

Abstract
Terminases comprise essential components of molecular motors required to package viral DNA into capsids in a variety of DNA virus systems. Previous studies indicated that the herpes simplex virus type 1 UL15 protein (pUL15) interacts with the pUL28 moiety of a pUL28-pUL33 complex to form the likely viral terminase. In the current study, a novel temperature-sensitive mutant virus was shown to contain a mutation in UL33 codon 61 predicted to change threonine to proline. At the nonpermissive temperature, this virus, designated ts8-22, replicated viral DNA and produced capsids that became enveloped at the inner nuclear membrane but failed to form plaques or to cleave or package viral DNA. Incubation at the nonpermissive temperature also precluded coimmunoprecipitation of UL33 protein with its normal interaction partners encoded by UL28 and UL15 in ts8-22-infected cells and with pUL28 in transient-expression assays. Moreover, a temperature-sensitive mutation in UL15 precluded coimmunoprecipitation of pUL15 with the UL28 and UL33 proteins at the nonpermissive temperature. We conclude that interactions between putative terminase components are tightly linked to successful viral DNA cleavage and packaging.
During infection with all herpesviruses, viral DNA accumulates as concatemers in the nuclei of infected cells. This DNA is then cleaved into monomeric lengths and inserted into preformed capsids through a unique portal vertex (reviewed in reference 4). An enzyme called terminase comprises an essential part of the molecular motor that drives DNA into the capsid. By analogy to bacteriophage systems that require similar functions, the herpesvirus terminase is believed to bind specific DNA sequences near concatameric ends, cleave the DNA precisely, dock with the portal vertex, and provide the considerable energy (through hydrolysis of ATP) needed to overcome the self-repulsive forces of DNA as it is tightly packed within the capsid.
In herpes simplex virus type 1 (HSV-1), the UL6, UL15, UL17, UL28, UL32, and UL33 genes are dispensable for assembly of capsids but required for viral-DNA packaging (2, 5, 7, 10, 15, 16, 19, 21). In cells infected with viral mutants lacking any one of these DNA-packaging proteins, DNA is not cleaved into genomic lengths from concatameric viral DNA. As viewed by electron microscopy, cells infected with such mutants lack DNA-containing capsids with electron-dense cores but contain capsids composed of two concentric shells. These capsids morphologically resemble B capsids, which lack DNA and are believed to be dead-end products of normal infection (13).
The roles that the respective DNA-packaging proteins play in the DNA-packaging reaction have been the focus of much recent study. Whereas the portal vertex is encoded by UL6 (14), the terminase is believed to comprise components encoded by UL15, UL28, and UL33. The products of these genes interact initially in the cytoplasm, followed by their importation into the nucleus through a nuclear localization signal within pUL15 (24). Evidence supporting the role of this protein complex as the terminase includes the following observations. (i) Mutations in UL28 that partly or completely disrupt the interaction between pUL15 and pUL33 preclude DNA cleavage and packaging (9, 21, 23). (ii) UL15 encodes a likely Walker box, which comprises a critical part of the active site of a subset of helicase-like ATPases and bacteriophage viral terminases (8, 12). Mutation of this site in HSV-1 precludes DNA cleavage and packaging (25). (iii) The UL28 protein has been shown to bind DNA known to be required for the formation of proper genomic termini (1). (iv) The UL15 protein has been shown to interact with the portal protein encoded by UL6 both in vitro and by coimmunoprecipitation from infected cells (22, 24). Thus, there is evidence supporting the DNA binding, ATP hydrolysis, and portal binding functions of the pUL15-pUL28-pUL33 complex, as would be expected of a viral terminase.
The studies reported here were undertaken to further characterize the phenotypes of viral mutants containing temperature-sensitive mutations in UL15 and UL33. The data provide insight into the nature of the pUL15-pUL28-pUL33 interactions and add further credence to the hypothesis that formation of the complex is essential for HSV viral DNA cleavage and packaging.
MATERIALS AND METHODS
Cells and viruses.
The wild-type HSV-1 strain [HSV-1(mP)] and ts66.4, containing a mutation in the UL15 gene that precludes viral DNA cleavage and packaging at the nonpermissive temperature, were described previously (16). Viral stocks were propagated using Vero cells. Vero and rabbit skin cells were maintained as previously described (24).
Plasmids, DNA sequencing, and Southern blotting.
Plasmid pJB112 containing UL28 downstream of the human cytomegalovirus promoter in pcDNA3 has been described previously (23). The entire UL33 gene was amplified by PCR from HSV-1(mP) and ts8-22 DNAs and cloned into the HindIII and EcoRI sites of pcDNA3. The resulting constructs were designated pJB546 and pJB547, respectively. The UL33 genes in these plasmids were sequenced in their entirety using techniques described below. The viral genes UL15 exon I and exon II, UL28, and UL33 were also amplified by PCR from HSV-1(mP) and ts8-22 viral DNAs and sequenced.
DNA sequencing was performed by the Cornell University DNA Sequencing and Genotyping Core Facility using a mixture of BigDye terminator v3.1 and dGTP BigDye terminator v3.0 in a 3:1 ratio for cycle sequencing (Applied Biosystems, Foster City, CA), followed by dye terminator removal using Performa DTR gel filtration plates (Edge Biosystems, Gaithersburg, MD) and automated data collection by capillary electrophoresis using 3730xl DNA Analyzers (Applied Biosystems, Foster City, CA). Base calling was done using the ABI base caller, and quality scores were assigned using associated software. The generally accepted error rate for Sanger dideoxy sequencing is less than 0.1% in the optimal range of a given run.
Southern blotting was performed as described previously (5). Briefly, Vero cells were infected with 5.0 to 10.0 PFU per cell of the indicated viruses. At approximately 16 to 20 h after infection, viral DNA was purified, digested with BamHI, electrophoretically separated, denatured at high pH, and transferred to nitrocellulose. The blots were probed with radiolabeled BamHI S fragment of HSV-1 DNA, representing genomic ends. After being extensively washed, the bound probe DNA was visualized by autoradiography.
Marker rescue.
The marker rescue experiments were performed as described previously (16). Briefly, HSV-1 DNA maintained in vector plasmids was obtained by restriction enzyme digestion, followed by extraction and purification from agarose gels. Rabbit skin cells grown in 25-cm2 flasks were cotransfected with HSV-1(mP) and ts8-22 viral DNAs and the various purified HSV-1 DNA fragments indicated in Tables 1 and 2. The cells were maintained at 34°C and were harvested when almost all cells exhibited cytopathic effects. Viruses obtained from each transfection were titered at 34°C and 39°C on Vero cells. Several viruses obtained from these experiments (listed in Table 2) that replicated at 39°C were plaque purified four times, and the stocks were expanded in Vero cells. The rescued viruses were characterized by efficiency of plating at 34°C and 39°C and the ability to cleave viral DNA as shown in Results.
TABLE 1.
Marker rescue of HSV-1(mP ts8-22)a
| Expt | DNA fragmentb | PFU at 39°C vs PFU at 34°C |
|---|---|---|
| A | None | <5.0 × 10−7 |
| XbaI C | <6.3 × 10−7 | |
| XbaI D | <4.8 × 10−7 | |
| XbaI E | 1.8 × 10−2 | |
| XbaI F | <5.6 × 10−7 | |
| B | XbaI E-BglII (1)c | 1.1 × 10−2 |
| BglII-XbaI E (2)c | <2.0 × 10−5 | |
| C | BamHI (C′) | 2.0 × 10−3 |
| BamHI D | <2.0 × 10−5 | |
| BamHI H | <1.4 × 10−5 | |
| Bam,HI H′ | <1.1 × 10−5 | |
| BamHI (O) | <1.4 × 10−5 | |
| D1 | BamHI (C′) | 3.9 × 10−3e, 10−2f |
| XbaI E-NarI (3)d | 2.1 × 10−2e, 2.1 × 10−3f | |
| Nar1-BamHI (4)d | <6.3 × 10−6f | |
| D2 | BamHI (C′) | 3.9 × 10−3e, 6.2 × 10−3f |
| XbaI E-NarI (3)d | 1.2 × 10−2e, 4.3 × 10−3f | |
| NarI-BamHI (4)d | <1.5 × 10−6e, <6.3 × 10−6f |
Rabbit skin cells were cotransfected with the indicated genomic DNA fragments and HSV-1(mP ts8-22) viral DNA at 34°C. Virus was harvested and used to infect Vero cell monolayers at 34 or 39°C. The yields of virus at the two temperatures were determined by plaque assay, and the ratio of the yield obtained at 39°C divided by the yield at 34°C is presented in the rightmost columns.
Fragments were derived from HSV-1(F) in all experiments except D2, in which they were derived from HSV-1(KOS).
Fragments 1 and 2 are diagrammed in Fig. 1, line 2.
Fragments 3 and 4 are diagrammed in Fig. 1, line 4.
Experiment 1.
Experiment 2.
TABLE 2.
Designations and derivations of rescued viruses
Electron microscopy.
Vero cells were infected with the ts8-22 virus at less than 1.0 PFU per cell and were fixed 48 h later. The cells were embedded, sectioned, and examined by electron microscopy as previously described (5).
Antibodies and coimmunoprecipitations.
Rabbit antisera used for immunoprecipitations were directed against the N-terminal 35 amino acids of pUL15, the full-length pUL28, or the UL33 protein as previously described (6, 17, 20). The same antisera were used for immunoblotting, except that antibody directed against the C terminus of UL15, was used in lieu of the antibody directed against the N terminus of UL15 (5). The procedures for immunoprecipitation and immunoblotting were performed as described previously (23).
RESULTS
Mapping and sequencing of a novel temperature-sensitive mutation in UL33.
As revealed by screening of plaque-purified viruses derived from HSV-1(mP) for the ability to replicate at 34 and 39 to 40°C in a previous study, a series of spontaneously arising temperature-sensitive mutants of HSV-1 were identified (16). One of these (ts66.4) was previously mapped to codon 653 of UL15, encoding a change from serine to proline, and precluded both DNA cleavage and packaging at the nonpermissive temperature (16). In the same screen, another such spontaneously arising mutant was identified and was designated ts8-22.
Several series of experiments were conducted to narrow the exact location of the mutation in ts8-22. In the first series of experiments, individual fragments from a library of cloned XbaI DNA fragments encompassing the entire HSV-1(F) genome were tested for the ability to rescue replication at 39°C using methods described previously (16). As shown in Table 1, the XbaI E fragment (diagrammed in Fig. 1, line 2) rescued the temperature-sensitive phenotype, whereas other fragments did not have such an effect (Fig. 1, line 2, and Table 1, experiment A). To narrow the site of the mutation further, fragments derived from HSV-1(F) DNA and spanning the XbaI E region were tested similarly, thereby localizing the temperature-sensitive mutation to the BamHI C′ fragment (Fig. 1, lines 2 to 5, and Table 1, experiments B and C), which contains the UL33 and UL34 coding sequences. In the next series of experiments, smaller DNA fragments derived from both HSV-1(F) and HSV-1(KOS) DNA (Table 1, experiments D1 and D2, respectively) were used in the marker rescue assay. The results indicated that the fragment containing the UL33 open reading frame was sufficient to rescue the ts8-22 mutation (Table 2).
FIG. 1.

Colinear schematic diagram of DNA fragments used to map a novel lethal temperature-sensitive mutation in the ts8-22 viral genome. (Line 1) Diagram of the HSV-1 genome and XbaI E fragment. The open rectangles represent repeated sequences flanking the ends of unique long (UL) or unique short (US) sequences. The approximate site of the XbaI E fragment in the UL segment is indicated. (Line 2) Expanded diagram of the XbaI E fragment showing XbaI/BglII fragments screened for the ability to rescue the temperature-sensitive phenotype. The fragments are designated 1 and 2 as shown and in Table 1. (Line 3) Expanded fragment 1, as shown in line 2, illustrating BamHI sites. Fragment 1 contains the entire BamHI D, H, and H′ fragments and truncated C′ and O fragments. Truncations of the last two fragments are indicated by brackets. (Line 4) Expansion of the truncated BamHI C′ fragment. The position of a NarI site used to divide the fragment into fragments 3 and 4 is shown. B, BamHI; Bg, BglII; N, NarI; X, XbaI.
To determine the precise location of the temperature-sensitive lesion, the UL33 genes of HSV-1(mP) and ts8-22 were sequenced and compared to UL33 of HSV-1 strain 17 determined previously (11). The results are shown in Fig. 2. At codon 109, HSV-1(17) encoded alanine whereas both HSV-1(mP) and ts8-22 encoded arginine. More importantly, the only deduced amino acid difference between UL33 of HSV-1(mP) and ts8-22 was located at codon 61, where threonine was changed to proline. No mutations in the UL15 or UL28 gene, encoding proteins known to interact with pUL33, were noted upon comparison of ts8-22 and HSV-1(mP) sequences (data not shown).
FIG. 2.

Deduced amino acid sequences of UL33 from HSV-1(17), HSV-1(mP), and ts8-22. Bold characters indicate differences between sequences.
Plating and replication efficiencies of the wild-type, mutant, and rescued viruses at the nonpermissive temperature.
All assays were done on Vero cells incubated at 34°C or 39°C, and the results of this study are summarized in Table 3. The results show that whereas the parent virus replicates and forms more plaques at 39°C than at 34°C, the mutant virus replicates and forms plaques with a much lower efficiency at 39°C than at 34°C. The replication and plaque formation of the rescued viruses resembled those of the wild-type virus, indicating that the mutation within UL33 was solely responsible for the growth defects at the nonpermissive temperature.
TABLE 3.
Plating efficiencies and yields of HSV-1(mP), ts8-22, and rescued viruses at 34°C and 39°C after infection at 1.0 and 0.1 PFU/cell
| Virus | Plating efficiencya (39°C/34°C) | Yieldb at 1 PFU/cell
|
Ratio of yields (39°C/34°C) | Yieldb at 0.1 PFU/cell
|
Ratio of yields (39°C/34°C) | ||
|---|---|---|---|---|---|---|---|
| 34°C | 39°C | 34°C | 39°C | ||||
| HSV-1(mP) | 0.97 | 1.7 × 107 | 3.2 × 107 | 1.9 | 3.0 × 106 | 1.0 × 107 | 3.3 |
| HSV-1(mP) ts8-22 | 1.3 × 107 | 5.5 × 102 | 4.2 × 10−5 | 1.0 × 106 | 8.8 × 107 | 8.8 × 10−5 | |
| HSV-1(mP) R93 | 0.86 | 4.4 × 106 | 9.3 × 106 | 2.1 | 6.0 × 105 | 7.8 × 105 | 1.3 |
| HSV-1 (mP) R142 | 0.9 | 7.9 × 106 | 2.0 × 107 | 2.5 | 1.0 × 106 | 4.0 × 106 | 4.0 |
Calculated as the ratio of the numbers of plaques produced by the same virus inoculum.
In PFU/ml.
Effect of the UL33 temperature-sensitive mutation on viral-DNA cleavage and packaging.
Given previous observations that insertion and temperature-sensitive mutations in UL33 can preclude viral DNA cleavage and packaging (2, 7), it was of interest to determine if the same was true of the mutation in ts8-22. To test this possibility, replicate Vero cell monolayers were infected at 34°C or 39°C with 10 PFU per cell of HSV-1(mP), ts8-22, or rescued viruses derived from ts8-22. Cells were harvested at 15.5 h after infection, and purified viral DNA was digested with BamHI, subjected to electrophoresis in a 0.7% agarose gel, transferred to a zeta-probe blotting membrane, and reacted with a denatured 32P-labeled BamHI S fragment, which is specific for genomic ends and the junction between the unique long and unique short components. The results (Fig. 3A) showed that (i) the junction fragment BamHI SP and a ladder of faster-migrating BamHI S fragments accumulated in cells infected with either wild-type or the ts8-22 mutant virus at the permissive temperature. (ii) At the nonpermissive temperature, both BamHI SP and S fragments accumulated in cells infected with the wild-type virus, whereas cells infected with ts8-22 accumulated the BamHI SP fragment only (i.e., no fragments indicative of cleaved genomes were detected). (iii) The BamHI SP and S fragments were readily detected in cells infected with several revertant viruses obtained from marker rescue experiments using BamHI C′ subfragments (Fig. 3B and C) and maintained at the nonpermissive temperature. We conclude that in cells infected with ts8-22 and maintained at the nonpermissive temperature, viral DNA accumulates in an endless, concatemeric form and is not cleaved into mature, genomic full-length molecules.
FIG. 3.
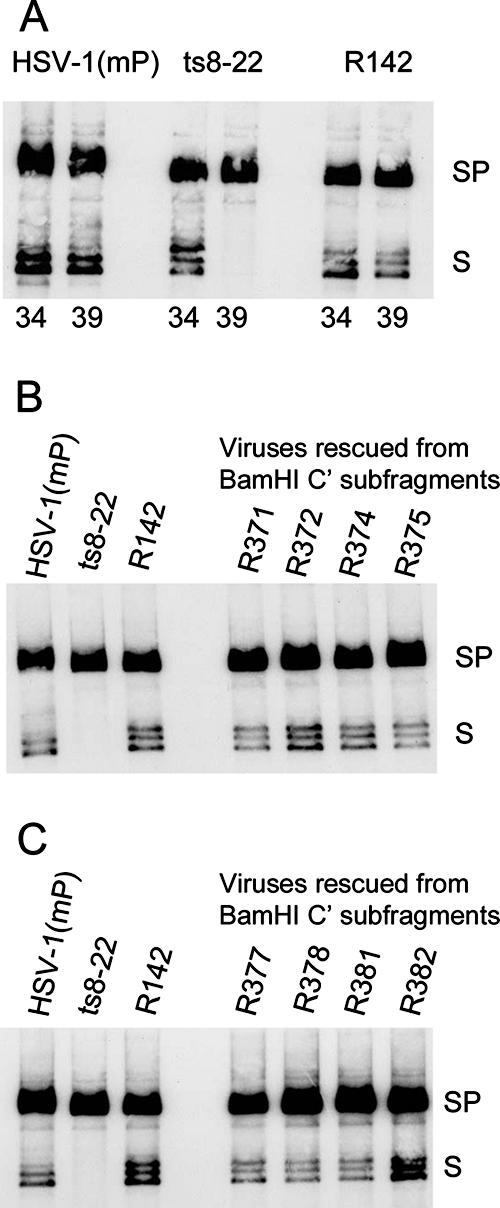
Analyses of HSV-1(mP), ts8-22, or rescued virus genomic DNAs at permissive and nonpermissive temperatures. Vero cells were infected with the indicated viruses at 10 PFU per cell at 34°C or 39°C. Genomic viral DNAs were purified, digested with BamHI, transferred to nitrocellulose, and probed with radiolabeled BamHI S fragment, which is specific for sequences at genomic ends (fragments S), and internal junction fragments (fragments SP). (A) Cells were infected with the indicated viruses at 34 or 39°C as shown. (B and C) Cells were infected at 39°C with the indicated viruses. The precise origins of the rescued viruses are indicated in Table 2.
To further characterize the restricted phenotype, Vero cells were infected with the ts8-22 mutant virus at 34°C and 39°C and prepared for electron microscopy. Representative electron micrographs of thin sections of the infected cells are shown in Fig. 4. Capsids with morphology similar to that of B capsids were present in the nucleoplasm of cells held at 39°C, but in contrast to the appearance of cells held at 34°C, none contained electron-dense cores indicative of packaged viral DNA. In addition, although enveloped capsids were readily apparent in the cytoplasm or perinuclear space of cells held at 39°C, they contained a roughly circular and lightly staining inner core also resembling those of B capsids. Thus, none of the enveloped cytoplasmic capsids or unenveloped nuclear capsids at 39°C contained electron-dense cores (Fig. 4, compare capsids in panel A containing electron-dense cores with those in panels B, C, and D). We conclude that the UL33 mutation precludes packaging of viral DNA at the nonpermissive temperature but does not preclude assembly or envelopment of B-like capsids.
FIG. 4.

Electron microscopic images of thin sections of Vero cells infected with virus ts8-22 at permissive and nonpermissive temperatures. Vero cells were infected and were fixed 48 h later, embedded, sectioned, and stained with uranyl acetate. (A) Infection at 34°C. Virions with densely stained inner cores indicative of packaged viral DNA are present in the perinuclear space. (B to D) Images of cells infected at 39°C. Enveloped and unenveloped capsids lack densely staining inner cores. As a size standard, HSV-1 capsids are 125 nm in diameter.
Effects of UL15 and UL33 temperature-sensitive mutations on interactions with the other putative terminase component, pUL28.
To further clarify the effects of the temperature-sensitive mutations on viral DNA cleavage and packaging, Vero cells were infected with a mutant containing a temperature-sensitive mutation in UL15, designated ts66.4; ts88-22 (described above); or HSV-1(mP), from which the mutant viruses were derived. The infected cells were held at 34 or 39°C for 16 h, at which time the UL15, UL28, and UL33 proteins were immunoprecipitated from cellular lysates. Whether the UL15, UL28, and UL33 proteins were present in the immunoprecipitates was determined by immunoblotting.
As shown in Fig. 5, pUL15, pUL28, and pUL33 were readily immunoprecipitated with their cognate antibodies from lysates of cells infected with HSV-1(mP), ts8-22, and ts66.4 at the permissive and nonpermissive temperatures (Fig. 5A to C, lanes 2 to 7). As expected, the UL15, UL28, and UL33 proteins were not immunoprecipitated from mock-infected cells (lanes 1). Less pUL15 was immunoprecipitated from lysates of cells infected with ts66.4 and held at the nonpermissive temperature than from ts66.4-infected cells maintained at 34°C or from cells infected with HSV-1(mP) held at either temperature (Fig. 5A, compare lane 5 with lanes 2 to 4). Similarly, less pUL33 was coimmunoprecipitated from lysates of cells infected with ts8-22 and held at the nonpermissive temperature than from cells infected with wild-type virus or ts8-22 and held at 34°C (Fig. 5C, lanes 6 and 7). These data indicate that while incubation at the nonpermissive temperature reduced immunoprecipitation of some pUL15 and pUL33 with their cognate antibodies, the higher temperature did not completely abrogate this immunoprecipitation.
FIG. 5.

Immunoblots of terminase components immunoprecipitated from lysates of infected cells. (A) Cells were mock infected or infected at 34 or 39°C. At 16 h after infection, the cells were lysed and the clarified lysates were reacted with pUL15-specific antibody. Antigen-antibody complexes were purified, denatured, electrophoretically separated, transferred to nitrocellulose, and probed for the presence of pUL15 (top), pUL28 (middle), or pUL33 (bottom) by immunoblotting. Bound immunoglobulin was revealed using enhanced chemiluminescence and X-ray film (Amersham). (B) Same as panel A, except that cell lysates were reacted with pUL28-specific antibody. (C) Same as panel A, except that cell lysates were reacted with pUL33-specific antibody.
Similar to previous results using HSV-1(F), antibody directed against pUL15 coimmunoprecipitated the UL28 and UL33 proteins from lysates of cells infected with HSV-1(mP) and ts66.4 when the cells were held at the permissive temperature (Fig. 5A, lanes 2 and 4, respectively). In contrast, incubation of cells infected with ts66.4 at the nonpermissive temperature precluded coimmunoprecipitation of pUL15 with either pUL28 or pUL33 (Fig. 5A, lane 5), despite the observation that both pUL28 and pUL33 were readily detectable in these cells when reacted with pUL28- and pUL33-specific antisera (Fig. 5, lanes 5). Moreover, pUL28 and pUL33 coimmunoprecipitated with one another at both permissive and nonpermissive temperatures (Fig. 5B and C, lanes 4 and 5). These data indicate that the temperature-sensitive lesion in pUL15 precludes interaction with a complex of pUL28 and pUL33 at the nonpermissive temperature.
The effects of the pUL33 temperature-sensitive lesion on interactions between putative terminase components were as follows. (i) At the permissive temperature, pUL33-specific antibody readily coimmunoprecipitated both pUL28 and pUL15 (Fig. 5C, lane 6). Moreover, levels of pUL15 coimmunoprecipitated with pUL28 antibody were increased in cells infected with ts8-22 and held at the permissive temperature compared to similar reactions using lysates of cells infected with HSV-1(mP) or ts66.4 (Fig. 5B). (ii) Incubation of cells at the nonpermissive temperature precluded pUL15 and pUL28 from coimmunoprecipitation with pUL33 antibody (Fig. 5C). (iii) Very low levels of pUL28 were immunoprecipitated with the anti-pUL28 antisera from cells infected with ts8-22 and held at the nonpermissive temperature (Fig. 5B, lane 7). (iv) Smaller but readily detectable amounts of pUL15 were immunoprecipitated from lysates of cells infected with ts8-22 held at the nonpermissive temperature (Fig. 5A, lane 7), but the higher temperature precluded coimmunoprecipitation of pUL28 and pUL33 with pUL15 antibody (Fig. 5A, lanes 7). Despite the low levels of immunoprecipitable pUL28 in this lysate, the pUL28 antibody coimmunoprecipitated small amounts of pUL15 (Fig. 5B, lane 7).
Together, these data suggest that the temperature-sensitive lesion in UL33 precludes interaction of pUL33 with other terminase components at the nonpermissive temperature. The data also suggest roles for pUL33 in increasing steady-state levels of pUL28 and enhancing the interaction between pUL28 and pUL15.
Efforts were also made to assess the effects of the UL33 mutation on interaction with pUL28 in the absence of other viral proteins. Thus, CV1 cells were transfected with expression plasmids encoding pUL28 and pUL33 from either HSV-1(mP) or ts8-22. The cells were held for 24 h at 34 or 39°C, at which time they were lysed and subjected to immunoprecipitation with pUL28-specific antibody, followed by immunoblotting with pUL28- and pUL33-specific antibodies. The results, shown in Fig. 6, indicated that pUL28 antibody readily coimmunoprecipitated transiently expressed pUL33 of HSV-1(mP) in lysates held at both 34 and 39°C. Although the mutant pUL33 from ts8-22 was coimmunoprecipitated with pUL28-specific antibody at 34°C, these proteins coimmunoprecipitated very poorly at 39°C. These data indicate that the lesion in pUL33 largely precludes coimmunoprecipitation with pUL28 at the nonpermissive temperature and that this effect occurs independently of other viral functions.
FIG. 6.

Immunoblots of transiently expressed pUL28 and pUL33 immunoprecipitated with pUL28-specific antibody. CV1 cells were transfected with pcDNA (lane 1) or UL28 (lanes 2 through 5) and UL33 expression plasmids as indicated. The cells were lysed 24 h later and reacted with pUL28-specific antisera. Purified immune complexes were electrophoretically separated on a denaturing gel and transferred to nitrocellulose, and the presence of pUL33 (top) and pUL28 (bottom) was determined by immunoblotting. pJB546 contains UL33 of HSV-1(mP); pJB pJB547 contains UL33 of ts8-22.
DISCUSSION
These studies further support the contention that interactions between the UL15, UL28, and UL33 proteins are critical for cleavage and packaging of HSV-1 DNA. Specifically, under conditions in which the temperature-sensitive mutations in both UL15 and UL33 preclude DNA cleavage and packaging, the interaction of pUL15 and pUL33 with pUL28 is substantially decreased. Thus, these data may provide a mechanistic reason for the lethal phenotype conferred by these mutations.
Normally, DNA-containing capsids are preferentially enveloped in HSV-1-infected cells, and envelopment of B capsids lacking DNA is uncommon (18). In contrast to this situation, we have shown that although the mutation in UL33 precludes DNA cleavage and packaging at the nonpermissive temperature, it does not preclude envelopment of B capsids at the inner nuclear membrane (Fig. 4). The ultrastructural appearance of these cells resembles that of Vero cells infected with UL15 and UL28 null mutants, in which ample enveloped capsids lacking DNA were also noted in the cytoplasm (3, 21). Thus, putative terminase components function in some way to preclude DNA-negative capsids from budding at the nuclear membrane.
The mutation identified in this study, UL33 (Thr-61-Pro), conferring a temperature-sensitive phenotype, was different from a previously described temperature-sensitive mutation in UL33 (Ile-17-Asn) that also precluded DNA cleavage and packaging at the nonpermissive temperature (2). Coincidentally, the temperature-sensitive mutation in UL15 also involves a change to proline (from serine 653) (3). In the absence of structural information, the reason why these mutations confer temperature-sensitive phenotypes must remain speculative. It is conceivable that the proline residues in pUL33 and UL15 would destabilize alpha helices or change local polarity, and these effects would be exacerbated to a critical level at higher temperatures. Along these lines, the sequences between positions 52 and 66 of pUL33 have a high probability of forming an alpha helix, according to the algorithm of Garnier and Robson, whereas the proline near amino acid 653 of pUL15 is predicted to decrease the otherwise high charge ratio in this region (data not shown).
We have previously shown that both pUL33 and pUL15 interact with pUL28 in the cytoplasm, with subsequent import of the complex into the nucleus (24). The failure of terminase components to coimmunoprecipitate at the nonpermissive temperature in cells infected with the mutants described here argues that the lethal effects of the mutations are attributable to an early failure to assemble the terminase complex at the nonpermissive temperature. Whether these conditions also affect nuclear import in a later step remains to be tested. The observation that the UL33 mutation did not preclude coimmunoprecipitation of pUL15 with pUL28 antibody at the nonpermissive temperature further supports previous conclusions that pUL28 and pUL15 interact directly, even in the absence of pUL33 (23). In contrast, pUL33 can interact with pUL15 only through its interaction with pUL28. This is potentially consistent with the observation that levels of pUL28 were reduced more significantly than levels of pUL15 in cells infected with the UL33 mutant at the nonpermissive temperature.
Previous results have shown that the presence of pUL33 augments the pUL15-pUL28 interaction and that pUL28 is necessary and sufficient for pUL33 stability (23). The current studies also suggest that an important role for pUL33 is to interact with pUL28 to allow DNA packaging to proceed. Whether this reflects a role in mediating correct pUL28 folding or as an essential part of the DNA binding component of the terminase awaits biochemical testing. Some data presented here may reflect a role for pUL33 in precluding dysfunctional aggregation of pUL28 and pUL15. Specifically, the data shown in Fig. 5 suggest that at the permissive temperature, more pUL15 is immunoprecipitated with pUL28-specific antibody from cells infected with the UL33 temperature-sensitive mutant ts8-22 than from cells infected with wild-type HSV-1(mP). Interestingly, the increased immunoprecipitation of pUL15 and pUL28 did not correspond to a greatly increased amount of coimmunoprecipitated pUL33 (Fig. 5B, lane 6), suggesting that some pUL15-pUL28 complexes lack pUL33 under these circumstances. We sequenced UL15, UL28, and UL33 in both HSV-1(mP) and ts8-22 and found no differences other than the mutation in codon 61 of UL33 already noted in Fig. 2. Thus, compensating mutations in UL15 and UL28 are not responsible for the enhanced coimmunoprecipitation of the encoded proteins. We speculate that the mutation in pUL33 compromises the normal interaction between pUL15 and pUL28 even at the permissive temperature but that normal complexes are also made that are sufficient to support DNA cleavage and packaging. Thus, some pUL15 may interact with pUL28 that has become misfolded in the absence of pUL33, and failure of pUL15 to dissociate from this misfolded pUL28 would be expected to cause enhanced coimmunoprecipitation at steady state. Although we favor this scenario, we cannot exclude the possibilities that the pUL33 mutation enhances production of pUL28 and pUL15 or increases formation of functional pUL15/pUL28 complexes at the permissive temperature.
Acknowledgments
We thank Shu-fen Chou for electron microscopy.
These studies were supported by public health services grants GM50740 (to J.D.B.) and CA 88860 (to B.R) from the National Institutes of Health.
Footnotes
Published ahead of print on 3 October 2007.
REFERENCES
- 1.Adelman, K., B. Salmon, and J. D. Baines. 2001. Herpes simplex DNA packaging sequences adopt novel structures that are specifically recognized by a component of the cleavage and packaging machinery. Proc. Natl. Acad. Sci. USA 983086-3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Kobaisi, M. F., F. J. Rixon, I. McDougall, and V. G. Preston. 1991. The herpes simplex virus UL33 gene product is required for the assembly of full capsids. Virology 180380-388. [DOI] [PubMed] [Google Scholar]
- 3.Baines, J. D., C. Cunningham, D. Nalwanga, and A. J. Davison. 1997. The UL15 gene of herpes simplex virus type 1 contains within its second exon a novel open reading frame that is translated in frame with the UL15 gene product. J. Virol. 712666-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baines, J. D., and C. Duffy. 2006. Nucleocapsid assembly and envelopment of herpes simplex virus, p. 175-204. In R. M. Sandri-Goldin (ed.), Alpha herpesviruses: pathogenesis, molecular biology and infection control. Caister Scientific Press, Norfolk, United Kingdom.
- 5.Baines, J. D., A. P. W. Poon, J. Rovnak, and B. Roizman. 1994. The UL15 gene of herpes simplex virus encodes two proteins and is required for cleavage of viral DNA. J. Virol. 688118-8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beard, P. M., N. S. Taus, and J. D. Baines. 2002. The DNA cleavage and packaging proteins encoded by genes UL28, UL15, and UL33 of herpes simplex virus type 1 form a complex in infected cells. J. Virol. 764785-4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunningham, C., and A. J. Davison. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197116-124. [DOI] [PubMed] [Google Scholar]
- 8.Davison, A. J. 1992. Channel catfish virus: a new type of herpesvirus. Virology 1869-14. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson, J. G., K. Yang, J. D. Baines, and F. L. Homa. 2006. Linker insertion mutations in the herpes simplex virus type 1 UL28 gene: effects on UL28 interaction with UL15 and UL33 and identification of a second-site mutation in the UL15 gene that suppresses a lethal UL28 mutation. J. Virol. 8012312-12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamberti, C., and S. K. Weller. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 722463-2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 691531-1574. [DOI] [PubMed] [Google Scholar]
- 12.Mitchell, M. S., S. Matsuzaki, S. Imai, and V. B. Rao. 2002. Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 304009-4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Newcomb, W. W., F. L. Homa, D. R. Thomsen, F. P. Booy, B. L. Trus, A. C. Steven, J. V. Spencer, and J. C. Brown. 1996. Assembly of the herpes simplex virus capsid: characterization of intermediates observed during cell-free capsid formation. J. Mol. Biol. 263432-446. [DOI] [PubMed] [Google Scholar]
- 14.Newcomb, W. W., R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller, and J. C. Brown. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 7510923-10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel, A. H., F. J. Rixon, C. Cunningham, and A. J. Davison. 1996. Isolation and characterization of herpes simplex virus type 1 mutants defective in the UL6 gene. Virology 217111-123. [DOI] [PubMed] [Google Scholar]
- 16.Poon, A. P. W., and B. Roizman. 1993. Characterization of a temperature-sensitive mutant of the UL15 open reading frame of herpes simplex virus 1. J. Virol. 674497-4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reynolds, A. E., Y. Fan, and J. D. Baines. 2000. Characterization of the UL33 gene product of herpes simplex virus 1. Virology 266310-318. [DOI] [PubMed] [Google Scholar]
- 18.Roizman, B., and D. Furlong. 1974. The replication of herpesviruses, p. 229-403. In H. Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virology. Plenum Press, New York, NY.
- 19.Salmon, B., C. Cunningham, A. J. Davison, W. J. Harris, and J. D. Baines. 1998. The herpes simplex virus 1 UL17 gene encodes virion tegument proteins that are required for cleavage and packaging of viral DNA. J. Virol. 723779-3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salmon, B., D. Nalwanga, Y. Fan, and J. D. Baines. 1999. Proteolytic cleavage of the amino-terminus of the UL15 gene product of herpes simplex virus type 1 UL15 protein is coupled with maturation of viral DNA into unit length genomes. J. Virol. 738338-8348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tengelsen, L. A., N. E. Pedersen, P. R. Shaver, M. W. Wathen, and F. L. Homa. 1993. Herpes simplex virus type 1 DNA cleavage and capsidation require the product of the UL28 gene: isolation and characterization of two UL28 deletion mutants. J. Virol. 673470-3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White, C. A., N. D. Stow, A. H. Patel, M. Hughes, and V. G. Preston. 2003. Herpes simplex virus type 1 portal protein UL6 interacts with the putative terminase subunits UL15 and UL28. J. Virol. 776351-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang, K., and J. D. Baines. 2006. The putative terminase subunit of herpes simplex virus 1 encoded by UL28 is necessary and sufficient to mediate interaction between pUL15 and pUL33. J. Virol. 805733-5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang, K., F. Homa, and J. D. Baines. 2007. Putative terminase subunits of herpes simplex virus type 1 form a complex in the cytoplasm and interact with portal protein in the nucleus. J. Virol. 816419-6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu, D., and S. K. Weller. 1998. Genetic analysis of the UL 15 gene locus for the putative terminase of herpes simplex virus type 1. Virology 24332-44. [DOI] [PubMed] [Google Scholar]