

Abstract
Targeted delivery of vaccine candidates to the gastrointestinal (GI) tract holds potential for mucosal immunization, particularly against mucosal pathogens like the human immunodeficiency virus (HIV). Among the different strategies for achieving targeted release in the GI tract, namely the small intestine, pH sensitive enteric coating polymers have been shown to protect solid oral dosage forms from the harsh digestive environment of the stomach and dissolve relatively rapidly in the small intestine by taking advantage of the luminal pH gradient. We developed an enteric polymethacrylate formulation for coating hydroxy-propyl-methyl-cellulose (HPMC) capsules containing lyophilized Adenoviral type 5 (Ad5) vectors expressing HIV-1 gag and a string of six highly-conserved HIV-1 envelope peptides representing broadly cross-reactive CD4+ and CD8+ T cell epitopes. Oral immunization of rhesus macaques with these capsules primed antigen-specific mucosal and systemic immune responses and subsequent intranasal delivery of the envelope peptide cocktail using a mutant cholera toxin adjuvant boosted cellular immune responses including, antigen-specific intracellular IFN-γ-producing CD4+ and CD8+ effector memory T cells in the intestine. These results suggest that the combination of oral adenoviral vector priming followed by intranasal protein/peptide boosting may be an effective mucosal HIV vaccination strategy for targeting viral antigens to the GI tract and priming systemic and mucosal immunity.
Keywords: oral delivery, enteric coatings, adenoviral vectors, HIV-1 peptides, mucosal vaccination
INTRODUCTION
Adenoviral type 5 (Ad5) vectors are potent gene delivery vehicles that are capable of stimulating systemic and mucosal immune responses [1]. Mucosal responses, including secretory IgA (sIgA) and mucosal-homing T cells have been shown to be important for protection of mucosal surfaces [2–4]. In fact, mucosal immunization is superior for generating long-lasting mucosal immunity [4–6]. However, studies have shown that oral mucosal vaccination produces much lower CD8+ T cell responses than systemic delivery [7] and requires much higher viral particle numbers for eliciting strong responses [8,9]. Moreover, oral vaccination studies with Ad vectors have shown that some animals do not mount an immune response, indicating that intestinal mucosal surfaces may not be reliably infected [8,10]. The difficulty of oral vaccination is not surprising considering that Ad5 is a respiratory pathogen and thus not adapted for survival in the intestinal tract. Moreover, the low pH and digestive enzymes of gastric fluid have been attributed to inactivation of Ad vectors in vitro [11]. A change in pH from 8 to 5 can decrease viral titer by 107-fold [12]. Since stomach acids can reach a pH as low as 1.2, there is strong rationale for the enteric protection of Ad vectors for oral delivery.
Previous studies have successfully used acid-resistant, biodegradable enteric polymer coatings to protect antigens for oral immunization [13–15]. Additionally, enteric coatings have been used with lyophilized Ad vaccines and recombinant Ad vectors for oral immunization [16–19]. These pH-dependent enteric polymers have proven effective for controlling drug delivery to the small intestine and the proximal colon [20–23]. The efficacy of any oral vaccine may depend on the delivery to regions where large aggregates of lymphocytes are located in the distal mammalian small intestine, the jejunum and ileum [24]. Therefore, we hypothesized that enteric-coated lyophilized Ad vaccine vectors could be protected from the harsh conditions of the digestive tract and controlled to release in immunologically relevant portions of the intestinal tract.
While there is prior art for applying enteric-coated adenoviruses, much of this work has been performed in an industrial setting where few methodological details were provided to guide other investigators to test the approach in small scale in a laboratory setting. Towards this end, we developed an enteric polymethacrylate formulation for coating hydroxy-propyl-methyl-cellulose (HPMC) capsules containing lyophilized Ad5 using a simplified coating apparatus. We verified the integrity of the coated capsules at varying pH and the activity of the lyophilized Ad vectors after capsule dissolution. A first generation recombinant Ad5 vector expressing HIV-1 gag (Ad-gag) and Ad5 expressing a string of six conserved CD4+ and CD8+ T cell epitopes of HIV-1 envelope (Ad-env-peptide) were lyophilized in HPMC capsules, coated using a non-industrial simplified tray technique with anionic hydrophilic 1:1 and 1:2 methacrylic acid and methylmethacrylate co-polymers (Eudragit®). Given commercial capsules are too large for testing in mice, the enteric-coated capsules were tested in a pilot study using rhesus macaques. Three adult rhesus macaques were immunized by oral priming with the enteric-coated capsules containing the Ad-gag and Ad-env-peptide constructs, and induction of antigen-specific humoral and cellular immune responses were observed. The peptide-specific cellular responses were boosted by intranasal delivery of the peptide-cocktail using a mutant cholera toxin that was effective as a mucosal adjuvant in our earlier murine studies [25,26]. These results in a non-human primate model provide proof of principle for a prime-boost HIV vaccination strategy employing Ad vectored antigens for priming and peptides/proteins in combination with the CT2* adjuvant for boosting in order to generate mucosal and systemic protective immunity.
MATERIALS & METHODS
Cells and Viruses
The human cervical carcinoma cell line (HeLa) and the human embryonic kidney cell line (293A) were maintained in Dulbecco’s modified Eagle’s Medium (DMEM, Hyclone, Logan, UT) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT) and 1% antibiotic-antimycotic (Gibco, Gaithersburg, MD).
Ad vectors used in this study are based on the AdEasy system (Q-BIOgene) and carry the full E1- and E3-deleted Ad5 genome. The stability studies used an Ad vector containing the firefly luciferase gene, an internal ribosome entry site, and the humanized Renilla GFP expressed from a cytomegalovirus (CMV) immediate-early promoter in the E1 region (Ad-Luc-IRES-hrGFP). The immunization studies used two Ad vectors containing two separate antigen genes expressed from a cytomegalovirus (CMV) immediate-early promoter in the E1 region. The first vector, Ad-gag, was generated by cloning a codon-optimized HIV-1 gag gene [27] downstream of a chimeric intron obtained from the pCI vector (Promega, Madison, WI). The second vector, Ad-env-peptide, was generated by gene building a synthetic construct (5’-CTCCTGAATTCAGATCTGACCATGGTACCGTCCTCCGTGTCCTGGGGCATCCTCCTGCTGGCCGGCCTGTGCTGCCTGGTCCCCGTCTCCCTGGCGGTACCCAAGCTGTGGGTTACCGTCTACTACGGGGTGCCCGTGTGGAAGGAGGCCATTATCAGCCTGTGGGACCAGTCCCTCAAGCCCTGCGTCAAGCTCACCCCCCTGTGCGTGTCCCTGTCGGTGATCACCCAAGCCTGTCCTAAGGTGAGTTTCGAGGGAACCGGGCCCTGTACCAACGTGTCTACCGTGCAGTGCACCCACGGTATCAAGCCGGTGCGGATCCAGCGCGGACCAGGCAGGACATTCGTAACGATCGGCAAATTTCTGGGCTTCCTCGGCGCCGCCGGGAGCACGATGGGCGCGGCAAGCCTGACTCTTACTGTCCAGGCTAGACAGTATTTGCGCGACCAGCAGCTGCTGGGCATCTGGGGCTGCGGACCGGTGTAAGCGGCC GCCTCGAGTCTAGATGAGTGAG-3’) from 14 overlapping 60-mer oligonucleotides. This construct expresses the six conserved HIV-1 envelope peptides, 104, 111, 113, 116, 63, and 61 as a “string of beads” fused to the c-terminus of the α-1 antitrypsin secretory leader and a linker to facilitate secretion and MHC II display. Virus was propagated, purified by CsCl gradient centrifugation, and viral particles (v.p.) were quantitated as described [28].
Animals
Three adult female rhesus macaques (Macaca mulatta) of Indian origin between the ages of 8–17 years, from the specific pathogen-free breeding colony at the Michael Keeling Center for comparative medicine and research of The University of Texas MD Anderson Cancer Center, Bastrop TX, were used in the study. The monkeys were maintained in animal facilities fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and the study was conducted according to National Institute of Health Guidelines on Care and Use of Laboratory Animals.
Peptides
A group of six HIV-1 envelope peptides, demonstrated in our previous studies to be effective antigens/immunogens for priming HIV-specific cellular immune responses in multiple animal models and humans, were employed [29–34]. The peptides were prepared in the institutional antigen-core facility utilizing FMOC solid phase chemistry on a PTI Symphony Peptide Synthesizer (Protein Technologies Inc., Tucson, AZ) and the purity of the peptides was determined to be >95% by high-pressure liquid chromatography and was validated by mass spectrometry.
Lyophilization of Adenoviral Vectors
A maximum of 20 μl per capsule of Ad vectors dissolved in potassium phosphate buffered saline (KPBS) with 1.0 M sucrose [35] were flash frozen in clear Quali-V® HPMC capsules (Size 2, Qualicaps Inc., Whitsett, NC) on dry ice, placed in 1.5-ml microcentrifuge tubes, wrapped with weighing paper, and immediately placed in a pre-chilled LabConco 300-ml flask. The contents were freeze-dried using a FreezeZone® 4.5 L Benchtop Freeze Dry System 77500 (LabConco, Kansas City, MO) for 2 hr at −50ºC with an initial vacuum of 133 × 10−3 mBar and a final vacuum of 3 × 10−3 mBar. Capsules were immediately stored desiccated at 4ºC.
Enteric Coating of HPMC Capsules
Prior to usage, HPMC capsules were evaluated for uniformity of weight and material integrity by light microscopy. Disintegration time of uncoated capsules was also evaluated in ddH20. Under constant stirring in a 500-ml sterile beaker, talc (Mg3Si4O10(OH)2, Sigma, St. Louis, MO) was added slowly and gradually to an aqueous solution of isopropyl alcohol (95.6% v/v). A 4:1 ratio (8.8% w/w) of Eudragit® L100 and S100 methacrylic acid co-polymers (Degussa Röhm Pharma Polymers, Darmstadt, Germany) was added very slowly to the talc suspension. The plasticizer triethyl citrate (TEC, Sigma) was added drop-wise to the suspension, covered with Parafilm, and mixed for an additional hour to ensure homogeneity. Both sides of the capsules were coated with three wet coats of the enteric polymer dispersion using a ProCoater (Torpac Inc., Fairfield, NJ).
Adenoviral Vector Stability Assay
Ad-Luc-IRES-hrGFP (5.0 × 109 v.p.) was lyophilized in HPMC capsules, enteric-coated, and stored desiccated at 4°C. Preparations were dissolved in triplicate at room temperature (RT) in 5 ml of phosphate-buffered saline (PBS, pH 7.2) in a 6-well plate until complete dissolution was observed. Next, HeLa cells were infected with reconstituted solution at 100 v.p./cell, assuming a maximum theoretical reconstituted concentration of 106 v.p./μl. After incubation at 37°C for 24 hr, cells were lysed, and luciferase activity (in lumens) was measured as described [36]. Capsule preparations were measured at 24 hr, 48 hr, and each week for four weeks post-lyophilization, and luciferase activity was compared at each time point to a stability control of freshly-diluted stock virus at 100 v.p./cell. Gene expression was also monitored by green fluorescence microscopy.
Integrity Test of Coated HPMC capsules
Prior to each dissolution test and oral delivery into rhesus macaques, enteric-coated HPMC capsules (n=6) were placed in a 0.1 N HCl solution at 37°C in a water bath with shaking for 2 hr. Afterwards, the capsules were dried at 25°C for 45 min., weighed, and visually observed by light microscopy.
In Vitro Capsule Dissolution and Stability Test
HPMC capsules were loaded with 100 mg of bromophenol blue sodium salt (Sigma) and enteric-coated as described. Capsules (n=6) were dissolved with shaking in sodium phosphate buffers of varying pH (6.0, 6.5, 7.0, and 7.5) and small samples were taken at varying time points until the capsules were completely dissolved. The absorbance was read and analyzed at 590 nm using an HTS 7000 Bio Assay Reader (PerkinElmer, Wellesley, MA). For Ad release, 5x109 vp of virus were lyophilized and coated as described above. Capsules were exposed to buffer at pH 6 or 7.5 and samples of the buffer were assayed by real time PCR of hexon. 5 μl sample of each virus was diluted 1/20 in PBS. 1 μl of Qiagen Proteinase K was added to the sample, and it was incubated at 55°C for 1hr and the Proteinase K was heat inactivated at 95°C for 20 minutes. The sample was then subjected to real-time PCR and viral genomes were calculated based on a standard curve of Ad virions.
Immunization of Rhesus Macaques
In a pilot study funded by the Center for AIDS Research (CFAR) at Baylor College of Medicine , 1.0 × 1011 v.p. of Ad-gag and 1.5 × 1010 v.p. of Ad-env-peptide, were lyophilized and enteric coated 24 hr prior to oral administration to rhesus monkeys. Macaques were anesthetized and enteric-coated capsules were inserted down the esophagus in to the stomach using a feeding tube. This procedure was repeated at 4, 8, and 12 weeks, making a total of four oral immunizations. At weeks 28 and 32, the macaques received intranasal immunizations with a mixture of the six HIV-1 envelope peptides (each at 100 μg/dose/monkey) along with the mucosal adjuvant CT2* (10 μg/dose/monkey) in 600 μl sterile PBS. Given the limited budget of the pilot study, control macaques were not included in the test. Instead, pre-immunization samples and historical comparisons were used to assess immunogenicity. Given that these macaques were specific-pathogen-free (SPF) and had not been manipulated previously, pre-immunization controls all generated only background levels of antibody and cellular responses.
Collection of Samples
All samples were collected at the indicated time points just prior to immunization. Blood samples were collected in sodium heparin from the femoral vein. Before the separation of peripheral blood mononuclear cells (PBMC) from the blood samples, plasma was collected and stored immediately at −80ºC until analyzed. The PBMC prepared from the blood samples by the standard ficoll-hypaque density-gradient centrifugation were used for various immune assays. Using a 3 cc syringe, saliva was collected from the pharyngeal area or in the cheek pouch. Vaginal wash samples were obtained from anesthetized animals by applying approximately 1 ml of sterile PBS into the vagina and aspirating the released fluid. Soon after collections, the various biological fluids were subjected to a short centrifugation, mixed with protease inhibitors and kept at −80ºC until assayed. At week 36, the study was terminated and various tissues were obtained from the three monkeys at necropsy for various immunological and immunohistochemistry analyses.
Anti-gag Enzyme-linked Immunosorbent Assay
Nunc-Immuno™ MaxiSorp™ (Nalge Nunc International, Rochester, NY) enzyme-linked immunosorbent assay (ELISA) plates (Corning Inc., Corning, N.Y.) were coated with 100 μl/well of 1 μg/ml of HIV-1 p55 (LAV/SF2 mix, NIH AIDS Research and Reference Reagent Program) in Plate Coating Solution (Hybridoma Subisotyping Kit, Calbiochem, San Diego, CA), washed thoroughly with PBS with 1% Tween (PBST) and then blocked with 5% milk in PBST at RT for 2 hr. Plates were then washed with PBST, and saliva and vaginal washes were diluted 1:2 in PBS. Diluted samples were incubated in triplicate at 100 μl/well at RT for 1 hr. Plates were washed with PBST and peroxidase-conjugated anti-monkey IgA goat antibody (Rockland Immunochemicals, Gilbertsville, PA) was incubated at RT for 1 hr. Plates were then washed with PBST, and bound antibody was detected by a colorimetric reaction with tetramethylbenzadine (Calbiochem). Reactions were stopped with 1M sulfuric acid, and absorbance was read at 450 nm. The oral (saliva) and vaginal responses were normalized by subtracting the background absorbance taken prior to immunization (week 0) and plotted against the number of weeks post-oral priming. Statistical significance was verified for each time point against the background using a Student’s paired t test and a p value < 0.05.
Assay for Neutralizing Antibodies Against Adenovirus
Samples were incubated in triplicate at the indicated dilutions with Ad5 expressing luciferase for 1 hour at 37°C. The resulting solution was then added to 293 cells at 250 viral particles per cell for 24 hours and luciferase activity was measured. Data is expressed as geometric mean titers that reduced Ad luciferase activity 50%.
Proliferation Assay
PBMC samples were obtained from animals at the indicated time points before each immunization. The proliferation of PBMC samples from the monkeys obtained at different time points during the study were determined by the standard [3H] thymidine incorporation assay, using the six HIV-1 envelope peptides (each at 10μg/ml final concentration) as a single mixture (designated as pep-mix) as well as the peptides corresponding to the gp41 and gp120 portions of the envelope as separate mixtures (designated as gp41 peptides and gp120 peptides, respectively), recombinant HIV-1 gag protein p55 (0.5 μg/ml), heat-inactivated purified preparation of the HIV-1IIIB (equivalent to 20,000 cpm of reverse transcriptase activity), and heat-inactivated control Ad vector (0.5 μg/ml). The culture medium and Con A (5 μg/ml) served as negative and positive controls, respectively. Additionally, in some experiments an unrelated control peptide (from human papilloma virus, HPV) at 10 μg/ml final concentration was used as negative control, and no differences were observed compared to culture medium as a negative control. Aliquots of the PBMC (105/well) were seeded in triplicate wells of 96-well plates. The proliferative response in terms of stimulation index (SI) was calculated as fold-increase in the radioactivity over that of the cells cultured in medium alone. The responses to antigens were considered positive when the SI values were 2.0.
ELISPOT Assay for Detecting Antigen-specific IFN-γ-producing Cells
Freshly-prepared PBMC were stimulated with the different HIV antigens as described above for the proliferation assay to determine the numbers of IFN-γ-producing cells by the ELISPOT assay using the methodology reported earlier [33,37,38]. Briefly, aliquots of PBMC (105/well) were seeded in triplicate wells of 96-well plates (polyvinylidene difluoride backed plates, MAIP S 45, Millipore, Bedford, MA) pre-coated with the primary IFN-γ antibody. After incubation for 36 hr at 37°C, the cells were removed and the wells were thoroughly washed with PBS. Subsequently, 100 μl of biotinylated secondary antibody to IFN-γ (detection antibody) was added to the wells for 3 hr at 37°C followed by avidin-peroxidase treatment for another 30 min. Purple colored spots representing individual cells secreting IFN-γ were developed using freshly-prepared substrate (0.3 mg/ml of 3-amino-9-ethyl-carbazole) in 0.1 M sodium acetate buffer, containing 0.015% hydrogen peroxide. Plates were washed to stop color development, and spots were counted by an independent agency (Zellnet Consulting, New Jersey, NJ) using the KS-ELISPOT automatic system (Carl Zeiss, Inc. Thornwood, NY) for the quantitative analysis of the number of IFN-γ spot forming cells (SFC) for 105 input PBMC. Responses were considered positive when the numbers of spot forming cells (SFC) with the test antigen were at least five and also were five above the background control values from cells cultured in the medium alone or with the negative control peptide.
Isolation of Intestinal Lymphocytes
The intraepithelial lymphocytes (IEL) from the jejunal sections of the macaques were isolated by using modifications of previously described procedures [39,40]. Briefly, samples of jejunum 10- to 20-cm long were collected within 30 min of necropsy. The tissue was cut into 0.5-cm2 pieces and rinsed in cold PBS. The mucosa were removed from the underlying muscularis layer and the tissue strips were treated with dithiothreitol (DTT, 1.5 mg/ml) in Hank’s balanced salt solution (HBSS) for 30 min at RT with agitation to remove intestinal mucus. The tissue strips were then treated with 0.75 mM EDTA in Ca2+- and Mg2+-free HBSS at RT for 60 min with stirring. After the treatment, the supernatants containing epithelial cells were removed and digested with 15 U of collagenase D and DNAse I in complete RPMI 1640 medium containing 100 U of penicillin and 100 U of streptomycin per ml, 5 ml L-glutamine, 5 ml HEPES buffer, and 10% FCS at 37°C with rapid shaking for 2 hr. The resulting cell suspensions were passed through stainless steel screen cups to remove the residual tissue fragments. The cells were washed and re-suspended in complete RPMI 1640 medium and enriched for lymphocytes by isotonic discontinuous Percoll (Sigma) density gradients (35 and 60% [vol/vol]) at 1,000 g for 20 min at 4°C. The lymphocyte band at the interface between the 35 and 60% Percoll layers was collected. Viability of lymphocytes was greater than 95% as determined by trypan blue exclusion staining. Aliquots of freshly-isolated cells were stained with fluorescently-labeled antibodies for flow cytometric analysis, while the remaining cells were washed in PBS and cryopreserved.
Assay for intracellular cytokine production
Lymphocyte populations were isolated from various tissues as previously described [41]. Cells were cultured in RPMI with 10% FBS, HEPES, and antibiotics at a density of 106 cells/ml in a 24-well dish at 37°C. To stimulate antigen-specific T cells, cultures were treated with or without pep-mix (each peptide at 1μg/ml) along with the costimulatory mAb, anti-human CD49d (9F10, 0.5 μg/ml). Golgistop (BD) was added to un-stimulated and stimulated cultures at a concentration of 1 μl/ml. Cells were harvested after 5 hr and stained with anti-CD8 Pacific Blue (RPA-T8), anti-CD4 PE-Cy7 (SK3), anti-CD95 APC (DX2). Cells were then fixed in 3% paraformaldehyde in PBS and stored overnight at 4°C. The next day, the cells were permeabilized by incubating in Perm/Wash solution (BD) for 20 min followed by incubation with anti-IFN-μ FITC (4S.B3) or control mouse IgG1 FITC (MOPC-21) for 30 min at 4°C. Cells were then washed once in Perm/Wash solution and fluorescence intensities were immediately measured on a CyAn™ flow cytometer (Dako, Carpinteria, CA). All antibodies used were purchased from BD Biosciences and used according to manufacturer’s instructions.
Immunohistochemistry analyses
Expression of nerve growth factor-b1 (NGF-b1) in the olfactory bulbs of rhesus macaques was used as a surrogate to determine retrograde transport of the cholera toxin mutant adjuvant CT2* according to the procedure described earlier [42]. Briefly, at the time of necropsy, the olfactory bulbs were obtained from each monkey and perfused with PBS at 25°C followed by Zamboni’s fixative (4% paraformaldehyde, 15% picric acid) in 0.1 M phosphate buffer. After overnight incubation at 4°C, the tissues were placed in fresh 4% paraformaldehyde and then transferred to a 30% sucrose solution at 4°C for 48 hr. The tissues were frozen in OCT compound, cut into thin sections (6 μm), and placed on microscope slides pre-coated with 10% BSA in saline. Frozen tissue sections were subjected to NGF-b1 antibody (Chemicon International, Temecula, CA) immunohistochemistry and immunofluoresence staining by reacting with avidin-biotin conjugate followed by incubation with streptavidin-HRP or streptavidin-Alexa Fluor 488 (Molecular Probes, Eugene, OR), respectively. Slides were examined with a fluorescence microscope (BX50/BXFLA; Olympus, Tokyo, Japan) equipped with a digital image capture system (Olympus) and photographed.
RESULTS
Adenoviral vector lyophilization and stability assay
Ad vectors have previously been lyophilized in various buffers using a multi-stage single shelf lyophilizer showing losses of one to five logs of activity compared to pre-lyophilized Ad [35]. We chose the optimal buffer of KPBS with 1 M sucrose in the Croyle et al. study for all Ad vectors requiring lyophilization. The sole purpose for lyophilization in this study was to allow Ad vectors to be encapsulated and subsequently enteric coated for determining feasibility of eliciting mucosal immune responses after oral vaccination. It is important to note that HPMC capsules placed in water will become pliable in a few seconds, completely lose integrity in less than 5 min, and completely dissolve in 10 min. Thus, Ad vectors were flash-frozen within the capsules immediately prior to lyophilization.
To assess vector viability after this process, Ad-Luc-IRES-hrGFP was lyophilized within HPMC capsules using a two-stage freeze drying process. Capsules containing lyophilized vectors stored at 4°C for varying times were completely dissolved in PBS and luciferase reporter gene expression was assessed on HeLa cells. Vector viability reduced dramatically after one week of storage and leveled off at around 5% through 4 weeks (Fig. 1). Since vector viability was highest after 24 hr of storage, all oral immunizations with enteric-coated capsules containing vaccine vectors were timed accordingly. Viability was also determined by evaluating hrGFP expression on HeLa cells by fluorescence microscopy with similar trends observed of decreased activity over time (data not shown).
Figure 1.

Stability of Lyophilized Ad Vectors. Enteric-coated capsules containing lyophilized Ad vectors were immediately stored in a dessicator at 4ºC and assayed for luciferase activity on HeLa cells upon reconstitution after storage of 24 hr., 48hr., and weekly for four weeks. Percent activity was determined by comparing to the luciferase activity of infection by the same number of particles of a freshly-diluted stability control vector as was used for lyophilization. Results are presented as the means of samples prepared in triplicate, and error bars indicate standard deviations.
Enteric polymer dissolution and stability test
The goal of the enteric coating is to remain solid and protect the contents of the capsule in the stomach, but then release its contents in an optimum time frame within the physiological pH range and the small intestinal transit time (SITT) of rhesus macaques. Survey of the literature suggests that macaque bowel segments have similar pH profiles as humans, but can have more variable transit times (Table 2). To assess the viability of the polymer, capsules were loaded with the dye bromphenol blue, coated with polymer, and the pH-specific release of the dye at 37°C was assessed over the range of pH found in stomach (1.5) and digestive tract of non-human primates and over the pH ranges at which the polymers are reported to be soluble (6.0 – 7.5). By this assay, it was shown that the polymer remained solid at pH 1.5 and prevented dye release (Fig. 2A). As pH was increased, dye release increased with near burst release occurring at pH 7.0 and 7.5. Similar dissolution curve analysis at 4°C and 25°C gave similar kinetics (data not shown). Moreover, the dissolution curves for capsules six months old held to the same trends (data not shown).
Table 2.
Comparison of G.I. Physiology of Human and Non-Human Primates. The distribution of pH for the human small intestine and the gastric residence time for each segment of the small intestine take into account an average between the fed and fasted state for travel of a solid meal.
| HUMAN | NON-HUMAN PRIMATE {Macaca Lacépède a:(mulattab, fascicularisb)} | ||||
|---|---|---|---|---|---|
| Small Intestine Region | pH Range | Small Intestine Transit Time (hr) | pH Range | Total Small Intestine Transit Time (hr)c | |
| Duodenum | 6.0 – 6.1 | 1 | 5.6 –5.8 | (Low Range) | (High Range) |
| Jejunum | 6.1 – 7.2 | 1 | 5.8 – 6.5 – 7.0 | 0.5 – 1.5 | 4.0 – 6.0 |
| Ileum | 7.0 – 7.8 | 2 | 7.0 – 7.4 | (Avg.) | (Avg.) |
| Caecum-Colon | 6.5 – 7.0 | 2 | 6.6 – 7.0 | 2.4 | 5.0 |
The total small intestine transit time shown for non-human primates are values shown as observed averages among rhesus and cynomolgus monkeys and represent the low and high range of time required for a liquid meal to pass through the entire small intestine and through the ileo-caecal junction into the proximal colon (agenus; bspecies; cdenotes total small bowel passage time not specific to an intestinal region) [Data compiled from 64,65–69].
Figure 2.
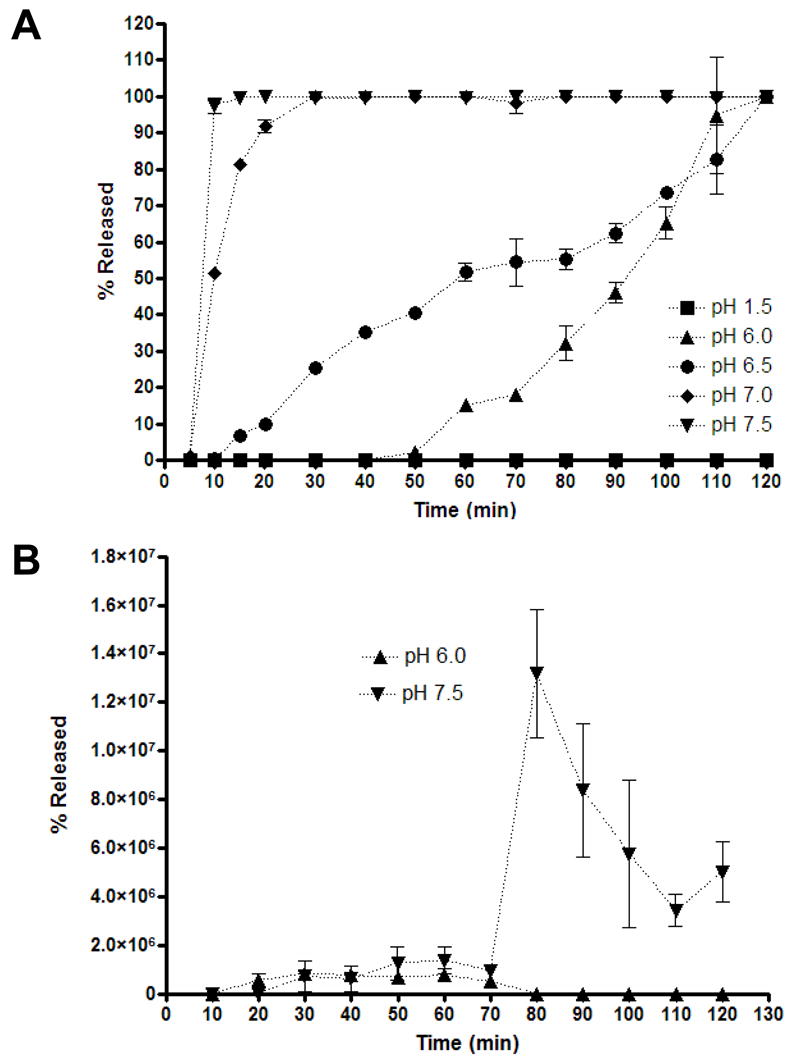
Enteric-coated HPMC capsule dissolution curve. Coated HPMC capsules with 13.2% w/w total solid polymer dispersion formulation of 4:1 Eudragit® L100 and Eudragit® S100 were subjected to dissolution test at 37ºC in 0.05 M sodium phosphate buffers of varying pH for the indicated times and the supernatant was assayed for A) dye release by spectrophotometry or B) virus release by real-time PCR. Results are presented as the means of samples performed in triplicate, and error bars represent the standard deviation.
A similar study was performed monitoring Ad release from the polymer-coated capsules. Ad transduction activity could not be used as a surrogate marker, since any virus released at low pH would be rapidly inactivated. Given this, we performed real-time PCR of viral genomes released into buffers at pH 6 and pH 7.5 (Fig. 2B). This assay demonstrated that the larger Ad virions were released more slowly than the small dye with near burst release after 80 minutes of exposure at pH 7.5. The real-time signal declined at later time points, perhaps due to DNA degradation or loss of virus or DNA to the plastic. Given data on monkey intestinal pH and transit times (Table 2), these data suggest that this particular enteric-coated capsule may begin dissolution in the jejenum and may mediate vector release prior to entry into the colon.
Immunization of Rhesus Macaques
Three rhesus macaques were immunized orally with 1.0 × 1011 v.p. of Ad-gag and 1.5 × 1010 v.p. of Ad-env-peptide lyophilized in enteric coated capsules at 0, 4, 8, and 12 weeks. Ad-gag expresses the codon-optimized HIV-1 gag gene. Ad-env-peptide expresses six conserved HIV-1 envelope peptides, 104, 111, 113, 116, 63, and 61 previously shown to elicit MHC I and II-restricted responses in multiple animal models and humans, were employed [29–34]. These peptides were expressed as a “string of beads” fusion protein fused to the c-terminus of the α-1 antitrypsin secretory leader and a linker to facilitate secretion and MHC II display. After oral adenovirus immunization, humoral and cellular immune responses were monitored against Ad, gag, and the envelope peptides through week 28. At weeks 28 and 32, the macaques received intranasal immunization 100 μg each of six HIV-1 envelope peptides mixed with the mucosal adjuvant CT2*. This immunization would boost the env-peptide-specific responses, but not responses against Ad or gag.
Humoral immune responses against HIV antigens after oral Ad immunization
To assess if mucosal priming of the humoral immune responses was being produced by oral immunization, saliva and vaginal secretions were collected from the macaques immediately before each immunization and were assayed by ELISA for antigen-specific sIgA against gag (the envelope peptides do not generate antibody responses [29]). Monkey numbers, 56 and H361, generated HIV-1 gag-specific sIgA in the saliva (Fig. 3A), and all monkeys produced vaginal anti-gag sIgA (Fig. 3B), indicating that gene delivery to the GI tract was successful. While anti-gag antibodies were observed, these were not persistent. Parallel testing of the samples for IgA, M, and G in vaginal and salivary samples generated a similar kinetic of antibody responses and decay (data not shown).
Figure 3.

Antigen-specific humoral immune responses. A and B, Antibody responses against HIV-1 gag in mucosal samples. C, Serum neutralizing responses against adenovirus. ELISA plates were coated with HIV-1 gag and 1:2 dilutions of saliva (A) and vaginal washes (B) were reacted. After reaction with secondary anti-IgA HRP conjugate and subsequent colorimetric substrate, data were presented as the O.D. values measured at 450 nm. Error bars indicate standard deviations of three measurements taken per time point. Statistical significance was verified for each time point against the background using a Student’s paired t test and a p value < 0.05. C) Serum samples take at the indicated times were incubated with Ad5 expressing luciferase for 1 hour at 37°C prior to addition to 293 cells at 250 viral particles per cel. 24 hours later, luciferase activity was measured and gene delivery was compared to untreated Ad5 vector. Data is expressed as geometric mean titers that reduced Ad luciferase activity 50%.
Humoral immune responses against adenovirus after oral Ad immunization
ELISA for anti-Ad5 antibodies in mucosal samples failed to detect anti-vector antibodies, except in the saliva of monkey H361 at week 17 (data not shown). In contrast, testing for neutralizing antibodies against Ad5 in the serum detected variable responses in the animals (Fig. 3C). Macaque 53 generated the strongest responses with geometric mean titers (GMT) of anti-Ad neutralizing antibodies above 160 at week 19 at then end of the Ad immunization sequence. These neutralizing antibodies remained at this level through week 36 or 4 months after last Ad immunization. Macaque 56 showed low, but detectable neutralizing antibody titers of 20 and 5 at weeks 19 and 28, but these declined to background by week 36. In contrast, macaque H361 showed no Ad neutralizing activity at any time point tested (Fig. 3C). The high levels of anti-vector serum humoral immune responses in macaque 53 could explain the attenuation of anti-HIV antibody responses with time. In contrast, the lack of serum neutralizing antibodies in H361 and low level response in 56 give a less obvious correlation for loss of anti-HIV antibodies in the mucosal tract.
PBMC proliferative responses after oral Ad immunization
Peripheral blood mononuclear cell (PBMC) samples were collected before any immunization and from the immunized macaques at varied times and were assayed for proliferation and IFN-γ production after stimulation with recombinant HIV-1 gag protein and mixtures of the cognate HIV-1 envelope peptides (Fig. 4). All three monkeys exhibited positive proliferation responses (SI values ≥ 2.0) to various combinations of the HIV-1 envelope peptides mainly during the priming phase between weeks 12 and 19 (Fig. 4A). This was true for the PBMC stimulated in vitro with all the six envelope peptides as a single mixture (pep-mix) as well as the gp41 and gp120 subsets (mixtures of 2 and 4 peptides respectively). With respect to Ad-gag used for priming, a low response to the recombinant HIV-1 gag protein (SI 7.7) was observed in the monkey #53 at week 12 (Fig. 4B). As a measure of success for oral delivery and release of capsule contents, monkeys #53 and #56 showed low proliferative responses to the adenoviral capsid at week 17 (SI 4.2 and 4.8, respectively), while all three monkeys exhibited strong responses at week 19 (SI 73.6, 92.5, and 25.5, respectively, Fig. 4B). This suggests that perhaps anti-vector cellular immune responses may have attenuated gene delivery to the GI tract and persistent anti-gag antibody responses (Fig. 3). Strong proliferative responses (average SI values ranging between 19.2 and 23.9) were observed with Con A in all three monkeys at all the time points tested indicating their immune competence (Fig. 4B).
Figure 4.



Antigen-specific cellular immune responses. The proliferative responses in terms of [3H] thymidine incorporation and IFN-γ producing cells by the ELISPOT method were determined using PBMC samples from rhesus macaques immunized by oral priming with enteric-coated capsules delivering Ad-gag and Ad-env-peptide and intranasal boosting with synthetic HIV-1 envelope peptides and CT2*, the mutant cholera toxin adjuvant. (A) Systemic T cell responses using PBMC were determined in terms of proliferation in response to mixtures of all six HIV-1 envelope peptides (pep-mix), two gp41 peptides, four gp120 peptides, and heat-inactivated HIV-1IIIB preparation. The fold-increases in proliferation responses to the different antigens were calculated by comparing the values from cells in the culture medium. (B) Proliferation responses of PBMC were determined in response to recombinant HIV-1 gag protein (p55 gag), heat-inactivated Ad5, and Con A. (C) ELISPOT analyses for antigen-specific IFN-γ-producing cells for 105 input PBMC in each monkey, in terms of spot-forming-cells (SFC/105 PBMC) at the indicated times after oral Ad vector mediated priming and intranasal boosting with peptide-cocktail (6 peptides, 100 μg each/dose) mixed with mutant cholera toxin, CT2* (10 μg/dose). The horizontal line in each panel indicates the cut-off value for positive response (SI of 2.0 for the proliferation assay and SFC of 5 for the ELISPOT assay).
PBMC ELISPOT responses after oral Ad immunization
Measurement of IFN-γ-producing cells within the PBMC samples showed no detectable IFN-γ production in response to stimulation with the mixture of all six HIV-1 envelope peptides (pep-mix) or with the gp41 and gp120 subset peptide mixtures (Fig. 4C). Similar results were obtained when the heat-inactivated HIV-1IIIB was used as the antigen for in vitro stimulation in this assay. Similarly, no IFN-γ-producing cells were observed when the PBMC from the animals were stimulated either with recombinant p55 HIV-1 gag protein or with adenovirus, even though strong responses were obtained with Con A stimulation. The proliferation and ELISPOT data suggest that the oral mucosal immunization crossed over only moderately into the systemic compartment from the mucosal compartment.
Intranasal boosting with synthetic peptides adjuvanted with inactivated cholera toxin
After all oral Ad immunizations were completed and humoral and cellular responses due to this were assessed, the animals were boosted with protein vaccine by the intranasal route. At weeks 28 and 32, the macaques received intranasal immunization 100 μg each of six HIV-1 envelope peptides mixed with the mucosal adjuvant CT2* [25,26]. In this case, the conserved envelope peptides were to boost the env-specific responses primed by oral capsule Ad vaccination. While env responses should be boosted, gag and Ad responses would not.
While all three monkeys had positive proliferation responses after priming, boosting with the peptides mediated only slight increase in these responses against env when PBMCs were stimulated with the same peptides in vitro (Fig. 4A). In contrast, very strong proliferative responses were observed against heat-inactivated HIV-1IIIB preparation only after intranasal boosting whereas only negligible levels of responses were observed during the priming phase. In contrast, ELISPOT IFN-γ responses from PBMCs against the six HIV-1 envelope peptides, the gp41 and gp120 subset peptides, and against inactivated HIV were markedly increased after intranasal boost (Fig. 4C). No IFN-γ-producing cells were observed when the PBMC from the animals were stimulated either with the recombinant p55 HIV-1 gag protein or adenovirus antigens, even though strong responses were obtained with Con A stimulation (Table 3). Since recombinant gag protein was not used during the boosting phase, it is difficult to determine whether the Ad-gag priming was effective in priming any precursor populations. Similarly, given that we did not have a separate group of animals immunized with the peptide-cocktail in the presence of the CT2* adjuvant due to the limited funding of this pilot project, it is difficult to rule out that the IFN-γ responses were solely due to the intranasal immunization regimen.
Table 3.
Cumulative Summary of Antigen-specific Proliferation and IFN-γ ELISPOT Responses in Monkeys Immunized with Oral Adenoviral DNA-priming and Intranasal Peptide-boosting.
| Response to | Monkey 53 | Monkey 56 | Monkey H361 | |||
|---|---|---|---|---|---|---|
| Proliferation (at week) | IFN-g ELISPOT (at week) | Proliferation (at week) | IFN-g ELISPOT (at week) | Proliferation (at week) | IFN-g ELISPOT (at week) | |
| gp41 peptide- mixture | 12, 30, 34 | 34 | 19, 34 | 34 | 17, 34 | 34 |
| gp120 peptide- mixture | 12, 19, 30, 34 | 34, 38 | 34 | 34 | 12, 17, 32 | 30, 34, 38 |
| gp41 + gp120 peptide-mixture | 12, 34 | 34, 38 | 12, 17 | 34, 38 | 17 | 30, 34, 38 |
| HIV-1 p55 gag protein | 12 | |||||
| Heat-inactivated HIV-1 | 17, 30, 34 | 38 | 30, 32, 34 | 38 | 17, 22, 30, 32 | 30, 38 |
| Heat-inactivated Ad5 | 17, 19 | 19 | 17, 19 | 19 | ||
| Con A | All time points tested | All time points tested | All time points tested | All time points tested | All time points tested | All time points tested |
Note: The weeks shown are for positive responses based on the cutoff values described in the methods section.
Post-mortem testing of tissue samples
Animals were euthanized 36 weeks after first immunization and samples were collected from the PBMCs, spleen (SP), mesenteric lymphnodes (MLN), and intestinal tissues to determine if any memory T cells were resident in these sites. The PBMCs and splenocytes would be representative of T cells in the systemic compartment whereas the mesenteric lymphnodes and intestinal tissue should be representative of mucosal T cells, particularly relevant to oral immunization. When T cells from these sites from macaque 53 were stimulated with the HIV-1 envelope peptides, IFN-γproducing CD8+ and CD4+ T cells were observed only in the intestine and not in other samples (Fig. 5). Previous testing with unimmunized macaques showed no responses by the same assay (data not shown). Similar assay on H361 failed to detect any responses (data not shown). Insufficient numbers of T cells were obtained from macaque 56 for the assay. Counterstaining the PBCMs from macaque 53 for cell surface CD95 to identify memory cells, revealed that again these cells were only resident in the intestinal samples. IFN-γ+ CD8+ and CD4+ T cells were not detected in the peripheral blood, spleen, and lymph nodes from macaque 53. Among the CD95+ T cells, 24% of the CD8+ T cells were IFN-γ + whereas 8% of the CD4+ T cells were IFN-γ+. The presence of peptide-specific CD8+ and CD4+ T cells in the intestine of macaque 53 suggest that the prime-boost immunization induced peptide-specific memory T cells in the intestine.
Figure 5.
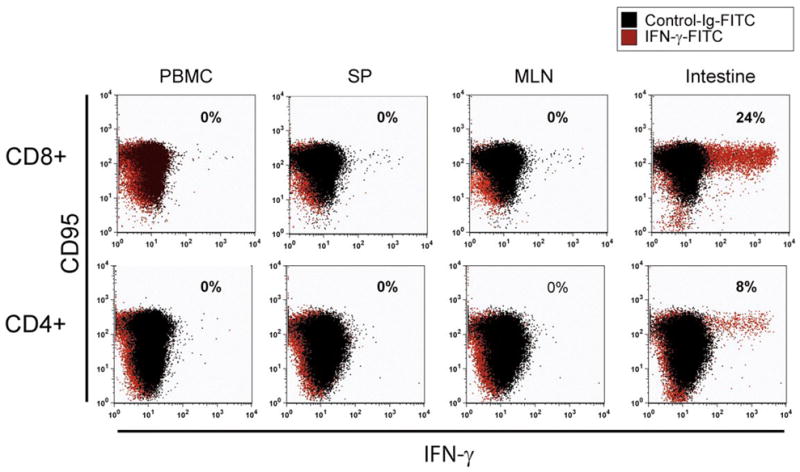
Antigen-specific intracellular IFN-γ production. Cells isolated from various tissues at necropsy of macaque 53 were used for determining intracellular IFN-γ producing T cells by immunofluorescence staining after stimulation with the HIV-1 envelope peptide mixture (pep-mix), cell fixation and permeabilization as described in the methods section. To define T cell subsets, lymphocytes were stained for cell surface CD4, CD8, and CD95, the later to distinguish memory-phenotype. Among the CD95+ T cells, IFN-γ + CD8+ and CD4+ T cells were detected in the intestinal lymphocytes, but not in peripheral blood, spleen, and lymph nodes.
Safety of CT2* as a mucosal adjuvant
We also attempted to address one important issue related to safety of using the cholera toxin mutant as mucosal adjuvant by the immunohistochemical analyses of brain tissue, specifically the olfactory bulb because antigens/adjuvants delivered by the intranasal route can be retrograde transported into the brain potentially causing complicaitons such as Bell’s palsy [43]. Based on literature reports on similar studies, we determined the expression of the nerve growth factor beta 1 (NGF-β1) as a sarrogate for toxicity in terms of retrograde transport to olfactory bulb [42]. As a control we used the tissue similarly obtained from another monkey (H341) from a different study where the monkey was dosed with the native form of the cholera toxin (nCT) that is known to casue retrograde transport to the brain and activation of NGF-b1 in the olfactory bulb [43]. The side-by-side immunohistochemial analyses revealed NGF-β1 expression only in the tissues of the monkey immunized using the nCT but not those where CT2* was used as adjuvant (Fig. 6). These results are similar to those reported by McGhee’s group [42] where another mutant of CT was observed to be non-toxic to rhesus macaques in terms of lack of NGF-b1 expression, when compared with that using nCT.
Figure 6.

Detection of NGF-b1 expression in the olfactory bulbs of rhesus macaques immunized by the intranasal route with the HIV-1 envelope peptide cocktail (pep-mix) using either the mucosal adjuvant mutant cholera toxin CT2* (the first three panels on the left-side indicated by monkey numbers, 53, 56, and H361) or native cholera toxin (nCT, right-side panel indicated by monkey number H341). Frozen tissue sections were subjected to anti-NGF-b1 antibody immunohistochemistry staining (upper panels) and immunofluoresence straining (lower panels) by reacting with avidin-biotin conjugate, followed by incubation with HRP-conjugated streptavidin or streptavidin-Alexa Fluor 488.
DISCUSSION
The goal of this study was to generate enteric-coated capsules for oral vaccines under laboratory conditions and provide these methods to the academic vaccine community. Towards this goal, we utilized reagents and instruments that can be purchased by the basic scientist including anionic hydrophilic polymers, HPMC capsules, and standard lyophilization procedures.
A simple lyophilization procedure was adapted from [35]. Ad vectors in KPBS and 1.0 M sucrose were flash frozen in capsules on dry ice to avoid dissolution of the water-soluble capsule and were lyophilized in a standard laboratory freeze-drying system. Vectors were directly loaded into capsules to avoid having to weigh μg masses of lyophilized virus powder to achieve relevant single doses of vaccine. By this approach, vectors lost 50% activity within a day of storage at 4°C and eventually stabilized to 95% lost activity by 3 weeks. The loss of activity for this buffer contrasts with Croyle et al., in which only 10% activity was lost over 10 days at 4°C [35]. In this case, a more intricate lyophilization procedure was followed rather than our simple one, which may explain the difference in virus stability. A more recent study of lyophilized replication-competent Ad in tablets reported 33% loss in activity with the same buffer, but after extended 3-day lyophilization [44]. Alternatively, this difference in stability could be due to our flash-freezing of the virus directly into capsules versus the lyophilization in glass vials [35]. If so, one could hypothetically lyophilize in a vial and then transfer to a capsule. However, weighing the relevant number of particles into a vial would likely be problematic. We anticipate the use of more effective stabilizing agents such as 0.4% Sucrose/0.4% Mannitol/0.001% Span 20 [35] and a more rigid lyophilization procedure would likely improve vector stability.
The polymers used were Eudragit® L100 and Eudragit® S100 which are methacrylic acid esters that are insoluble below a pH of 5.0. The enteric reagents were developed as a peroral dosage form with a step-wise release of the coated ingredient within the GI tract as a function of its luminal pH. Eudragit® L100 is a 1:1 co-polymer of methacrylic acid and methylmethacrylate and Eudragit® S100 a 1:2 copolymer. The gastroresistance of polymethacrylates can be attributed to the exposed ester side chains that are very resistant to hydrolysis, and the pH dependent solubility can be attributed to the step-wise alkaline hydrolysis of the exposed esterified terminal groups [45]. However, the Eudragit® polymers are not ionized and solubilized at a specific pH, but rather a range of pH. Since the ratio of carboxyl to ester groups in the polymer chains of Eudragit® S100 is 1:2 and 1:1 in Eudragit® L100, the lower ratio of carboxyl groups in S100 causes a lesser degree of ionization in an alkaline environment and therefore a decreased rate of solubilization [46]. By varying the type of Eudragit® polymer blend and ratio, the resulting drug release and dissolution kinetics in response to changing pH can be varied accordingly [47]. A ratio of 4:1 of Eudragit® L100 to Eudragit® S100 [48,49] was used in this study (Table 1) for the purpose of allowing the increased presence of Eudragit® L100 to cause pore formation, creating channels for alkaline media to penetrate the coating, causing dissolution of the polymer coat, and releasing the capsule contents.
Table 1.
Enteric Coating Formulation. Amounts reported are for making a 157-g coating dispersion for use with a Torpac Inc. Pro-Coater.
| Excipients | Quantity |
|---|---|
| ddH2O | 6.8 ml |
| Isopropyl Alcohol | 164.2 ml |
| Talc | 6.93 g |
| Eudragit® L100 | 11.08 g |
| Eudragit® S100 | 2.77 g |
| TEC | 0.809 ml |
Excipients are listed in the sequence in which they are mixed.
HPMC capsules were used in this study since they promote good polymer to polymer compatibility with anionic hydrophilic enteric polymers [21], they eliminate the need for pre-coating treatments [50], and they avoid incorporating additional adhesive polymer derivatives that may alter dissolution behavior and release kinetics [51]. In order to ensure the adhesion of the enteric coating polymer dispersion, the amount of plasticizer (TEC) was adjusted to manufacturer recommended specifications for enteric coatings (Table 1). Because pharmaceutical acrylic polymers are brittle, fragile and exhibit strong adhesiveness [52], the addition of plasticizers and glidants, such as talc (Table 1), ensures the favorable elasticity of the coatings by decreasing polymer tensile strength [52,53], lowering the film forming temperature without adverse mechanical effects [54–58], and facilitating GI transit through mucosal surfaces by avoiding polymer dispersion agglomeration.
When polymer-coated capsules were tested directly for dissolution kinetics, the polymer remained intact at low pH values and mediated burst release of small molecule dye at pH 7.0 and 7.5. Release of Ad from the capsules was more delayed, with near burst release after 80 minutes of exposure at pH 7.5. Published literature on monkey intestinal pH (Table 2) would suggest that these enteric-coated capsules should remain intact through the duodenum and begin dissolving in the jejenum. Given the 80 minute delay in Ad release at pH 7.5 and jejenum transit times from 0.5 to 6 hours, we anticipate that virus release from the capsules likely occurs prior to the colon. Whether this is indeed the case and to what degree variation in the site of Ad release affects mucosal immune responses will require additional studies.
After oral immunization with capsules bearing replication-defective Ad vectors expressing HIV-1 gag and HIV-1 envelope peptides, combined mucosal and systemic immune responses were observed. In particular, oral delivery of Ad generated detectable anti-gag IgA responses in saliva of two of the three animals and vaginal IgA in all of the animals, which declined rapidly after immunization. Production of higher affinity IgM or IgG antibodies against gag may have reduced detection of gag-specific IgA in these ELISA assays. However, ELISA with secondary antibodies against IgA, M, and G failed to detect more persistent antibodies in the samples (data not shown). This suggests, the formation of immune complexes may not explain the loss of anti-gag antibodies in the animals.
In contrast to the mucosal antibody responses against gag, anti-adenovirus antibodies were only detected in one sample from one animal by ELISA. This contrasted with assays for serum antibodies against Ad, where responses were detected in two animals. Macaque 53 had strong Ad neutralizing activity by the end of the Ad immunization series with GMT above 160 and these antibody levels persisted through 36 weeks. In contrast, macaque 56 had weaker serum neutralizing activity (GMT =20) and H361 had no detectable neutralizing antibodies in the serum. Given that macaque H361 had the most persistent salivary anti-gag antibodies and macaque 53 had the most persistent vaginal antibodies, a direct correlation between anti-Ad antibodies and vaccine-generated antibody persistence is unclear. In contrast, robust cellular responses were observed in all of the animals at weeks 17 and 19 and these coincided with the loss of anti-gag IgA antibodies. These data suggest anti-vector T cell responses and possibly antibody responses may have a role in attenuating anti-HIV responses after oral delivery. Even if anti-vector immunity is to blame for the eventual reduction in IgA, it is significant that responses were observed in at least some of the animals after each oral immunization, and the data suggest that the oral tract may not be as sensitive to neutralization even after four exposures to adenovirus vectors.
T cell responses generated by Ad vaccination were only be tested from the systemic compartment during immunization by assay of PBMCs. Ad immunization drove detectable proliferative responses to various HIV-1 envelope peptides in all three animals and against gag only in macaque 53. In contrast, PBMC samples showed that oral Ad drove no detectable ELISPOT IFN-γ against envelope or gag antigens. These data suggest that the oral mucosal immunization crossed over only moderately into the systemic compartment. Previous work with the peptide vaccine showed that depletion of CD8 cells from PBMCs attenuates ELISPOT, but not proliferative responses (data not shown). Based on this, we speculate that oral Ad vaccination may drive production or crossover of helper T cells into the systemic compartment better than its ability to drive CD8 T cells into the systemic compartment from mucosal sites.
While attenuation of IgA responses was eventually observed, the ability to immunize multiple times appears effective as a priming regimen for other vaccines or for the use of other serotypes of Ad [59–62]. Proof of principle is demonstrated here where mucosal oral priming of macaques by oral Ad administration was followed by mucosal boosting with an intranasal peptide vaccine. In this case, boosting with synthetic envelope peptides adjuvanted with CT2* generated rapid T cell responses against env peptides and whole inactivated HIV virions. While we cannot formally exclude that these responses were not directly related to the priming by the peptide immunization alone, due to the limited scope of this pilot project, the rapid, strong kinetics of the responses within 2 weeks contrasts with previous work immunizing macaques with peptides alone [25]. Moreover, post-mortem T cell analysis of systemic and gut samples demonstrated robust T cell responses only in the intestinal tissues and not in PBMCs, the spleen, or in mesenteric lymph nodes. These data suggest, but do not prove, that these T cells were originally “educated” in the gut and “home” preferentially back to this site. Future work with larger groups of animals will compare different routes of systemic and mucosal vaccination in macaques to determine how this affects the establishment of memory responses at different sites.
In summary, this pilot study provides proof of principle for oral capsule delivery of replication-defective adenoviral vaccines. This simple vaccination route may provide an attractive means to deliver HIV and other vaccines across the globe if more stringent lyophilization and encapsulation approaches are perfected to maximize vector stability. Efforts to evade pre-existing and vector-induced anti-Ad immunity will likely enhance vaccine efficacy by this route. Nonetheless, the ability to deliver the same vector multiple times to the host and generate detectable responses suggests the oral route may help avoid immunity to the vector. This is consistent with previous work in mice with Ad vectors [63]. These data also suggest this route can be used for prime-boost strategies with other vaccines. Work is underway to determine the optimal combination of priming and boosting sites to afford the best barrier protection at mucosal sites for HIV-1 and other pathogens.
Acknowledgments
We thank Mary E. Barry for excellent technical assistance, members of the Barry Lab and Sastry Lab, and Dr. Pervez Firozi for many useful discussions and technical assistance, and Dr. Ashok Chopra, University of Texas Medical Branch, Galveston, for providing the purified mutant cholera toxin CT2*. We thank Rohm Degussa Pharma for providing samples of the Eudragit polymer coatings. This study was supported by a pilot grant from the Center for AIDS Research at Baylor College of Medicine funded by AI36211, grants AI42588 and AI067095 to M.A.B. from the National Institutes of Health, to K.J.S. from NIAID grants AI42694 and 46969, National Science Foundation IGERT Award DGE-0114264 (to G.T.M.). All the cell culture media were produced by the Central Media lab and all the synthetic peptides were prepared in the Synthetic Antigen Core Facility, both supported by funds from NIH grant CA 16672. HIV and SIV reagents were obtained from the NIH AIDS Research and Reagent Program, and the immunohistochemistry analyses were performed in the core facility at the Center for Cancer Immunology Research, the University of Texas MD Anderson Cancer Center, with expert technical support from Mrs. Yi Hong Wang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shiver JW, Emini EA. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu Rev Med. 2004;55:355–372. doi: 10.1146/annurev.med.55.091902.104344. [DOI] [PubMed] [Google Scholar]
- 2.Belyakov IM, Derby MA, Ahlers JD, et al. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc Natl Acad Sci U S A. 1998;95(4):1709–1714. doi: 10.1073/pnas.95.4.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belyakov IM, Ahlers JD, Brandwein BY, et al. The importance of local mucosal HIV-specific CD8(+) cytotoxic T lymphocytes for resistance to mucosal viral transmission in mice and enhancement of resistance by local administration of IL-12. J Clin Invest. 1998;102(12):2072–2081. doi: 10.1172/JCI5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallichan WS, Rosenthal KL. Long-term immunity and protection against herpes simplex virus type 2 in the murine female genital tract after mucosal but not systemic immunization. J Infect Dis. 1998;177(5):1155–1161. doi: 10.1086/515286. [DOI] [PubMed] [Google Scholar]
- 5.Gallichan WS, Rosenthal KL. Long-lived cytotoxic T lymphocyte memory in mucosal tissues after mucosal but not systemic immunization. J Exp Med. 1996;184(5):1879–1890. doi: 10.1084/jem.184.5.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaneko H, Bednarek I, Wierzbicki A, et al. Oral DNA vaccination promotes mucosal and systemic immune responses to HIV envelope glycoprotein. Virology. 2000;267(1):8–16. doi: 10.1006/viro.1999.0093. [DOI] [PubMed] [Google Scholar]
- 7.Pinto AR, Fitzgerald JC, Gao GP, Wilson JM, Ertl HC. Induction of CD8+ T cells to an HIV-1 antigen upon oral immunization of mice with a simian E1-deleted adenoviral vector. Vaccine. 2004;22(5–6):697–703. doi: 10.1016/j.vaccine.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 8.Sharpe S, Fooks A, Lee J, Hayes K, Clegg C, Cranage M. Single oral immunization with replication deficient recombinant adenovirus elicits long-lived transgene-specific cellular and humoral immune responses. Virology. 2002;293(2):210–216. doi: 10.1006/viro.2001.1281. [DOI] [PubMed] [Google Scholar]
- 9.Ertl HC. Immunological insights from genetic vaccines. Virus Res. 2005;111(1):89–92. doi: 10.1016/j.virusres.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 10.Flanagan B, Pringle CR, Leppard KN. A recombinant human adenovirus expressing the simian immunodeficiency virus Gag antigen can induce long-lived immune responses in mice. J Gen Virol. 1997;78(Pt 5):991–997. doi: 10.1099/0022-1317-78-5-991. [DOI] [PubMed] [Google Scholar]
- 11.Cheng X, Ming X, Croyle MA. PEGylated adenoviruses for gene delivery to the intestinal epithelium by the oral route. Pharm Res. 2003;20(9):1444–1451. doi: 10.1023/a:1025714412337. [DOI] [PubMed] [Google Scholar]
- 12.Nyberg-Hoffman C, Aguilar-Cordova E. Instability of adenoviral vectors during transport and its implication for clinical studies. Nat Med. 1999;5(8):955–957. doi: 10.1038/11400. [DOI] [PubMed] [Google Scholar]
- 13.Wong G, Kaattari SL, Christensen JM. Effectiveness of an oral enteric coated vibrio vaccine for use in salmonid fish. Immunol Invest. 1992;21(4):353–364. doi: 10.3109/08820139209069375. [DOI] [PubMed] [Google Scholar]
- 14.Jain SL, Barone KS, Flanagan MP, Michael JG. Activation patterns of murine B cells after oral administration of an encapsulated soluble antigen. Vaccine. 1996;14(1):42–48. doi: 10.1016/0264-410x(95)00158-w. [DOI] [PubMed] [Google Scholar]
- 15.Delgado A, Lavelle EC, Hartshorne M, Davis SS. PLG microparticles stabilised using enteric coating polymers as oral vaccine delivery systems. Vaccine. 1999;17(22):2927–2938. doi: 10.1016/s0264-410x(99)00140-1. [DOI] [PubMed] [Google Scholar]
- 16.Couch RB, Chanock RM, Cate TR, Lang DJ, Knight V, Huebner RJ. Immunization with Types 4 and 7 Adenovirus by Selective Infection of the Intestinal Tract. Am Rev Respir Dis. 1963;88(SUPPL):394–403. doi: 10.1164/arrd.1963.88.3P2.394. [DOI] [PubMed] [Google Scholar]
- 17.Top FH, Jr, Buescher EL, Bancroft WH, Russell PK. Immunization with live types 7 and 4 adenovirus vaccines. II. Antibody response and protective effect against acute respiratory disease due to adenovirus type 7. J Infect Dis. 1971;124(2):155–160. doi: 10.1093/infdis/124.2.155. [DOI] [PubMed] [Google Scholar]
- 18.Top FH, Jr, Grossman RA, Bartelloni PJ, et al. Immunization with live types 7 and 4 adenovirus vaccines. I. Safety, infectivity, antigenicity, and potency of adenovirus type 7 vaccine in humans. J Infect Dis. 1971;124(2):148–154. doi: 10.1093/infdis/124.2.148. [DOI] [PubMed] [Google Scholar]
- 19.Lubeck MD, Davis AR, Chengalvala M, et al. Immunogenicity and efficacy testing in chimpanzees of an oral hepatitis B vaccine based on live recombinant adenovirus. Proc Natl Acad Sci U S A. 1989;86(17):6763–6767. doi: 10.1073/pnas.86.17.6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukui E, Miyamura N, Uemura K, Kobayashi M. Preparation of enteric coated timed-release press-coated tablets and evaluation of their function by in vitro and in vivo tests for colon targeting. Int J Pharm. 2000;204(1–2):7–15. doi: 10.1016/s0378-5173(00)00454-3. [DOI] [PubMed] [Google Scholar]
- 21.Cole ET, Scott RA, Connor AL, et al. Enteric coated HPMC capsules designed to achieve intestinal targeting. Int J Pharm. 2002;231(1):83–95. doi: 10.1016/s0378-5173(01)00871-7. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez-Fuentes J, Fernandez-Arevalo M, Gonzalez-Rodriguez ML, Cirri M, Mura P. Development of enteric-coated timed-release matrix tablets for colon targeting. J Drug Target. 2004;12(9–10):607–612. doi: 10.1080/10611860400013501. [DOI] [PubMed] [Google Scholar]
- 23.Huyghebaert N, Vermeire A, Remon JP. In vitro evaluation of coating polymers for enteric coating and human ileal targeting. Int J Pharm. 2005;298(1):26–37. doi: 10.1016/j.ijpharm.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 24.Tamura A, Soga H, Yaguchi K, et al. Distribution of two types of lymphocytes (intraepithelial and lamina-propria-associated) in the murine small intestine. Cell Tissue Res. 2003;313(1):47–53. doi: 10.1007/s00441-003-0706-4. [DOI] [PubMed] [Google Scholar]
- 25.Lomada D, Gambhira R, Nehete PN, et al. A two-codon mutant of cholera toxin lacking ADP-ribosylating activity functions as an effective adjuvant for eliciting mucosal and systemic cellular immune responses to peptide antigens. Vaccine. 2004;23(4):555–565. doi: 10.1016/j.vaccine.2004.05.039. [DOI] [PubMed] [Google Scholar]
- 26.Manuri PR, Nehete B, Nehete PN, et al. Intranasal immunization with synthetic peptides corresponding to the E6 and E7 oncoproteins of human papillomavirus type 16 induces systemic and mucosal cellular immune responses and tumor protection. Vaccine. 2007 doi: 10.1016/j.vaccine.2007.01.010. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotsopoulou E, Kim VN, Kingsman AJ, Kingsman SM, Mitrophanous KA. A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J Virol. 2000;74(10):4839–4852. doi: 10.1128/jvi.74.10.4839-4852.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis AR, Wivel NA, Palladino JL, Tao L, Wilson JM. Construction of adenoviral vectors. Methods in Molecular Biology. 2000;135:515–523. doi: 10.1385/1-59259-685-1:515. [DOI] [PubMed] [Google Scholar]
- 29.Sastry KJ, Arlinghaus RB. Identification of T-cell epitopes without B-cell activity in the first and second conserved regions of the HIV Env protein. Aids. 1991;5(6):699–707. doi: 10.1097/00002030-199106000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Nehete PN, Satterfield WC, Matherne CM, Arlinghaus RB, Sastry KJ. Induction of human immunodeficiency virus-specific T cell responses in rhesus monkeys by synthetic peptides from gp160. AIDS Res Hum Retroviruses. 1993;9(3):235–240. doi: 10.1089/aid.1993.9.235. [DOI] [PubMed] [Google Scholar]
- 31.Nehete PN, Schapiro SJ, Johnson PC, Murthy KK, Satterfield WC, Sastry KJ. A synthetic peptide from the first conserved region in the envelope protein gp160 is a strong T-cell epitope in HIV-infected chimpanzees and humans. Viral Immunol. 1998;11(3):147–158. doi: 10.1089/vim.1998.11.147. [DOI] [PubMed] [Google Scholar]
- 32.Nehete PN, Lewis DE, Tang DN, Pollack MS, Sastry KJ. Presence of HLA-C-restricted cytotoxic T-lymphocyte responses in long-term nonprogressors infected with human immunodeficiency virus. Viral Immunol. 1998;11(3):119–129. doi: 10.1089/vim.1998.11.119. [DOI] [PubMed] [Google Scholar]
- 33.Nehete PN, Chitta S, Hossain MM, et al. Protection against chronic infection and AIDS by an HIV envelope peptide-cocktail vaccine in a pathogenic SHIV-rhesus model. Vaccine. 2001;20(5–6):813–825. doi: 10.1016/s0264-410x(01)00408-x. [DOI] [PubMed] [Google Scholar]
- 34.Nehete PN, Nehete BP, Manuri P, Hill L, Palmer JL, Sastry KJ. Protection by dendritic cells-based HIV synthetic peptide cocktail vaccine: preclinical studies in the SHIV-rhesus model. Vaccine. 2005;23(17–18):2154–2159. doi: 10.1016/j.vaccine.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 35.Croyle MA, Cheng X, Wilson JM. Development of formulations that enhance physical stability of viral vectors for gene therapy. Gene Ther. 2001;8(17):1281–1290. doi: 10.1038/sj.gt.3301527. [DOI] [PubMed] [Google Scholar]
- 36.Parrott MB, Adams KE, Mercier GT, Mok H, Campos SK, Barry MA. Metabolically biotinylated adenovirus for cell targeting, ligand screening, and vector purification. Mol Ther. 2003;8(4):688–700. doi: 10.1016/s1525-0016(03)00213-2. [DOI] [PubMed] [Google Scholar]
- 37.Nehete PN, Gambhira R, Nehete BP, Sastry KJ. Dendritic cells enhance detection of antigen-specific cellular immune responses by lymphocytes from rhesus macaques immunized with an HIV envelope peptide cocktail vaccine. J Med Primatol. 2003;32(2):67–73. doi: 10.1034/j.1600-0684.2003.00011.x. [DOI] [PubMed] [Google Scholar]
- 38.Nehete PN, Nehete BGR, Sastry KJ. Dendritic cells enhance detection of antigen-specific cellular immune responses by lymphocytes from rhesus macaques immunized with an HIV envelope peptide cocktail vaccine. Jr Medical Primatology. 2003 doi: 10.1034/j.1600-0684.2003.00011.x. In Press. [DOI] [PubMed] [Google Scholar]
- 39.Davies MD, Parrott DM. Preparation and purification of lymphocytes from the epithelium and lamina propria of murine small intestine. Gut. 1981;22(6):481–488. doi: 10.1136/gut.22.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.James SP, Graeff AS, Zeitz M. Predominance of helper-inducer T cells in mesenteric lymph nodes and intestinal lamina propria of normal nonhuman primates. Cell Immunol. 1987;107(2):372–383. doi: 10.1016/0008-8749(87)90245-0. [DOI] [PubMed] [Google Scholar]
- 41.Shacklett BL, Yang O, Hausner MA, et al. Optimization of methods to assess human mucosal T-cell responses to HIV infection. J Immunol Methods. 2003;279(1–2):17–31. doi: 10.1016/s0022-1759(03)00255-2. [DOI] [PubMed] [Google Scholar]
- 42.Yoshino N, Lu FX, Fujihashi K, et al. A novel adjuvant for mucosal immunity to HIV-1 gp120 in nonhuman primates. J Immunol. 2004;173(11):6850–6857. doi: 10.4049/jimmunol.173.11.6850. [DOI] [PubMed] [Google Scholar]
- 43.van Ginkel FW, Jackson RJ, Yuki Y, McGhee JR. Cutting edge: the mucosal adjuvant cholera toxin redirects vaccine proteins into olfactory tissues. J Immunol. 2000;165(9):4778–4782. doi: 10.4049/jimmunol.165.9.4778. [DOI] [PubMed] [Google Scholar]
- 44.Gomez-Roman VR, Grimes GJ, Jr, Potti GK, et al. Oral delivery of replication-competent adenovirus vectors is well tolerated by SIV- and SHIV-infected rhesus macaques. Vaccine. 2006;24(23):5064–5072. doi: 10.1016/j.vaccine.2006.03.048. [DOI] [PubMed] [Google Scholar]
- 45.Lehmann K. Chemistry and Application Properties of Polymethacrylate Coating Systems. In: McGinity JW, editor. Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms. Marcel Dekker, Inc; 1996. pp. 101–174. [Google Scholar]
- 46.Chourasia MK, Jain SK. Pharmaceutical approaches to colon targeted drug delivery systems. J Pharm Pharm Sci. 2003;6(1):33–66. [PubMed] [Google Scholar]
- 47.Lecomte F, Siepmann J, Walther M, MacRae RJ, Bodmeier R. pH-Sensitive polymer blends used as coating materials to control drug release from spherical beads: elucidation of the underlying mass transport mechanisms. Pharm Res. 2005;22(7):1129–1141. doi: 10.1007/s11095-005-5421-2. [DOI] [PubMed] [Google Scholar]
- 48.Khan MZ, Stedul HP, Kurjakovic N. A pH-dependent colon-targeted oral drug delivery system using methacrylic acid copolymers. II. Manipulation of drug release using Eudragit L100 and Eudragit S100 combinations. Drug Dev Ind Pharm. 2000;26(5):549–554. doi: 10.1081/ddc-100101266. [DOI] [PubMed] [Google Scholar]
- 49.Khan MZ, Prebeg Z, Kurjakovic N. A pH-dependent colon targeted oral drug delivery system using methacrylic acid copolymers. I. Manipulation Of drug release using Eudragit L100-55 and Eudragit S100 combinations. J Control Release. 1999;58(2):215–222. doi: 10.1016/s0168-3659(98)00151-5. [DOI] [PubMed] [Google Scholar]
- 50.Thoma K, Bechtold K. Influence of aqueous coatings on the stability of enteric coated pellets and tablets. Eur J Pharm Biopharm. 1999;47(1):39–50. doi: 10.1016/s0939-6411(98)00086-1. [DOI] [PubMed] [Google Scholar]
- 51.Murthy KS, Enders NA, Mahjour M, Fawzi MB. A comparitive evaluation of aqueous enteric polymers in capsule coating. Pharm Technol. 1986;(10):36–46. [Google Scholar]
- 52.Lee BJ, Ryu SG, Cui JH. Controlled release of dual drug-loaded hydroxypropyl methylcellulose matrix tablet using drug-containing polymeric coatings. Int J Pharm. 1999;188(1):71–80. doi: 10.1016/s0378-5173(99)00204-5. [DOI] [PubMed] [Google Scholar]
- 53.Lee BJ, Ryu SG, Cui JH. Formulation and release characteristics of hydroxypropyl methylcellulose matrix tablet containing melatonin. Drug Dev Ind Pharm. 1999;25(4):493–501. doi: 10.1081/ddc-100102199. [DOI] [PubMed] [Google Scholar]
- 54.McGinity JW. Aqueous Polymeric Coatings for Pharmaceutical Dosage Forms. Marcel Dekker; NY: 1987. [Google Scholar]
- 55.Wang CC, Zhang G, Shah NH, Infeld MH, Malick AW, McGinity JW. Influence of plasticizers on the mechanical properties of pellets containing Eudragit RS 30D. Int J Pharm. 1997;152:153–163. [Google Scholar]
- 56.Wang CC, Zhang G, Shah NH, Infeld MH, Malick AW, McGinity JW. Mechanical properties of single pellets containing acrylic polymers. Pharm Dev Technol. 1996;1(2):213–222. doi: 10.3109/10837459609029896. [DOI] [PubMed] [Google Scholar]
- 57.Repka MA, Gerding TG, Repka SL, McGinity JW. Influence of plasticizers and drugs on the physical-mechanical properties of hydroxypropylcellulose films prepared by hot melt extrusion. Drug Dev Ind Pharm. 1999;25(5):625–633. doi: 10.1081/ddc-100102218. [DOI] [PubMed] [Google Scholar]
- 58.Gutierrez-Rocca JC, McGinity JW. Influence of water soluble and insoluble plasticizers on the physical mechanical properties of acrylic resin copolymers. Int J Pharm. 1994;103:293–301. [Google Scholar]
- 59.Fitzgerald JC, Gao GP, Reyes-Sandoval A, et al. A simian replication-defective adenoviral recombinant vaccine to HIV-1 gag. J Immunol. 2003;170(3):1416–1422. doi: 10.4049/jimmunol.170.3.1416. [DOI] [PubMed] [Google Scholar]
- 60.Pinto AR, Fitzgerald JC, Giles-Davis W, Gao GP, Wilson JM, Ertl HC. Induction of CD8+ T cells to an HIV-1 antigen through a prime boost regimen with heterologous E1-deleted adenoviral vaccine carriers. J Immunol. 2003;171(12):6774–6779. doi: 10.4049/jimmunol.171.12.6774. [DOI] [PubMed] [Google Scholar]
- 61.Barouch DH, Pau MG, Custers JH, et al. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172(10):6290–6297. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 62.Lemckert AA, Sumida SM, Holterman L, et al. Immunogenicity of heterologous prime-boost regimens involving recombinant adenovirus serotype 11 (Ad11) and Ad35 vaccine vectors in the presence of anti-ad5 immunity. J Virol. 2005;79(15):9694–9701. doi: 10.1128/JVI.79.15.9694-9701.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiang ZQ, Gao GP, Reyes-Sandoval A, Li Y, Wilson JM, Ertl HC. Oral vaccination of mice with adenoviral vectors is not impaired by preexisting immunity to the vaccine carrier. J Virol. 2003;77(20):10780–10789. doi: 10.1128/JVI.77.20.10780-10789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kararli TT. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm Drug Dispos. 1995;16(5):351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 65.Avraham R. The Digestive System. Chelsea House; 2000. [Google Scholar]
- 66.Gray H. The Anatomy of the Human Body. Lea & Febinger; 1985. [Google Scholar]
- 67.Wilson DEMRD. Mammal Species of the World: A Taxonomic and Geographic Reference, II. Smithsonian Institution Press; Washington, D.C: 1993. [Google Scholar]
- 68.Kondo H, Takahashi Y, Watanabe T, Yokohama S, Watanabe J. Gastrointestinal transit of liquids in unfed cynomolgus monkeys. Biopharm Drug Dispos. 2003;24(3):131–140. doi: 10.1002/bdd.348. [DOI] [PubMed] [Google Scholar]
- 69.Kondo H, Watanabe T, Yokohama S, Watanabe J. Effect of food on gastrointestinal transit of liquids in cynomolgus monkeys. Biopharm Drug Dispos. 2003;24(4):141–151. doi: 10.1002/bdd.349. [DOI] [PubMed] [Google Scholar]