

Abstract
Hyperglycemia-induced overproduction of superoxide by mitochondrial electron-transport chain triggers several pathways of injury involved in the pathogenesis of diabetic complications [protein kinase C (PKC), hexosamine and polyol pathway fluxes, advanced glycation end product (AGE) formation] by inhibiting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity. Increased oxidative and nitrosative stress activates the nuclear enzyme, poly(ADP-ribose) polymerase-1 (PARP). PARP activation, on the one hand, depletes its substrate, NAD+, slowing the rate of glycolysis, electron transport, and ATP formation. On the other hand, it inhibits GAPDH by poly(ADP-ribosy)lation. These processes result in acute endothelial dysfunction in diabetic blood vessels, which importantly contributes to the development of various diabetic complications. Accordingly, hyperglycemia-induced activation of PKC isoforms, hexosaminase pathway flux, and AGE formation is prevented by blocking PARP activity. Furthermore, inhibition of PARP protects against diabetic cardiovascular dysfunction in preclinical models. PARP activation is present in microvasculature of human diabetic subjects. The oxidative/nitrosative stress–PARP pathway leads to diabetes-induced endothelial dysfunction, which may be an important underlying mechanism for the pathogenesis of other diabetic complications (cardiomyopathy, nephropathy, neuropathy, and retinopathy). This review focuses on the role of PARP in diabetic complications and the unique therapeutic potential of PARP inhibition in the prevention or reversal of diabetic complications.
INTRODUCTION
POLY(ADP-RIBOSE) POLYMERASE (PARP) is a nuclear DNA repair enzyme with multiple regulatory functions (23–25, 38, 49, 55, 85, 95, 96, 99, 114). Overactivation of PARP represents an important mechanism of tissue damage in various pathological conditions associated with oxidative and nitrosative stress, including myocardial reperfusion injury (107, 120), heart transplantation (106), heart failure (70, 71), stroke (31, 45), circulatory shock (42, 68, 69, 89, 93, 98), and autoimmune β-cell destruction associated with diabetes mellitus (10, 80). Activation of PARP and beneficial effect of various PARP inhibitors have been demonstrated in various forms of endothelial dysfunction, such as those associated with circulatory shock, hypertension, atherosclerosis, pre-eclampsia, and aging (41, 54, 73, 74, 98). Furthermore, recent evidence suggests that activation of PARP importantly contributes to the development of endothelial dysfunction in various experimental models of diabetes and also in humans (33, 72, 91, 102). In addition, it has recently been demonstrated that PARP activation plays a pathogenetic role in diabetic nephropathy, neuropathy, and retinopathy. The following review will discuss the role of PARP activation in the pathogenesis of diabetic complications with special focus on endothelial dysfunction, as a common underlying theme.
THE PROCESS OF PARP ACTIVATION
Poly(ADP-ribose) polymerase-1 (PARP-1; EC 2.4.2.30) [also known as poly(ADP-ribose) synthetase (PARS) or poly(ADP-ribose) transferase (ADPRT)] is a member of the PARP enzyme family consisting of PARP-1 and an increasing number of additional, recently identified poly(ADP-ribosyl)ating enzymes (minor PARP isoforms). PARP-1, the major PARP isoform, is one of the most abundant proteins in the nucleus. PARP-1 is a 116-kDa protein that consists of three main domains: the N-terminal DNA-binding domain containing two zinc fingers, the automodification domain, and the C-terminal catalytic domain. The primary structure of the enzyme is highly conserved in eukaryotes with the catalytic domain showing the highest degree of homology between different species. The structure and functions of PARP have been the subject of several recent overviews and monographs (95, 96, 114, 118). For the purpose of the current review, it is important to note that PARP-1 is considered the major isoform of PARP in intact cells, and remains commonly termed as “PARP.”
PARP-1 plays an important role in multiple physiological functions, as well as in the pathophysiology of many diseases. This has been a subject of several recent reviews and monograph (24, 25, 49, 95, 98). PARP-1 functions as a DNA damage sensor and signaling molecule binding to both single- and double-stranded DNA breaks. Upon binding to damaged DNA (mainly through the second zinc finger domain), PARP-1 forms homodimers and catalyzes the cleavage of nicotinamide adenine dinucleotide (NAD+) into nicotinamide and ADP-ribose and uses the latter to synthesize branched nucleic acid-like polymers of poly(ADP-ribose) covalently attached to nuclear acceptor proteins. The size of the branched polymer varies from a few to 200 ADP-ribose units. Due to its high negative charge, covalently attached ADP-ribose polymer dramatically affects the function of target proteins. In vivo the auto-poly(ADP-ribosyl)ation represents a major regulatory mechanism for PARP-1, resulting in the down-regulation of the enzyme activity. In addition to PARP-1, histones are also considered as major acceptors of poly(ADP-ribose). Poly(ADP-ribosy)lation confers negative charge to histones, leading to electrostatic repulsion between DNA and histones. This process has been implicated in chromatin remodeling, DNA repair, and transcriptional regulation. Several transcription factors, DNA replication factors, and signaling molecules [nuclear factor-κB (NFκB), activator protein-1 (AP-1), Oct-1, YY1, TEF-1, DNA-PK, p53] have also been shown to become poly(ADP-ribosyl)ated by PARP-1. The effect of PARP-1 on the function of these proteins is carried out by noncovalent protein–protein interactions and by covalent poly(ADP-ribosyl)ation (for review, see 114).
Poly(ADP-ribosyl)ation is a dynamic process as indicated by the short half-life of the polymer. Two enzymes, poly(ADP-ribose) glycohydrolase (PARG) and ADP-ribosyl protein lyase, are involved in the catabolism of poly(ADP-ribose) with PARG cleaving ribose–ribose bonds of both linear and branched portions of poly(ADP-ribose) and the lyase removing the protein proximal ADP-ribose monomer (23). PARP-1 plays a role in DNA repair and maintenance of genomic integrity (55, 85) and also regulates the expression of various proteins at the transcriptional level. Of special importance is the regulation by PARP-1 of the production of inflammatory mediators such as inducible nitric oxide synthase (iNOS), intercellular adhesion molecule-1 (ICAM-1), and major histocompatibility complex class II (30, 38, 90, 99, 120). NF-κB is a key transcription factor in the regulation of this set of proteins, and PARP has been shown to act as a coactivator in the NF-κB-mediated transcription. Poly(ADP-ribosyl)ation can loosen up the chromatin structure, thereby making genes more accessible for the transcriptional machinery (37, 48, 67, 79, 81, 88).
PARP-1 activation has been proposed to represent a cell elimination pathway whereby severely damaged cells are removed from tissues. PARP-1-mediated cell death occurs in the form of necrosis, which is the least desirable form of cell death. During necrotic cell death, the cellular content is released into the tissue exposing neighboring cells to harmful attacks by proteases and various proinflammatory intracellular factors, and triggering positive feedback pathways of inflammatory tissue injury.
Recently, it has been shown that poly(ADP-ribose) polymer can also serve as an emergency source of energy used by the base excision machinery to synthesize ATP (66). Furthermore, poly(ADP-ribose) may also serve as a signal for protein degradation in oxidatively injured cells (109).
Under pathophysiological conditions, reactive species (such as hydrogen peroxide, hydroxyl radical, and peroxynitrite) trigger DNA single-strand breakage and PARP activation (97, 100). Peroxynitrite is considered a key trigger of DNA strand breakage because (as opposed to hydroxyl radical, for instance) it can travel significant distances and readily crosses cell membranes. When activated by DNA single-strand breaks, PARP initiates an energy-consuming cycle by transferring ADP ribose units from NAD+ to nuclear proteins. This process results in rapid depletion of the intracellular NAD+ and ATP pools, slowing the rate of glycolysis and mitochondrial respiration, eventually leading to cellular dysfunction and cell death (for review, see 114). It is noteworthy that, in addition to the process of NAD+ depletion and the induction of cellular dysfunction, part of the PARP overactivation-induced cell dysfunction and necrosis is related to intra-cellular acidification. Part of this process is related to inhibition of sodium/hydrogen exchange in energy-depleted cells (37). Another part of this process is due to a direct acidification: when PARP catabolizes NAD+, in addition to ADP-ribose and nicotinamide a “by-product” of the reaction is H+, which directly induces intracellular acidification, with direct consequences for cell viability (1).
The PARP-mediated pathway of cell necrosis and the PARP-mediated pathway of inflammatory signal transduction and gene expression may be interrelated in pathophysiological conditions. Oxidant stress can generate DNA single-strand breaks. DNA strand breaks then activate PARP, which in turn potentiates NF-κB activation and AP-1 expression, resulting in greater expression of the AP-1- and NF-κB-dependent genes, such as the gene for ICAM-1, as well as chemokines such as macrophage inflammatory protein-1α and cytokines such as tumor necrosis factor-α. Chemokine generation, in combination with increased endothelial expression of ICAM-1, recruits more activated leukocytes to inflammatory foci, producing greater oxidant stress. It is possible that a low-level, localized inflammatory response may be beneficial in recruiting mononuclear cells to an inflammatory site. However, in many pathophysiological states, the above-described feedback cycles amplify themselves beyond control.
ROLE OF PARP ACTIVATION IN THE PATHOGENESIS OF ENDOTHELIAL DYSFUNCTION
The contribution of the PARP pathway to the development of endothelial dysfunction was proposed in 1997 (98), using an endotoxic shock model in the rat. This model is known to induce severe oxidative and nitrosative stress in the vicinity of the vascular endothelium, due to the up-regulation of iNOS, as well as the activation of various superoxide-generating sources, including NADPH oxidase. In vascular rings taken from rats subjected to endotoxic shock, there was a loss of the endothelium-dependent relaxations, and these alterations were prevented by pharmacological inhibition of PARP with 3-aminobenzamide (98). In in vitro studies, vascular rings exposed to peroxynitrite also exhibited reduced endothelium-dependent relaxations in response to acetylcholine, and the development of this endothelial dysfunction was ameliorated by 3-aminobenzamide (98). These findings were consistent with previous in vitro data demonstrating that PARP inhibition protects against the metabolic suppression and death of oxidatively (3, 41, 44, 46, 108) or nitrosatively (98) injured endothelial cells. These findings were also consistent with studies where endothelial cells were incubated in vitro with various pathophysiologically relevant factors that induce oxidative stress, including homocysteine (a model of a variety of cardiovascular diseases) (7) or elevated glucose concentrations (a model of diabetic vascular complications) (33).
Over the last 5 years, the list of pathophysiological conditions where the endothelial dysfunction has been demonstrated to be dependent on PARP activation has increased. The list, in addition to various forms of shock (20, 42, 52, 69, 98), now includes complement-mediated endothelial injury (22), myocardial infarction and various forms of myocardial reperfusion injury, and heart transplantation (106, 120), as well as the endothelial dysfunction associated with chronic heart failure (70, 71), aging (73), hypertension (74), and diabetes mellitus (33, 72, 91, 92, 102).
ENDOTHELIAL DYSFUNCTION IN EXPERIMENTAL MODELS OF DIABETES: THE ROLE OF PARP ACTIVATION
Endothelial dysfunction has been documented in various forms of diabetes, and even in prediabetic individuals (11, 12, 14, 19, 86, 102). The pathogenesis of this endothelial dysfunction involves many components, including increased polyol pathway flux, altered cellular redox state, increased formation of diacylglycerol and the subsequent activation of specific protein kinase C (PKC) isoforms, and accelerated nonenzymatic formation of advanced glycation end products (AGEs) (5, 8, 15, 25, 34, 36, 58). Many of these pathways, in concert, trigger the production of oxygen- and nitrogen-derived oxidants and free radicals, such as superoxide anion and peroxynitrite, which play a significant role in the pathogenesis of the diabetes-associated endothelial dysfunction and other diabetic complications. The cellular sources of reactive oxygen species such as superoxide anion are multiple and include AGEs, NADH/NADPH oxidases, the mitochondrial respiratory chain, xanthine oxidase, the arachidonic acid cascade (lipoxygenase and cyclooxygenase), and microsomal enzymes (8, 15, 25, 34).
In a recent study, we have shown that high glucose-induced oxidative and nitrosative stress leads to DNA single-strand breakage and PARP activation in murine and human endothelial cells (33) (Fig. 1). The involvement of oxyradicals and nitric oxide (NO)-derived reactive species in PARP activation and the evidence for nitrated tyrosine residues both suggested that peroxynitrite may be one of the final mediators responsible for single-strand breakage and subsequent PARP activation (33). The role of hyperglycemia-induced oxidative stress in producing DNA damage is also supported by recent findings showing that increased amounts of 8-hydroxyguanine and 8-hydroxydeoxyguanosine (markers of oxidative damage to DNA) can be found in both the plasma and tissues of streptozotocin (STZ) diabetic rats (78). Importantly, various forms of oxidant-induced DNA damage (base modifications as well as DNA strand breaks) have also been demonstrated in diabetic patients (2, 4, 26, 53, 87).
FIG. 1. Reactive nitrogen species generation, DNA breakage, and PARP activation in diabetic blood vessels.
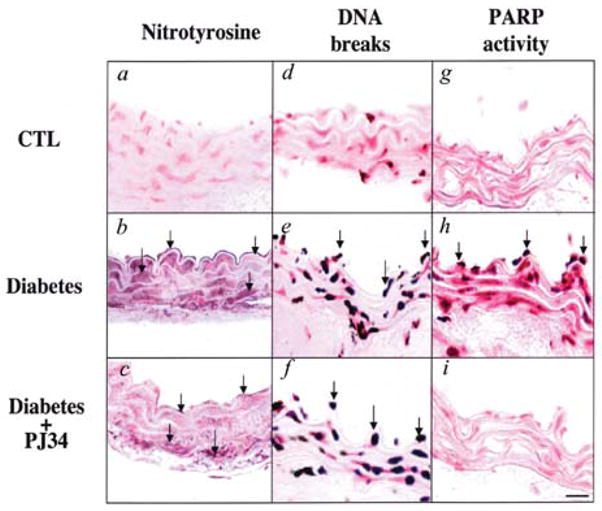
(a–c) Immunohistochemical staining for nitrotyrosine in control rings (a), in rings from diabetic mice treated with vehicle at 8 weeks (b), and in rings from diabetic mice treated with PJ34 (c). (d–f) Terminal deoxyribonucleotidyl transferase-mediated dUTP nick-end labeling, an indicator of DNA-strand breakage, in control rings (d), in rings from diabetic mice treated with vehicle at 8 weeks (e), and in rings from diabetic mice treated with PJ34 (f). (g–i) Immunohistochemical staining for poly(ADP-ribose), an indicator of PARP activation, in control rings (g), in rings from diabetic mice treated with vehicle at 8 weeks (h), and in rings from diabetic mice treated with PJ34 (i). Reproduced with permission from 33.
In a STZ-induced murine model of type I diabetes, we observed that the diabetes-associated loss of endothelial function is not only preventable, but also rapidly reversible with PARP inhibition (33, 91). Intravascular PARP activation (seen primarily in endothelial cells, as well as in vascular smooth muscle cells) was already apparent 2 weeks after the onset of diabetes, and thus it slightly preceded the occurrence of the endothelial dysfunction, which developed between the 2nd and the 4th week of diabetes (33, 91) (Fig. 2). Delayed treatment with the PARP inhibitor, starting at 1 week after STZ, ameliorated vascular poly(ADP-ribose) accumulation and restored normal vascular function without altering systemic glucose levels, plasma glycated hemoglobin levels, or pancreatic insulin content (33, 91). Furthermore, delayed treatment of the animals with the PARP inhibitor restored the already established diabetic endothelial dysfunction (Fig. 3), and even in vitro incubation of diabetic blood vessels with PARP inhibitors of various structural classes significantly enhanced their endothelium-dependent relaxant responsiveness (91) (Fig. 4). The development of the endothelial dysfunction and its reversibility by pharmacological inhibition of PARP have also been demonstrated in an autoimmune model of diabetes (72) (Fig. 5). The endothelial dysfunction was associated with a simultaneous loss of NAD+ and NADPH in the vasculature, and PARP inhibition reversed these changes. Based on these observations, and the known fact that endothelial nitric oxide synthase (eNOS; the NOS isoform present in the vascular endothelial cells) is dependent on NADPH and is sensitively regulated by this cofactor, we hypothesized that the endothelial dysfunction in diabetes is dependent on a PARP-mediated, reversible cellular NADPH deficiency (33, 91). In fact, previous in vitro work demonstrated that the NADPH depletion in oxidatively stressed cells is dependent on PARP activation (18, 39, 47). It is interesting to note that other groups have demonstrated that diabetic endothelial dysfunction is also associated with direct oxidation and consequent cellular depletion of other cofactors of eNOS, such as tetrahydrobiopterin (32, 33, 35, 75, 117). As in the absence of tetrahydrobiopterin a functional uncoupling of eNOS occurs and the enzyme produces superoxide and peroxynitrite, rather than NO (114), the consequences of these processes are increased free radical and oxidant production, oxidative damage, and further exacerbation of the endothelial dysfunction.
FIG. 2. Reversal of diabetes-induced endothelial dysfunction by pharmacological inhibition of PARP.
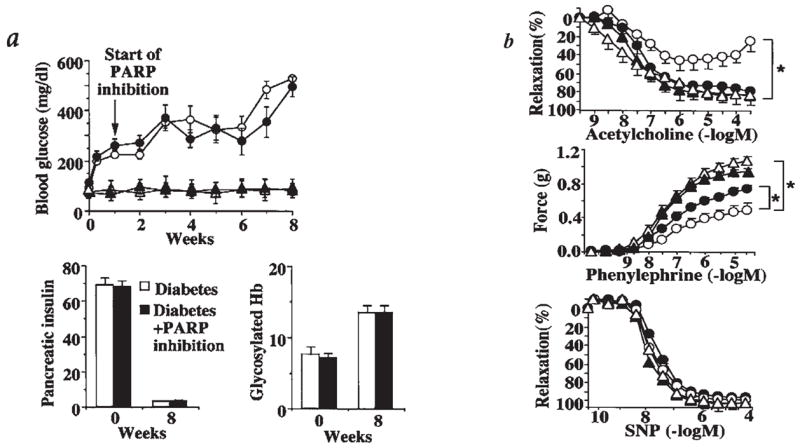
The following symbols were used for the respective groups: animals that received no STZ injection (Δ), nondiabetic control animals at 8 weeks treated with PJ34 between week 1 and 8 (▴), diabetic animals at 8 weeks treated with vehicle (▲), diabetic animals at 8 weeks treated with PJ34 between week 1 and 8 (●). (a) Blood glucose levels, pancreatic insulin content (ng of insulin/mg of pancreatic protein), and blood glycosylated hemoglobin (Hb) (expressed as % of total Hb) at 0–8 weeks in nondiabetic, control male BALB/c mice, and at 0–8 weeks after STZ treatment (diabetic) in male BALB/c mice. PARP inhibitor treatment, starting at 1 week after STZ and continuing until the end of week 8, is indicated by the arrow. Pancreatic insulin and glycated hemoglobin levels are shown at 8 weeks in vehicle-treated and STZ-treated animals, in the presence or absence of PJ34 treatment. (b) Acetylcholine-induced, endothelium-dependent relaxations, phenylephrine-induced contractions, and sodium nitroprusside (SNP)-induced endothelium-independent relaxations. *p < 0.05 for vehicle-treated diabetic versus PJ34-treated diabetic mice (n = 8 per group). Reproduced with permission from 33.
FIG. 3. Pharmacological inhibition of PARP restores impaired endothelium-dependent relaxant ability of the diabetic vessels.
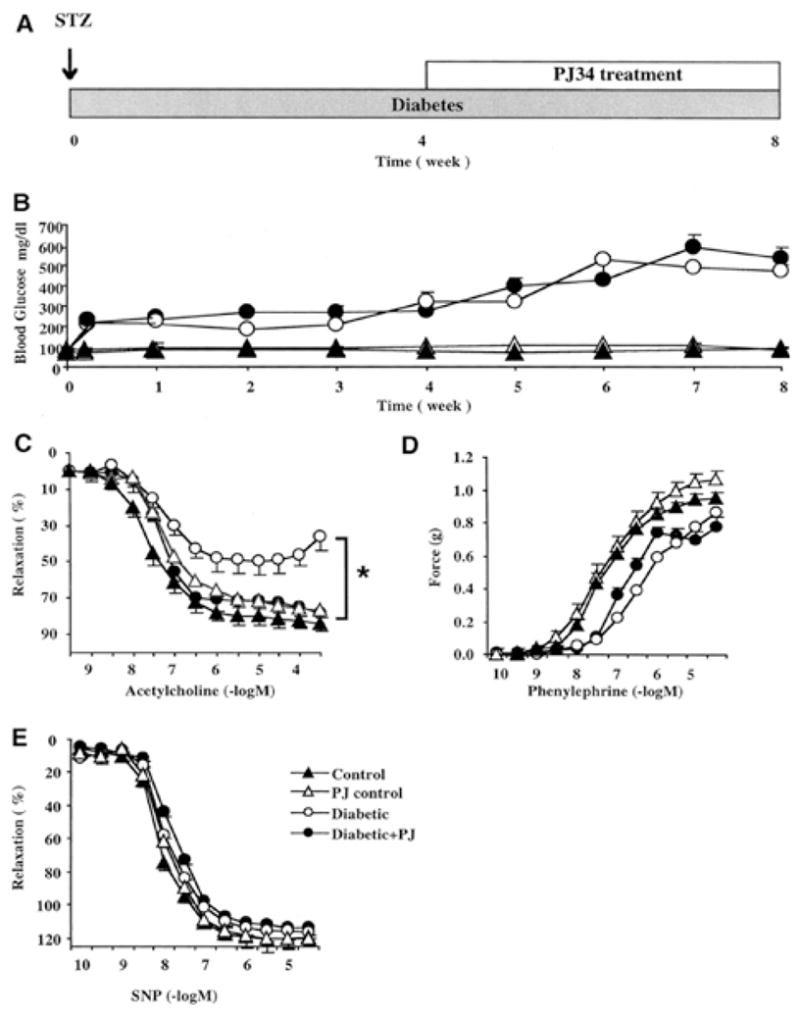
Blood glucose levels and vascular responsiveness are presented. Endothelium-dependent relaxations were induced by acetylcholine, contractions induced by phenylephrine, and endothelium-independent relaxations induced by sodium nitroprusside (SNP) in control (nondiabetic) male Balb/c mice and 1, 4, and 8 weeks after STZ-induced diabetes. Vehicle or PARP inhibitor (PJ34, 10 mg/kg oral gavage once a day) treatment started at 4 weeks after STZ and continued until 8 weeks (the end of the experimental period). There was a marked and selective impairment of the endothelium-dependent relaxant ability of the vascular rings in diabetes at 4 and 8 weeks. Treatment with the PARP inhibitor between weeks 4 and 8 restored to normal the endothelium-dependent relaxant ability of the diabetic vessels despite the persistence of hyperglycemia. *p < 0.05 for differences between experimental groups, as indicated. n = 8 per group. Reproduced with permission from 91.
FIG. 4. In vitro treatment with all PARP inhibitors improved the endothelium-dependent relaxant ability of the diabetic vessels.
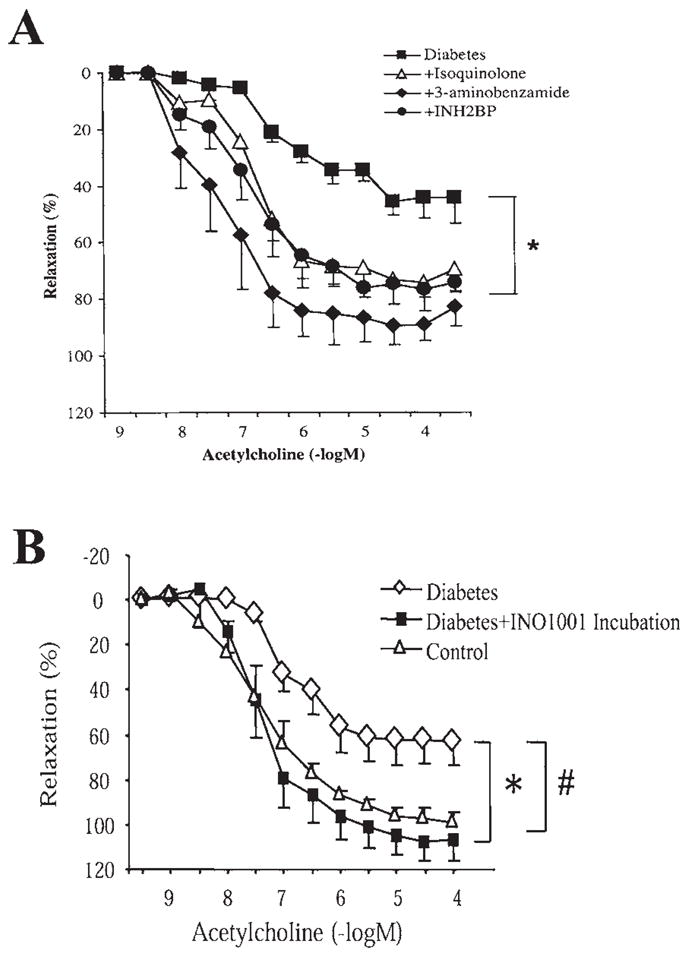
(A) Endothelium-dependent relaxations induced by acetylcholine in control (nondiabetic) male Balb/c mice and 4 weeks after STZ-induced diabetes. In a subgroup of the vascular rings, evaluation of vascular responsiveness was preceded by 1-h incubation with three structurally different PARP inhibitors: 3-aminobenzamide (3 mmol/L), 5-iodo-6-amino-1,2-benzopyrone (INH2BP) (100 μmol/L), or 1,5-dihydroxyisoquinoline (Isoquinolone) (30 μmol/L). There was a marked and selective impairment of the endothelium-dependent relaxant ability of the vascular rings in diabetes at 4 weeks. In vitro treatment with all PARP inhibitors improved the endothelium-dependent relaxant ability of the diabetic vessels. *p < 0.05 for differences between experimental groups, as indicated. n = 8 per group. Reproduced with permission from 91. (B) Endothelium-dependent relaxations induced by acetylcholine in control (nondiabetic) male Balb/c mice and 6 weeks after STZ-induced diabetes. In a subgroup of the vascular rings, evaluation of vascular responsiveness was preceded by 1-h incubation with the novel potent PARP inhibitor, INO1001 (3 μmol/L). There was a marked and selective impairment of the endothelium-dependent relaxant ability of the vascular rings in diabetes at 6 weeks. In vitro treatment with all PARP inhibitors improved the endothelium-dependent relaxant ability of the diabetic vessels. #, *p < 0.05 for differences between experimental groups, as indicated. n = 8 per group.
FIG. 5. Reversal of diabetes-induced endothelial dysfunction by pharmacological inhibition of PARP in diabetic NOD mouse vascular rings.
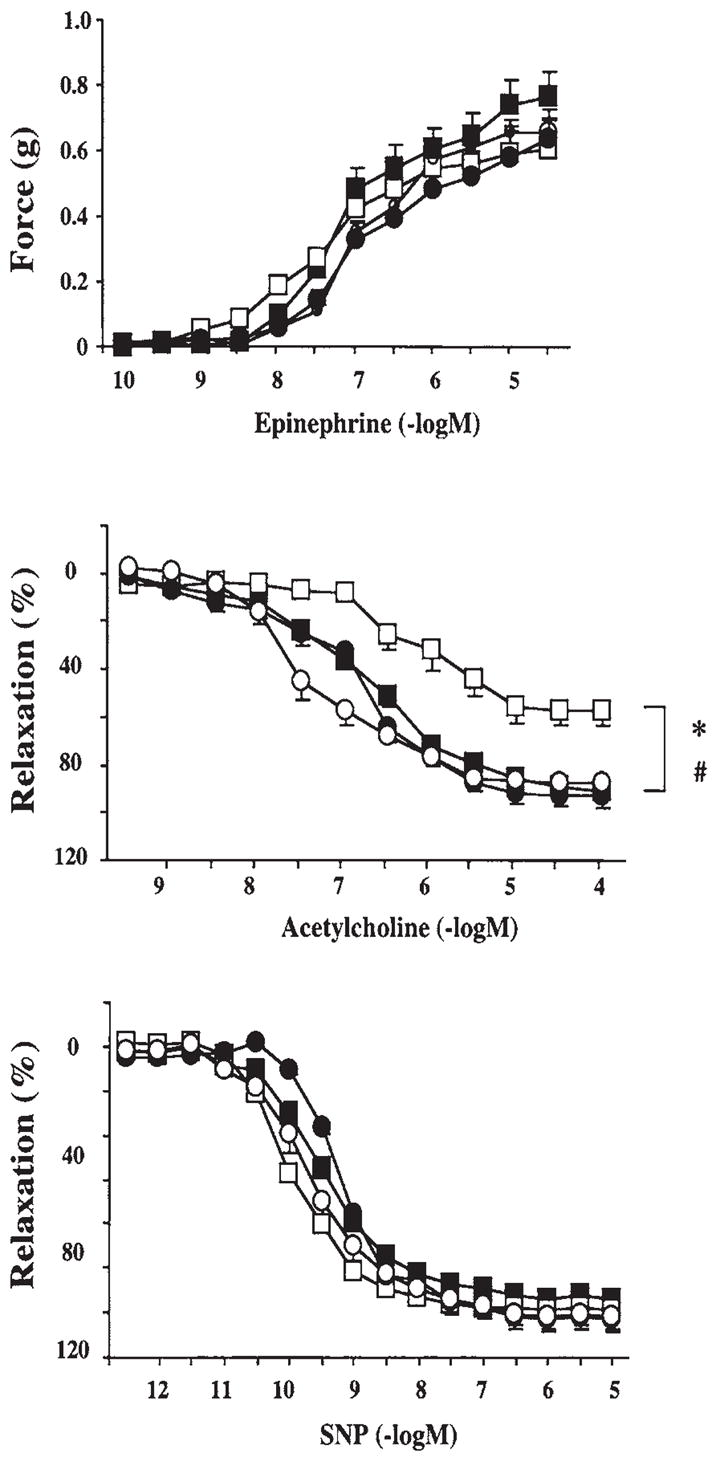
Epinephrine-induced contractions (upper panel), acetylcholine-induced endothelium-dependent relaxation (middle panel), and sodium nitroprusside (SNP)-induced endothelium-independent relaxations (lower panel).■, control; ○, control + PJ34; □, diabetes; ●, diabetes + PJ34. Each point of the curve represents the mean ± SE of five to eight experiments in vascular rings. *p < 0.05 versus control; #p < 0.05 versus diabetes. Reproduced with permission from 72.
The mode of the protective action of PARP inhibitors on the vascular endothelium in vivo likely involves the conservation of cellular energetic pools, as well as a prevention of the up-regulation of various proinflammatory pathways [cytokines, adhesion molecules (ICAM-1, VCAM-1, and E-selectin) mononuclear cell infiltration] triggered by hyperglycemia (16, 33, 92). This latter mechanism may represent an important additional pathway whereby PARP activation can contribute to vascular dysfunction via the up-regulation of adhesion molecules. As mentioned earlier, PARP regulates the activation of a variety of signal transduction pathways, and some of these pathways regulate the expression of cell surface and soluble adhesion molecules. Recent preliminary data indicate that pharmacological inhibition of PARP can suppress this process (16). Intermittent high/low glucose induces a more pronounced expression of adhesion molecules than constant high glucose (82), and PARP inhibition suppresses NF-κB activation and the expression of adhesion molecules both under constant high glucose and under intermittent high/low glucose conditions in cultured endothelial cells in vitro (16).
Brownlee and colleagues have demonstrated that the hyperglycemia-induced overproduction of superoxide by mitochondrial electron-transport chain activated major pathways of hyperglycemic damage found in aortic endothelial cells (activation of PKC isoforms, hexosamine pathway flux, and AGE formation) by inhibiting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity (28, 29, 83). Importantly, the hyperglycemia-induced GAPDH inhibition was found to be a consequence of poly(ADP-ribosyl)ation of GAPDH by PARP, which was activated by DNA strand breaks produced by reactive species generated by hyperglycemia. One of the likely DNA-damaging factors is peroxynitrite, which is generated when mitochondrial superoxide reacts with NO produced by the constitutive eNOS. Both the hyperglycemia-induced decrease in activation of GAPDH and its poly (ADP-ribosyl)ation can be prevented by overexpression of either uncoupling protein-1 (UCP-1) or manganese superoxide dismutase (MnSOD), which decrease hyperglycemia-induced superoxide generation. Overexpression of UCP-1 or MnSOD also prevented hyperglycemia-induced DNA strand breaks and activation of PARP (28). Similarly, administration of the mitochondrial uncoupler 2,4-dinitrophenol to endothelial cells exposed to high glucose blocked glucose-induced DNA strand breakage and PARP activation (76). Importantly, the hyperglycemia-induced activation of PKC isoforms, hexosaminase pathway flux, and AGE formation was prevented by blocking PARP activity with various structurally unrelated inhibitors of the enzyme (28).
An additional factor to be considered in the context of PARP activation and the pathogenesis of endothelial dysfunction and diabetic complications is angiotensin II. Angiotensin II is a known factor in the pathogenesis of diabetic complications, perhaps most importantly in nephropathy, cardiomyopathy, and retinopathy. Recent studies indicate that the protective effects of angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists may go beyond the blood pressure-lowering effects of these agents (6, 9, 50). In this context, it is noteworthy that angiotensin II can induce direct, prooxidative effects on the vascular endothelium. These effects are, at least in part, mediated by intraendothelial reactive species formation via a new family of NAD(P)H oxidase subunits, known as the nonphagocytic NAD(P)H oxidase proteins. Reactive oxidant species produced following angiotensin II-mediated stimulation of NAD(P)H oxidases can exert direct oxidative effects, but can also signal through pathways such as mitogen-activated protein kinases, tyrosine kinases, and transcription factors, and lead to events such as inflammation, hypertrophy, remodeling, and angiogenesis (13). Recent work demonstrates that angiotensin II can also induce intraendothelial peroxynitrite formation (56, 116), as well as PARP activation (104). Administration of angiotensin II triggers the activation of PARP in cultured endothelial cells in vitro. The in vitro PARP activation is dose-dependently inhibited by PARP inhibitors of various structural classes, as well as by the compound apocynin, indicating that NAD(P)H oxidase-generated superoxide anion accounts for the generation of the reactive species that trigger DNA single strand breakage and PARP activation (104). Angiotensin-induced PARP activation is also inhibited by Nω-nitro-L-arginine methyl ester and diphenyleneiodonium (104). Thus, angiotensin triggers the endothelial generation of reactive oxygen species from NAD(P)H oxidase, and these with constitutively produced NO produce peroxynitrite and other reactive nitrogen species, which induce DNA breakage and activate PARP in the vascular endothelium, leading to the development of endothelial dysfunction. This pathway is also operative in vivo, as chronic infusion of subpressor doses of angiotensin infusion triggers endothelial dysfunction in vivo, which can be prevented or reversed by PARP inhibition (103). Future work needs to establish the importance of this pathway in the context of diabetic complications.
ENDOTHELIAL DYSFUNCTION IN DIABETIC AND PREDIABETIC PATIENTS: THE POTENTIAL ROLE OF PARP ACTIVATION
A recent study of forearm skin biopsies from healthy subjects extended our knowledge on the role of PARP activation in the development of diabetic endothelial dysfunction in human subjects. Analysis of dermal biopsy samples from healthy individuals with parental history of type 2 diabetes (T2DM), subjects with impaired glucose tolerance, and a group of type 2 diabetic patients indicated that the percentage of PARP-positive endothelial nuclei was higher in the group of parental history of T2DM and diabetic patients when compared with the controls (102). In addition, significant correlations were observed between the percentage of PARP-positive endothelial nuclei and fasting blood glucose, resting skin blood flow, maximal skin vasodilatory response to the iontophoresis of acetylcholine (which indicates endothelium-dependent vasodilation), and nitrotyrosine immunostaining intensity. Nitrotyrosine immunoreactivity [a marker of reactive nitrogen species (chiefly peroxynitrite) formation] was also higher in the diabetic patients when compared with all other groups (102). Significant correlations were observed between nitrotyrosine immunostaining intensity and fasting blood glucose, glycosylated hemoglobin (HbA1c), ICAM, and VCAM. No differences in the expression of eNOS and RAGE were found among all four groups. The polymorphism of the eNOS gene was also studied and was not found to influence eNOS expression or microvascular functional measurements. Thus, in humans, PARP activation is present in healthy subjects at risk of developing diabetes, as well as in established type 2 diabetic patients, and it correlates with impairments in the vascular reactivity in the skin microcirculation (102). As interventional studies with PARP inhibitors in humans with diabetic endothelial dysfunction have not yet been conducted, it remains to be seen whether PARP activation in diabetic or prediabetic humans can be seen as a predictor or early marker for the development of diabetic vascular complications.
THE ROLE OF PARP ACTIVATION IN THE PATHOGENESIS OF DIABETIC CARDIOMYOPATHY
It is well established that the superoxide–peroxynitrite–PARP pathway plays a pivotal role in various models of myocardial ischemia–reperfusion injury (a condition in which oxidative and nitrosative stress plays a key pathogenetic role) (106, 107, 120). Recent data demonstrate that the PARP pathway also plays a pathogenetic role in the development of diabetic cardiomyopathy (72). Cardiac dysfunction and PARP activation in the cardiac myocytes and the coronary vasculature were observed in both STZ-induced and genetic (nonobese diabetic) models of diabetes mellitus in rats and mice. Furthermore, treatment with the phenanthridinone-based PARP inhibitor PJ34, starting 1 week after the onset of diabetes, restored normal vascular responsiveness and significantly improved cardiac function in diabetic mice and rats, despite the persistence of severe hyperglycemia. The beneficial effect of PARP inhibition persisted even after several weeks of the discontinuation of the PARP inhibitor treatment (72).
It is conceivable that the diabetic endothelial PARP pathway and the diabetic cardiomyopathy are interrelated: the impairment of the endothelial function may lead to global or regional myocardial ischemia, which may secondarily impair cardiac performance. The beneficial effect of PARP inhibition on myocardial function, however, is not related to an anabolic effect because PJ34 treatment did not influence the body and heart weight loss in diabetic animals, whereas it dramatically improved cardiac function. It is noteworthy that the protective effect of PARP inhibition against diabetic cardiac dysfunction extends several weeks beyond the discontinuation of treatment; this observation may have important implications for the design of future clinical trials with PARP inhibitors. The prolonged protective effect may be related to the permanent interruption by the PARP inhibitor of positive feedback cycles of cardiac injury. Indeed, previous studies in various pathophysiological conditions have demonstrated that PARP inhibitors suppress positive feedback cycles of adhesion receptor expression and mononuclear cell infiltration, as well as cellular oxidant generation (16, 106, 120). The mode of the PARP inhibitors’ cardioprotective action involves a conservation of myocardial energetics, as well as a prevention of the up-regulation of various proinflammatory pathways (cytokines, adhesion receptors, mononuclear cell infiltration) triggered by ischemia and reperfusion (106, 120). It is conceivable that PARP inhibition exerts beneficial effects in experimental models of diabetic cardiomyopathy by affecting both above-referenced pathways of injury, and also by suppressing positive feedback cycles initiated by them.
THE ROLE OF PARP ACTIVATION IN THE PATHOGENESIS OF DIABETIC RETINOPATHY, NEPHROPATHY, AND NEUROPATHY
Although most of the studies on the role of PARP in the pathogenesis of diabetic endothelial dysfunction were conducted in macrovessels (see above), there is circumstantial evidence that similar processes are operative for the pathogenesis of diabetic microvascular injury (which is an important underlying mechanism for the pathogenesis of retinopathy, nephropathy, and neuropathy). In fact, there is now evidence of PARP activation in the microvessels and ganglionic layer of the diabetic retina (64, 105). The causative role of PARP in diabetic retinopathy is now supported by two independent interventional preclinical studies. In one report (119), a long-term (9-month) study was used to investigate the role of PARP in hyperglycemia-induced cell death in vitro and in the development of diabetic retinopathy in vivo. STZ-diabetic Lewis rats were treated with vehicle or the PARP inhibitor PJ34. Diabetes was found to increase activity of PARP in retina measured at 2 months, and PJ34 inhibited this increase. PARP activation was detectable also in a subset of nuclei from retinal capillary endothelial cells and pericytes. Diabetes of 9 months duration significantly increased the number of both TUNEL-positive capillary cells and acellular capillaries (a marker of degenerate capillaries), and PJ34 significantly inhibited these alterations without influencing glycemic control. PJ34 also inhibited a diabetes-induced up-regulation of ICAM and leukostasis within the retinal vasculature. In a complementary in vitro study, bovine retinal endothelial cells and pericytes were incubated in 5 mM (normal) and 25 mM (elevated) glucose for 5 days with or without PJ34. High glucose significantly increased death of retinal capillary endothelial cells, and PARP inhibition prevented this cell death. In a second, independent study (119), male C57/BL6 mice were rendered diabetic with a single injection of STZ. Diabetic mice, treated with the PARP inhibitor PJ34 for 6 months, were investigated for experimental retinopathy by using retinal digest preparations and quantitative retinal morphometry. Diabetes over 6 months induced pericyte loss and increased the number of acellular capillaries. Treatment with PJ34 inhibited both the loss of pericytes and the formation of acellular capillaries. These data, taken together, suggest that hyperglycemia-induced PARP activation affects predominantly the retinal vasculature and is susceptible to pharmacological PARP inhibition.
As far as the role of PARP in diabetic nephropathy goes, the presence of glomerular depositions (mesangial distribution) of IgG was significantly reduced in STZ-diabetic rats treated with the PARP inhibitor nicotinamide for 6 months (115). In agreement with these results, we have recently provided evidence that PARP activation is present in the tubuli of STZ-induced diabetic rats. This PARP activation is attenuated by two unrelated PARP inhibitors, 3-aminobenzamide and 1,5-isoquinolinediol, which also counteracted the overexpression of endothelin-1 and endothelin receptors in the renal cortex (57).
It has recently been suggested that the oxidative/nitrosative stress–PARP pathway PARP also plays a key role in the development of diabetic neuropathy: the progressive slowing of sensory and motor neuron conductance in diabetic rats and mice is preventable by PARP inhibition or PARP deficiency, and this is associated with maintained neuronal phosphocreatine levels, as well as improved endoneurial blood flow (17, 51, 61, 62, 76). Importantly, pharmacological PARP inhibition is not only a preventive option; it can also restore sensory and motor neuronal conduction in already established diabetic neuropathy, at least in murine models of the disease (51).
Additional studies, utilizing potent and specific inhibitors of PARP, are needed to further delineate the role of PARP in the pathogenesis of diabetic retinopathy, neuropathy, and nephropathy. It is important to reemphasize that, although the above conditions are generally considered as separate patho-physiological entities, there is good evidence that, at least in part, they all develop on the basis of endothelial (vascular) dysfunction (59, 60). As diabetic erectile dysfunction is also known to develop on the basis of diabetic endothelial dysfunction and diabetic neuropathy (84), the potential role of PARP activation in this condition must also be explored in future studies.
CONCLUSIONS AND IMPLICATIONS
Based on the evidence reviewed herein, we conclude that the PARP pathway plays very important regulatory roles in the pathogenesis of vascular endothelial dysfunction in patho-physiological conditions associated with oxidative stress, including diabetes (20, 75, 76, 110). It remains to be studied whether various clinical therapeutic or experimental therapeutic interventions, which are known to have some vascular protective effects in diabetes (antioxidant therapies, peroxisome proliferator-activated receptor agonists, etc.), are able to suppress the activation of PARP in the cardiovascular system. It is noteworthy in this respect that, in preclinical studies, administration of the aldose reductase inhibitors sorbinil or fidarestat to diabetic rats not only corrected diabetes-induced depletion of glutathione and ascorbate, down-regulation of superoxide dismutase activity, and accumulation of lipid peroxidation products in the peripheral nerve, counteracted superoxide formation in vasa nervorum, and was effective against multiple indices of diabetes-associated retinal oxidative and nitrosative stress, but also inhibited poly(ADP-ribose) accumulation (a marker of PARP activation) in diabetic nerve and retina (65). Similar results were obtained with FP15, a novel peroxynitrite decomposition catalyst compound (63, 76, 101). In a murine study, sciatic motor nerve conduction velocity and hind-limb digital sensory conduction velocity were reduced in diabetic mice versus controls, and both indices were normalized by FP15, which also ameliorated the accumulation of poly(ADP-ribose) accumulation in diabetic nerves (63).
The pathogenetic role of the oxidative/nitrosative stress–PARP pathway (Fig. 6) is not limited to the diabetes-induced vascular dysfunction, but it has also been demonstrated in various animal models of other diabetic complications, including cardiomyopathy, nephropathy, neuropathy, and retinopathy. PARP activation, thus, is a unique checkpoint in the development and progression of various diabetic complications. PARP inhibition may emerge as a novel approach for the prevention or reversal of diabetic complications. The benefits and potential risks associated with chronic administration of PARP inhibitors are discussed in a recent review (94). The comparative therapeutic utility of PARP inhibition for the experimental therapy of diabetic complications should be explored by additional preclinical and subsequent clinical investigations.
FIG. 6. Overview of the role of PARP in regulating multiple components of hyperglycemia-induced endothelial dysfunction.
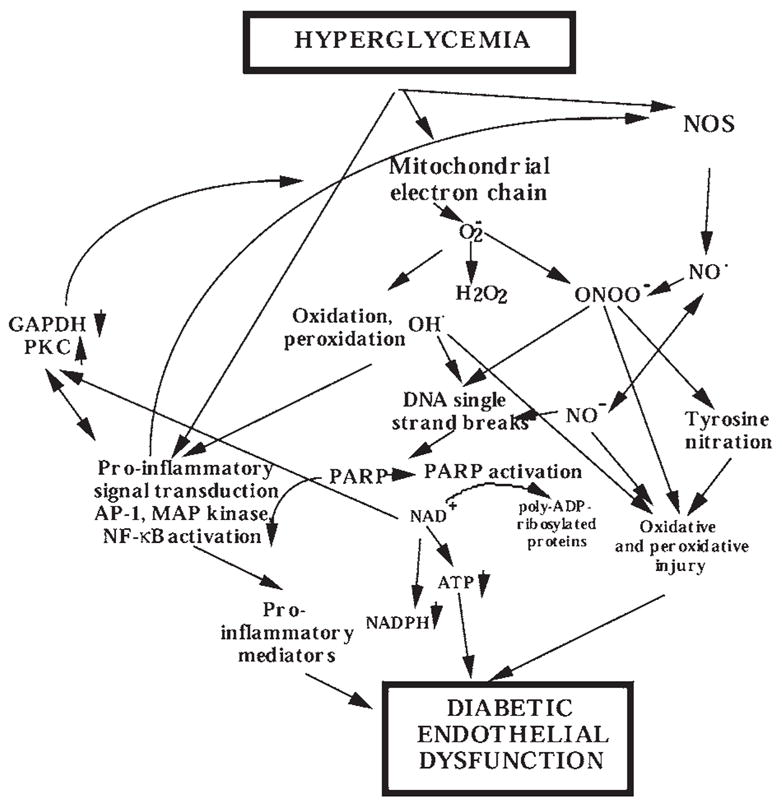
High circulating glucose interacts with the vascular endothelium where it triggers the release of oxidant mediators from the mitochondrial electron transport chain, as well as from NADH/NADPH oxidase and other sources. NO, in turn, combines with superoxide (O2− to yield peroxynitrite (ONOO−). Hydroxyl radical (OH·) (produced from superoxide via the iron-catalyzed Haber–Weiss reaction) and peroxynitrite or peroxynitrous acid induce the development of DNA single-strand breakage, with consequent activation of PARP. Depletion of the cellular NAD+ leads to inhibition of cellular ATP-generating pathways leading to cellular dysfunction. The PARP-triggered depletion of cellular NADPH directly impairs the endothelium-dependent relaxations. The effects of elevated glucose are also exacerbated by increased aldose reductase activity leading to depletion of NADPH and generation of reactive oxidants. NO alone does not induce DNA single-strand breakage, but may combine with superoxide (produced from the mitochondrial chain or from other cellular sources) to yield peroxynitrite. Under conditions of low cellular L-arginine, NOS may produce both superoxide and NO, which then can combine to form peroxynitrite. PARP activation, via a not yet characterized fashion, can promote the activation of nuclear factor-κB, AP-1, mitogen-activated protein (MAP) kinases, and the expression of proinflammatory mediators, adhesion molecules, and iNOS. PARP activation contributes to the activation of PKC. PARP activation also leads to the inhibition of cellular GAPDH activity, at least in part via the direct poly(ADP-ribosyl)ation of GAPDH. PARP-independent, parallel pathways of cellular metabolic inhibition can be activated by NO, hydroxyl radical, superoxide, and peroxynitrite.
ABBREVIATIONS
- AGE
advanced glycation end product
- AP-1
activator protein-1
- eNOS
endothelial nitric oxide synthase
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- ICAM-1
inter-cellular adhesion molecule-1
- iNOS
inducible nitric oxide synthase
- MnSOD
manganese superoxide dismutase
- NAD+
nicotinamide adenine dinucleotide
- NF-κB
nuclear factor-κB
- NO
nitric oxide
- PARG
poly(ADP-ribose) glycohydrolase
- PARP/PARS
poly(ADP-ribose) polymerase/synthase
- PJ34
potent water-soluble phenanthridinone-derived PARP inhibitor
- PKC
protein kinase C
- STZ
streptozotocin
- T2DM
type 2 diabetes mellitus
- UCP-1
uncoupling protein-1
- VCAM-1
vascular cellular adhesion molecule-1
References
- 1.Affar EB, Shah RG, Dallaire AK, Castonguay V, Shah GM. Role of poly(ADP-ribose) polymerase in rapid intracellular acidification induced by alkylating DNA damage. Proc Natl Acad Sci U S A. 2002;99:245–250. doi: 10.1073/pnas.012460399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson D, Yu TW, Wright J, Ioannides C. An examination of DNA strand breakage in the comet assay and antioxidant capacity in diabetic patients. Mutat Res. 1998;398:151–161. doi: 10.1016/s0027-5107(97)00271-6. [DOI] [PubMed] [Google Scholar]
- 3.Andreoli SP. Mechanisms of endothelial cell ATP depletion after oxidant injury. Pediatr Res. 1989;25:97–101. doi: 10.1203/00006450-198901000-00021. [DOI] [PubMed] [Google Scholar]
- 4.Astley S, Langrish-Smith A, Southon S, Sampson M. Vitamin E supplementation and oxidative damage to DNA and plasma LDL in type 1 diabetes. Diabetes Care. 1999;22:1626–1631. doi: 10.2337/diacare.22.10.1626. [DOI] [PubMed] [Google Scholar]
- 5.Beckman JA. Inhibition of protein kinase Cbeta prevents impaired endothelium-dependent vasodilation caused by hyperglycemia in humans. Circ Res. 2002;90:107–111. doi: 10.1161/hh0102.102359. [DOI] [PubMed] [Google Scholar]
- 6.Bell DS. Heart failure: the frequent, forgotten, and often fatal complication of diabetes. Diabetes Care. 2003;26:2433–2441. doi: 10.2337/diacare.26.8.2433. [DOI] [PubMed] [Google Scholar]
- 7.Blundell G, Jones BG, Rose FA, Tudball N. Homocysteine mediated endothelial cell toxicity and its amelioration. Atherosclerosis. 1996;122:163–172. doi: 10.1016/0021-9150(95)05730-7. [DOI] [PubMed] [Google Scholar]
- 8.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 9.Bui BV, Armitage JA, Tolcos M, Cooper ME, Vingrys AJ. ACE inhibition salvages the visual loss caused by diabetes. Diabetologia. 2003;46:401–408. doi: 10.1007/s00125-003-1042-7. [DOI] [PubMed] [Google Scholar]
- 10.Burkart V, Wang ZQ, Radons J, Heller B, Herceg Z, Stingl L, Wagner EF, Kolb H. Mice lacking the poly(ADP-ribose) polymerase gene are resistant to pancreatic beta-cell destruction and diabetes development induced by streptozocin. Nat Med. 1999;5:314–319. doi: 10.1038/6535. [DOI] [PubMed] [Google Scholar]
- 11.Caballero AE, Arora S, Saouaf R, Lim SC, Smakowski P, Park JY, King GL, LoGerfo FW, Horton ES, Veves A. Microvascular and macrovascular reactivity is reduced in subjects at risk for type 2 diabetes. Diabetes. 1999;48:1856–1862. doi: 10.2337/diabetes.48.9.1856. [DOI] [PubMed] [Google Scholar]
- 12.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 13.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 14.Calles-Escandon J, Cipolla M. Diabetes and endothelial dysfunction: a clinical perspective. Endocr Rev. 2001;22:36–52. doi: 10.1210/edrv.22.1.0417. [DOI] [PubMed] [Google Scholar]
- 15.Ceriello A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care. 2003;26:1589–1596. doi: 10.2337/diacare.26.5.1589. [DOI] [PubMed] [Google Scholar]
- 16.Ceriello A, Piconi L, Quagliaro L, Ros RD, Marini C, Giugliano D, et al. Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical endothelial cells in culture: the role of poly(ADP-ribose) polymerase. FASEB J. 2003;17:A260. doi: 10.1111/j.1538-7836.2004.00835.x. [DOI] [PubMed] [Google Scholar]
- 17.Cheng C, Zochodne DW. Sensory neurons with activated caspase-3 survive long-term experimental diabetes. Diabetes. 2003;52:2363–2371. doi: 10.2337/diabetes.52.9.2363. [DOI] [PubMed] [Google Scholar]
- 18.Cole KK, Perez-Polo JR. Poly(ADP-ribose) polymerase inhibition prevents both apoptotic-like delayed neuronal death and necrosis after H2O2 injury. J Neurochem. 2002;82:19–29. doi: 10.1046/j.1471-4159.2002.00935.x. [DOI] [PubMed] [Google Scholar]
- 19.Cosentino F, Luscher TF. Endothelial dysfunction in diabetes mellitus. J Cardiovasc Pharmacol. 1998;32:S54–S61. [PubMed] [Google Scholar]
- 20.Csiszar A, Pacher P, Kaley G, Ungvari Z. Role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr Vasc Pharmacol. 2005;3:285–291. doi: 10.2174/1570161054368616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuzzocrea S, Zingarelli B, Costantino G, Szabó A, Salzman AL, Caputi AP, Szabó C. Beneficial effects of 3-aminobenzamide, an inhibitor of poly (ADP-ribose) synthetase in a rat model of splanchnic artery occlusion and reperfusion. Br J Pharmacol. 1997;121:1065–1074. doi: 10.1038/sj.bjp.0701234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cuzzocrea S, Zingarelli B, Caputi AP. Role of peroxynitrite and poly (ADP-ribosyl) synthetase activation in cardiovascular derangement induced by zymosan in the rat. Life Sci. 1998;63:923–933. doi: 10.1016/s0024-3205(98)00350-6. [DOI] [PubMed] [Google Scholar]
- 23.Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- 24.De Murcia G, Shall S, editors. From DNA Damage and Stress Signaling to Cell Death; Poly ADP-Ribosylation Reactions. Oxford: Oxford University Press; 2000. [Google Scholar]
- 25.De Murcia G, Schreiber V, Molinete M, Saulier B, Poch O, Masson M, Niedergang C, Menissier de Murcia J. Structure and function of poly(ADP-ribose) polymerase. Mol Cell Biochem. 1994;138:15–24. doi: 10.1007/BF00928438. [DOI] [PubMed] [Google Scholar]
- 26.De Vriese AS, Verbeuren TJ, Van de Voorde J, Lameire NH, Vanhoutte PM. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dincer Y, Akcay T, Ilkova H, Alademir Z, Ozbay G. DNA damage and antioxidant defense in peripheral leukocytes of patients with type I diabetes mellitus. Mutat Res. 2003;527:49–55. doi: 10.1016/s0027-5107(03)00073-3. [DOI] [PubMed] [Google Scholar]
- 28.Du X, Matsumura T, Szabó C, Edelstein D, Brownlee M. Hyperglycemia-induced superoxide activates PKC, the hexosamine pathway, AGE formation, and NF-κB via poly(ADP-ribose) polymerase inhibition of GAPDH. Diabetes. 2002;51(Suppl 2):A175. [Google Scholar]
- 29.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehrlich W, Huser H, Kroger H. Inhibition of the induction of collagenase by interleukin-1 beta in cultured rabbit synovial fibroblasts after treatment with the poly(ADP-ribose)-polymerase inhibitor 3-aminobenzamide. Rheumatol Int. 1995;15:171–172. doi: 10.1007/BF00301776. [DOI] [PubMed] [Google Scholar]
- 31.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 32.Fukuda Y, Teragawa H, Matsuda K, Yamagata T, Matsuura H, Chayama K. Tetrahydrobiopterin restores endothelial function of coronary arteries in patients with hypercholesterolaemia. Heart. 2002;87:264–269. doi: 10.1136/heart.87.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia Soriano F, Virág L, Jagtap P, Szabó É, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ, Szabo C. Diabetic endothelial dysfunction: the role of poly (ADP-ribose) polymerase activation. Nat Med. 2001;7:108–113. doi: 10.1038/83241. [DOI] [PubMed] [Google Scholar]
- 34.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19:257–267. doi: 10.2337/diacare.19.3.257. [DOI] [PubMed] [Google Scholar]
- 35.Guzik TJ, West NE, Black E, McDonald D, Ratnatunga C, Pillai R, Channon KM. Vascular superoxide production by NAD(P)H oxidase: association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86:E85–E90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- 36.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–1662. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 37.Herceg Z, Wang ZQ. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat Res. 2001;477:97–110. doi: 10.1016/s0027-5107(01)00111-7. [DOI] [PubMed] [Google Scholar]
- 38.Hiromatsu Y, Sato M, Yamada K, Nonaka K. Nicotinamide and 3-aminobenzamide inhibit recombinant human interferon-gamma-induced HLA-DR antigen expression, but not HLA-A, B, C antigen expression, on cultured human thyroid cells. Clin Endocrinol. 1992;36:91–95. doi: 10.1111/j.1365-2265.1992.tb02907.x. [DOI] [PubMed] [Google Scholar]
- 39.Hoyt DG, Lazo JS. Acute pneumocyte injury, poly(ADP-ribose) polymerase activity, and pyridine nucleotide levels after in vitro exposure of murine lung slices to cyclophosphamide. Biochem Pharmacol. 1994;48:1757–1765. doi: 10.1016/0006-2952(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 40.Hu Q, Xia Y, Corda S, Zweier JL, Ziegelstein RC. Hydrogen peroxide decreases pH in human aortic endothelial cells by inhibiting Na+/H+ exchange. Circ Res. 1998;83:644– 651. doi: 10.1161/01.res.83.6.644. [DOI] [PubMed] [Google Scholar]
- 41.Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ. Hypoxia–reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res. 2002;90:1274–1281. doi: 10.1161/01.res.0000024411.22110.aa. [DOI] [PubMed] [Google Scholar]
- 42.Jagtap P, Soriano FG, Virág L, Liaudet L, Mabley J, Szabó E, Haskó G, Marton A, Lorigados CB, Gallyas F, Jr, Sumegi B, Hoyt DG, Baloglu E, VanDuzer J, Salzman AL, Southan GJ, Szabó C. Novel phenanthridinone inhibitors of poly (adenosine 5′-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit Care Med. 2002;30:1071–1082. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 43.Junod AF, Jornot L, Petersen H. Differential effects of hyperoxia and hydrogen peroxide on DNA damage, polyadenosine diphosphate-ribose polymerase activity, and nicotinamide adenine dinucleotide and adenosine triphosphate contents in cultured endothelial cells and fibroblasts. J Cell Physiol. 1989;140:177–185. doi: 10.1002/jcp.1041400121. [DOI] [PubMed] [Google Scholar]
- 44.Kirkland JB. Lipid peroxidation, protein thiol oxidation and DNA damage in hydrogen peroxide-induced injury to endothelial cells: role of activation of poly(ADP-ribose)polymerase. Biochim Biophys Acta. 1991;1092:319–325. doi: 10.1016/s0167-4889(97)90007-0. [DOI] [PubMed] [Google Scholar]
- 45.Komjáti K, Jagtap P, Baloglu E, VanDuzer J, Salzman AL, Szabó C. Poly(ADP-ribose) polymerase inhibition in stroke: establishment of the therapeutic window of intervention and delineation of its role in the pathogenesis of white matter damage. FASEB J. 2002;16:A599. [Google Scholar]
- 46.Kossenjans W, Rymaszewski Z, Barankiewicz J, Bobst A, Ashraf M. Menadione-induced oxidative stress in bovine heart microvascular endothelial cells. Microcirculation. 1996;3:39–47. doi: 10.3109/10739689609146781. [DOI] [PubMed] [Google Scholar]
- 47.Landmesser U, Hornig B, Drexler H. Endothelial dysfunction in hypercholesterolemia: mechanisms, patho-physiological importance, and therapeutic interventions. Semin Thromb Hemost. 2000;26:529–537. doi: 10.1055/s-2000-13209. [DOI] [PubMed] [Google Scholar]
- 48.Lautier D, Lageux J, Thibodeau J, Ménard L, Poirier GG. Molecular and biochemical features of poly (ADP-ribose) metabolism. Mol Cell Biochem. 1993;122:171–193. doi: 10.1007/BF01076101. [DOI] [PubMed] [Google Scholar]
- 49.Le Rhun Y, Kirkland JB, Shah GM. Cellular responses to DNA damage in the absence of poly(ADP-ribose) polymerase. Biochem Biophys Res Commun. 1998;245:1–10. doi: 10.1006/bbrc.1998.8257. [DOI] [PubMed] [Google Scholar]
- 50.Lewis EJ, Lewis JB. Treatment of diabetic nephropathy with angiotensin II receptor antagonist. Clin Exp Nephrol. 2003;7:1–8. doi: 10.1007/s101570300000. [DOI] [PubMed] [Google Scholar]
- 51.Li F, Szabo C, Pacher P, Southan GJ, Abatan OI, Charniauskaya T, Stevens MJ, Obrosova IG. Evaluation of orally active poly(ADP-ribose) polymerase inhibitor in streptozotocin-diabetic rat model of early peripheral neuropathy. Diabetology. 2004;47:710–717. doi: 10.1007/s00125-004-1356-0. [DOI] [PubMed] [Google Scholar]
- 52.Liaudet L, Soriano FG, Szabó E, Virág L, Mabley JG, Salzman AL, Szabó C. Protection against hemorrhagic shock in mice genetically deficient in poly(ADP-ribose)polymerase. Proc Natl Acad Sci U S A. 2000;97:10203–10208. doi: 10.1073/pnas.170226797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lorenzi M, Montisano DF, Toledo S, Wong HC. Increased single strand breaks in DNA of lymphocytes from diabetic subjects. J Clin Invest. 1987;79:653–656. doi: 10.1172/JCI112863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation. 2002;106:927–932. doi: 10.1161/01.cir.0000026393.47805.21. [DOI] [PubMed] [Google Scholar]
- 55.Menissier-de Murcia J, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, Oliver FJ, Masson M, Dierich A, LeMeur M, Waltzinger C, Chambon P, de Murcia G. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci U S A. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mihm MJ, Wattanapitayakul SK, Piao SF, Hoyt DG, Bauer JA. Effects of angiotensin II on vascular endothelial cells: formation of receptor-mediated reactive nitrogen species. Biochem Pharmacol. 2003;65:1189–1197. doi: 10.1016/s0006-2952(03)00012-1. [DOI] [PubMed] [Google Scholar]
- 57.Minchenko AG, Stevens MJ, White L, Abatan OI, Komjáti K, Pacher P, Szabo C, Obrosova IG. Diabetes-induced overexpression of endothelin-1 and endothelin receptors in the rat renal cortex is mediated via poly(ADP-ribose) polymerase activation. FASEB J. 2003;17:1514–1516. doi: 10.1096/fj.03-0013fje. [DOI] [PubMed] [Google Scholar]
- 58.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- 59.Nitenberg A. Vascular endothelium: a target organ for diabetes mellitus. Ann Endocrinol (Paris) 2002;63:S13–S17. [PubMed] [Google Scholar]
- 60.Obrosova IG. Update on the pathogenesis of diabetic neuropathy. Curr Diab Rep. 2003;3:439–445. doi: 10.1007/s11892-003-0005-1. [DOI] [PubMed] [Google Scholar]
- 61.Obrosova IG, Li F, Abatan OI, Komjáti K, Pacher P, Szabo C, Stevens MJ. Poly(ADP-ribose) polymerase (PARP) activation in the development of diabetic neuropathy [Abstract] FASEB J. 2003;17:A261. [Google Scholar]
- 62.Obrosova IG, Li F, Abatan OI, Forsell MA, Komjáti K, Pacher P, Szabo C, Stevens MJ. Role of poly(ADP-ribose) polymerase activation in diabetic neuropathy. Diabetes. 2004;53:711–720. doi: 10.2337/diabetes.53.3.711. [DOI] [PubMed] [Google Scholar]
- 63.Obrosova IG, Mabley JG, Zsengeller Z, Charniauskaya T, Abatan OI, Groves JT, Szabó C. Role for nitrosative stress in diabetic neuropathy: evidence from studies with a peroxynitrite decomposition catalyst. FASEB J. 2005;19:401–403. doi: 10.1096/fj.04-1913fje. [DOI] [PubMed] [Google Scholar]
- 64.Obrosova IG, Minchenko AG, Frank RN, Seigel GM, Zsengeller Z, Pacher P, Stevens MJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor (VEGF) overexpression. Int J Mol Med. 2004;14:55–64. [PubMed] [Google Scholar]
- 65.Obrosova IG, Pacher P, Szabó C, Zsengeller Z, Hirooka H, Stevens MJ, Yorek MA. Aldose reductase inhibition counteracts oxidative/nitrosative stress and poly(ADP-ribose) polymerase activation in tissue sites for diabetic complications. Diabetes. 2005;54:234–242. doi: 10.2337/diabetes.54.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oei SL, Ziegler M. ATP for the DNA ligation step in base excision repair is generated from poly(ADP-ribose) J Biol Chem. 2000;275:23234–23239. doi: 10.1074/jbc.m002429200. [DOI] [PubMed] [Google Scholar]
- 67.Oikawa A, Tohda H, Kanai M, Miwa M, Sugimura T. Inhibitors of poly(adenosine diphosphate ribose) polymerase induce sister chromatid exchanges. Biochem Biophys Res Commun. 1980;97:1311–1316. doi: 10.1016/s0006-291x(80)80009-x. [DOI] [PubMed] [Google Scholar]
- 68.Oliver FJ, Menissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pacher P, Cziraki A, Mabley JG, Liaudet L, Papp L, Szabo C. Role of poly(ADP-ribose) polymerase activation in endotoxin-induced cardiac collapse in rodents. Biochem Pharmacol. 2002;64:1785–1791. doi: 10.1016/s0006-2952(02)01421-1. [DOI] [PubMed] [Google Scholar]
- 70.Pacher P, Liaudet L, Bai P, Virag L, Mabley J, Hasko G, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to the development of doxorubicin-induced heart failure. J Pharmacol Exp Ther. 2002;300:862–687. doi: 10.1124/jpet.300.3.862. [DOI] [PubMed] [Google Scholar]
- 71.Pacher P, Liaudet L, Mabley J, Komjati K, Szabo C. Pharmacologic inhibition of poly(adenosine diphosphate-ribose) polymerase may represent a novel therapeutic approach in chronic heart failure. J Am Coll Cardiol. 2002;40:1006–1016. doi: 10.1016/s0735-1097(02)02062-4. [DOI] [PubMed] [Google Scholar]
- 72.Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabó É, Szabó C. The role of poly(ADP-ribose) polymerase in the development of myocardial and endothelial dysfunction in diabetes mellitus. Diabetes. 2002;51:514–521. doi: 10.2337/diabetes.51.2.514. [DOI] [PubMed] [Google Scholar]
- 73.Pacher P, Mabley JG, Soriano FG, Liaudet L, Komjati K, Szabo C. Endothelial dysfunction in aging animals: the role of poly(ADP-ribose) polymerase activation. Br J Pharmacol. 2002;135:1347–1350. doi: 10.1038/sj.bjp.0704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pacher P, Mabley JG, Soriano FG, Liaudet L, Szabo C. Activation of poly(ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int J Mol Med. 2002;9:659–664. [PubMed] [Google Scholar]
- 75.Pacher P, Schulz R, Liaudet L, Szabo C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol Sci. 2005;26:302–310. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pacher P, Obrosova IG, Mabley JG, Szabo C. Role of nitrosative stress and peroxynitrite in the pathogenesis of diabetic complications. Emerging new therapeutical strategies. Curr Med Chem. 2005;12:267–275. doi: 10.2174/0929867053363207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pannirselvam M, Verma S, Anderson TJ, Triggle CR. Cellular basis of endothelial dysfunction in small mesenteric arteries from spontaneously diabetic (db/db −/−) mice: role of decreased tetrahydrobiopterin bioavailability. Br J Pharmacol. 2002;136:255–263. doi: 10.1038/sj.bjp.0704683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Park KS, Kim JH, Kim MS, Kim JM, Kim SK, Choi JY, Chung MH, Han B, Kim SY, Lee HK. Effects of insulin and antioxidant on plasma 8-hydroxyguanine and tissue 8-hydroxydeoxyguanosine in streptozotocin-induced diabetic rats. Diabetes. 2001;50:2837–2841. doi: 10.2337/diabetes.50.12.2837. [DOI] [PubMed] [Google Scholar]
- 79.Park SD, Kim CG, Kim MG. Inhibitors of poly(ADP-ribose) polymerase enhance DNA strand breaks, excision repair, and sister chromatid exchanges induced by alkylating agents. Environ Mutagen. 1983;5:515–525. doi: 10.1002/em.2860050402. [DOI] [PubMed] [Google Scholar]
- 80.Pieper AA, Brat DJ, Krug DK, Watkins CC, Gupta A, Blackshaw S, Verma A, Wang ZQ, Snyder SH. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96:3059–3064. doi: 10.1073/pnas.96.6.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A. 1982;79:3423–3427. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quagliaro L, Piconi L, Assaloni R, Martinelli L, Motz E, Ceriello A. Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of protein kinase C and NAD(P)H-oxidase activation. Diabetes. 2003;52:2795–2804. doi: 10.2337/diabetes.52.11.2795. [DOI] [PubMed] [Google Scholar]
- 83.Reusch JE. Diabetes, microvascular complications, and cardiovascular complications: what is it about glucose? J Clin Invest. 2003;112:986–988. doi: 10.1172/JCI19902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richardson D, Vinik A. Etiology and treatment of erectile failure in diabetes mellitus. Curr Diab Rep. 2002;2:501–509. doi: 10.1007/s11892-002-0120-4. [DOI] [PubMed] [Google Scholar]
- 85.Rudat V, Kupper JH, Weber KJ. Trans-dominant inhibition of poly(ADP-ribosyl)ation leads to decreased recovery from ionizing radiation-induced cell killing. Int J Radiat Biol. 1998;73:325–330. doi: 10.1080/095530098142428. [DOI] [PubMed] [Google Scholar]
- 86.Ruderman NB, Williamson JR, Brownlee M. Glucose and diabetic vascular disease. FASEB J. 1992;6:2905–2914. doi: 10.1096/fasebj.6.11.1644256. [DOI] [PubMed] [Google Scholar]
- 87.Sardas S, Yilmaz M, Oztok U, Cakir N, Karakaya AE. Assessment of DNA strand breakage by comet assay in diabetic patients and the role of antioxidant supplementation. Mutat Res. 2001;490:123–129. doi: 10.1016/s1383-5718(00)00157-1. [DOI] [PubMed] [Google Scholar]
- 88.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 89.Shimoda K, Murakami K, Enkhbaatar P, Traber LD, Cox RA, Hawkins HK, Schmatstieg FC, Kemjati K, Mabley JG, Szabo C, Salzman AL, Traber DL. Effect of poly(ADP ribose) synthetase inhibition on burn and smoke inhalation injury in sheep. Am J Physiol Lung Cell Mol Physiol. 2003;285:L240–L249. doi: 10.1152/ajplung.00319.2002. [DOI] [PubMed] [Google Scholar]
- 90.Simbulan-Rosenthal CM, Ly DH, Rosenthal DS, Konopka G, Luo R, Wang ZQ, Schultz PG, Smulson ME. Misregulation of gene expression in primary fibroblasts lacking poly(ADP-ribose) polymerase. Proc Natl Acad Sci U S A. 2000;97:11274–11279. doi: 10.1073/pnas.200285797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soriano FG, Mabley JG, Pacher P, Liaudet L, Szabó C. Rapid reversal of the diabetic endothelial dysfunction by pharmacological inhibition of poly(ADP-ribose) polymerase. Circ Res. 2001;89:684–691. doi: 10.1161/hh2001.097797. [DOI] [PubMed] [Google Scholar]
- 92.Soriano FG, Virag L, Szabo C. Diabetic endothelial dysfunction: role of reactive oxygen and nitrogen species production and poly(ADP-ribose) polymerase activation. J Mol Med. 2001;79:437–448. doi: 10.1007/s001090100236. [DOI] [PubMed] [Google Scholar]
- 93.Soriano FG, Liaudet L, Szabo E, Virag L, Mabley JG, Pacher P, Szabo C. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock. 2002;17:286–292. doi: 10.1097/00024382-200204000-00008. [DOI] [PubMed] [Google Scholar]
- 94.Southan GJ, Szabo C. Poly(ADP-ribose) polymerase inhibitors. Curr Med Chem. 2003;10:321–340. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- 95.Szabó C. Cell Death: The role of PARP. Boca Raton, FL: CRC Press; 2000. [Google Scholar]
- 96.Szabó C, Dawson VL. Role of poly (ADP-ribose) synthetase activation in inflammation and reperfusion injury. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 97.Szabó C, Zingarelli B, O’Connor M, Salzman AL. DNA strand breakage, activation of poly-ADP ribosyl synthetase, and cellular energy depletion are involved in the cytotoxicity in macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Szabo C, Cuzzocrea S, Zingarelli B, O’Connor M, Salzman AL. Endothelial dysfunction in a rat model of endotoxic shock: importance of the activation of poly (ADP-ribose) synthetase by peroxynitrite. J Clin Invest. 1997;100:723–735. doi: 10.1172/JCI119585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Szabó C, Wong H, Bauer PI, Kirsten E, O’Connor M, Zingarelli B, et al. Regulation of components of the inflammatory response by 5-iodo-6-amino-1,2-benzopyrone, an inhibitor of poly (ADP-ribose) synthetase and pleiotropic modifier of cellular signal pathways. Int J Oncol. 1997;10:1093–1104. doi: 10.3892/ijo.10.6.1093. [DOI] [PubMed] [Google Scholar]
- 100.Szabó C, Virág L, Cuzzocrea S, Scott GS, Hake P, O’Connor MP, Zingarelli B, Salzman A, Kun E. Protection against peroxynitrite-induced fibroblast injury and arthritis development by inhibition of poly (ADP-ribose) synthetase. Proc Natl Acad Sci U S A. 1998;95:3867–3872. doi: 10.1073/pnas.95.7.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Szabo C, Mabley JG, Moeller SM, Shimanovich R, Pacher P, Virag L, Soriano FG, Van Duzer JH, Williams W, Salzman AL, Groves JT. Pathogenetic role of peroxynitrite in the development of diabetes and diabetic vascular complications: studies with FP15, a novel potent peroxynitrite decomposition catalyst. Mol Med. 2002;8:571–580. [PMC free article] [PubMed] [Google Scholar]
- 102.Szabó C, Zanchi A, Komjati K, Pacher P, Krolewski AS, Quist WC, LoGerfo FW, Horton ES, Veves A. Poly(ADP-ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002;106:2680–2686. doi: 10.1161/01.cir.0000038365.78031.9c. [DOI] [PubMed] [Google Scholar]
- 103.Szabó C, Pachaer P, Komjati K, Mabley JG, Benko R, Killai M. Poly(ADP-ribose) polymerase (PARP) activation is an early event in angiotensin-induced cardiovascular disorders [Abstract] FASEB J. 2003;17:A803. [Google Scholar]
- 104.Szabó C, Pacher P, Zsengeller Z, Vaslin A, Komjati K, Benko R, Chen M, Mabley JG, Kollai M. Angiotensin II-mediated endothelial dysfunction: role of poly(ADP-ribose) polymerase activation. Mol Med. 2004;10:28–35. doi: 10.2119/2004-00001.szabo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Szabó E, Kern TS, Virag L, Mabley J, Szabó C. Evidence for poly(ADP-ribose) polymerase activation in the diabetic retina. FASEB J. 2001;15:A942. [Google Scholar]
- 106.Szabo G, Bahrle S, Stumpf N, Sonnenberg K, Szabo EE, Pacher P, Csont T, Schulz R, Dengler TJ, Liaudet L, Jagtap PG, Southan GJ, Vahl CF, Hagl S, Szabo C. Poly(ADP-ribose) polymerase inhibition reduces reperfusion injury after heart transplantation. Circ Res. 2002;90:100–106. doi: 10.1161/hh0102.102657. [DOI] [PubMed] [Google Scholar]
- 107.Thiemermann C, Bowes J, Myint FP, Vane JR. Inhibition of the activity of poly(ADP ribose) synthetase reduces ischemia–reperfusion injury in the heart and skeletal muscle. Proc Natl Acad Sci U S A. 1997;94:679–683. doi: 10.1073/pnas.94.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thies RL, Autor AP. Reactive oxygen injury to cultured pulmonary artery endothelial cells: mediation by poly(ADP-ribose) polymerase activation causing NAD depletion and altered energy balance. Arch Biochem Biophys. 1991;286:353–363. doi: 10.1016/0003-9861(91)90051-j. [DOI] [PubMed] [Google Scholar]
- 109.Ullrich O, Ciftci O, Hass R. Proteasome activation by poly-ADP-ribose-polymerase in human myelomonocytic cells after oxidative stress. Free Radic Biol Med. 2000;29:995–1004. doi: 10.1016/s0891-5849(00)00399-3. [DOI] [PubMed] [Google Scholar]
- 110.Ungvari Z, Gupte SA, Recchia FA, Batkai S, Pacher P. Role of oxidative-nitrosative stress and downstream pathways in various forms of cardiomyopathy and heart failure. Curr Vasc Pharmacol. 2005;3:221–229. doi: 10.2174/1570161054368607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Verrotti A, Trotta D, Salladini C, Laura M, Chiarelli F. Preventing microvascular diabetic complications in children and adolescents: looking beyond glycaemic control. Expert Opin Pharmacother. 2003;4:525–532. doi: 10.1517/14656566.4.4.525. [DOI] [PubMed] [Google Scholar]
- 113.Vinik AI, Vinik E. Prevention of the complications of diabetes. Am J Manag Care. 2003;9(3 Suppl):S63–S80. [PubMed] [Google Scholar]
- 114.Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 115.Wahlberg G, Carlson LA, Wasserman J, Ljungqvist A. Protective effect of nicotinamide against nephropathy in diabetic rats. Diabetes Res. 1985;2:307–312. [PubMed] [Google Scholar]
- 116.Wattanapitayakul SK, Weinstein DM, Holycross BJ, Bauer JA. Endothelial dysfunction and peroxynitrite formation are early events in angiotensin-induced cardiovascular disorders. FASEB J. 2000;14:271–278. doi: 10.1096/fasebj.14.2.271. [DOI] [PubMed] [Google Scholar]
- 117.Werner-Felmayer G, Golderer G, Werner ER. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Curr Drug Metab. 2002;3:159–173. doi: 10.2174/1389200024605073. [DOI] [PubMed] [Google Scholar]
- 118.Zhang J, editor. Therapeutic Implications of PARP Inhibition. Boca Raton, FL: CRC Press; 2002. [Google Scholar]
- 119.Zheng L, Szabo C, Kern TS. Poly(ADP-ribose) polymerase is involved in the development of diabetic retinopathy via regulation of nuclear faction-kappaB. Diabetes. 2004;53:2960–2967. doi: 10.2337/diabetes.53.11.2960. [DOI] [PubMed] [Google Scholar]
- 120.Zingarelli B, Salzman AL, Szabó C. Genetic disruption of poly (ADP ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia–reperfusion injury. Circ Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]