.Schweinsberg, S., K. Hagstrom, D. Gohl, P. Schedl, R. P. Kumar, R. Mishra, and F. Karch. 2004. The enhancer-blocking activity of the Fab-7 boundary from the Drosophila bithorax complex requires GAGA-factor-binding sites. Genetics 1681371-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schweinsberg, S. E., and P. Schedl. 2004. Developmental modulation of Fab-7 boundary function. Development 1314743-4749. [DOI] [PubMed] [Google Scholar]
- 30.Valenzuela, L., and R. T. Kamakaka. 2006. Chromatin insulators. Annu. Rev. Genet. 40107-138. [DOI] [PubMed] [Google Scholar]
- 31.Zhao, K., C. M. Hart, and U. K. Laemmli. 1995. Visualization of chromosomal domains with boundary element-associated factor BEAF-32. Cell 81879-889. [DOI] [PubMed] [Google Scholar]
- 32.Zhou, J., S. Barolo, P. Szymanski, and M. Levine. 1996. The Fab-7 element of the bithorax complex attenuates enhancer-promoter interactions in the Drosophila embryo. Genes Dev. 103195-3201. [DOI] [PubMed] [Google Scholar]