

X-ray crystallographic analysis of human inosine triphosphate pyrophosphohydrolase provided the secondary structure and active-site structure at 1.6 Å resolution in an orthorhombic crystal form. The structure gives a framework for future structure–function studies employing site-directed mutagenesis and for the identification of substrate/product-binding sites.
Keywords: inosine triphosphate pyrophosphohydrolase
Abstract
The structure of human inosine triphosphate pyrophosphohydrolase (ITPA) has been determined using diffraction data to 1.6 Å resolution. ITPA contributes to the accurate replication of DNA by cleansing cellular dNTP pools of mutagenic nucleotide purine analogs such as dITP or dXTP. A similar high-resolution unpublished structure has been deposited in the Protein Data Bank from a monoclinic and pseudo-merohedrally twinned crystal. Here, cocrystallization of ITPA with a molar ratio of XTP appears to have improved the crystals by eliminating twinning and resulted in an orthorhombic space group. However, there was no evidence for bound XTP in the structure. Comparison with substrate-bound NTPase from a thermophilic organism predicts the movement of residues within helix α1, the loop before α6 and helix α7 to cap off the active site when substrate is bound.
1. Introduction
Accurate replication of DNA is a prerequisite for genome stability and the prevention of cancer. One component of high-fidelity replication is the cleansing of dNTP pools of unwanted and potentially mutagenic nucleotide analogs by specific triphosphatases (Mathews, 2006 ▶). Classical examples are dUTP-triphosphatase (Hochhauser & Weiss, 1978 ▶) and MutT protein (Maki & Sekiguchi, 1992 ▶), which remove dUTP and 8-oxoguanineTP from nucleotide pools, respectively. We originally cloned the yeast HAM1 gene that prevents base-analog hydroxylamino-purine (HAP) mutagenesis, demonstrated that it is present in virtually all species and predicted it to be a novel triphosphatase that provides protection from adenine analogs (Noskov et al., 1996 ▶; Kozmin et al., 1998 ▶). A crystal structure of the HAM1 homolog from the thermostable bacterium Methanococcus jannaschii (Mj0226) has been determined and its biochemical activity has been characterized (Hwang et al., 1999 ▶; Chung et al., 2001 ▶). The enzyme catalyzed the pyrophosphohydrolysis of ITP/dITP and xanthosine triphosphate (XTP) to the corresponding monophosphates, preventing the incorporation of these noncanonical nucleotides into RNA and DNA. The orthologous rdgB gene in Escherichia coli and the human ITPA gene encoded proteins with similar activities (Chung et al., 2002 ▶; Lin et al., 2001 ▶). We will refer to human inosine triphosphate pyrophosphohydrolase as ITPA throughout this paper. Recently, the crystal structure of another type of E. coli enzyme, a triphosphatase converting ITP to IDP encoded by the yjjX gene, was determined, showing some structural features similar to those of the Mj0226 enzyme (Chung et al., 2002 ▶; Zheng et al., 2005 ▶).
Genetic analysis has demonstrated that the sanitization of nucleotide pools of unwanted purine analogs is essential to genome stability. Defects in the pyrophosphatase encoded by the rdgB gene in bacteria leads to incompatibility with mutations abolishing homologous recombination (Bradshaw & Kuzminov, 2003 ▶; Burgis et al., 2003 ▶) owing to the accumulation of double-strand breaks and chromosome fragmentation (Lukas & Kuzminov, 2006 ▶). There is an association between polymorphisms in the ITPA gene in humans and anomalous sensitivity to anti-inflammatory drugs, e.g. azathioprine (Cao & Hegele, 2002 ▶; Zelinkova et al., 2006 ▶). In order to understand the molecular mechanisms of ITPA transactions in humans, we have determined the crystal structure of human ITPA.
2. Materials and methods
2.1. Expression and purification
The ITPA gene from a HeLa cDNA library was amplified by PCR and subcloned into pET-15b expression plasmid (EMD Biosciences, Novagen) using XhoI–NdeI restriction sites to contain an N-terminal His tag and thrombin site. ITPA was expressed in E. coli Rosetta 2 utilizing ampicillin–chloramphenicol selection. The bacterial culture was grown at 310 K in 10 l Terrific Broth supplemented with 40 ml glycerol in a New Brunswick Fermentor/Bioreactor (20 l air per minute and 250 rev min−1 agitation). At mid-log stage, the culture was induced with 1 mM IPTG. After 4 h, cells were harvested and resuspended in 20 mM bicine pH 8.5, 2 mM β-mercaptoethanol (βME) buffer (buffer A), lysed with an Emulisflex cell homogenizer and centrifuged.
For protein purification, the clarified lysate was applied onto an Ni2+-charged HisTrap HP column (GE Healthcare Bio-Sciences, Corp.). The column was washed and eluted with a gradient of 40 mM–1 M imidazole in buffer A on an AKTA Explorer chromatography system (GE Healthcare Bio-Sciences, Corp.). The protein eluted at approximately 300 mM imidazole. Peak fractions were pooled together and the His tag was cleaved by incubation at room temperature overnight with 1 unit of restriction-grade thrombin (EMD Biosciences, Novagen). The thrombin was then removed by treatment with 200 µl benzamidine resin (Amersham Biosciences) per millilitre of protein solution. After centrifugation, the supernatant was dialyzed against buffer A. The sample was then applied onto a Hi-Trap Q-FF anion-exchange column (GE Healthcare Bio-Sciences, Corp.), which was washed and eluted with a gradient of 0–1 M NaCl in buffer A. The protein eluted at approximately 300 mM NaCl. After SDS–PAGE analysis, the peak fractions were pooled and the buffer exchanged back to buffer A and the protein was concentrated using an Amicon Ultra 5 000 molecular-weight cutoff centrifugal filter device (Millipore). The activity of the protein towards XTP and ITP was confirmed by thin-layer chromatography in 1 M LiCl–formic acid–phosphate buffer (Pavlov et al., 2004 ▶).
2.2. Crystallization
The protein sample was concentrated to 10 mg ml−1. The protein concentration was measured using the BioRad protein assay with bovine serum albumin as the reference standard. Initial crystallization conditions were found by setting up 15 commercially available 96-well format crystallization screens (Hampton Research, Emerald BioSystems and Nextal Biotechnologies). For 96-well crystallization experiments, a Phoenix robot (Art Robbins Instruments) was used to rapidly pipette 400 nl drops (200 nl protein + 200 nl reservoir) over 70 µl reservoirs in a sitting-drop format, which was covered with tape. Plates were incubated at ambient temperature and automatically imaged with a Rock Imager 54 system (Formulatrix). Hits were found in several drops containing PEG 3000 and PEG 3350 (Fig. 1 ▶ a) from Index HT (Hampton Research) and Wizard I and II (Emerald Biosystems) screens. For optimization, a protein sample with XTP was prepared by combining 1 µl 10 mM XTP with 21.7 µl 10 mg ml−1 ITPA to give a 1:1 molar ratio. Optimizations were then set up in 24-well trays by the hanging-drop vapor-diffusion method. Two drops (500 nl protein + 500 nl reservoir) on one cover slip, one with XTP and one without XTP, were suspended over a 500 µl reservoir solution composed of 23–28% PEG 3350, 100 mM HEPES pH 7.5 and 2 mM β-ME. The crystals grew within 2 d of set-up at a PEG concentration of 27%.
Figure 1.

(a) One of the initial crystal hits (with crystals resembling a sheaf of wheat and a dove) grown from 96-well screening using Wizard I and II screens; the reservoir solution contained 0.1 M CHES pH 9.5 with 30% PEG 3000. (b) The final crystal used for data collection. Although they were visually imperfect, a fragment of one of these crystals was successfully used for data collection (Table 1 ▶).
2.3. Data collection and processing
A plate-like crystal 0.50 mm in length was cryoprotected by dragging it through a solution of 50/50(v/v) Paratone/mineral oil (Fig. 1 ▶ b). Cryocooled X-ray diffraction data were collected at 100 K with a Rigaku FR-E Cu Kα rotating-anode generator operating at 45 kV and 45 mA and equipped with an R-AXIS IV++ detector and Xstream 2000 low-temperature system (Rigaku). A full native data set to 1.6 Å was collected as 180 images with 1° oscillation at a crystal-to-distance of 100 mm with 5 min exposure times. The data were processed using HKL-2000 (Otwinowski & Minor, 2001 ▶) (Table 1 ▶).
Table 1. Crystallographic statistics.
The X-ray data were processed using HKL-2000 (Otwinowski & Minor, 2001 ▶). Values in parentheses are for the highest resolution bin.
| PDB code | 2i5d |
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 31.1, b = 105.0, c = 49.9 |
| Z (No. of molecules per ASU) | 1 |
| VM (Å3 Da−1) | 1.89 |
| Solvent content† (%) | 34.96 |
| Resolution range (Å) | 105–1.63 (1.69–1.63) |
| No. of observed/unique reflections | 123793/20838 |
| Completeness (%) | 98.1 (98.2) |
| Multiplicity | 5.9 (5.3) |
| Rsym‡ (%) | 4.8 (44.2) |
| I/σ(I) | 35.7 (3.3) |
| Rcryst | 19.0 |
| Rfree§ | 24.0 |
| No. of atoms | 1888 |
| No. of water atoms | 228 |
| R.m.s.d. bonds (Å) | 0.013 |
| R.m.s.d. angles (°) | 1.39 |
| Ramachandran analysis¶ (%) | |
| Residues in most favorable region | 98.2 |
| Residues in allowed regions | 1.2 |
| Residues in disallowed regions | 0.6 |
| Average B (Å2) | 20.8 |
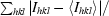
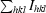
2.4. Structure solution and model refinement
A molecular-replacement search model was created by deleting all solvent and dual conformations from the A monomer of monoclinic human ITPA (PDB code 2car). Molecular replacement was performed at 3 Å resolution with MOLREP from the CCP4 suite (Collaborative Computational Project, Number 4, 1994 ▶; Vagin & Teplyakov, 1997 ▶) and resulted in an R value of 38% and a correlation coefficient of 0.67. The structure was refined with REFMAC5 to 1.6 Å resolution with R = 26.3% and R free = 30.6% (Murshudov et al., 1997 ▶). In order to eliminate model bias in our electron-density maps, OMIT maps were calculated by omitting 20 amino-acid sections (with a 6 Å sphere also omitted); the remaining structure was randomly perturbed by 0.3 Å displacements using MOLEMAN2 (Kleywegt, 2000 ▶) and refined and maps were calculated with REFMAC5. The following residues were modeled in two conformations: Glu44, Gln55, Glu93, Glu108, Asp124, Pro125, Ser126, Gln127, Arg130, Arg133, Arg135, Ser137, Arg139, Cys146, Cys154, Glu184 and Glu187. The occupancies of dual conformers were adjusted until there was no significant F o − F c density (i.e. >±3σ). Thrombin cleavage left a Gly-Ser-His sequence on the N-terminus that could not clearly be identified in any electron-density maps. Only the main-chain atoms of the His were observed and were modeled as Ala. There is a cis-peptide bond between Leu83 and Pro84. Finally, 228 water molecules were modeled into F o − F c density >3σ and refined.
3. Results and discussion
The crystallization of human ITPA was apparently enhanced and improved by the inclusion of XTP in an equimolar ratio to the enzyme. After we had collected the 1.6 Å X-ray diffraction data from human ITPA crystals using our home source, a similar unpublished structure was deposited in the Protein Data Bank as entry 2car. Although of high resolution (1.1 Å), this structure resulted from a monoclinic and pseudo-merohedrally twinned crystal. Therefore, we checked our diffraction data using the The Merohedral Crystal Twinning Server and found no evidence of twinning (Yeates, 1997 ▶). Also, the possibility of a monoclinic space group was ruled out by a comparison of parallel refinements. We speculate that the inclusion of XTP in the crystallization experiments may have improved the crystals by eliminating twinning and resulting in the higher symmetry space group.
Crystallographic refinement resulted in a final R value of 19% with an R free of 24% and the structure refined with reasonable stereochemistry (Table 1 ▶). Two residues, both with excellent electron density, have positive ϕ/ψ values (Fig. 2 ▶): Gln65 is located in the left-handed helical region of the Ramachandran plot (Ramachandran & Sasisekharan, 1968 ▶) and Lys96 has ϕ = 66° and ψ = 168° and is located just below a generously allowed region as defined by PROCHECK (Laskowski et al., 1993 ▶). After superposition of Cα atoms, the r.m.s.d. between 2car monomer A and our coordinates is 0.48 Å, with the largest difference being at loop 124–127, which has differences of up to 5 Å. When the superposition was performed omitting this loop, the r.m.s.d. was only 0.2 Å. The structure has two lobes (Fig. 2 ▶ b). The top lobe is composed of helices α4, α5, α6, strand β7 and helix α7. The bottom lobe is composed of helices α0, α1 and α2 and strands β1, β2, β3, β4, β5 and β6. The active-site cleft lies between the two lobes. Under physiological conditions, NTPases are most likely to exist as a homodimer (Hwang et al., 1999 ▶; Lin et al., 2001 ▶). A putative homodimer can be extracted from crystallographic symmetry that is very similar to the homodimer found in the 2car asymmetric unit (Fig. 3 ▶ a). The active sites are located on opposite sides of the homodimer.
Figure 2.

(a) Annotated protein sequence and secondary-structure analysis using JOY software (Mizuguchi et al., 1998 ▶). The figure is organized as follows: line 1, residue numbers; line 2, an asterisk indicates putative active-site residues that were identified by comparison with the M. jannaschii NTPase structure complexed with the ATP analogue adenylyl imidodiphosphate (AMPPNP; PDB code 2mjp); line 3, JOY annotation of the protein sequence, where α-helices are red, β-strands are blue and 310-helices are maroon, solvent-accessible residues are in lower case, solvent-inaccessible residues are in upper case, residues making hydrogen bonds to main-chain amides are in bold, those making hydrogen bonds to main-chain carbonyls are underlined and those with positive ϕ torsion angles are in italics; line 4, α-helices are shown as red rectangles, 310-helices as maroon rectangles, β-sheets as blue arrows and the type of tight β-turns are indicated as I or II; line 5, major secondary-structure elements are named as defined for M. jannaschii NTPase (Hwang et al., 1999 ▶). (b) Cross-eyed stereo diagram of the overall structure of human ITPA. This figure was produced using PyMOL (DeLano, 2002 ▶).
Figure 3.

Cross-eyed stereo pairs of the physiological dimer of ITPA. (a) Ribbon diagrams for each monomer are colored in a rainbow with blue at the N-terminus varying to red at the C-terminus. (b) The same drawing rotated 90° about the vertical axis to show the relative orientation of the active-site clefts, which are indicated by arrows. The dimer was generated by crystallographic symmetry operators on the monomeric asymmetric unit.
As noted above, the inclusion of XTP during crystallization was beneficial to the crystals. We were curious to see if we could identify an XTP or possibly XMP bound to ITPA. After refinement of the protein structure was completed, F o − F c and 2F o − F c electron-density maps were closely examined (Fig. 4 ▶ a). There was no apparent density for bound ligand. M. jannaschii NTPase was solved with AMPPNP bound (PDB code 2mjp). Superposition of Cα coordinates between ITPA and 2mjp (r.m.s.d. of 1.3 Å) shows the types of structural movements that can be predicted on ligand binding of human ITPA. Structural movement (towards AMPPNP) that seems to be associated with ligand binding includes helix α1, the loop before α6 and helix α7. These regions include active-site residues (indicated by an asterisk in Fig. 2 ▶ a). These residues appear to close in and cap off the substrate from the solvent. In order to verify these predictions, future work will include increasing the concentration of XTP or other substrates in the crystallization experiments. With this structure of wild-type human ITPA completed, we are well positioned to perform detailed structure–function studies.
Figure 4.

(a) Cross-eyed stereo pair of the electron density surrounding the active site of human ITPA. The REFMAC/σA-weighted 2mF o − DF c map is displayed at 1σ. Active-site residues are highlighted with violet bonds and water molecules are shown as spheres (Murshudov et al., 1997 ▶; Read, 1997 ▶). Although the protein was crystallized with a 1:1 molar ratio of XTP, the active site appears to be empty. (b) Cross-eyed stereo pair of the superposition of ITPA (blue) onto monomer A of the M. jannaschii enzyme (green; PDB code 2mjp). The AMPPNP molecule bound to the M. jannaschii enzyme is shown in ball-and-stick format. This view is rotated 180° about the vertical relative to Fig. 2 ▶(b).
Supplementary Material
PDB reference: human inosine triphosphate pyrophosphatase, 2i5d, r2i5dsf
Acknowledgments
This project was funded by the Nebraska Research Initiative funding of the Nebraska Center for Structural Biology. We thank Paul Sorgen, Jeff Lovelace and Admir Kellezi for useful discussions and technical assistance.
References
- Bradshaw, J. S. & Kuzminov, A. (2003). Mol. Microbiol.48, 1711–1725. [DOI] [PubMed] [Google Scholar]
- Brünger, A. T. (1993). Acta Cryst. D49, 24–36. [DOI] [PubMed] [Google Scholar]
- Burgis, N. E., Brucker, J. J. & Cunningham, R. P. (2003). J. Bacteriol.185, 3101–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, H. & Hegele, R. A. (2002). J. Hum. Genet.47, 620–622. [DOI] [PubMed] [Google Scholar]
- Chung, J. H., Back, J. H., Park, Y. I. & Han, Y. S. (2001). Nucleic Acids Res.29, 3099–3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, J. H., Park, H. Y., Lee, J. H. & Jang, Y. (2002). J. Biochem. Mol. Biol.35, 403–408. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA.
- Hochhauser, S. J. & Weiss, B. (1978). J. Bacteriol.134, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, K. Y., Chung, J. H., Kim, S.-H., Han, Y. S. & Cho, Y. (1999). Nature Struct. Biol.6, 691–696. [DOI] [PubMed] [Google Scholar]
- Kleywegt, G. J. (2000). Acta Cryst. D56, 249–265. [DOI] [PubMed] [Google Scholar]
- Kozmin, S. G., Schaaper, R. M., Shcherbakova, P. V., Kulikov, V. N., Noskov, V. N., Guetsova, M. L., Alenin, V. V., Rogozin, I. B., Makarova, K. S. & Pavlov, Y. I. (1998). Mutat. Res.402, 41–50. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst.26, 283–291. [Google Scholar]
- Lin, S., McLennan, A. G., Ying, K., Wang, Z., Gu, S., Jin, H., Wu, C., Liu, W., Yuan, Y., Tang, R., Xie, Y. & Mao, Y. (2001). J. Biol. Chem.276, 18695–18701. [DOI] [PubMed] [Google Scholar]
- Lukas, L. & Kuzminov, A. (2006). Genetics, 172, 1359–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki, H. & Sekiguchi, M. (1992). Nature (London), 355, 273–275. [DOI] [PubMed] [Google Scholar]
- Mathews, C. K. (2006). FASEB J.20, 1300–1314. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Mizuguchi, K., Deane, C. M., Blundell, T. L., Johnson, M. S. & Overington, J. P. (1998). Bioinformatics, 14, 617–623. [DOI] [PubMed] [Google Scholar]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Noskov, V. N., Staak, K., Shcherbakova, P. V., Kozmin, S. G., Negishi, K., Ono, B. C., Hayatsu, H. & Pavlov, Y. I. (1996). Yeast, 12, 17–29. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (2001). International Tables for Crystallography, Vol. F, edited by M. G. Rossmann & E. Arnold, pp. 226–235. Dordrecht: Kluwer Academic Publishers.
- Pavlov, Y. I., Maki, S., Maki, H. & Kunkel, T. A. (2004). BMC Biol.2, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran, G. N. & Sasisekharan, V. (1968). Adv. Protein Chem.23, 283–438. [DOI] [PubMed] [Google Scholar]
- Read, R. J. (1997). Methods Enzymol.277, 110–128. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst.30, 1022–1025. [Google Scholar]
- Yeates, T. O. (1997). Methods Enzymol.276, 344–358. [PubMed] [Google Scholar]
- Zelinkova, Z., Derijks, L. J., Stokkers, P. C., Vogels, E. W., van Kampen, A. H., Curvers, W. L., Cohn, D., van Deventer, S. J. & Hommes, D. W. (2006). Clin. Gastroenterol. Hepatol.4, 44–49. [DOI] [PubMed] [Google Scholar]
- Zheng, J., Singh, V. K. & Jia, Z. (2005). Structure, 13, 1511–1520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: human inosine triphosphate pyrophosphatase, 2i5d, r2i5dsf