

Abstract
Due to the limited coding capacity of their small genomes, human papillomaviruses (HPV) rely extensively on host factors for the completion of their life cycles. Accordingly, most HPV proteins, including the replicative helicase E1, engage in multiple protein interactions. The fact that conserved regions of E1 have not yet been ascribed a function prompted us to use tandem affinity protein purification (TAP) coupled to mass spectrometry to identify novel targets of this helicase. This method led to the discovery of a novel interaction between the N-terminal 40 amino acids of HPV type 11 (HPV11) E1 and the cellular WD repeat protein p80 (WDR48). We found that interaction with p80 is conserved among E1 proteins from anogenital HPV but not among cutaneous or animal types. Colocalization studies showed that E1 can redistribute p80 from the cytoplasm to the nucleus in a manner that is dependent on the E1 nuclear localization signal. Three amino acid substitutions in E1 proteins from HPV11 and -31 were identified that abrogate binding to p80 and its relocalization to the nucleus. In HPV31 E1, these substitutions reduced but did not completely abolish transient viral DNA replication. HPV31 genomes encoding two of the mutant E1 proteins were not maintained as episomes in immortalized primary keratinocytes, whereas one encoding the third mutant protein was maintained at a very low copy number. These findings suggest that the interaction of E1 with p80 is required for efficient maintenance of the viral episome in undifferentiated keratinocytes.
Papillomaviruses (PV) are small, nonenveloped, double-stranded DNA viruses that induce hyperproliferative lesions of cutaneous and mucosal stratified squamous epithelia. They can infect the majority of higher vertebrates, with humans being the host for more than 100 different PV types. Based on oncogenic propensity, human PV (HPV) types that infect the anogenital region are categorized as either low-risk types, such as HPV type 6 (HPV6) and HPV11, which cause genital warts, or high-risk types, like HPV16, -18, and -31, which induce dysplasic lesions that can progress to cancer. The viral replication cycle is tightly linked to the differentiation program of stratified epithelia, the expression of viral proteins being modulated by intracellular changes occurring as cells differentiate toward the upper layers of the epithelium. The viral replication cycle begins with the infection of an undifferentiated basal cell in which the viral genome is established as an extrachromosomal episome in the nucleus at 50 to 100 copies, a number that is maintained until infected keratinocytes reach the upper cell layers (reviewed in reference 15). In those differentiated cells, the viral genome is amplified to a high copy number, probably as a result of increased expression of the viral replicative helicase E1, brought about by differentiation-dependent activation of the late promoter (20, 34). The viral structural proteins, which are also expressed from the late promoter, are produced in the upper layers of the epithelium, where the viral cycle is completed by the encapsidation of the viral genome and the release of newly assembled infectious virions.
In addition to the cellular DNA replication machinery, only two virally encoded proteins, E1 and E2, are required for viral DNA replication (4, 22, 52, 54). E2 is a sequence-dependent DNA binding protein that recognizes specific sequences in the viral origin of replication (ori). E2 also directly interacts with E1 to help recruit it to the origin. E1, the most conserved protein among PV and the only one with enzymatic activity, belongs to helicase superfamily 3 (21). Although this initiator protein is recruited to the ori in its monomeric form, it must assemble into a double hexamer to unwind DNA ahead of the bidirectional replication fork (reviewed in reference 45).
E1 is comprised of three functional domains. The C-terminal domain possesses the helicase/ATPase activity and includes sequences involved in the oligomerization of the protein (50). The central portion of the protein contains the origin binding domain (OBD), which recognizes distinct sequences in the ori, albeit with low affinity and moderate sequence specificity (44, 49, 50). Not surprisingly, both the OBD and the helicase domain are required for DNA replication in vitro and in vivo (1). Unlike with these two domains, the N-terminal portion of E1 is required for viral genome replication only in vivo, not in vitro (11, 46), thus suggesting that it has a regulatory function. Accordingly, the N-terminal domain has been shown to contain functional nuclear localization signal (NLS) and nuclear export sequences, a conserved cyclin-binding motif (CBM) needed for interaction with cyclin A/cyclin E-cyclin-dependent kinase 2 (Cdk2), and several phosphorylation sites for this kinase and others (9, 16, 24).
In agreement with its central role in viral DNA replication, E1 has been reported to interact with several cellular replication factors. These include the polymerase α-primase complex, which synthesizes the RNA primer essential for lagging-strand synthesis and which is recruited to the viral replication fork through its association with the C-terminal helicase domain of E1 (1, 6, 32, 51), the single-stranded DNA binding protein replication protein A (14, 30), and topoisomerase I (Topo I). The interaction of E1 with Topo I increases both the activity of Topo I, which is needed for releasing the torsional stress generated by DNA unwinding, and the binding of E1 to the ori (5, 17). The molecular chaperones Hsp40 and Hsp70 have also been found to promote the binding of E1 to the ori as well as its subsequent assembly into active double hexamers (28, 29). E1 also interacts with the cell cycle regulatory kinase complexes cyclin A/cyclin E-Cdk2 through a highly conserved CBM in the N-terminal region of the protein (7, 16, 27, 31). The phosphorylation by Cdk2 of specific serine residues in the N-terminal region of HPV11 E1 abrogates its nuclear export, thus promoting its nuclear accumulation (7, 9, 25, 31). In addition to interacting with replication factors and regulatory proteins, E1 was reported to interact with two proteins that could favor replication by modifying chromatin structure: histone H1, which binds nucleosome-organized chromatin to create an ordered, more compact DNA structure and which is dislodged from DNA upon its interaction with E1 (47), and Ini1/SNF5 (23), a subunit of the SWI/SNF chromatin remodeling complex. Finally, E1 also interacts with E1-BP, a putative ATPase of unknown function (55). Mutations in E1 that prevent its association with E1-BP are unable to support viral DNA replication, suggesting a role for E1-BP in this process (55).
Small DNA tumor viruses, including PV, encode only a few proteins and thus must rely extensively on the infected cell proteome for the completion of their life cycles. Accordingly, most PV proteins are multifunctional and have been shown to engage in multiple protein interactions with cellular factors to carry out their functions. The fact that highly conserved regions of E1 have not yet been ascribed a function provides a rationale for thinking that novel cellular targets of this helicase remain to be discovered. We used an approach of tandem affinity protein purification (TAP) to identify novel functions and interaction partners of E1. We report here that the WD repeat protein p80 associates with the N-terminal portion of E1 from low- and high-risk anogenital HPV types and that this interaction is required for efficient maintenance of the viral episome in undifferentiated primary human keratinocytes.
MATERIALS AND METHODS
Plasmid constructions and mutagenesis.
The Xpress-p80 expression vector was a kind gift from Jae U. Jung (Harvard University, Boston, MA) and has been described previously (36). All HPV11 E1 constructs contained the previously reported mutations inactivating the major splicing site (8). The corresponding splicing site in HPV31 E1 was also inactivated by using two silent mutations (GCA GGT to GCT GGC). The proper expression of full-length, splice site-mutated HPV31 E1 was confirmed by Western blot analysis, and its nuclear localization was confirmed by confocal microscopy (data not shown).
(i) TAP fusions.
HPV11 E1 sequences were amplified by PCR and cloned into the vector pMzi (38) between the XhoI and NotI sites. For the TAP-HPV11 E1(353-649) construct, the simian virus 40 (SV40) nuclear localization signal sequence (5′-CCG AAG AAG AAA AGG AAG GTG-3′) was also inserted into pMzi between the XbaI and XhoI sites.
(ii) GST fusions.
PCR-amplified E1 fragments were inserted between the BamHI and EcoRI sites of plasmid AB-401, a modified version of pGEX-4T-1 (GE Healthcare) in which a sequence encoding a six-histidine tag has been inserted downstream of the glutathione S-transferase (GST) coding region. The N-terminal domains of different PV types were defined by amino acid sequence alignment with HPV11 E1(1-191) and are as follows: HPV1 E1(1-159), HPV6 E1(1-191), HPV16 E1(1-190), HPV18 E1(1-197), HPV31 E1(1-170), BPV1 E1(1-149), and CRPV E1(1-148).
(iii) Fluorescent protein fusions.
For the HPV11 E1N-green fluorescent protein (GFP) construct, the DNA sequence encoding amino acids (aa) 1 to 191 was PCR amplified and inserted into pGFP2-N1 (PerkinElmer) between the EcoRI and BamHI sites. The GFP-HPV11 E1(1-649) expression vector was obtained by cloning the full-length HPV11 E1 open reading frame (ORF) into pQBI25-FC1 (Qbiogene) between the BamHI and HindIII sites. The enhanced yellow fluorescent protein (EYFP)-HPV31 E1 plasmid was obtained by inserting the HPV31 E1 ORF between the XhoI and BamHI sites of the pEYFP-C1 vector (Clontech). To obtain the red fluorescent protein (RFP)-p80 expression plasmid, monomeric RFP1 (3) was first cloned between the KpnI and NotI sites of pcDNA3 and the p80-amplified ORF was inserted between the NotI and XbaI sites. Site-directed mutagenesis was performed by using the QuikChange site-directed mutagenenis kit (Stratagene). All DNA constructs were verified by sequencing. Sequences of primers and additional details on the construction of these plasmids are available upon request.
Cell culture and transfections.
The monkey epithelial cell line COS-7 as well as the human epithelial kidney cell line 293 (HEK293) and its derivative EcR-293, which expresses the ecdysone receptor, were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 2 mM l-glutamine. For the growth of stably transfected cell lines, the culture medium was supplemented with 30 μg/ml bleocin (for EcR-293 cells) and 300 μg/ml Geneticin (for EcR-293 cells expressing TAP-tagged versions of E1). Human foreskin keratinocytes (HFK) were maintained in keratinocyte growth medium (KGM; Clonetics) or in E medium in the presence of fibroblast feeders treated with mitomycin C (Boehringer Mannheim). Transfections of COS-7 and EcR-293 cells were performed by using the Lipofectamine 2000 reagent (Invitrogen), unless indicated otherwise. HFK were transfected by using the FuGENE 6 reagent (Roche) according to the manufacturer's protocol.
TAP.
TAP was performed essentially as described previously (19). Briefly, EcR-293 cells were transfected with TAP-tagged expression plasmids by using the calcium phosphate method and stable clones were selected with G418. Individual clones were tested for TAP-tagged protein expression after 24 h of induction with 3 μM ponasterone A (Invitrogen), an ecdysone analogue, by Western blotting using an anti-Rpb1 antibody (note that the specificity of this antibody is irrelevant; rather, it is the fact that it is an immunoglobulin G [IgG] antibody that makes it bind to the protein A portion of the TAP tag). For purification, whole-cell extracts (WCE) were prepared from about 7 to 8 g of cells as described previously (19) and dialyzed overnight (10 mM HEPES-NaOH, pH 7.9, 0.1 mM EDTA, 0.1 mM dithiothreitol [DTT], 0.1 mM potassium acetate, 10% glycerol). Purification on IgG Sepharose beads (GE Healthcare) was performed in buffer containing 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.1% Triton X-100, and 10% glycerol, and the protein complexes were eluted overnight with tobacco etch virus (TEV) protease (Invitrogen) (30 U/g of cells) in TEV buffer (10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.1% Triton X-100, 0.5 mM EDTA, 10% glycerol, 1 mM DTT). Binding to calmodulin-Sepharose beads (Stratagene) was performed in buffer containing 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM imidazole, 1 mM Mg acetate, 2 mM CaCl2, 0.1% Triton X-100, 10% glycerol, and 10 mM β-mercaptoethanol. Final elution was performed in 10 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM imidazole, 1 mM Mg acetate, 2 mM EGTA, and 10 mM β-mercaptoethanol.
Protein identification by MS.
Proteins copurifying with E1 were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with silver or Sypro Ruby (Bio-Rad), and gel slices were excised and digested with trypsin as previously described (19). Tryptic peptides were identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) with microcapillary reversed-phase high-pressure liquid chromatography coupled to a LCQ DecaXP (Thermo Finnigan) or a LTQ (Thermo Electron) quadrupole ion trap mass spectrometer with a nanospray interface. Resulting peptide MS/MS spectra were interpreted by using Mascot (Matrix Science) software and searched against proteins in the NCBI nonredundant protein database or the UniRef protein database (2). Many peptide sequences were confirmed by manual inspection of the spectrum.
Coimmunoprecipitation assays.
Transfected cells were lysed 48 h posttransfection in coimmunoprecipitation (co-IP) buffer {50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 5 mM EDTA, 1% Triton X-100, protease inhibitor cocktail, 1 mM DTT, 1 mM AEBSF [4-(2-aminoethyl)-benzenesulfonyl fluoride]} for 30 min. Protein concentration of cleared WCE was determined by using the Bio-Rad Bradford assay, and then 1 mg of WCE was incubated with 1 μg anti-GFP or anti-Xpress antibodies for 90 min. Proteins were precipitated for 3 h with 25 to 30 μl protein G-Sepharose (GE Healthcare). The beads were washed four times with co-IP buffer, and the bound proteins were analyzed by SDS-PAGE and Western blotting.
Antibodies and Western blotting.
The rabbit antiserum against the C-terminal domain of p80 was kindly provided by Jae U. Jung (Harvard University). The mixture of two mouse monoclonal antibodies against GFP was purchased from Roche, the mouse monoclonal β-tubulin antibody from Sigma-Aldrich, and the mouse monoclonal antibody against the Xpress epitope and the Alexa Fluor 633-conjugated antibody from Invitrogen. For Western blot analysis, proteins were transferred onto polyvinylidene difluoride membranes and detected by using horseradish peroxidase-conjugated donkey anti-rabbit (GE Healthcare), sheep anti-mouse (GE Healthcare), or goat anti-mouse (Sigma-Aldrich) secondary antibody and an enhanced chemiluminescence detection kit (GE Healthcare).
Confocal microscopy.
COS-7 cells were grown on coverslips. Twenty-four hours posttransfection, cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 when required and the background was quenched with 50 mM NH4Cl-phosphate-buffered saline (PBS). DNA was stained with TO-PRO-3 (Molecular Probes). For immunofluorescence studies, cells were incubated with anti-Xpress antibody diluted 1:500 in PBS-2% milk for 1.5 h, followed by 1 h of incubation with a 1:1,000 dilution of Alexa Fluor 633-conjugated secondary antibody. Cells were washed with PBS after each step. Cells were mounted by using Vectashield mounting medium (Vector Laboratories). Images were acquired by using an LSM 510 confocal laser coupled to an Axiovert 100M inverted scanning microscope (Zeiss, Toronto, Canada) and analyzed by using LSM Image Browser, version 3.2.0.70 (Zeiss, Toronto, Canada).
Purification of GST fusion proteins and pulldown assays.
GST fusion proteins were produced in Escherichia coli BL21(DE3) (Novagen) as previously described (42). GST pulldown assays were performed as described previously (42). GST-E1(2-332) fusion proteins were cleaved with thrombin to remove the GST moiety and further purified as described previously (12). All protein concentrations were determined by Bradford analysis.
Fluorescence polarization DNA binding assay.
Binding assays were performed as described previously (12) by using 15 nM fluorescein-labeled probe and the indicated concentrations of protein in the following buffer: 20 mM Tris-HCl (pH 7.6), 50 mM NaCl, 0.01% NP-40, and 1 mM DTT. Fluorescence readings were taken after an hour of incubation at room temperature by using a Wallac 1420 multilabel HTS counter (Victor3V) equipped with a 485-nm/535-nm filter set. Background fluorescence from the buffer was subtracted, and polarization values (P) were defined with the following equation: P = [I‖ − I(perp)]/[I‖ + I(perp)], where I‖ and I(perp) are the fluorescence intensities recorded in the parallel and perpendicular orientations respective to the excitation polarizer. Fluorescein-labeled oligonucleotides were purchased from Invitrogen, with the fluorophore attached at the 5′ end by a six-carbon linker. Duplex DNA probes were prepared by annealing each fluorescein-labeled oligonucleotide to a complementary oligonucleotide in a ratio of 1:1.5 as previously described (12).
Transient HPV DNA replication assay.
Short-term replication assays were performed essentially as described previously (56). Two days prior to transfection, C33A cells were seeded at 6 × 105 in a 10-cm dish. Cells were transfected with 3 μg of pSG5-E1, 1 μg of pSG5-E2, and 1 μg of pGL3-E1Luc, which contains the viral ori. Low-molecular-weight DNA was harvested 72 h posttransfection by the Hirt extraction method and subsequently digested with KpnI to linearize the origin-containing vector as well as with DpnI to digest any unreplicated DNA. Southern blot analysis was performed, and replicated DNAs were detected by hybridization with a 32P-labeled HPV31 E1Luc DNA probe, followed by autoradiography.
HPV31 genome maintenance and amplification assays.
Wild-type (WT) and mutant HPV31 genomes (accession number J04353) (13) were released from the pBR322 min plasmid (18) by restriction enzyme digestion, followed by unimolecular ligation with T4 DNA ligase (New England Biolabs). The genomes were then precipitated in 35% isopropyl alcohol and 10% NaCl and resuspended in TE (10 mM Tris-HCl, 1 mM EDTA, pH 7.5). Genomes were cotransfected with pSV2neo vector in HFK cells grown to 30% confluence in 10-cm dishes. After 24 h, the transfected cells were trypsinized and replated onto tissue culture dishes containing E medium with epidermal growth factor and J2 fibroblast feeder cells. After G418 selection was completed, HFK were suspended in 1.5% methylcellulose-containing E medium (methylcellulose solution was prepared as previously described [10]) to induce differentiation. Fibroblast feeders were removed prior to cell harvesting at 0, 24, and 48 h by 2 min of treatment with PBS containing 0.5 mM EDTA. Following centrifugation and washing, cells were resuspended in DNA lysis buffer (400 mM NaCl, 10 mM Tris-HCl, pH 7.4, 10 mM EDTA, 50 μg/ml RNase A, and 0.2% SDS) and incubated overnight at 37°C. Samples were then passed through an 18-gauge needle 10 times to shear the DNA, which was next extracted by phenol-chloroform, followed by ethanol precipitation. TE-resuspended DNA was finally analyzed by Southern blotting as previously described (48) to detect HPV31 genomic DNA.
RESULTS
Identification of the WDR48, the WD repeat protein p80, as a novel cellular interacting partner of the HPV11 E1 helicase.
TAP has become the method of choice for efficient purification of cellular protein complexes under native conditions. Therefore, a TAP procedure adapted to human cells (19) was used to isolate cellular proteins that associate with the HPV11 E1 helicase expressed in HEK293 cells, a cell line capable of supporting transient HPV DNA replication (4, 8, 46). The TAP tag used in these experiments consisted of two IgG binding domains from Staphylococcus aureus protein A and a calmodulin-binding peptide separated by a TEV protease cleavage site. This TAP tag was fused at the N or C terminus of the complete HPV11 E1 helicase, and both fusion proteins were stably expressed in EcR-293 cells under the control of an ecdysone-inducible promoter (Fig. 1A). Since we anticipated that the full-length HPV11 E1 (11E1FL) might be difficult to purify under native conditions, functional domains of the protein were also expressed in EcR-293 cells. Specifically, for subsequent purification, we chose to express the N-terminal regulatory domain alone (aa 1 to 191), a fragment spanning both the N-terminal domain and the OBD (aa 1 to 353), and the C-terminal helicase domain (aa 353 to 649); the latter was fused to the SV40 NLS to ensure its proper nuclear localization (Fig. 1A).
FIG. 1.

Identification of cellular proteins that interact with HPV11 E1. (A) EcR-293 stable cell lines expressing different TAP-tagged constructs of HPV11 E1 under the control of an ecdysone-inducible promoter. Protein expression was analyzed by Western blotting after a 24-h induction with (+) or without (−) 3 μM ponasterone A (Pon. A), an ecdysone analogue. The structures of the different TAP-tagged E1 proteins are diagrammed on the left. The amino acid boundaries of the N-terminal (N), OBD, and helicase domains are indicated. The location of the CBM is indicated by dark gray bars. The hatched bar in HPV11 E1(353-649) represents the SV40 nuclear localization sequence that was attached to this construct. (B) TAP-purified protein complexes from cells expressing HPV11 E1(1-191) and E1(1-353). Purified proteins were separated by SDS-PAGE and stained with Sypro Ruby. Major protein bands indicated by arrows were excised, trypsin digested, and identified by LC-MS/MS analysis. Some leaky expression of both TAP-tagged constructs led to the purification of E1 and its associated proteins even in uninduced EcR-293 cells, but in much smaller amounts. (C) Amino acid sequence of p80. The seven putative WD domains are aligned and shown in black boxes. A potential eight-WD repeat is highlighted in gray. Tryptic peptides identified by MS are in italics.
The levels of expression of the different E1 constructs before and after a 24-h induction with 3 μM of the ecdysone analogue ponasterone A are shown in Fig. 1A. Western blot analysis of HPV11 E1(1-353)-TAP consistently revealed three bands that were observed in all stable clones tested. The larger one was of the expected molecular weight for HPV11 E1(1-353)-TAP, whereas the two lower bands were smaller and probably corresponded to proteolytic fragments, since they were observed only upon induction with ponasterone A. TAP of E1-containing protein complexes was then attempted from all cell lines, under induced and noninduced conditions (see Materials and Methods). Purified protein complexes were separated by SDS-PAGE and visualized by Sypro Ruby staining. Unfortunately, we have been unable to purify 11E1FL or its C-terminal domain by using the standard TAP procedure. Although the exact reason for this is unclear, our results are consistent with our previous observation (53) and that of others (39) that recombinant HPV11 E1 is difficult to purify unless high concentrations of salt are used throughout all chromatographic steps. In order to avoid disrupting the integrity of E1-containing protein complexes, we did not consider the use of high-salt concentrations during the TAP procedure. Fortunately, TAP by using the N-terminal domain of HPV11 E1 (11E1N) or using this domain together with the OBD was successful. The compositions of the purified complexes analyzed by SDS-PAGE are shown in Fig. 1B. Major protein bands were cut, digested by trypsin, and identified by LC-MS/MS. This analysis led to the identification of two E1-associated proteins, namely, Cdk2 and the WD repeat protein p80 (WDR48 or p80). Cdk2, in complex with cyclin E or A, is known to interact with the N-terminal domain of HPV11 E1 through a conserved cyclin-binding motif, RxL, located between aa 123 and 127 (9, 31). The identification of Cdk2 validates the efficacy of our TAP procedure for the purification of E1-associated proteins. As for p80, little is known about its function. Structurally, its main distinguishing feature is the presence of seven or eight WD repeats in its N-terminal region (Fig. 1C). Functionally, p80 was first identified as an interaction partner of the herpesvirus saimiri tyrosine kinase-interacting protein (Tip), an association that appears to facilitate lysosomal vesicle formation (36). The amino acid sequence of p80 with the tryptic peptides identified by MS (corresponding to 40% of the whole protein sequence) is shown in Fig. 1C.
To confirm the interaction of E1 with p80, we investigated whether both proteins could be coimmunoprecipitated in transient-transfection experiments. Immunoprecipitations (IP) were performed on WCE made from HEK293 cells expressing 11E1N or 11E1FL fused to GFP, together with Xpress-tagged p80 (Fig. 2). In these experiments, E1 was precipitated by using a monoclonal antibody against GFP and the presence of p80 in the precipitate assessed by Western blotting with an anti-p80 polyclonal antibody. As shown in Fig. 2A, p80 was coimmunoprecipitated by 11E1N, confirming that both proteins do interact in vivo. p80 was also coimmunoprecipitated with 11E1FL, though at lower levels, presumably due to its lower levels of expression than those of 11E1N. Furthermore, the co-IP of 11E1FL with p80 as well as that of 11E1N with p80 was clearly detectable when p80 was immunoprecipitated with an anti-Xpress monoclonal antibody (Fig. 2B). Thus, our TAP procedure allowed the identification of the WD repeat protein p80 as a new cellular binding partner of HPV11 E1, an interaction that was confirmed by co-IP.
FIG. 2.
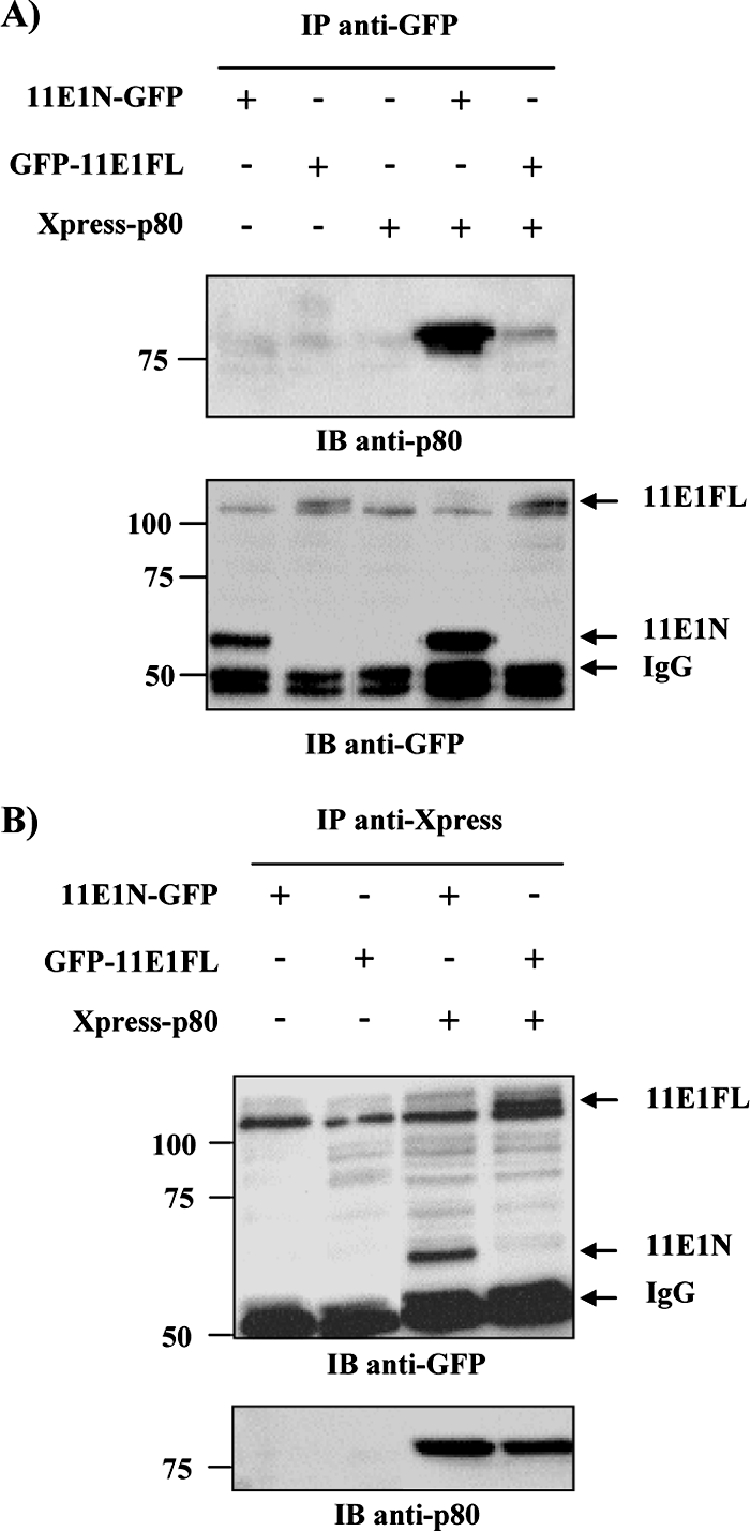
p80 interacts with HPV11 E1 in co-IP assays. co-IP of Xpress-fused p80 with GFP-fused HPV11 E1(1-191) (11E1N) and full-length HPV11 E1(1-649) (11E1FL). HEK293 cells were transfected with the indicated combination of expression vectors and harvested 48 h posttransfection, and WCE were immunoprecipitated by using either anti-GFP (A) or anti-Xpress (B) antibodies. IB, immunoblot; −, absence of; +, presence of.
Intracellular colocalization of p80 and HPV11 E1.
While E1 is primarily a nuclear protein, p80 has been reported to be in the cytoplasm and to partially localize to the late endosomal/lysosomal compartments (36). This report led us to investigate the cellular colocalization of these proteins by confocal microscopy using GFP fusions of 11E1N and 11E1FL and Xpress-tagged p80. The expression and migration of these fusion proteins at their predicted molecular weights were first verified by Western blot analysis using anti-GFP or anti-Xpress antibodies (Fig. 3A). Localization studies were done by using three different cell lines (HEK293, C33A, and COS-7) with comparable results. Figure 3B shows the pictures obtained for COS-7 cells. As anticipated, both the N-terminal domain, which retains the bipartite NLS, and 11E1FL were predominantly nuclear. Also, as previously reported (36), p80 was observed almost exclusively in the cytoplasm, although it did not present the reported punctate distribution of a protein localizing to endosomal compartments (Fig. 3B). We also obtained the same diffuse cytoplasmic pattern by using p80 fused to RFP, rather than Xpress, in HeLa, HEK293, and COS-7 cells. Furthermore, we did not observe the colocalization of p80 with lysosome-associated membrane protein 1 (LAMP-1), a marker of late endosomes (data not shown). Thus, under our experimental conditions, p80 is located throughout the cytoplasm.
FIG. 3.

Intracellular localization of p80 and HPV11 E1 in COS-7 cells. (A) Expression levels of GFP-E1 and p80 fusion proteins. COS-7 cells were transiently transfected with either 11E1N-GFP, GFP-11E1FL, or Xpress-p80 expression vector. Cells were harvested 24 h posttransfection, and WCE were submitted to Western blot analysis. (B) Cytoplasmic localization of p80 and nuclear localization of 11E1N-GFP and GFP-11E1FL in COS-7 cells. DNA was stained with TO-PRO-3. (C) Cellular colocalization of Xpress-p80 with 11E1N-GFP, GFP-11E1FL, or 11E1NmNLS-GFP in COS-7 cells. IB, immunoblot.
11E1N was found to colocalize with p80 in the three cell lines tested. Strikingly, whereas p80 is cytoplasmic in the absence of E1, it relocalized to the nucleus when coexpressed with 11E1N (Fig. 3C). In fact, in cells where 11E1N was expressed, p80 could be found exclusively in the nucleus. This result does not necessarily imply that all of p80 is translocated to the nucleus during HPV infection, as this may depend on the amount of E1 produced. It does suggest, however, that the interaction of 11E1N with p80 is neither weak nor transient. The redistribution of p80 by 11E1N was also observed in two epithelial cell lines, HeLa (HPV18 positive) and C33A (HPV negative) (data not shown). Importantly, the relocalization of p80 was also observed for 11E1FL (Fig. 3C), although some p80 remained in the cytoplasm. The fact that 11E1FL is expressed at much lower levels than is 11E1N (Fig. 2A and data not shown) is likely the reason why some p80 was not relocalized to the nuclear compartment. We surmised that the E1-p80 complex forms in the cytoplasm and is then imported into the nucleus via the E1 NLS. To validate this hypothesis, we showed that an 11E1N protein with a mutant NLS (K83G/R84G) was unable to relocalize p80 to the nucleus (Fig. 3C). Collectively, these studies provided further evidence that E1 and p80 form a complex in vivo, which is imported into the nucleus via the E1 NLS.
p80 binds to a short 30-aa sequence in the N-terminal domain of HPV11 E1.
To further confirm the interaction of E1 with p80 and develop a rapid method to map the region of E1 implicated in p80 binding, we tested whether both proteins could interact in GST pulldown assays. These experiments were initially performed by using three different fragments of HPV11 E1 fused to GST (Fig. 4A). Affinity columns were prepared by immobilizing GST and GST-E1 fusion proteins on glutathione-Sepharose beads at a concentration of 4 mg/ml and then loaded with EcR-293 WCE and washed, and the bound proteins were eluted with buffer containing reduced glutathione. These eluates were then analyzed by Western blotting for the presence of p80 and Cdk2 as a control (Fig. 4A). These experiments confirmed that 11E1N (aa 1 to 191) interacts specifically with p80, in contrast to the interaction of the OBD (aa 191 to 353), which was used as a negative control. Interestingly, a fusion protein containing the N-terminal domain and the OBD, but lacking the first 72 aa, did not interact with p80, although it retained the ability to bind Cdk2. This finding suggested that the p80 binding site on E1 is contained, at least in part, within these first 72 aa. GST pulldown assays were also used to verify that the N-terminal domain of HPV11 E1 (aa 1 to 191) and the corresponding region of HPV31 E1 (aa 1 to 170) can bind p80 from the two cervical carcinoma cell lines HeLa (HPV18 positive) and C33A (HPV negative) (Fig. 4B).
FIG. 4.

Mapping of a p80 binding domain on E1 and interaction of p80 with E1 from different PV types. (A) p80 interacts with the N-terminal domain of HPV11 E1. The indicated HPV11 E1 fragments were expressed as GST fusions in bacteria and used in pulldown assays using EcR-293 WCE. Bound proteins were separated by SDS-PAGE and analyzed by Western blotting using antibodies directed against p80 or against Cdk2 as a positive control. The bottom panel shows a Coomassie blue-stained SDS-PAGE gel of the purified GST-E1 proteins that were first immobilized and then eluted from the beads in these pulldown experiments. The sizes of molecular weight markers (in thousands) are indicated on the left. (B) Binding of p80 from HeLa and C33A cells to the N-terminal domains of HPV11 and HPV31 E1. The indicated HPV11 and corresponding HPV31 E1 fragments were expressed as GST fusions and used in pulldown assays using HeLa and C33A WCE, as indicated. Bound proteins were separated by SDS-PAGE and analyzed by Western blotting using antibodies directed against p80. The bottom panel shows an SDS-PAGE gel of the GST-E1 proteins eluted from the beads. (C) Mapping of p80 interaction domain on HPV11 E1. Short N-terminal (N) peptides of HPV11 E1 were expressed as GST fusions in bacteria and tested for p80 binding in pulldown assays as indicated above. The location of the p80 interaction domain on E1 is illustrated. The bottom panel shows an SDS-PAGE gel of the purified GST-E1 proteins eluted from the beads. (D) Interaction of p80 with E1 from different PV types. E1 N-terminal domains of one cutaneous HPV type (1), two genital low-risk HPV (types 6 and 11), three genital high-risk HPV (types 16, 18, and 31), BPV1, and CRPV were expressed as GST fusions and tested for p80 binding in pulldown assays as described above. The bottom panel shows an SDS-PAGE gel of the purified GST-E1 proteins eluted from the beads.
To delineate further the minimal region of E1 required for p80 binding, a series of C-terminal truncations, starting at aa 80, were constructed and tested in pulldown assays for their abilities to bind p80 out of an extract (Fig. 4C). These studies indicated that aa 1 to 40 of 11E1 are sufficient for binding p80. Weak binding to p80 was also observed for 11E1(1-35). Note that in all of these experiments and subsequent ones, the eluted proteins were analyzed by SDS-PAGE and stained with Coomassie blue to ascertain that a lack of interaction was not due to a failure of the GST fusion proteins to be properly eluted (Fig. 4). Similarly, a series of N-terminal truncations in 11E1(1-40) were created (Fig. 4C). Binding to p80 was retained when the first 10 residues were deleted, but it was lost upon removal of the first 15 residues (Fig. 4C). Thus, the minimal p80-interacting region on HPV11 E1 spans aa 10 to 40. This region is predicted to fold as a short β-sheet (spanning aa 17 to 25) by the PROF secondary structure prediction program (40, 41).
The interaction between E1 and p80 is conserved among genital HPV types.
The GST pulldown assay described above was used to test whether the interaction of E1 with p80 was conserved among different types of PV (Fig. 4D). Specifically, we tested whether p80 could bind to the E1 N-terminal domains from one cutaneous type (HPV1), two genital low-risk types (HPV6 and -11), three high-risk types (HPV16, -18, and -31), and two prototypical animal types (bovine PV type 1 [BPV1] and cottontail rabbit PV [CRPV]). As shown in Fig. 4D, the E1 proteins from all genital HPVs tested, including those from low- and high-risk types, bound to p80 (Fig. 4D). In contrast, p80 binding was detected with neither HPV1 nor BPV1 or CRPV E1. This result is consistent with the sequence of these E1 proteins being substantially different from those of the anogenital types in the region corresponding to the p80 binding domain (Fig. 5A). In addition to differences in primary sequence, both HPV1 and CRPV E1 also lack residues at the N terminus of the p80 binding domain that were shown to be required for binding in the deletion analysis presented above (Fig. 4C). Thus, the interaction between E1 and p80 appears to be a conserved and specific feature of HPVs infecting genital mucosal epithelia.
FIG. 5.

Amino acid substitutions in HPV11 E1 that abrogate its interaction with p80. (A) The top panel shows amino acid sequence alignment of the p80 interaction domains of E1 from different anogenital HPV types, and the lower panel shows the corresponding region from three cutaneous PV types (HPV1, CRPV, and BPV) which do not bind p80. Boxed amino acids were mutated to alanine. (B) Double-alanine substitutions were introduced into GST-11E1(1-40), and the resulting mutant proteins were used in pulldown assays with EcR-293 WCE. Bound proteins were separated by SDS-PAGE and analyzed by Western blotting using a polyclonal anti-p80 antibody. (C) Alanine mutants in GFP-11E1FL abrogate its interaction with p80 in co-IP assays. Plasmids containing GFP-fused WT or mutant 11E1FL were cotransfected with Xpress-p80 expression vector in HEK293 cells. WCE were prepared 48 h posttransfection and submitted to IP by using an anti-Xpress antibody. Precipitated proteins were analyzed by Western blot analysis using anti-GFP antibody or anti-p80 antibody as an IP control. The lower panel shows the amount of input GFP-E1 proteins present in the WCE prior to IP. IB, immunoblot; −, absence of; +, presence of.
Identification of amino acid substitutions in HPV11 E1 that abrogate its interaction with p80.
For the purpose of testing the biological significance of the E1-p80 interaction, we set out to identify mutant E1 proteins specifically defective in p80 binding. Residues of the E1 p80-binding domain that are highly conserved among genital HPV types were changed to alanine, and the resulting mutant proteins were tested for their abilities to bind p80. Five double-amino-acid substitutions (Fig. 5A) were introduced into GST-11E1(1-40) and tested for p80 binding in pulldown assays. As shown in Fig. 5B, three of the five double-amino-acid substitutions (WF, VE, and IV) severely impaired p80 binding. Additionally, co-IP experiments were performed to confirm that these three substitutions do indeed affect p80 binding (Fig. 5C). Plasmids expressing GFP-11E1FL (either the WT or one of the three mutant proteins) were cotransfected with the Xpress-p80 expression vector in HEK293 cells. IP of p80 was carried out 48 h posttransfection by using an anti-Xpress antibody, and the precipitates were analyzed for the presence of E1 as well as p80 protein as a control by Western blotting. As shown in Fig. 5C, none of the three 11E1FL mutant proteins coprecipitated with p80 compared to WT E1, indicating that they are also unable to associate with p80 in vivo. Finally, we investigated the effect of these three substitutions on the ability of 11E1N-GFP to relocalize RFP-p80 from the cytoplasm to the nucleus (Fig. 6). p80 was found to remain cytoplasmic in cells expressing the three mutant E1 proteins in contrast to the situation in cells expressing WT 11E1N, where it was relocalized to the nucleus. From a mechanistic point of view, this result provided strong evidence that the relocalization of p80 by 11E1N depends on an interaction between both proteins.
FIG. 6.
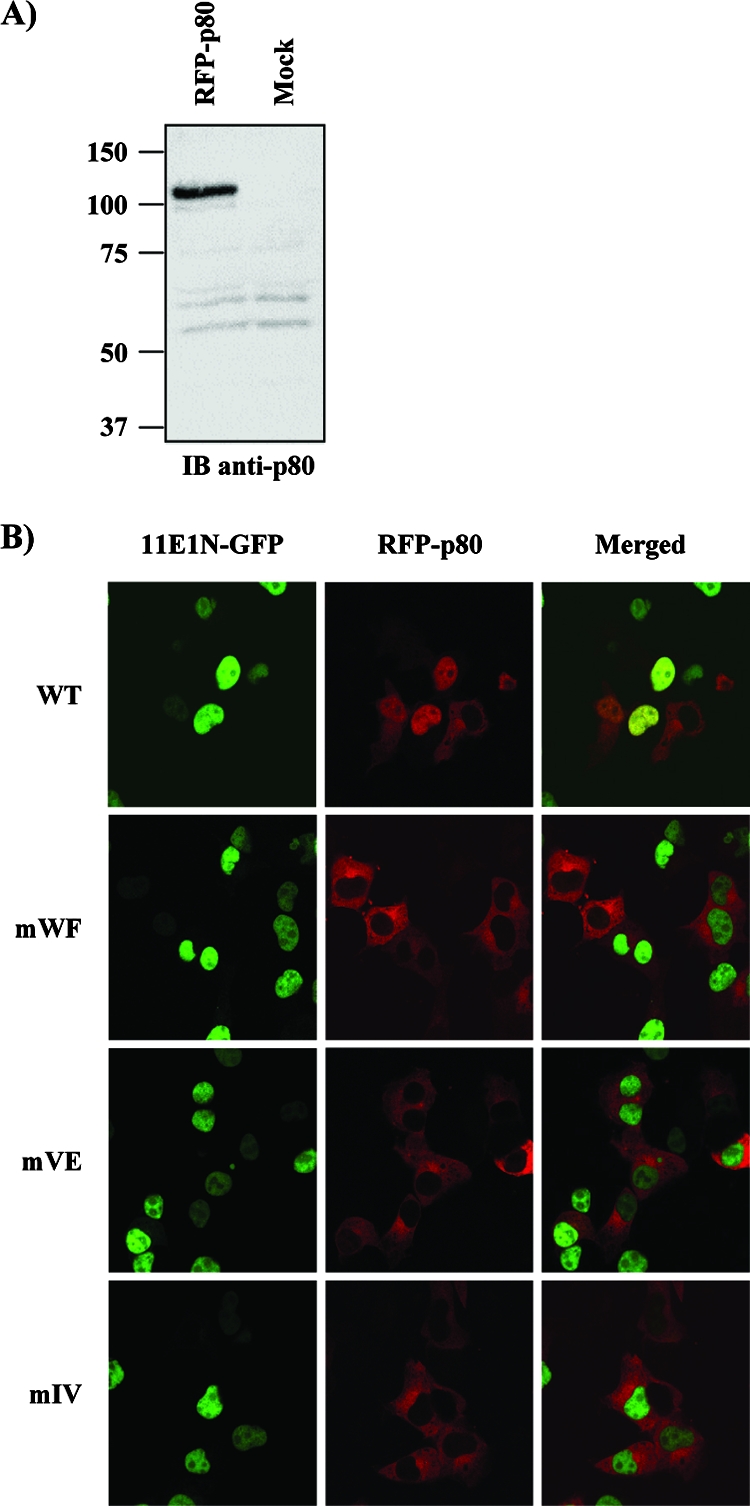
Mutant E1 proteins fail to relocalize p80 from the cytoplasm to the nucleus. (A) Expression of RFP-p80 fusion protein. COS-7 cells were transiently transfected with the indicated expression vectors and harvested 24 h posttransfection, and WCE were submitted to Western blot analysis with an anti-p80 antibody. (B) COS-7 cells were cotransfected with RFP-p80 and WT or mutant 11E1N-GFP expression vectors for cellular localization studies.
Construction of mutant HPV31 E1 proteins defective for p80 binding.
Because functional assays involving the immortalization of primary keratinocytes with cloned HPV genomes can be performed only with high-risk HPV types, we engineered the same three amino acid substitutions in HPV31 E1 as described above. First, we introduced them in the N-terminal domain of HPV31 (aa 1 to 170) and confirmed by GST pulldown assays that they also affect binding to p80 in this context (Fig. 7A). As a control, we showed that the mutant proteins retained the ability to bind Cdk2 (Fig. 7A), thereby indicating that their overall structure is not grossly altered. Next, we performed a series of confocal microscopy studies to test the effects of these substitutions on the intracellular localization of full-length HPV31 E1 (31E1FL) fused to EYFP in COS-7 cells. As anticipated, the mutant E1 proteins still accumulated in the nucleus, like in their WT counterpart (data not shown and Fig. 7B). Satisfyingly, we found that WT EYFP-31E1FL could relocalize p80 to the nucleus, whereas the mutant proteins could not (Fig. 7B). Together, these results indicate that these three amino acid substitutions do affect the ability of HPV31 E1 to interact with p80 in vitro as well as in vivo.
FIG. 7.
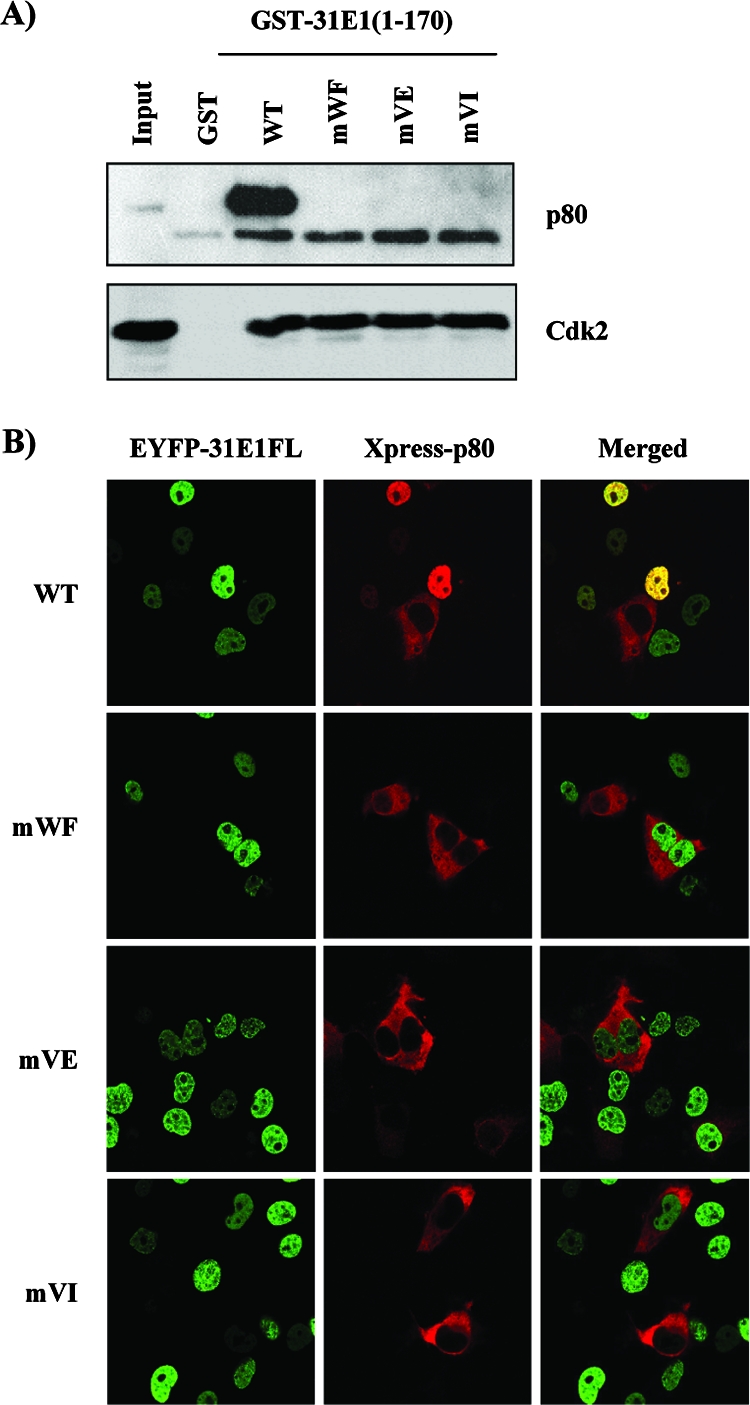
HPV31 E1 proteins defective for p80 interaction. (A) The three E1 double-amino-acid substitutions previously shown in HPV11 E1 to abrogate interaction with p80 were introduced into a fusion of the HPV31 E1 N-terminal domain to GST. The resulting mutant proteins were purified from bacteria and used in pulldown experiments with EcR-293 WCE. Bound proteins were analyzed by Western blot analysis using a polyclonal anti-p80 antibody. Western blot analysis using antibody against Cdk2 is shown as control. (B) Effect of E1 substitutions on p80 cellular localization. The substitutions were introduced into pEYFP-HPV31 E1 expression vector. WT or mutant EYFP-31E1 plasmids were cotransfected with Xpress-p80 in COS-7 cells, and the cellular localization of the encoded proteins was analyzed by confocal microscopy.
Transient replication activity of HPV31 E1 proteins defective for p80 binding.
To investigate whether the E1-p80 interaction is needed for viral DNA replication, we tested whether the p80 binding-defective HPV31 E1 mutant proteins described above were capable of supporting transient HPV DNA replication in C33A cells. As shown in Fig. 8A, all three mutant proteins could support transient replication, albeit at a reduced level compared to that of the WT protein. These results suggested that the interaction of E1 with p80 is not essential for transient DNA replication, although it may contribute to its overall efficiency (see Discussion). To investigate whether this decrease in replication was due to a reduced accumulation of the mutant proteins, we determined their steady-state levels in C33A cells relative to that of the WT. However, since there are no anti-E1 antibodies suitable for such studies, we investigated the accumulation of WT and mutant E1 fused to EYFP, which can be visualized by Western blotting with an anti-GFP antibody. These studies showed that all three mutant proteins were expressed at levels comparable to that of the WT at 24, 48, and 72 h posttransfection (Fig. 8B), indicating that their replication defects are not due to reduced accumulation. In fact, we noted consistently that the WF mutant and, to a lesser extent, the VI mutant accumulated at slightly higher levels than WT E1 did. We also considered the possibility that the three double-amino-acid substitutions in HPV31 E1 reduce transient replication by affecting the binding of E1 to the origin. This possibility was tested in vitro. We expressed and purified from bacteria a fragment of HPV31 E1 (aa 2 to 332) encompassing both the N-terminal domain and the OBD (Fig. 9A) as well as the corresponding mutant fragments and tested them for their abilities to bind DNA in a fluorescence polarization assay (12). We monitored the binding of these proteins to two different fluorescein-labeled DNA probes, one containing and the other lacking two E1 binding sites (E1BS). As can be seen in Fig. 9C, little binding of these proteins was observed to the mutant probe, as expected. More importantly, all three mutant fragments could bind to the 2E1BS probe as well as or slightly better than the WT E1 fragment could. These results suggest that the replication defect imposed by the p80-binding substitutions is not due to a reduced affinity for the origin. Collectively, the results presented above suggest that the approximately 50% reduction in transient replication observed for the three E1 mutant proteins is due neither to their failure to accumulate in vivo nor to their failure to bind to the origin. The simplest interpretation is that their reduced replication capacities are due, at least in part, to their inabilities to bind to p80. This result would suggest that the interaction of E1 with p80, although not essential for transient DNA replication, makes this process more efficient.
FIG. 8.
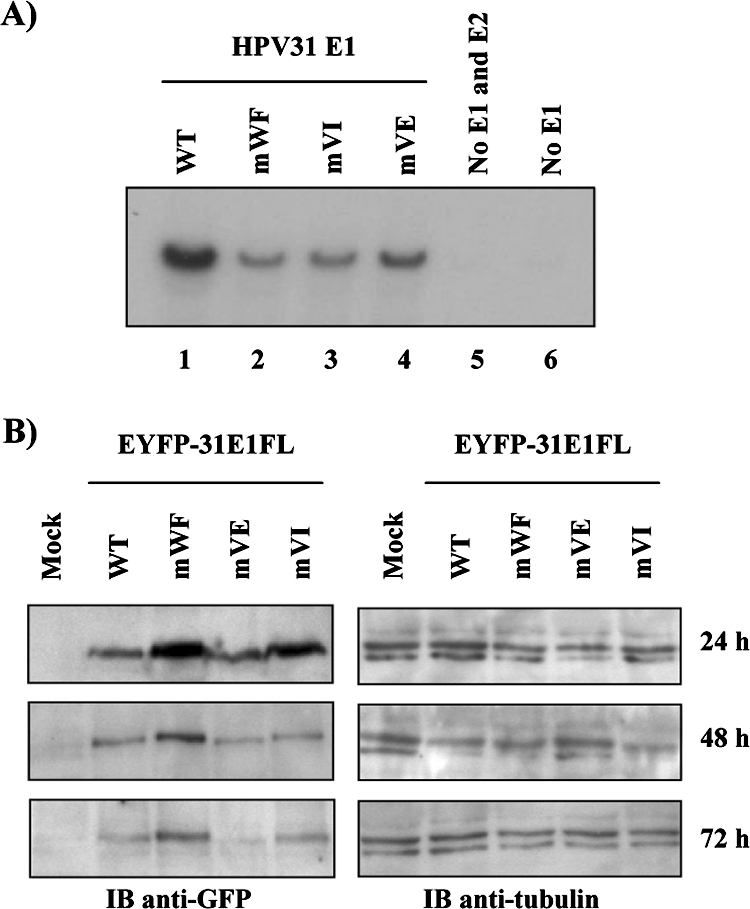
Transient replication activity of mutant HPV31 E1 proteins. (A) Transient HPV DNA replication. C33A cells were transfected with 3 μg of the WT (lane 1) or the indicated mutant E1 (lanes 2 to 4) expression vector plus 1 μg of the E2 expression vector and 1 μg of the HPV31Luc vector, which contains the ori. As negative controls, cells were transfected with the origin-containing vector alone (no E1 and E2 [lane 5]) or with the E2 and HPV31Luc vectors (no E1 [lane 6]). (B) Accumulation of WT and mutant EYFP-HPV31 E1. The levels of the indicated E1 proteins in transfected C33A cells were detected by Western blotting with an anti-GFP antibody 24, 48, and 72 h posttransfection (left panels). Mock-transfected cells were used as a negative control. Western blot analysis with an anti-β-tubulin antibody (right panels) was performed to ensure that equal amounts of total cellular proteins were loaded in each lane.
FIG. 9.

Effect of amino acid substitutions that abrogate the p80 interaction on binding of HPV31 E1 to the origin in vitro. (A) Schematic representations of the full-length HPV31 E1 protein and of the 332-aa-long fragment that was expressed and purified from E. coli either in WT form or containing the WF, VE, or VI substitution. The right panel shows a Coomassie blue-stained gel of the purified fragments (3 μg of each were loaded on the gel). The band migrating at about 70 kDa is likely DnaK. The positions and sizes (in thousands) of molecular weight markers (MWM) are shown on the left. N, N-terminal domain. (B) Nucleotide sequences of the two fluorescein-labeled probes containing, respectively, two E1 binding sites (2E1BS, underlined and bolded) and two mutated sites. Only the sequence of the labeled DNA strand is shown. (C) Fluorescence polarization binding isotherms were determined with 15 nM of the indicated probe and increasing concentrations of the indicated E1 fragment. Each isotherm was determined in triplicate; errors bars (indicating standard deviations) are barely visible on the graph as they are smaller than the symbols. Polarization values were determined as described in Materials and Methods.
Ability of the mutant HPV31 E1 proteins to support maintenance and amplification of the viral episome in primary human keratinocytes.
To further investigate the significance of the E1-p80 interaction in the HPV life cycle, we examined whether the three amino acid substitutions in E1 that abrogate p80 binding had any effect on the maintenance of the HPV31 episome in HFK. To do this, we transfected circular WT and mutant HPV31 genomes into primary HFK, along with a G418 resistance marker, and subsequently selected and expanded drug-resistant immortalized cell clones. Southern blotting was then used to determine the status of the viral genome in these cell clones prior to or following 24 or 48 h of growth in methylcellulose to induce cellular differentiation and amplification of the viral episome. As shown in Fig. 10, two of the mutant genomes, mWF and mVE, were not present in episomal form, whereas the third one, mVI, was present at a much reduced copy number compared to that of the WT. For the mVI mutant, some levels of amplification were detected upon differentiation in methylcellulose. Similar results were obtained with HFKs from a different donor (data not shown). Collectively, these results suggest that the E1-p80 interaction is important for maintaining WT levels of episomes in undifferentiated cells but may not be essential for the differentiation-dependent amplification of the genome.
FIG. 10.

Maintenance and amplification of mutant HPV31 genomes in primary human keratinocytes. Southern blot analysis showing the status of the viral genome in HFK immortalized with either WT HPV31 (WT) or mutant derivatives bearing the indicated E1 amino acid substitutions that abrogate p80 binding. HPV31-immortalized HFK were induced to differentiate in methylcellulose-containing medium for 24 and 48 h, as indicated.
DISCUSSION
In this study, we used a TAP approach coupled to MS to identify cellular interacting partners of the E1 helicase from HPV11. This led us to the discovery of a novel interaction between the N-terminal 40 aa of HPV11 E1 and the WD repeat protein p80, an interaction that is specific to low- and high-risk genital HPV types. Our study of E1 mutants defective in p80 binding suggests that this interaction is important for maintenance of the viral episome in undifferentiated keratinocytes.
Identification of p80 as a new cellular target of the E1 helicase from genital HPV.
The purification of E1-associated cellular proteins yielded p80 as a new interaction partner of E1, a finding confirmed by co-IP and colocalization studies as well as by protein affinity chromatography (GST pulldowns). TAP of E1 also identified Cdk2, which was already known to interact with E1 in association with cyclin A or E through a conserved CBM, thus validating our approach. p80 is a protein whose cellular function remains largely unknown. It is expressed in most tissues, including skin. Furthermore, we have shown that the endogenous p80 protein can be purified by GST-E1 affinity chromatography not only from HEK293 cells but also from the HeLa and C33A cell lines (Fig. 4B). The main structural features of p80 are the presence of seven or eight WD repeats in the N-terminal half of the protein and of a putative coiled-coil domain in the C-terminal part. WD repeat domains are found in 1 or 2% of eukaryotic proteins of many diverse functions. Most structurally characterized WD repeat proteins contain seven such repeats, although not all are readily identifiable by sequence alignment. These repeats fold into a seven-bladed β-propeller that serves as a stable platform for protein interactions (reviewed in references 26 and 43). Accordingly, our preliminary mapping data suggest that the WD repeats of p80 are mediating its interaction with E1 (data not shown).
Characterization of the E1-p80 interaction.
We determined that the minimal p80 binding domain on E1 spans aa 10 to 40. This region contains many nonpolar residues consistent with it being a hydrophobic protein interaction interface. By mutagenesis of residues conserved between genital HPV types, we identified three amino acid doublets critical for p80 interaction that lie in a region predicted to fold as a β-sheet (40, 41). Collectively, these observations suggest that E1 binds to p80 through a hydrophobic β-sheet. Our data also point toward this interaction being relatively strong and stable. To begin with, p80 was copurified with E1 by TAP in nearly stoichiometric amounts (Fig. 1B). Furthermore, substantial quantities of p80 could also be purified from WCE by GST-E1 affinity chromatography (Fig. 4 and 7 and data not shown). p80 is in fact the predominant protein in E1 affinity column eluates analyzed by SDS-PAGE and silver staining (data not shown). Lastly, the fact that p80 can be efficiently translocated from the cytoplasm to the nucleus (Fig. 6 and 7) also argues for a stable interaction. The relocalization of p80 by the 11E1N was highly efficient such that almost all of p80 was nuclear in cells expressing this E1 fragment. If the interaction was transient, we would expect a fraction of p80 to remain cytoplasmic. We have begun to address the mechanism by which p80 is redistributed to the nuclear compartment. Thus far, we have gathered evidence that the interaction of E1 with p80 is needed for this redistribution, as mutant E1 proteins defective in p80 binding do not relocalize it to the nucleus. We have also shown that the integrity of the E1 NLS is important for p80 relocalization. Thus, our current model is that the E1-p80 complex forms in the cytoplasm and is imported to the nucleus via the E1 NLS. As HPV E1 has been shown to shuttle between the nucleus and cytoplasm (9), there might be many opportunities for E1 to interact with p80 and efficiently bring it into the nucleus.
Known functions of p80.
p80 was first isolated as a cellular protein that associates with the tyrosine kinase-interacting protein, Tip, from the lymphotropic herpesvirus saimiri (36). Tip, as its name suggests, also associates with a kinase, Lck, involved in T-cell receptor (TCR) signaling (35, 36). Interestingly, the interaction of Tip with Lck and p80, through separate domains, results in lower cell surface expression of the TCR and of CD4, respectively, perhaps as a means of immune evasion. The interaction of Tip with p80 was found to induce the formation of large cytoplasmic vesicles into which the TCR is recruited for downregulation (35, 36). The group which performed these studies also reported that p80 is localized to the late endosome and lysosome. Specifically, they showed by immunofluorescence that Xpress-tagged p80 displays a punctate staining in COS-1, HEK293T, and Jurkat T cell and colocalizes significantly with LAMP-1 in Jurkat T cells. However, in our hands, the same Xpress-tagged p80, or p80 fused to RFP, was distributed throughout the cytoplasm in all the cell lines tested (HeLa, C33A, HEK293, and COS-7 cells) and did not colocalize with LAMP-1 (data not shown). Although we do not know the reason for this discrepancy, it may be related to different levels of p80 expression, which we kept as low as possible in our experiments.
Possible functions of the E1-p80 interaction in the HPV life cycle.
To address the function of the E1-p80 interaction, we took the approach of identifying amino acid substitutions in E1 that specifically abrogate p80 binding and examined their effects on the maintenance and differentiation-dependent amplification of the HPV31 episome in immortalized HFK. Three double-amino-acid substitutions were isolated, within a 10-aa stretch of E1, which abrogated its interaction with p80 but had no effect on its binding to Cdk2 or nuclear localization. Thus, all three substitutions appeared to specifically impair p80 binding. Mutant E1 proteins bearing these substitutions were capable of supporting transient replication, albeit at an approximately 50% reduced level. This result indicated that the interaction of E1 with p80 is not essential for viral DNA replication, although it likely contributes to its overall efficiency. The suggestion that the interaction of E1 with p80 is not essential for replication is consistent with our previous finding that a mutant HPV11 E1 protein lacking the N-terminal 166 aa, and thus missing the p80 binding site, is almost as active as WT E1 is in supporting HPV DNA replication in vitro (1). The fact that the N-terminal domain is dispensable in vitro (1), albeit not in vivo (46; this study), is consistent with this region playing a regulatory rather than a catalytic role. Accordingly, this region has been shown to contain NLS and nuclear export sequences as well as a binding motif for cyclin A/cyclin E-Cdk2 and phosphorylation sites for this kinase (9, 16, 24). Here we provide evidence that it also contains a binding site for p80.
Our finding that p80 interacts with the E1 proteins of anogenital HPV types, but not those of cutaneous PV types, is intriguing. An inspection of the amino acid sequences of the cutaneous E1s revealed that they differ substantially from those of anogenital types in the region of the p80-binding site (Fig. 5A), thereby accounting for their inabilities to interact with p80. The fact that cutaneous types can replicate even though their E1 proteins do not interact with p80 further supports the notion that the E1-p80 interaction is not critical for viral DNA replication but rather likely plays a regulatory role in the case of anogenital types. If so, this role suggests that the E1 proteins of anogenital and cutaneous types are regulated somewhat differently. This notion is not unprecedented. For example, it has been found that phosphorylation by cyclinA/cyclinE-Cdk2 inhibits the nuclear export of HPV11 E1 (9), whereas it promotes the export of the BPV protein (16). It is intriguing to speculate that differences in the regulation of E1 may reflect, in part, the heterogeneity observed between the life cycles of anogenital HPVs and those of cutaneous types, including HPV1, CRPV, and BPV. For instance, while anogenital types tend to initiate viral genome amplification and other late events only in the upper layers of the epithelium, cutaneous types do so in the lower layers (37).
To investigate the role of the E1-p80 interaction in the HPV life cycle, we first tested whether mutant HPV31 genomes bearing the p80-binding substitutions could be maintained in primary keratinocytes. These studies revealed that two of the mutant genomes (WF and VE) could not be maintained in detectable amounts, whereas the third one (VI) was present only at a low copy number in immortalized keratinocytes. It is entirely possible that the maintenance defect of these genomes is a direct consequence of the reduced replication capacity of the encoded E1. Indeed, it would not be surprising that optimal viral DNA replication would be necessary for the long-term maintenance of the episome in infected cells and, thus, that a 50% reduction in transient replication activity would be deleterious over several cell generations. However, at this point we cannot rule out the possibility that the E1-p80 interaction plays additional roles in episomal maintenance, such as in the segregation of the episome. Regardless, our results suggest that the E1-p80 interaction is important for long-term genome maintenance. From this suggestion, we surmise that interfering with the E1-p80 interaction would result in the loss of the viral episome from infected cells over time and, hence, might be a valuable antiviral strategy for the treatment of HPV infections.
It is important to realize that our assessment of the role of p80 in the HPV life cycle is currently limited to the analysis of specific amino acid substitutions in E1 that prevent p80 binding. To the extent that they have been characterized, these E1 substitutions abrogate primarily p80 binding and do not have a major effect on the steady-state accumulation of E1 or its binding to the origin (Fig. 8 and 9). However, we cannot be absolutely certain that they do not affect another function of E1 needed for episomal maintenance. Ideally, we would have liked to address the importance of p80 in the HPV life cycle by downregulating its expression by using RNA interference and determining how this downregulation would affect the maintenance of the episome in keratinocytes. However, our preliminary findings indicate that the downregulation of p80 with short hairpin RNAs impairs cellular proliferation of both HPV-positive (HeLa) and HPV-negative (C33A) cells (M. Lehoux and J. Archambault, unpublished data), suggesting that p80 may be encoded by an essential gene. The negative effect of p80 short hairpin RNAs on cell proliferation would obviously complicate the interpretation of any effect these short hairpin RNAs may have on episomal maintenance in long-term or transient-transfection studies, as the downregulation of any essential gene may affect viral DNA replication indirectly. Future studies in our laboratories will be aimed at determining why p80 is needed for cellular proliferation, with the hope that this determination may also provide insights on its role in the HPV life cycle.
Interestingly, we have shown recently that E1 contains a conserved and functional DxxD caspase-3 cleavage site located immediately C-terminal of the p80-binding domain and that caspase-3 is activated in HPV31-containing keratinocytes upon cellular differentiation (33). These and other findings led us to propose a model where capase-3 would cleave E1 in differentiated keratinocytes, resulting in a truncated protein lacking the p80-binding domain. Interestingly, we found that an HPV31 genome carrying a mutation in the E1 caspase cleavage site could still be maintained at WT levels in undifferentiated keratinocytes but could not be amplified upon differentiation. These observations suggested that cleavage of E1 by caspase-3, resulting in the truncation of the p80-binding site, is needed for amplification of the viral genome. From these results, one could speculate that caspase-mediated removal of the p80-binding site is important for viral genome amplification. Characterization of the truncated E1 protein and how it differs from full-length E1 is ongoing in our laboratories, as it may shed light on the mechanism of genome amplification and, indirectly, on the function of the p80-binding site.
Acknowledgments
We thank S. Bourassa and I. Kelly from the Eastern Quebec Proteomics Center for the identification of proteins by MS as well as Jae U. Jung (Harvard University) for the gift of p80 antibodies and expression vectors.
This work was supported by grants from the Canadian Institutes for Health Research to J.A. and B.C. and from the National Cancer Institute to L.A.L. A.C.-M. was supported by a studentship from the Fonds de la Recherche en Santé du Québec (FRSQ). C.M. was supported by a postdoctoral fellowship from the American Cancer Society. G.G.P. holds a Canada Research Chair in Proteomics. J.A. is a senior scholar from the FRSQ.
Footnotes
Published ahead of print on 21 November 2007.
REFERENCES
- 1.Amin, A. A., S. Titolo, A. Pelletier, D. Fink, M. G. Cordingley, and J. Archambault. 2000. Identification of domains of the HPV11 E1 protein required for DNA replication in vitro. Virology 272137-150. [DOI] [PubMed] [Google Scholar]
- 2.Bairoch, A., R. Apweiler, C. H. Wu, W. C. Barker, B. Boeckmann, S. Ferro, E. Gasteiger, H. Huang, R. Lopez, M. Magrane, M. J. Martin, D. A. Natale, C. O'Donovan, N. Redaschi, and L. S. Yeh. 2005. The Universal Protein Resource (UniProt). Nucleic Acids Res. 33D154-D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 997877-7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiang, C. M., M. Ustav, A. Stenlund, T. F. Ho, T. R. Broker, and L. T. Chow. 1992. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. USA 895799-5803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clower, R. V., J. C. Fisk, and T. Melendy. 2006. Papillomavirus E1 protein binds to and stimulates human topoisomerase I. J. Virol. 801584-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Conger, K. L., J. S. Liu, S. R. Kuo, L. T. Chow, and T. S. Wang. 1999. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase alpha/primase. J. Biol. Chem. 2742696-2705. [DOI] [PubMed] [Google Scholar]
- 7.Cueille, N., R. Nougarede, F. Mechali, M. Philippe, and C. Bonne-Andrea. 1998. Functional interaction between the bovine papillomavirus virus type 1 replicative helicase E1 and cyclin E-Cdk2. J. Virol. 727255-7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng, W., G. Jin, B. Y. Lin, B. A. Van Tine, T. R. Broker, and L. T. Chow. 2003. mRNA splicing regulates human papillomavirus type 11 E1 protein production and DNA replication. J. Virol. 7710213-10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng, W., B. Y. Lin, G. Jin, C. G. Wheeler, T. Ma, J. W. Harper, T. R. Broker, and L. T. Chow. 2004. Cyclin/CDK regulates the nucleocytoplasmic localization of the human papillomavirus E1 DNA helicase. J. Virol. 7813954-13965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fehrmann, F., D. J. Klumpp, and L. A. Laimins. 2003. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 772819-2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferran, M. C., and A. A. McBride. 1998. Transient viral DNA replication and repression of viral transcription are supported by the C-terminal domain of the bovine papillomavirus type 1 E1 protein. J. Virol. 72796-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fradet-Turcotte, A., C. Vincent, S. Joubert, P. A. Bullock, and J. Archambault. 2007. Quantitative analysis of the binding of simian virus 40 large T antigen to DNA. J. Virol. 819162-9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldsborough, M. D., D. DiSilvestre, G. F. Temple, and A. T. Lorincz. 1989. Nucleotide sequence of human papillomavirus type 31: a cervical neoplasia-associated virus. Virology 171306-311. [DOI] [PubMed] [Google Scholar]
- 14.Han, Y., Y. M. Loo, K. T. Militello, and T. Melendy. 1999. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A. J. Virol. 734899-4907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hebner, C. M., and L. A. Laimins. 2006. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 1683-97. [DOI] [PubMed] [Google Scholar]
- 16.Hsu, C. Y., F. Mechali, and C. Bonne-Andrea. 2007. Nucleocytoplasmic shuttling of bovine papillomavirus E1 helicase downregulates viral DNA replication in S phase. J. Virol. 81384-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu, Y., R. V. Clower, and T. Melendy. 2006. Cellular topoisomerase I modulates origin binding by bovine papillomavirus type 1 E1. J. Virol. 804363-4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hubert, W. G., T. Kanaya, and L. A. Laimins. 1999. DNA replication of human papillomavirus type 31 is modulated by elements of the upstream regulatory region that lie 5′ of the minimal origin. J. Virol. 731835-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeronimo, C., M. F. Langelier, M. Zeghouf, M. Cojocaru, D. Bergeron, D. Baali, D. Forget, S. Mnaimneh, A. P. Davierwala, J. Pootoolal, M. Chandy, V. Canadien, B. K. Beattie, D. P. Richards, J. L. Workman, T. R. Hughes, J. Greenblatt, and B. Coulombe. 2004. RPAP1, a novel human RNA polymerase II-associated protein affinity purified with recombinant wild-type and mutated polymerase subunits. Mol. Cell. Biol. 247043-7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klumpp, D. J., and L. A. Laimins. 1999. Differentiation-induced changes in promoter usage for transcripts encoding the human papillomavirus type 31 replication protein E1. Virology 257239-246. [DOI] [PubMed] [Google Scholar]
- 21.Koonin, E. V. 1993. A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res. 212541-2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo, S. R., J. S. Liu, T. R. Broker, and L. T. Chow. 1994. Cell-free replication of the human papillomavirus DNA with homologous viral E1 and E2 proteins and human cell extracts. J. Biol. Chem. 26924058-24065. [PubMed] [Google Scholar]
- 23.Lee, D., H. Sohn, G. V. Kalpana, and J. Choe. 1999. Interaction of E1 and hSNF5 proteins stimulates replication of human papillomavirus DNA. Nature 399487-491. [DOI] [PubMed] [Google Scholar]
- 24.Lentz, M. R., D. Pak, I. Mohr, and M. R. Botchan. 1993. The E1 replication protein of bovine papillomavirus type 1 contains an extended nuclear localization signal that includes a p34cdc2 phosphorylation site. J. Virol. 671414-1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lentz, M. R., S. M. Stevens, Jr., J. Raynes, and N. Elkhoury. 2006. A phosphorylation map of the bovine papillomavirus E1 helicase. Virol. J. 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, D., and R. Roberts. 2001. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 582085-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin, B. Y., T. Ma, J. S. Liu, S. R. Kuo, G. Jin, T. R. Broker, J. W. Harper, and L. T. Chow. 2000. HeLa cells are phenotypically limiting in cyclin E/CDK2 for efficient human papillomavirus DNA replication. J. Biol. Chem. 2756167-6174. [DOI] [PubMed] [Google Scholar]
- 28.Lin, B. Y., A. M. Makhov, J. D. Griffith, T. R. Broker, and L. T. Chow. 2002. Chaperone proteins abrogate inhibition of the human papillomavirus (HPV) E1 replicative helicase by the HPV E2 protein. Mol. Cell. Biol. 226592-6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu, J. S., S. R. Kuo, A. M. Makhov, D. M. Cyr, J. D. Griffith, T. R. Broker, and L. T. Chow. 1998. Human Hsp70 and Hsp40 chaperone proteins facilitate human papillomavirus-11 E1 protein binding to the origin and stimulate cell-free DNA replication. J. Biol. Chem. 27330704-30712. [DOI] [PubMed] [Google Scholar]
- 30.Loo, Y. M., and T. Melendy. 2004. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J. Virol. 781605-1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma, T., N. Zou, B. Y. Lin, L. T. Chow, and J. W. Harper. 1999. Interaction between cyclin-dependent kinases and human papillomavirus replication-initiation protein E1 is required for efficient viral replication. Proc. Natl. Acad. Sci. USA 96382-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masterson, P. J., M. A. Stanley, A. P. Lewis, and M. A. Romanos. 1998. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase α-primase p68 subunit. J. Virol. 727407-7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moody, C. A., A. Fradet-Turcotte, J. Archambault, and L. A. Laimins. Human papillomaviruses activate caspases upon differentiation to promote viral genome amplification. Proc. Natl. Acad. Sci. USA, in press. [DOI] [PMC free article] [PubMed]
- 34.Ozbun, M. A., and C. Meyers. 1998. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J. Virol. 722715-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park, J., N. H. Cho, J. K. Choi, P. Feng, J. Choe, and J. U. Jung. 2003. Distinct roles of cellular Lck and p80 proteins in herpesvirus saimiri Tip function on lipid rafts. J. Virol. 779041-9051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park, J., B. S. Lee, J. K. Choi, R. E. Means, J. Choe, and J. U. Jung. 2002. Herpesviral protein targets a cellular WD repeat endosomal protein to downregulate T lymphocyte receptor expression. Immunity 17221-233. [DOI] [PubMed] [Google Scholar]
- 37.Peh, W. L., K. Middleton, N. Christensen, P. Nicholls, K. Egawa, K. Sotlar, J. Brandsma, A. Percival, J. Lewis, W. J. Liu, and J. Doorbar. 2002. Life cycle heterogeneity in animal models of human papillomavirus-associated disease. J. Virol. 7610401-10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rigaut, G., A. Shevchenko, B. Rutz, M. Wilm, M. Mann, and B. Seraphin. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 171030-1032. [DOI] [PubMed] [Google Scholar]
- 39.Rocque, W. J., D. J. Porter, J. A. Barnes, E. P. Dixon, D. C. Lobe, J. L. Su, D. H. Willard, R. Gaillard, J. P. Condreay, W. C. Clay, C. R. Hoffman, L. K. Overton, G. Pahel, T. A. Kost, and W. C. Phelps. 2000. Replication-associated activities of purified human papillomavirus type 11 E1 helicase. Protein Expr. Purif. 18148-159. [DOI] [PubMed] [Google Scholar]
- 40.Rost, B., P. Fariselli, and R. Casadio. 1996. Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci. 51704-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rost, B., and C. Sander. 1993. Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232584-599. [DOI] [PubMed] [Google Scholar]
- 42.Sénéchal, H., G. G. Poirier, B. Coulombe, L. A. Laimins, and J. Archambault. 2007. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology 35810-17. [DOI] [PubMed] [Google Scholar]
- 43.Smith, T. F., C. Gaitatzes, K. Saxena, and E. J. Neer. 1999. The WD repeat: a common architecture for diverse functions. Trends Biochem. Sci. 24181-185. [DOI] [PubMed] [Google Scholar]
- 44.Stenlund, A. 2003. E1 initiator DNA binding specificity is unmasked by selective inhibition of non-specific DNA binding. EMBO J. 22954-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stenlund, A. 2003. Initiation of DNA replication: lessons from viral initiator proteins. Nat. Rev. Mol. Cell Biol. 4777-785. [DOI] [PubMed] [Google Scholar]
- 46.Sun, Y., H. Han, and D. J. McCance. 1998. Active domains of human papillomavirus type 11 E1 protein for origin replication. J. Gen. Virol. 791651-1658. [DOI] [PubMed] [Google Scholar]
- 47.Swindle, C. S., and J. A. Engler. 1998. Association of the human papillomavirus type 11 E1 protein with histone H1. J. Virol. 721994-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas, J. T., W. G. Hubert, M. N. Ruesch, and L. A. Laimins. 1999. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. Proc. Natl. Acad. Sci. USA 968449-8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Titolo, S., K. Brault, J. Majewski, P. W. White, and J. Archambault. 2003. Characterization of the minimal DNA binding domain of the human papillomavirus E1 helicase: fluorescence anisotropy studies and characterization of a dimerization-defective mutant protein. J. Virol. 775178-5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Titolo, S., A. Pelletier, A. M. Pulichino, K. Brault, E. Wardrop, P. W. White, M. G. Cordingley, and J. Archambault. 2000. Identification of domains of the human papillomavirus type 11 E1 helicase involved in oligomerization and binding to the viral origin. J. Virol. 747349-7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Titolo, S., A. Pelletier, F. Sauve, K. Brault, E. Wardrop, P. W. White, A. Amin, M. G. Cordingley, and J. Archambault. 1999. Role of the ATP-binding domain of the human papillomavirus type 11 E1 helicase in E2-dependent binding to the origin. J. Virol. 735282-5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ustav, M., and A. Stenlund. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 10449-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White, P. W., A. Pelletier, K. Brault, S. Titolo, E. Welchner, L. Thauvette, M. Fazekas, M. G. Cordingley, and J. Archambault. 2001. Characterization of recombinant HPV6 and 11 E1 helicases: effect of ATP on the interaction of E1 with E2 and mapping of a minimal helicase domain. J. Biol. Chem. 27622426-22438. [DOI] [PubMed] [Google Scholar]
- 54.Yang, L., R. Li, I. J. Mohr, R. Clark, and M. R. Botchan. 1991. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature 353628-632. [DOI] [PubMed] [Google Scholar]
- 55.Yasugi, T., M. Vidal, H. Sakai, P. M. Howley, and J. D. Benson. 1997. Two classes of human papillomavirus type 16 E1 mutants suggest pleiotropic conformational constraints affecting E1 multimerization, E2 interaction, and interaction with cellular proteins. J. Virol. 715942-5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ziegler, K., T. Bui, R. J. Frisque, A. Grandinetti, and V. R. Nerurkar. 2004. A rapid in vitro polyomavirus DNA replication assay. J. Virol. Methods 122123-127. [DOI] [PubMed] [Google Scholar]