

Abstract
Heteromultimerization of Kir4.1 and Kir5.1 leads to a channel with distinct functional properties. The heteromeric Kir4.1-Kir5.1 channel is expressed in the eye, kidney and brainstem and has CO2/pH sensitivity in the physiological range, suggesting a candidate molecule for the regulation of K+ homeostasis and central CO2 chemoreception. It is known that K+ transport in renal epithelium and brainstem CO2 chemosensitivity are subject to modulation by hormones and neurotransmitters that activate distinct intracellular signaling pathways. If the Kir4.1-Kir5.1 channel is involved in pH-dependent regulation of cellular functions, it may also be regulated by some of the intracellular signaling systems. Therefore, we undertook studies to determine whether PKC modulates the heteromeric Kir4.1-Kir5.1 channel. The channel expressed using a Kir4.1-Kir5.1 tandem dimer construct was inhibited by the PKC activator PMA in a dose-dependent manner. The channel inhibition was produced via reduction of the Popen. The effect of PMA was abolished by specific PKC inhibitors. In contrast, exposure of oocytes to forskolin (a PKA activator) had no significant effect on Kir4.1-Kir5.1 currents. The channel inhibition appeared to be independent of PIP2 depletion and PKC-dependent internalization. Several consensus sequences of potential PKC phosphorylation sites were identified in the Kir4.1 and Kir5.1 subunits by sequence scan. Although the C terminal peptides of both Kir4.1 and Kir5.1 were phosphorylated in vitro, site-directed mutagenesis of individual residues failed to reveal the PKC phosphorylation sites suggesting that the channel may have multiple phosphorylation sites. Taken together, these results suggest that the Kir4.1-Kir5.1 but not the homomeric Kir4.1 channel is strongly inhibited by PKC activation.
Keywords: Kir4.1-Kir5.1, protein kinase C, phosphorylation, endocytosis
INTRODUCTION
The inward rectifier K+ channels (Kir) control membrane potential and K+ homeostasis. Unlike other Kir family members, heteromultimerization of inter-subfamily members Kir4.1 and Kir5.1 leads to a channel with distinct functional properties [1,2,3,4,5,6,7]. Of particular interest in these newly emerging properties is the enhanced sensitivity to intracellular pH (pKa 7.45), allowing the heteromeric Kir4.1-Kir5.1 channel to detect pH changes at physiological levels [3,6,7,8]. This as well as the fact that both Kir4.1 and Kir5.1 subunits are expressed in the kidney, eye and brainstem suggests that the channel may be a candidate molecule for the regulation of K+ homeostasis and central CO2 chemoreception [6,8,9,10].
The Kir4.1 and Kir5.1 subunits are expressed in the proximal convoluted tubule, the distal convoluted tubule and the cortical collecting duct of the kidney [4,5,11]. Immunoprecipitation studies show that the Kir4.1 and Kir5.1 co-immunoprecipitate together consistent with the idea that the heteromeric Kir4.1-Kir5.1 channel plays a role in the absorption of high extracellular K+, a process known as K+ “buffering” [11]. Since K+ secretion in the kidney is tightly linked to systemic pH levels, the high pH sensitivity of the Kir4.1-Kir5.1 may allow the channel to control K+ secretion according to systemic acid-base status. The K+ buffering may be mainly produced by the Kir4.1-Kir5.1 channel as the homomeric Kir4.1 channel is not as sensitive to changes in intracellular pH in the physiological range. Müller cells that are responsible for buffering and recycling extracellular K+ released by retinal neurons in the eye also show differential expressions of the Kir4.1 and Kir5.1 subunits [12]. Whereas Kir4.1 alone exists at the end-feet, both Kir4.1 and Kir5.1 are detected in the cell body [12]. Genetic disruption of Kir4.1 cause abnormalities in the Müller cells and affects K+ buffering in the retina [13]. In the brainstem, mRNAs for both Kir4.1 and Kir5.1 subunits have been detected together in neurons in several CO2 chemosensitive areas suggesting a role of the heteromeric Kir4.1-Kir5.1 channel in central CO2 chemoreception, an important brainstem function for respiratory and cardiovascular controls [10].
It is known that the central CO2 chemoreception and K+ transport in renal epithelium are regulated by neurotransmitters, hormones and pharmacological agents through specific intracellular signaling systems [14,15,16]. Therefore, it is possible that the heteromeric Kir4.1-Kir5.1 channel is regulated by certain second messenger systems. Such a modulation may enable cells expressing the Kir4.1-Kir5.1 channel to respond to PCO2 and pH changes according to systemic needs. One common intracellular signaling system activated by neurotransmitter and hormone is mediated by protein kinase C (PKC). Indeed, a number of consensus sequences of potential PKC phosphorylation sites exist in the Kir4.1 and Kir5.1 subunits. Supporting the idea of PKC phosphorylation are also previous reports indicating that other Kir channels such as Kir3.1-Kir3.4, Kir2.2, Kir2.3, and Kir6.2 are modulated by PKC with several phosphorylation sites identified in these channel proteins [17,18,19,20,21,22,23,24,25,26]. Therefore, we performed experiments to test the possibility that PKC regulates the Kir4.1-Kir5.1 channel. Our results showed that the Kir4.1-Kir5.1 channel was strongly inhibited by the activation of PKC.
MATERIALS AND METHODS
Oocytes preparation and injection
Frogs were anesthetized by bathing them in distilled water containing 0.3% 3-aminobenzoic acid ethyl ester. A few lobes of ovaries were removed through a small abdominal incision (∼5 mm). Then, the surgical incision was closed and the frogs were allowed to recover from the anesthesia. Xenopus oocytes were treated with 1 mg/ml of collagenase (Type IA, Sigma Chemicals) in the OR2 solution containing (in mM): NaCl 82, KCl 2, MgCl2 1 and HEPES 5, pH 7.4 for 50 min at room temperature. After five washes (10 min each) of the oocytes with the OR2 solution, cDNAs (25–50 ng or 5-10 femtomoles in 50 nl water) were injected into the oocytes. The oocytes were then incubated at 18 °C in the ND-96 solution containing (in mM): NaCl 96, KCl 2, MgCl2 1, CaCl2 1.8, HEPES 5, and sodium pyruvate 2.5 with 100 mg/l geneticin and 50 mg/l tetracycline added (pH 7.4).
Molecular biology
Rat Kir4.1 cDNA (GenBank #X83585) and rat Kir5.1 cDNA (GenBank #X83581) are gifts from Dr. John Adelman (Oregon Health Science University, Portland, OR). These cDNAs were subcloned into the eukaryotic expression vector pcDNA3.1 (Invitrogen, Carlsbad, CA) and used for Xenopus oocyte expression without cRNA synthesis. For co-expression of Kir4.1 and Kir5.1, a tandem dimer of these two cDNAs was constructed using the overlapping extension technique, in which full length Kir4.1 and Kir5.1 sequences were obtained using Pfu DNA polymerase (Stratagene, La Jolla, CA) chain reaction (PCR). The PCR products were joined to each other at the 3′ end of Kir4.1 and 5′ end of Kir5.1. Correct constructions were confirmed with DNA sequencing. Site-specific mutations were made using a site-directed mutagenesis kit (Stratagene, La Jolla, CA). Correct constructions and mutations were confirmed with DNA sequencing. Dynamin II (aa) wild-type and Dynamin II (aa) K44A are gifts from Dr. Mark McNiven at the Mayo Clinic. These two cDNAs were subcloned into the eukaryotic expression vector pEGFP-N1 creating an N-terminal GFP-Dynamin fusion and used directly for expression in Xenopus oocytes.
Expression and purification of MBP and C-terminal-MBP fusion proteins
The membrane and C-terminal cytoplasmic junction for the Kir4.1 (GenBank #X83585) and Kir5.1 (GenBank #X83581) was determined by ScanProsite. The C-terminal fragments were amplified by PCR and inserted into the pMALc2x vector (New England Biolabs) using EcoRI and XbaI. The PCR fragments contained these restriction enzyme sites as well as a stop codon to terminate transcription after the C-terminus. To express the maltose binding protein (MBP) alone a stop codon was introduced to prevent expression of part of the LacZ gene present in the pMALc2x vector. Correct constructions were confirmed with DNA sequencing.
Expression and purification of the fusion proteins were carried out in Eschericia coli BL21 competent cells. The cells were grown overnight at 37 °C. The next morning the cultures were diluted in LB broth medium and grown to an A600 ∼1 at 37 °C. Protein induction was achieved by the addition of 1 mM isopropylthiogalactoside (IPTG) to the cultures which were then incubated at 25 °C for 2 hours. The cells were harvested by centrifugation (4,000 × g, 15 min, 4 °C). The bacterial pellet was washed with STE (150 mM NaCl; 10 mM Tris, pH 8.0; 1 mM EDTA) plus protease inhibitors (EDTA-free) (Roche Molecular Biochemicals) once and then resuspended in the same buffer. The cell membranes were lysed by the addition of lysozyme (100 μg/ml) added immediately following resuspension. The samples were incubated on ice for 15 min. Dithiothreitol (DTT) (5 mM) was added to the samples and the cell membranes were further lysed by sonication (4 × 20 s) followed by centrifugation (4,000 × g, 15 min, 4 °C). The resulting supernatant was collected and Triton X-100 was added to a final concentration of 1%. The supernatant was then loaded onto amylose resin (New England Biolabs) pre-equilibrated with protein elution buffer (PEB) (50 mM Tris, pH 8.0; 1 mM EDTA) and incubated on ice for 45 minutes. The amylose resin was centrifuged and the supernatant was discarded. The resin was washed 7 times with 2 volumes of PEB. Elution of the maltose binding protein (MBP) and MBP fusion proteins was achieved by incubating the amylose resin with 1 volume of PEB containing 10 mM maltose. All fractions containing the fusion proteins were pooled and concentrated using centricon YM-10 centrifugal units (Millipore, Bedford, MA). Protein purity was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10%) and subsequent coomassie blue staining. The protein concentration was determined using a UV spectrophotometer at A280.
In vitro kinase assay
One μg of purified proteins (Kir4.1-C-terminus-MBP, Kir5.1-C-terminus-MBP, MBP, Histone III-S) was added to a reaction mixture consisting of the catalytically active PKC subunit (20 ng) (BioMol, Plymouth Meeting, PA) 10 μCi of 32P-γ-ATP (Perkin Elmer, Waltham, MA), Magnesium/ATP cocktail (75 mM MgCl2 and 500 μM ATP in 20 mM MOPS, pH 7.2, 25 mM β-glycerol phosphate, 5 mM EGTA, 1 mM sodium orthovanadate and 1 mM dithiothreitol) and 5x reaction buffer (containing: 125 mM Tris-HCl, pH 7.5 and 0.1 mM EGTA) in a total volume of 25 μl. The samples were incubated at 30 °C for 1 hour and 5x loading buffer was added to stop the reaction. The samples were then analyzed by running a symmetric 10% SDS-PAGE gel. The gel was cut down the middle and one half was stained with Coomassie brilliant blue solution and destained and the other half was dried and subjected to autoradiography. The autoradiograph was taken using the Fujifilm BAS 2500 phosphor imager and analyzed with the Multigauge software.
Electrophysiology
Whole-cell currents were studied on oocytes 2–4 days post cDNA injection. Two-electrode voltage clamp was performed using an amplifier (Geneclamp 500, Axon Instruments Inc., Foster City, CA) at room temperature (∼24 °C). The extracellular solution contained (in mM): KCl 90, MgCl2 3, and HEPES 5 (pH 7.4). The recording pipettes were filled with 3 M KCl. Cells were impaled with these electrodes. The potential leakage of KCl from the recording electrodes was not corrected because of the large volume of oocytes. One of the electrodes (1.0-2.0 MΩ) served as the voltage recording which was connected to the HS-2 ×1L headstage (input resistance=1011 Ω) and the other (0.3-0.6 MΩ) was used for current recording connected to the HS-2 ×10MG headstage (maximum current=130 μA). Oocytes were accepted for further study if they did not show leakage in membrane currents. Current records were low-pass filtered (Bessel, 4-pole filter, 3 dB at 1 kHz), digitized at 5 kHz (12-bit resolution) and stored on a computer disk for later analysis.
Single-channel Kir4.1-Kir5.1 currents were studied in cell-attached patches using an Axopatch 200B amplifier (Axon Instruments). The bath solution contained (in mM): KCl 40, potassium gluconate 75, potassium fluoride 5, sodium vanadate 0.1, potassium pyrophosphate 10, ethylene glycol-bis-β-aminoethylether-N,N,N′,N′-tetraacetic acid (EGTA) 1, adenosine diphosphate (ADP) 0.2, piperazine-N,N′-bis-2-ethanesulfonic acid (Pipes) 10, glucose 10 and spermine 0.1 (FVPP solution, pH 7.4). The pipette was filled with the same FVPP solution used in the bath or a solution containing (mM): KCl 40, potassium gluconate 110, ADP 0.2, EGTA 1, Hepes 10, glucose 10 and MgCl2 2 (pH 7.4). The pipette tip was ∼2 μm for cell-attached patch. The oocytes were prepared for patch clamp recordings by mechanically removing the vitelline membranes after being exposed to a hypertonic solution (400 mOsm) for 10 min. The stripped oocytes were placed in a petri dish containing the regular bath solution (see above). The open-state probability (NPo) was calculated by counting all active channels from a given patch.
Human embryonic kidney cells (HEK293, CRL-1573, Batch #2187595, ATCC, Rockville, MD) were chosen to express the Kir4.1-Kir5.1 channel. The HEK293 Cells were cultured as a monolayer in the DMEM-F12 medium with 10% fetal bovine serum and penicillin/streptomycin added. Cultured at 37 °C with 5% CO2 in atmosphere, the cells were split twice weekly. The HEK293 cells were transfected using Lipofectamine2000 (Invitrogen, Carlsbad, CA) with 3.5μg Kir4.1-Kir5.1 dimer cDNA. To facilitate the identification of positively transfected cells, 0.5μg green fluorescent protein (GFP) cDNA (pEGFP-N2, Clontech, Palo Alto, CA) was added to the cDNA mixture. Cells were disassociated from the monolayer using 0.25% trypsin ∼24 hrs post-transfection. A few drops of the cell suspension were added on to 5×5 mm cover slips in a 35mm petri dish. The cells were then incubated at 37 °C for an additional 24-48 hours before experiments.
Single-electrode whole-cell voltage-clamp was performed on HEK293 cells at room temperature as described previously [27,28,29]. In brief, fire-polished patch pipettes were made from 1.2 mm borosilicate capillary glass (Sutter Instruments, Novato, CA). Tight seals (>1 gigaohm before breaking into the whole-cell mode) were obtained with the transfected cells. The patch electrodes had an open tip resistance of 0.5-1.0 MΩ. The series resistance during recording varied from 5 to 10 MΩ and was not compensated. Recordings were terminated whenever significant increase (>20%) in series resistance occurred. Current records were low-pass filtered (2 kHz, Bessel, 4-pole filter, -3 dB), digitized (20 kHz, 16-bit resolution), and stored on computer disk for later analysis using the pCLAMP 9 software (Axon Instruments Inc, Foster City, CA). Recordings were performed using solutions containing equal concentrations of K+ applied to the bath and recording pipettes. This solution contained (in mM): KCl 40, potassium gluconate 75, potassium fluoride 5, sodium vanadate 0.1, potassium pyrophosphate 10, ethylene glycolbis-β-aminoethylether-N,N,N′,N′-tetraacetic acid (EGTA) 1, adenosine diphosphate (ADP) 0.2, piperazine-N,N′-bis-2-ethanesulfonic acid (Pipes) 10, glucose 10 and spermine 0.1 (FVPP solution, pH 7.4). 100 nM thymeleatoxin and 15 nM PMA dissolved in DMSO as well as the same concentration of DMSO alone was applied to the bath solution. PKCi was added to the pipette solution at a final concentration of 10 μM.
Immunocytochemistry
Cultured HEK293 cells were transfected with the Kir4.1-Kir5.1 dimer cDNA together with GFP cDNA as described above. Three days later, the cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) (0.1 M, pH 7.4) for 30 minutes. The cells were washed three times with PBS and then blocked for 30 min in PBS containing 1% bovine serum albumin (BSA), 10% normal donkey serum (NDS), and 0.3% Triton X-100. The cultures were then incubated overnight at 4 °C in the primary antibody of goat anti-Kir5.1 (1:200) (Santa Cruz Biotechnology, Santa Cruz, CA) diluted in antibody dilution solution (ADS), containing 0.1% gelatin, 0.1% NaN3, and 0.3% Triton X-100 in PBS. The goat anti-Kir5.1 antibody was used to detect antigen of human, rat, and mouse origin. After washing five times with ADS (5 minutes each), the cultured cells were incubated at 25 °C with Alexa Fluor 594 donkey anti-goat IgG (Molecular Probes, Eugene, OR) diluted to 1:400 in ADS. The fluorescence reaction was first visualized using a Zeiss axioscope 200 fluorescence microscope (Zeiss, Oberkochen, Germany). Subsequently, fluorescence imaging was performed with a confocal microscope (LSM 510) (Zeiss, Jena, Germany). The confocal images were taken using a 40x oil immersion objective lens.
Chemical administration and exposure
4-α-Phorbol 12-myristate 13-acetate (PMA), 4α-phorbol-12,13-didecanoate (4α-PDD) and PKC inhibitor peptide 19-31 (PKCi) were purchased from Calbiochem (La Jolla, CA). 1-Oleoyl-2-acetyl-sn-glycerol (OAG), thymeleatoxin, chelerythrine chloride and calphostin-C were purchased from Sigma (St Louis, MO). Genistein and wortmannin were purchased from Tocris (Ellisville, MO). PMA, OAG, thymeleatoxin, genistein, wortmannin, chelerythrine, calphostin-C and 4α-PDD were dissolved in dimethylsulfoxide (DMSO) as stocks and mixed with a recording solution reaching a final concentration as indicated in the results. The final DMSO concentration was ≤0.1%. Other chemicals were dissolved in double-distilled water or experimental solutions. Exposures to these chemicals were done after baseline currents were stabilized.
Data analysis
Data are presented as means ± s.e. (standard error). Student t test or single-factor ANOVA was used to examine differences of chemical effects before versus during chemical exposures. The differences were considered to be statistically significant if P ≤ 0.05.
RESULTS
Inhibition of the Heteromeric Kir4.1-Kir5.1 Channel by PMA
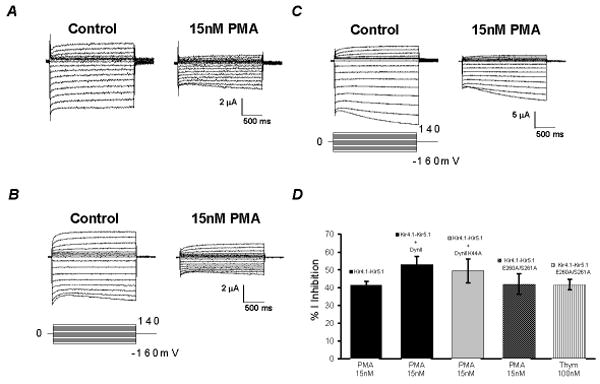
In two-electrode voltage clamp, inward rectifying K+ currents were recorded from Xenopus oocytes 2-3 days post injection of the Kir4.1-Kir5.1 cDNA using a bath solution containing 90 mM K+. Typical Kir4.1-Kir5.1 currents were revealed: small outward currents and large inward currents with slow activation at highly negative membrane potentials (Fig. 1A). Following stabilization of the baseline currents, PMA, a specific and potent PKC activator, was added to the bath solution. The PMA exposure resulted in a strong inhibition of the Kir4.1-Kir5.1 currents. This inhibition was dose-dependent (Fig. 2A, B): Evident channel inhibition was seen with PMA concentrations as low as 5 nM; maximum effect occurred with 1 μM PMA at which the Kir4.1-Kir5.1 currents were inhibited by 93.3±1.4% (n=4); and 15 nM PMA inhibited the Kir4.1-Kir5.1 currents by 41.4±2.2% (n=13). In comparison, the homomeric Kir4.1 channel showed no response to 15 nM PMA (Fig. 1B), and was slightly inhibited (8.4±8.6%, n=5) by 100 nM PMA. Numerous attempts were made to express the homomeric Kir5.1 channel. Unfortunately, we were unable to detect inward rectifying K+ currents that were sensitive to micromolar concentrations of barium in Xenopus oocytes and HEK293 cells. Therefore, we were unable to determine the effect of PMA on the Kir5.1 channel.
Fig 1. The Kir4.1-Kir5.1 channel but not homomeric Kir4.1 is sensitive to PMA.
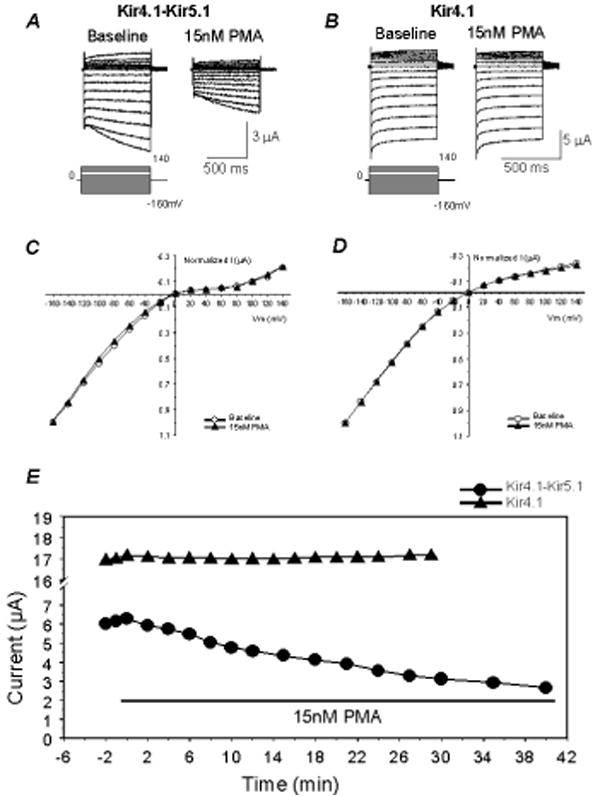
A. Using TEVC whole-cell Kir4.1-Kir5.1 currents were recorded from an oocyte 3 days post-injection of the Kir4.1-Kir5.1 dimer cDNA. With 90 mM K+ in the extracellular solution inward rectifying currents were recorded at baseline. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −160 mV to 140 mV with a 20-mV increment was applied to the cell. Note that in highly negative membrane potentials, there was slow activation of the currents. Exposure to 15 nM PMA (a specific and potent PKC activator) inhibited the Kir4.1-Kir5.1 currents by 40%. B. The same experiment was carried out with the homomeric Kir4.1. Exposure to 15 nM PMA failed to inhibit this channel. C, D. When baseline and peak PMA affected currents were scaled to the same magnitude at -160 mV, the I/V relationship of the currents recorded under these two conditions were superimposed, suggesting that the effects were voltage-independent. E. The time profile showed that the Kir4.1-Kir5.1 current amplitude decreased rapidly when PMA was present in the bath solution, and reached maximum inhibition in ∼35 min. The Kir4.1 currents remained constant for 30 minutes during exposure to PMA.
Fig 2. Concentration-dependent response of PMA.
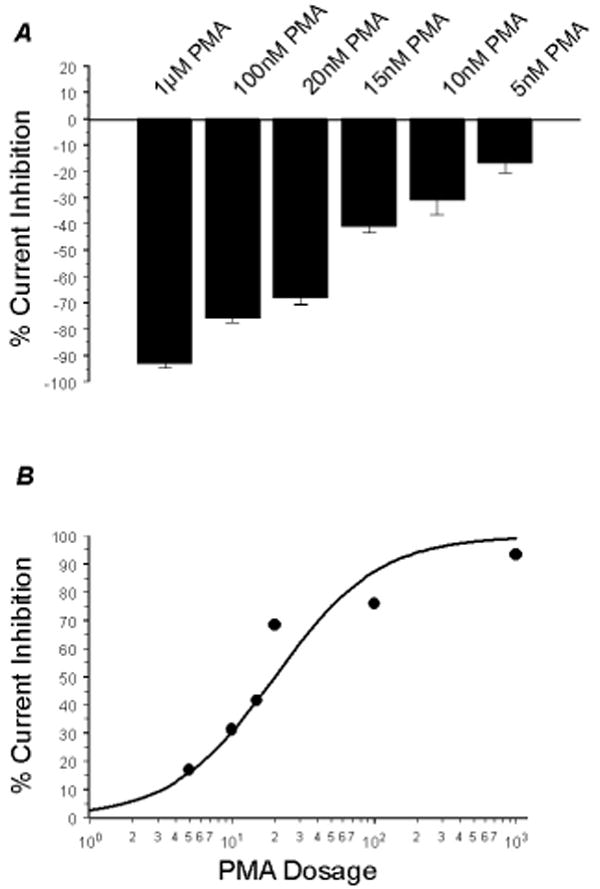
A. The effect of PMA on the Kir4.1-Kir5.1 channel was clearly concentration-dependent. The Kir4.1-Kir5.1 channel was inhibited by very low concentrations of PMA. Exposure to 5 nM PMA suppressed Kir4.1-Kir5.1 channels by 16.9±3.8% (n=5). 1 μM PMA maximally inhibited the Kir4.1-Kir5.1 currents by 93.3±1.4% (n=4). n≥4 for each concentration tested. B. The concentration-dependent response fits a sigmoid curve. This curve was used to more accurately obtain the IC50 concentration which was 15 nM PMA.
The voltage dependence was analyzed using the current-voltage (I-V) plot. The currents recorded at -160 mV were normalized to the same level for both baseline and PMA treatment. When plotted in the I-V curve, the curve of the PMA treatment overlapped that obtained from the baseline suggesting that the inhibition of the Kir4.1-Kir5.1 currents by PMA is voltage independent (Fig. 1C). The same analysis on the Kir4.1 showed an identical effect (Fig. 1D).
When the time course of the Kir4.1-Kir5.1 channel inhibition by 15 nM PMA was plotted, the inhibition started 2 min after exposure to PMA and reached the maximum in ∼30 min (Fig. 1E). Thus, all PMA experiments were done with a PMA exposure of 20-40 min. Higher concentrations of PMA resulted in a more rapid inhibition of the Kir4.1-Kir5.1 currents (Online Fig. 3E).
Involvement of PKC
To demonstrate that PKC activation underscores the PMA inhibition of Kir4.1-Kir5.1, we took advantage of several other specific kinase activators and inhibitors. Exposure of the oocytes to phorbol ester (OAG), another PKC activator, resulted in a similar inhibition as PMA (Fig 3A, F). Thymeleatoxin (100 nM), that selectively activates the conventional PKC isoforms (α, β, & γ), brought about an inhibition of the Kir4.1-Kir5.1 channel (47.8±2.5%, n=5) to the same degree as PMA (P>0.05), suggesting that at least one of the conventional PKC isoforms is involved (Fig. 3B, F). In contrast, the homomeric Kir4.1 was not inhibited by OAG and thymeleatoxin, consistent with the results from PMA (Fig. 3D-F). An inactive analogue of PMA (4α-PDD, 15 and 20 nM) failed to inhibit the Kir4.1-Kir5.1 channel (Fig. 3F; Online Fig. 3B, D, E).
Fig 3. PKC activation inhibits the Kir4.1-Kir5.1 channel.
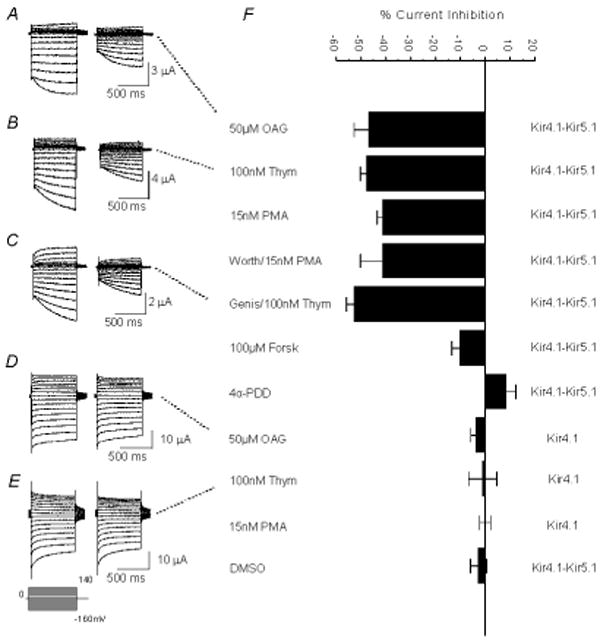
A. Currents were recorded from an oocyte in the same condition as in Figure 1A. These currents were strongly inhibited (46.3%) by exposure to 50 μM OAG (another potent activator of PKC). B. Exposure of an oocyte expressing the Kir4.1-Kir5.1 channel to 100 nM thymeleatoxin (activator of the conventional PKC isoforms α, β, γ) resulted in a similar inhibition (48.9%). C. Currents were recorded from an oocyte in the same condition as in 3B. Prior to application of 100 nM thymeleatoxin the cell was pre-incubated with 100 μM Genistein (a potent and general tyrosine kinase inhibitor) for 1 hour. Blocking activation of tyrosine kinases failed to reduce the inhibition by thymeleatoxin. D, E. The same experiments in 3A and 3B were carried out on oocytes expressing the homomeric Kir4.1 channel. This channel was barely inhibited by 50 μM OAG and 100 nM thymeleatoxin 8.9%, 6.8%, respectively. F. The effect of PMA appears to be via activation of PKC as Forskolin (a PKA activator) shows very little inhibition. Also, neither DMSO alone nor 4α-PDD (an inactive analog of PMA) had a significant effect. n≥4 for each experiment.
The cAMP dependent protein kinase A (PKA) has been shown to modulate Kir channels [19,20,26,30]. We therefore used forskolin that activates the enzyme adenylyl cyclase leading to increases in the cAMP concentration and activation of PKA. With 100 μM forskolin, oocytes expressing the Kir4.1-Kir5.1 channel showed a modest inhibition of the Kir4.1-Kir5.1 currents (10.2±3.3%, n=8; Fig. 3F), suggesting that the PMA-induced channel inhibition is not a result of PKA activation.
Specific PKC inhibitors were studied, in which oocytes were pre-incubated in chelerythrine or calphostin-C prior to PMA application to the recording solution. The pre-incubation of cells with 3 μM calphostin-C for 2 hours resulted in a complete elimination of the PMA-induced inhibition (Fig. 5A, B). A similar effect was seen with chelerythrine (50 μM for 1 hour), though to a lesser degree (Fig. 5B). Pre-incubation with genistein, a potent inhibitor of tyrosine kinases did not block the PMA effect (Fig. 3C, F). These results strongly suggest that the PMA inhibition of the Kir4.1-Kir5.1 channel is mediated by PKC, but neither by PKA nor by tyrosine kinase activation.
Fig 5. PKC is necessary for inhibition of Kir4.1-Kir5.1 by PMA.
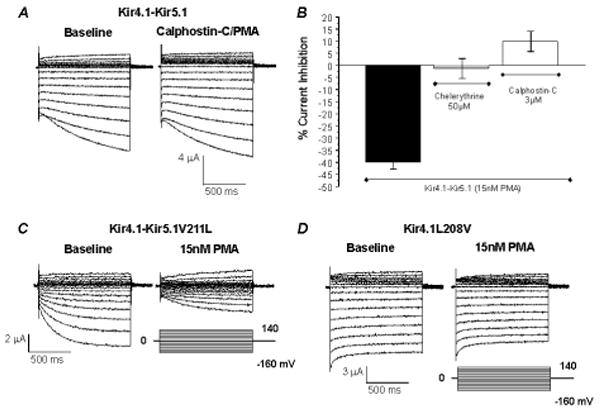
A. Using TEVC whole-cell Kir4.1-Kir5.1 currents were recorded from an oocyte 3 days post-injection of the Kir4.1-Kir5.1 dimer cDNA. With 90 mM K+ in the extracellular solution inward rectifying currents were recorded at baseline. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −160 mV to 140 mV with a 20-mV increment was applied to the cell. Before exposure to 15 nM PMA the oocyte was pre-incubated with 3 μM Calphostin-C (a potent and specific inhibitor of PKC) for 2 hours. Inhibition of PKC drastically attenuated the effect of PMA. PMA exposure after incubation with calphostin-C slightly activated the Kir4.1-Kir5.1 currents by 9.8%. B. Pre-incubation of oocytes with specific PKC inhibitors (50 μM chelerythrine or 3 μM calphostin-C) strongly attenuated the effect of 15 nM PMA (n≥4). C. Whole-cell currents were recorded from an oocyte 3 days post injection of the Kir4.1-Kir5.1V211L mutant channel recorded under the same condition as in A. This mutant is suggested to have increased PIP2 affinity. Exposure of the oocytes to 15 nM PMA inhibited the Kir4.1-Kir5.1V211L currents by 57.6%. D. The converse mutant was created on the Kir4.1 channel (Kir4.1L208V) and tested in an oocyte under the same experimental conditions as for Figure 4C. Similarly as for the wild-type channel this mutant failed to be inhibited by 15 nM PMA.
Inhibition of the Kir4.1-Kir5.1 Channel by PKC activation in HEK293 cells
The Kir4.1-Kir5.1 channel was transiently expressed in HEK293 cells. Whole-cell voltage clamp was performed on GFP-positive cells. Both the bath and pipette solutions contained 145 mM K+. The Kir4.1-Kir5.1 positive transfected cells showed typical Kir4.1-Kir5.1 currents that were relatively big at the basal level upon the formation of the whole-cell configuration. Without any treatment the currents remained at this level for more than 5 min. The cells were held at a membrane potential of 0 mV. Command pulses of -80 mV were applied to the cell every 3 seconds and the Kir4.1-Kir5.1 currents were recorded. Application of 15 nM PMA strongly inhibited the Kir4.1-Kir5.1 channel by 39.3±2.1% (n=10) (Fig. 4A, D). The inhibition by 15 nM PMA was greatly diminished [1.40±2.6% (n=6)] in the presence of the PKC inhibitor peptide (10 μM PKCi) that was added to the pipette solution (Fig. 4B, D). 100 nM thymeleatoxin similar to 15 nM PMA also inhibited the Kir4.1-Kir5.1 channel [44.7±2.6% (n=10)] expressed in HEK293 cells. However, an inactive analogue of PMA (4α-PDD) only inhibited the Kir4.1-Kir5.1 currents in HEK293 cell by 10.7±2.7% (n=9) (Fig. 4D). Taken together, these results suggest that the Kir4.1-Kir5.1 channel is inhibited by PKC activation in HEK293 cells in a similar manner as in Xenopus oocytes.
Fig 4. PKC activation inhibits the Kir4.1-Kir5.1 channel in HEK293 cells.
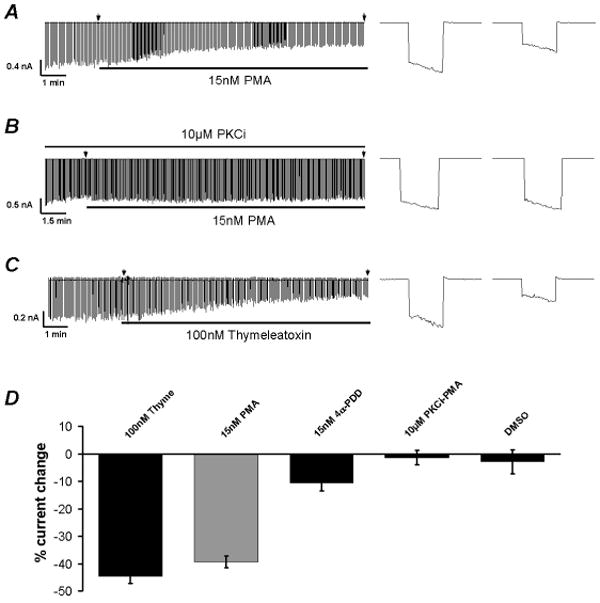
A. Whole-cell currents were recorded from an HEK293 cell transfected with the Kir4.1-Kir5.1 dimer cDNA with a holding potential at 0mV and command pulses of −80mV in every 3 seconds. After the whole-cell configuration was formed, the cell was perfused with the extracellular solution for a 2-5 min period of baseline recording. The currents were inhibited by exposure to 15nM PMA. The arrow represents the magnification of the current at the time point and shown on the right. B. In the presence of the PKC inhibitory peptide (10μM PKCi), 15nM PMA failed to inhibit the Kir4.1-Kir5.1 channel. C. Inhibition of the Kir4.1-Kir5.1 channel was also seen with exposure to 100nM thymeleatoxin. D. Bargraph showing that PKC activation results in inhibition of the Kir4.1-Kir5.1 channel. In each experiment n≥6.
Single-channel Properties Affected
The effect of PMA on the single-channel biophysical properties was studied in Xenopus oocytes using cell-attached patches with 145 mM K+ applied to the extracellular solution at a membrane potential of -80 mV. Only patches containing ≤4 active channels were considered in the experiment. Inward rectifying currents with single-channel conductance of ∼40 pS were recorded from oocytes that were injected with the Kir4.1-Kir5.1 cDNA 2-3 days prior to recordings. These currents were inhibited with an exposure of the cells to 15 nM PMA (Fig. 6A). The current inhibition was mainly produced by a suppression of the channel open-state probability (Popen), while the single-channel conductance was barely effected (Fig. 6).
Fig 6. Effects of PMA on the single channel properties of the Kir4.1-Kir5.1.
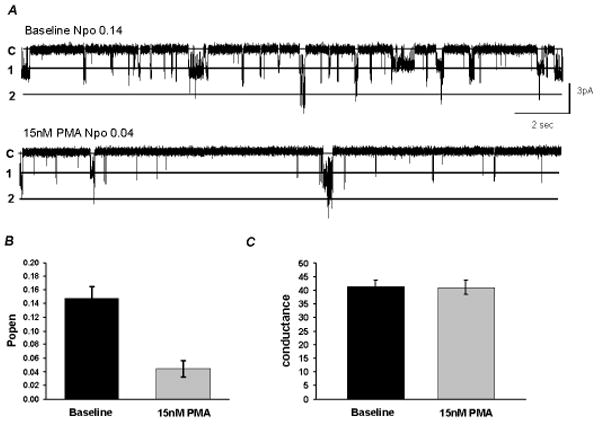
A. Single Kir4.1-Kir5.1 currents were recorded from an oocyte in a cell-attached patch configuration with 145 mM K+ in the patch pipette. At Vm of -80 mV, two active channels are seen at pH 7.4. Following stabilization, exposure to 15 nM PMA reduced the channel activity by a decrease in the NPo. Labels on the left: c, closure; 1, the first opening; 2, the second opening; etc. B, C. Bargraph showing that 15 nM PMA drastically reduced the Po, but did not affect the single-channel conductance (n≥6).
Independence of PIP2
A number of Kir channels are regulated by membrane lipids including phosphatidylinositol bisphosphate (PIP2). The presence of this phospholipid has been shown to increase Kir channel activity, and consequently depletion of PIP2 leads to channel inhibition. To test the possibility that the PMA-induced Kir4.1-Kir5.1 channel inhibition is related to PIP2 depletion, we mutated the potential PIP2 binding sites in the channel to increase or decrease the channel affinity for the phospholipid based on a sequence alignment with other Kir channels [21,31,32]. One mutant R178Q, that has previously been reported to increase PIP2 binding affinity [7], was created in the Kir5.1 and expressed with wild-type Kir4.1. This mutant channel showed a similar PMA sensitivity as the wild-type (Table 1). Another mutant Kir4.1-Kir5.1V211L, that has also been shown to increase the PIP2 sensitivity, did not affect the channel inhibition by 15 nM PMA (41.2±5.7%, n=10) (Fig. 5C). A converse mutation, exchanging these residues at a different site on the Kir4.1 (Kir4.1L208V) had no effect.
Table 1.
Effect of 15 nM PMA on wild-type Kir4.1, Kir4.1-Kir5.1 and mutant channels
| Baseline (μA) | PMA (μA) | % effect | N | |
|---|---|---|---|---|
| Kir4.1-Kir5.1 (Dim) | 6.5±0.6 | 3.8±0.43 | -41.4±1.9 | 13 |
| Kir4.1+Kir5.1 (Co-inj) | 8.9±2.1 | 5.2±1.2 | -40.7±1.9 | 6 |
| Kir4.1(homomer) | 15.3±2.3 | 15.23±2.2 | -0.04±2.2 | 6 |
| Kir4.1K67M-Kir5.1 | 20.53±3.0 | 12.3±2 | -41.35±3.23 | 6 |
| Kir4.1T178A-Kir5.1 | 3.4±0.7 | 2.22±0.43 | -33.1±2.9 | 6 |
| Kir4.1S206A-Kir5.1 | 6.0±1.3 | 3.8±0.84 | -36.7±3.7 | 13 |
| Kir4.1T214A-Kir5.1 | 3.3±0.2 | 2.1±0.2 | -37.2±4.6 | 11 |
| Kir4.1T262A-Kir5.1 | 16.96±1.5 | 11.1±1.3 | -34.9±3.5 | 5 |
| Kir4.1T262S-Kir5.1 | 14.1±0.7 | 9.7±0.6 | -31.4±2.9 | 5 |
| Kir4.1S263A-Kir5.1 | 9.7±1.0 | 6.2±1.0 | -36.2±3.9 | 4 |
| Kir4.1S299A-Kir5.1 | 8.8±1.6 | 5.4±1.0 | -38.6±2.9 | 7 |
| Kir4.1S320A-Kir5.1 | 4.7±0.6 | 3.0±0.6 | -39.0±5.0 | 7 |
| Kir4.1T346A-Kir5.1 | 4.8±0.4 | 2.75±0.48 | -44.1±7.1 | 6 |
| Kir4.1S360A-Kir5.1 | 4.0±0.4 | 2.3±0.23 | -40.3±5.8 | 4 |
| Kir4.1S373A-Kir5.1 | 4.1±0.4 | 2.46±0.23 | -38.1±7.3 | 7 |
| Kir4.1-Kir5.1T68A | 11.9±0.8 | 8.2±0.5 | -30.5±1.5 | 7 |
| Kir4.1-Kir5.1T174A | 3.1±0.3 | 1.9±0.27 | -38.1±4.1 | 5 |
| Kir4.1-Kir5.1T174D | 16.5±3.9 | 15.38±3.3 | -0.9±7.2 | 8 |
| Kir4.1-Kir5.1T174K | 5.1±1.1 | 2.94±0.61 | -40.1±9.6 | 7 |
| Kir4.1-Kir5.1R178Q | 6.2±0.5 | 3.88±0.54 | -37.8±4.0 | 7 |
| Kir4.1-Kir5.1T181A | 3.8±0.3 | 2.08±0.31 | -45.8±4.7 | 5 |
| Kir4.1-Kir5.1S185A | 10.1±0.6 | 6.28±0.64 | -37.8±5.1 | 4 |
| Kir4.1-Kir5.1T215A | 3.6±0.2 | 2.08±0.17 | -40.8±7.2 | 5 |
| Kir4.1-Kir5.1T232A | 4.0±0.5 | 2.3±0.27 | -41.4±5.6 | 4 |
| Kir4.1-Kir5.1S298A | 10.2±0.4 | 6.12±0.82 | -39.4±8.5 | 4 |
| Kir4.1-Kir5.1T370A | 3.6±0.4 | 2.26±0.25 | -36.6±5.2 | 7 |
| Kir4.1T32A-S299A-Kir5.1 | 22.5±3.3 | 14.8±3.17 | -36.4±5.8 | 6 |
| Kir4.1K67M-Kir5.1T68A | 8.5±3.3 | 5.95±2.6 | -35.8±5.2 | 4 |
| Kir4.1T262A-Kir5.1T68A | 11.04±0.6 | 7.1±0.27 | -35.3±3.4 | 5 |
| Kir4.1-Kir5.1V211L | 4.5±0.6 | 2.6±0.4 | -41.2±5.7 | 10 |
| Kir4.1L208V | 10.4±2.0 | 9.45±1.9 | -10.1±4.7 | 7 |
| Kir4.1L208I | n.f. | n.f. | ||
| Kir4.1L208V-Kir5.1 | 7.8±1.46 | 4.77±0.8 | -37.8±2.9 | 6 |
| Kir4.1L208I-Kir5.1 | n.f. | n.f. |
Abbreviations: n, number of observation; n.f., nonfunctional
To further assess the effect of PIP2 depletion in the channel inhibition, wortmannin that inhibits the phosphoinositide kinase-3 (PI3K) and thus the resynthesis of PIP2 from PIP3 was used to treat the oocytes prior to PMA exposure. Following incubation of oocytes with 10 μM wortmannin, PMA continued to inhibit the Kir4.1-Kir5.1 channel by 41.4±8.8% (n=5) (Fig. 3F). Taken together these results suggest that PIP2 depletion does not seem to underlie the PMA-induced Kir4.1-Kir5.1 channel inhibition.
Lack of Evidence for Endocytosis
Several recent studies indicate that protein kinase activation can lead to endocytosis and thus the suppression of surface Kir channel activity [33,34,35,36,37]. To evaluate the role of endocytosis in the inhibition of the Kir4.1-Kir5.1 channel by PKC activation, we carried out experiments with dynamin II and its dominant negative mutant where lysine at position 44 is mutated to alanine (K44A). We reasoned that co-expression of this dominant-negative dynamin II may abolish the effect of PKC if the Kir4.1-Kir5.1 channel inhibition by PKC results in dynamin dependent endocytosis. The Kir4.1-Kir5.1 channel was co-expressed with the wild-type dynamin II as well as the dynamin II K44A mutant. Exposure to 15 nM PMA led to inhibition of the currents by 53.2±4.4% (n=4) in the cells expressing the wild-type dynamin II (Fig. 7A, D) and by 49.5±6.8% (n=5) in cells expressing the dynamin II K44A mutant (Fig. 7B, D).
Fig 7. PKC activation inhibits Kir4.1-Kir5.1 currents independent of endocytosis.
A. Whole-cell currents were recorded from an oocyte 3 days following injection of the Kir4.1-Kir5.1+ dynamin II cDNAs (co-injection) under the same conditions as described for Figure 1A. PMA exposure inhibited the Kir4.1-Kir5.1+ dynamin II currents by 53%. B. Whole-cell Kir4.1-Kir5.1 currents were recorded from an oocyte 3 days following injection of the Kir4.1-Kir5.1 + dynamin II K44A mutant cDNAs (co-injection) using TEVC. Exposure to 15 nM PMA also inhibited the Kir4.1-Kir5.1+ dynamin II K44A (-49%). C. Whole-cell currents were recorded from an oocyte 3 days following injection of the Kir4.1-Kir5.1E260A/S261A mutant channel. This mutant has lost the recognition site for CCV endocytosis. Exposure of the oocyte to 15 nM PMA inhibited the Kir4.1-Kir5.1E260A/S261A currents by 42%. D. Bargraph summarizing panels A, B, and C. Note that the Dynamin K44A mutant and the Kir4.1-Kir5.1E260A/S261A mutant showed no significant difference to the Kir4.1-Kir5.1 in respect to inhibition by PMA. n≥4 for each experiment.
Another mechanism of endocytosis involves clathrin-coated pits. Proteins that undergo clathrin-dependent endocytosis contain an internalization recognition motif in at least one of the intracellular domains, usually in the C-terminal. This motif [(Y/F)(D/E)NPXY] shares high homology in many proteins. A primary scan of the Kir4.1 amino acid sequence failed to show any similar recognition motif. However, a scan of the Kir5.1 led to the identification of such a motif. The second and third amino acids of the motif have been found to be critical for endocytosis of Kir1.1 [37]. Therefore, we mutated these residues in Kir5.1. Consistent with the dynamin experiment we found that the Kir4.1-Kir5.1E260A/S261A mutant remained highly sensitive to PMA (Fig. 7C, D).
To further assess the possibility of channel endocytosis, whole cell currents were recorded from HEK293 cells before and during a treatment with thymeleatoxin. The whole cell capacitance (CAP) and series resistance (SR) were measured. If the Kir4.1-Kir5.1 channel inhibition by PKC activation was a result of endocytosis, the whole cell capacitance would decrease following application of thymeleatoxin to the bath solution. The percent change in the whole cell capacitance of HEK293 cells expressing the Kir4.1-Kir5.1 channel exposed to 100 nM thymeleatoxin for 15 min was 5.9±8.2% (n=4). This change in capacitance did not significantly differ from that of cells exposed to DMSO only (3.1±9.9%, n=4) (Online Fig. 1). The series resistance did not show a significant difference between cells exposed to DMSO versus cells exposed to 100 nM thymeleatoxin (Online Fig. 1).
We also performed immunocytochemistry to demonstrate membrane expression of the Kir4.1-Kir5.1 channel. Both the Kir4.1 and Kir5.1 antibodies are commercially available. By taking advantage of these antibodies and using immunocytochemistry we decided to look at membrane expression of the Kir4.1-Kir5.1 channel before and after exposure to PMA. HEK293 cells were transfected with the Kir4.1-Kir5.1 dimer. Immunocytochemistry was performed as described in the methods section. Prior to PMA exposure the channel showed membrane surface expression (Online Fig. 2A1-A3). After exposure to 15 nM PMA for 25 min the membrane expression was retained (Online Fig. 2B1-B3). Taken together these results suggest that the Kir4.1-Kir5.1 channel inhibition does not seem to be a result of endocytosis.
Phosphorylation Sites
To identify the PKC phosphorylation site, a sequence alignment of amino acids was made for both the Kir4.1 and Kir5.1 channels. Numerous putative PKC phosphorylation sites were found in Kir4.1 and Kir5.1, according to the consensus sequence, i.e., R(K)-X0-2-T/S-X0-2-R(K). Four different phosphorylation prediction programs were also used to predict potential PKC phosphorylation sites (NetPhos, Scansite, ScanProsite, Phosphobase). We performed systematic screening of these predicted sites according to the prediction programs in both Kir4.1 and Kir5.1 by single mutation of the serines or threonines to an alanine residue. The effect of PMA on these mutants was tested subsequently in Xenopus oocytes. The mutant channels were grouped: the first group of mutants consisted of the potential phosphorylation sites that were predicted using all prediction programs and had the highest score for potential phosphorylation by PKC; the second group consisted of those residues that had a lower phosphorylation prediction score; and the third group was residues not predicted by the phosphorylation prediction programs, but was found in a sequence similar to the consensus sequence for PKC. We tested 30 mutant channels with a single mutation representing each group and 9 channels with double mutations. All single mutants tested except for Kir4.1-Kir5.1T174D remained highly sensitive to PMA (Online-Fig. 4A, B, Table 1). Further analysis of the Kir4.1-Kir5.1T174 position suggests that it is involved in another channel function as mutants carrying an alanine or lysine at this position remained sensitive to PMA (Online-Fig. 4G, Table 1). Also the Kir4.1-Kir5.1T174D mutant also failed to respond to 15% CO2. Single channel analysis showed that the T174D mutant has an increased Popen (Online-Fig. 4D). Taken together we concluded that the T174 position is not a PKC phosphorylation site. The double mutants were created by comparing the PMA effect of the single mutants and selecting those that had a slightly lower PMA sensitivity. When tested, the double mutants also showed a PMA sensitivity similar to the wild-type. Therefore, the PKC phosphorylation sites remain to be demonstrated.
Direct Phosphorylation of Kir4.1 and Kir5.1
The failure to demonstrate PKC phosphorylation sites raises a question as the whether the channel proteins are indeed phosphorylated by PKC. Thus, we performed in-vitro phosphorylation experiments. Since most of the predicted PKC sites are located in the C-terminus of Kir4.1 and Kir5.1, MBP fusion peptides with the Kir4.1-C-terminus and Kir5.1C-terminus were produced as described in the Materials and Methods. After extraction and purification, the fusion peptides were incubated with 32P-γ-ATP in the presence of the catalytically active subunit of PKC (20ng). Such a treatment led to positive phosphorylation of these peptides suggesting that both Kir4.1 and Kir5.1 are directly phosphorylated by PKC (Fig. 8).
Fig 8. Phosphorylation of MBP fusion proteins.
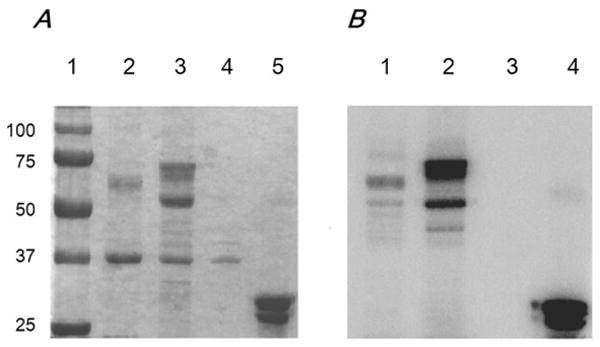
A. 10% SDS-PAGE gel showing the presence of the Kir4.1-C-terminus-MBP (∼65 kDa) (lane 2), Kir5.1-C-terminus-MBP (∼70 kDa) (Lane 3), MBP alone (∼42 kDa) (Lane 4) and Histone III-S (Lane 5). The size is indicated by the ladder (Lane 1) (precision plus protein standards) (Biorad). B. Autoradiograph after 12 hours showing phosphorylation of Kir4.1-C-terminus-MBP, Kir5.1-C-terminus-MBP, MBP, and Histone III-S. The proteins were incubated in the presence of 32P-γ-ATP and the catalytically active PKC subunit.
DISCUSSION
Protein phosphorylation is an important process in regulating cellular functions. Several inward rectifying K+ channels have been shown to be regulated by protein kinase phosphorylation [17,18,19,20,21,22,23,24,25,26,30,38]. For example, the heteromeric GIRK1/GIRK4 channel has previously been shown to be phosphorylated by protein kinase C at specific serine residues in the channel protein affecting channel gating [21]. This phosphorylation underscored the effect of substance-P on this channel allowing the channel to partake in the cellular response to this neurotransmitter.
In the present study we have presented evidence for the inhibition of the heteromeric Kir4.1-Kir5.1 channel by PKC. The Kir4.1-Kir5.1 channel is inhibited by nanomolar concentrations of PMA in a dose-dependent manner. Several experiments show that the effect of PMA is specific. Pre-incubation of the oocytes with specific PKC blockers (chelerythrine and calphostin-C) abolishes the PMA effect. Exposure of oocytes to 4α-PDD (an analogue of PMA incapable of activating PKC) fails to inhibit the Kir4.1-Kir5.1 channel. On the other hand, two other PKC activators (OAG & thymeleatoxin) strongly inhibit the channel. The biophysical mechanism for the inhibition of the Kir4.1-Kir5.1 channel by PKC appears to be mediated by reducing the channel open state probability without affecting the single channel conductance. The Kir4.1-Kir5.1 channel inhibition does not seem to be produced by PKA and tyrosine kinase as forskolin and genistein have rather small effects.
The PKC induced inhibition of the Kir4.1-Kir5.1 channel is independent of PIP2 and does not seem to be mediated by endocytosis. Since PIP2 has been shown to regulate many Kir channels it is possible that the PKC induced inhibition of the Kir4.1-Kir5.1 was a result of PIP2 depletion. The above experiments show that PMA inhibits the Kir4.1-Kir5.1 channel independent of PIP2 depletion. Similarly, mutant channels with increased PIP2 binding remains to be inhibited by PMA exposure to the same degree as the wild-type. Also, pre-incubation of oocytes with worthmannin (a PI3K inhibitor that reduces PIP2) fails to abolish the PMA induced inhibition of the channel, suggesting that the channel inhibition is not a result of PIP2 depletion. Immunocytochemistry experiments demonstrating surface expression in addition to experiments using a defective dynamin II mutant show that the effect of PMA is not a result of endocytosis. Although there may be other mechanisms involved, these results suggest that activation of PKC inhibits the Kir4.1-Kir5.1 channel by changing the channel gating mechanism.
Our biochemical experiments suggest a direct phosphorylation of the channel protein as both C-termini of Kir4.1 and Kir5.1 can be phosphorylated by PKC. To find the PKC phosphorylation sites, we screened more than 30 serine / threonine residues (some identified as phosphorylation sites by prediction software programs) by mutation to a non-phosphorylatable amino acid. None of the single mutations led to identification of the phosphorylation site, as all single mutants tested except for Kir5.1T174D remained highly sensitive to PMA. The Kir5.1T174 is not a PKC phosphorylation site either, as the Kir5.1T174A and Kir5.1T174K mutants are strongly inhibited by PMA. The effect of the T174D mutation suggests that this residue or its surrounding area may play a role in the channel gating mechanism and/or regulation by other intracellular factors. This mutant channel lost the sensitivity to both CO2 and PKC. Since this area contains several charged residues it is possible that a change in electrostatic charge in this area by the T174D affects the process of channel gating. Several double mutations based on single sites shown by phosphorylation prediction programs were also tested. None of these double mutations affects the PMA sensitivity. Therefore, we believe that the heteromeric combination of the Kir4.1-Kir5.1 channel may introduce multiple phosphorylation sites whose phosphorylation collectively may result in a change in channel activity. Removal of one or two of the phosphorylation sites may not be adequate to abolish the channel inhibition by PKC. This is further supported by the in vitro phosphorylation experiments that shows both Kir4.1 and Kir5.1 C-termini are directly phosphorylated by PKC. The Kir4.1 channel can express as a homomeric channel in several tissues [12,39,40,41,42,43]. However, this channel has low pH sensitivity [6,44,45] and is insensitive to phosphorylation by PKC according to our current studies. The Kir5.1 channel on the other hand is believed to only function in nature through heteromultimerization with either Kir4.1 or Kir4.2. Indeed, there is a study showing the existence of mRNA for both Kir4.1 and Kir5.1 channels in neurons of the brainstem suggesting that the heteromeric combination exists in these neurons [10]. In addition to brainstem expression, the Kir4.1-Kir5.1 channel has CO2/pH sensitivity in the physiological range [6,8,9]. This channel directly couples the CO2/pH sensitivity to membrane excitability [8]. All of these properties of the Kir4.1-Kir5.1 allows for its consideration as a CO2 central chemoreceptor molecule in the brainstem.
Brainstem neurons in addition to being regulated by pH are modulated by numerous hormones and neurotransmitters. This receptor ligand interaction is often used to identify certain neurons in the brainstem. Although receptor activation leads to multiple second messenger cascades, a common receptor activated cascade appears to be activation of phospholipase-C β that cleaves PIP2 into IP3 and DAG. The PIP2 derivative DAG activates PKC and IP3 increases intracellular calcium which is also necessary for PKC activation. Therefore, it would be very interesting to determine if the Kir4.1-Kir5.1 channel can be modulated by neurotransmitters and hormones through activation of GPCRs that are expressed in neurons of the brainstem and whether activation of PKC is involved in this modulation. For example, neurons of the locus coeruleus (expressing the NK1R) that are regulated by the neurokinin substance-P have been shown to contain mRNA for both Kir4.1 and Kir5.1 [10]. Therefore, further studies are needed to understand the modulation of the Kir4.1-Kir5.1 channel by G-protein coupled receptors that lead to PKC activation.
Supplementary Material
Whole-cell currents were recorded from HEK293 cells 3 days post-transfection of the Kir4.1-Kir5.1 cDNA. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −120 mV to 80 mV was applied to the cell and the corresponding currents were recorded. Following stabilization 100 nM thymeleatoxin was applied to the bath solution and inhibited the Kir4.1-Kir5.1 currents by 51.4±2.6% (n=4). Application of DMSO alone barely inhibited the Kir4.1-Kir5.1 currents. During the experiments with thymeleatoxin and DMSO, the whole cell capacitance and series resistance were monitored. Neither capacitance nor series resistance changed significantly upon application of 100 nM thymeleatoxin or DMSO to the bath solution.
Immunocytochemistry was performed on HEK293 cells 48 hours post-transfection of the Kir4.1-Kir5.1 dimer cDNA together with the GFP cDNA. A. Prior to PMA exposure the channel was mainly expressed on the surface of the HEK293 cells, suggesting membrane expression. B. Following application of 15 nM PMA for 25 minutes the channel maintained expression on the cell surface. ∧ Denotes GFP fluorescence imaging. * Denotes Kir5.1 antibody was used as the primary antibody. GFP was used as an indicator of transfection efficiency. Note that all HEK293 cells expressing GFP also expressed the Kir4.1-Kir5.1 channel although the GFP intensity was compromised by the fixation process. Bar=10 μm.
A. Using TEVC whole-cell Kir4.1-Kir5.1 currents were recorded from an oocyte 3 days post-injection of the Kir4.1-Kir5.1 dimer cDNA. With 90 mM K+ in the extracellular solution inward rectifying currents were recorded at baseline. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −160 mV to 140 mV with a 20-mV increment was applied to the cell. Exposure to 100 nM PMA strongly inhibited the Kir4.1-Kir5.1 currents. B. The same experiment was carried out using the 4α-PDD (an inactive analogue of PMA) instead of PMA. Exposure to 20 nM 4α-PDD failed to inhibit the Kir4.1-Kir5.1 channel. C, D. Shows the I-V curve relationships for the experiments carried out in A and B. E. The time profile shows that the Kir4.1-Kir5.1 current amplitude decreased rapidly when PMA was present in the bath solution at a high concentration (100 nM), and reached maximum inhibition in ∼25 min.
A. Using TEVC whole-cell currents were recorded from an oocyte 3 days post-injection of the Kir4.1-Kir5.1T174D mutant. With 90 mM K+ in the extracellular solution inward rectifying currents were recorded at baseline. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −160 mV to 140 mV with a 20-mV increment was applied to the cell. Exposure to 15 nM PMA failed to inhibit the T174D currents. B. The same experiment was carried out using 1μM PMA. Exposure to 1μM PMA also failed to inhibit the Kir4.1-Kir5.1T174D channel. C. Unlike the wild-type Kir4.1-Kir5.1 channel that is strongly inhibited by 15% CO2, the T174D currents were unaffected by such an exposure. D, E. Single channel recordings and analyses shows that the T174D mutant has a higher basal Popen than the wild-type Kir4.1-Kir5.1 although the conductances are the very similar (n≥5). F. Shows the I-V curve relationships for the experiments carried out in A, B and C. G. Bargraph showing the effects of 15nM PMA on the T174 mutants. Note that the T174D mutant had a much larger basal current and failed to be inhibited by PMA in comparison to the wild-type and the other T174 mutants (n≥5).
Fig 9. Schematic for the inhibition of Kir4.1-Kir5.1 channels by PKC.
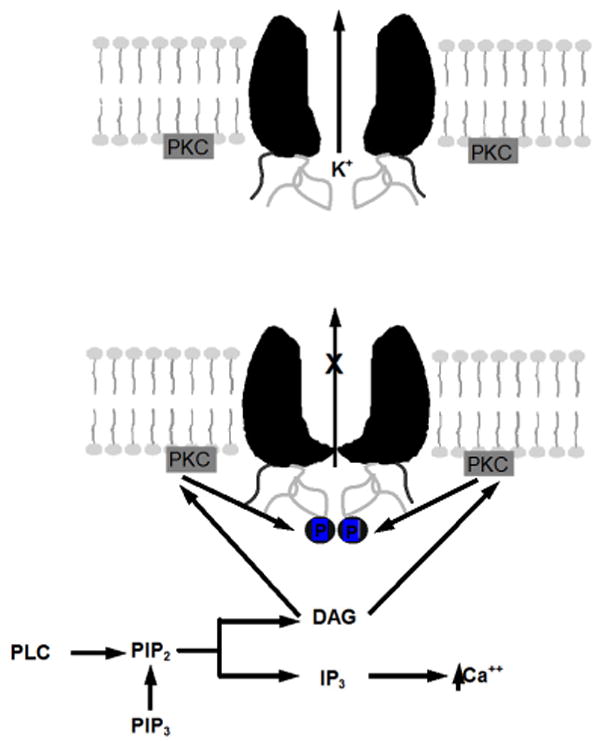
The inhibition of the Kir4.1-Kir5.1 channel is through the activation of PKC by way of DAG activation. The channel appears to be inhibited by PKC directly (phosphorylation) and not by a reduction of PIP2.
Acknowledgments
Special thanks to Ms. Dyanna Fountain and Ms. Carmen Adams for their technical assistance. We also thank Ms. Heena Tapankumar Dey and Mr. Malcolm E. Johns for their assistance with the in vitro phosphorylation experiments. We are grateful to Dr. John Adelman for the gifts of Kir4.1 and Kir5.1. We are also grateful to Dr. Mark McNiven for the gift of Dynamin II (aa) wild-type and K44A mutant. This work is supported by the NIH (HL067890).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Casamassima M, D'Adamo MC, Pessia M, Tucker SJ. Identification of a heteromeric interaction that influences the rectification, gating, and pH sensitivity of Kir4.1/Kir5.1 potassium channels. J Biol Chem. 2003;278:43533–43540. doi: 10.1074/jbc.M306596200. [DOI] [PubMed] [Google Scholar]
- 2.Konstas AA, Korbmacher C, Tucker SJ. Identification of domains that control the heteromeric assembly of Kir5.1/Kir4.0 potassium channels. Am J Physiol Cell Physiol. 2003;284:C910–C917. doi: 10.1152/ajpcell.00479.2002. [DOI] [PubMed] [Google Scholar]
- 3.Pessia M, Imbrici P, D'Adamo MC, Salvatore L, Tucker SJ. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol. 2001;532:359–367. doi: 10.1111/j.1469-7793.2001.0359f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanemoto M, Kittaka N, Inanobe A, Kurachi Y. In vivo formation of a proton-sensitive K+ channel by heteromeric subunit assembly of Kir5.1 with Kir4.1. J Physiol. 2000;525(Pt 3):587–592. doi: 10.1111/j.1469-7793.2000.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tucker SJ, Imbrici P, Salvatore L, D'Adamo MC, Pessia M. pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem. 2000;275:16404–16407. doi: 10.1074/jbc.C000127200. [DOI] [PubMed] [Google Scholar]
- 6.Xu H, Cui N, Yang Z, Qu Z, Jiang C. Modulation of kir4.1 and kir5.1 by hypercapnia and intracellular acidosis. J Physiol. 2000;524(Pt 3):725–735. doi: 10.1111/j.1469-7793.2000.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z, Xu H, Cui N, Qu Z, Chanchevalap S, Shen W, Jiang C. Biophysical and molecular mechanisms underlying the modulation of heteromeric Kir4.1-Kir5.1 channels by CO2 and pH. J Gen Physiol. 2000;116:33–45. doi: 10.1085/jgp.116.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui N, Giwa LR, Xu H, Rojas A, Abdulkadir L, Jiang C. Modulation of the heteromeric Kir4.1-Kir5.1 channels by P(CO(2)) at physiological levels. J Cell Physiol. 2001;189:229–236. doi: 10.1002/jcp.10021. [DOI] [PubMed] [Google Scholar]
- 9.Jiang C, Xu H, Cui N, Wu J. An alternative approach to the identification of respiratory central chemoreceptors in the brainstem. Respir Physiol. 2001;129:141–157. doi: 10.1016/s0034-5687(01)00301-2. [DOI] [PubMed] [Google Scholar]
- 10.Wu J, Xu H, Shen W, Jiang C. Expression and coexpression of CO2-sensitive Kir channels in brainstem neurons of rats. J Membr Biol. 2004;197:179–191. doi: 10.1007/s00232-004-0652-4. [DOI] [PubMed] [Google Scholar]
- 11.Tanemoto M, Abe T, Onogawa T, Ito S. PDZ binding motif-dependent localization of K+ channel on the basolateral side in distal tubules. Am J Physiol Renal Physiol. 2004;287:F1148–F1153. doi: 10.1152/ajprenal.00203.2004. [DOI] [PubMed] [Google Scholar]
- 12.Ishii M, Fujita A, Iwai K, Kusaka S, Higashi K, Inanobe A, Hibino H, Kurachi Y. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am J Physiol Cell Physiol. 2003;285:C260–C267. doi: 10.1152/ajpcell.00560.2002. [DOI] [PubMed] [Google Scholar]
- 13.Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Germann WJ, Stanfield CL. Principles of Human Physiology. Benjamin Cummings; San Francisco: 2005. The urinary system; fluid and electrolyte balance; pp. 618–631. [Google Scholar]
- 16.Richerson GB. Serotonergic neurons as carbon dioxide sensors that maintain pH homeostasis. Nat Rev Neurosci. 2004;5:449–461. doi: 10.1038/nrn1409. [DOI] [PubMed] [Google Scholar]
- 17.Fakler B, Brandle U, Glowatzki E, Zenner HP, Ruppersberg JP. Kir2.1 inward rectifier K+ channels are regulated independently by protein kinases and ATP hydrolysis. Neuron. 1994;13:1413–1420. doi: 10.1016/0896-6273(94)90426-x. [DOI] [PubMed] [Google Scholar]
- 18.Henry P, Pearson WL, Nichols CG. Protein kinase C inhibition of cloned inward rectifier (HRK1/KIR2.3) K+ channels expressed in Xenopus oocytes. J Physiol. 1996;495(Pt 3):681–688. doi: 10.1113/jphysiol.1996.sp021625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci U S A. 2000;97:9058–9063. doi: 10.1073/pnas.160068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin YF, Jan YN, Jan LY. Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. EMBO J. 2000;19:942–955. doi: 10.1093/emboj/19.5.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao J, Wang X, Chen F, Wang R, Rojas A, Shi Y, Piao H, Jiang C. Molecular basis for the inhibition of G protein-coupled inward rectifier K(+) channels by protein kinase C. Proc Natl Acad Sci U S A. 2004;101:1087–1092. doi: 10.1073/pnas.0304827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens EB, Shah BS, Pinnock RD, Lee K. Bombesin receptors inhibit G protein-coupled inwardly rectifying K+ channels expressed in Xenopus oocytes through a protein kinase C-dependent pathway. Mol Pharmacol. 1999;55:1020–1027. [PubMed] [Google Scholar]
- 23.Vorobiov D, Levin G, Lotan I, Dascal N. Agonist-independent inactivation and agonist-induced desensitization of the G protein-activated K+ channel (GIRK) in Xenopus oocytes. Pflugers Arch. 1998;436:56–68. doi: 10.1007/s004240050604. [DOI] [PubMed] [Google Scholar]
- 24.Zhang L, Lee JK, John SA, Uozumi N, Kodama I. Mechanosensitivity of GIRK channels is mediated by protein kinase C-dependent channel-phosphatidylinositol 4,5-bisphosphate interaction. J Biol Chem. 2004;279:7037–7047. doi: 10.1074/jbc.M307323200. [DOI] [PubMed] [Google Scholar]
- 25.Zhu G, Qu Z, Cui N, Jiang C. Suppression of Kir2.3 activity by protein kinase C phosphorylation of the channel protein at threonine 53. J Biol Chem. 1999;274:11643–11646. doi: 10.1074/jbc.274.17.11643. [DOI] [PubMed] [Google Scholar]
- 26.Zitron E, Kiesecker C, Luck S, Kathofer S, Thomas D, Kreye VA, Kiehn J, Katus HA, Schoels W, Karle CA. Human cardiac inwardly rectifying current Kir2.2 is upregulated by activation of protein kinase A. Cardiovasc Res. 2004;63:520–527. doi: 10.1016/j.cardiores.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Rojas A, Wu J, Jiang C. Disruption of glucose sensing and insulin secretion by ribozyme Kir6.2-gene targeting in insulin-secreting cells. Endocrinology. 2004;145:4408–4414. doi: 10.1210/en.2004-0285. [DOI] [PubMed] [Google Scholar]
- 28.Wu J, Cui N, Piao H, Wang Y, Xu H, Mao J, Jiang C. Allosteric modulation of the mouse Kir6.2 channel by intracellular H+ and ATP. J Physiol. 2002;543:495–504. doi: 10.1113/jphysiol.2002.025247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu G, Zhang Y, Xu H, Jiang C. Identification of endogenous outward currents in the human embryonic kidney (HEK 293) cell line. J Neurosci Methods. 1998;81:73–83. doi: 10.1016/s0165-0270(98)00019-3. [DOI] [PubMed] [Google Scholar]
- 30.Wischmeyer E, Karschin A. Receptor stimulation causes slow inhibition of IRK1 inwardly rectifying K+ channels by direct protein kinase A-mediated phosphorylation. Proc Natl Acad Sci U S A. 1996;93:5819–5823. doi: 10.1073/pnas.93.12.5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of kir channels by diverse modulators. J Biol Chem. 2004;279:37271–37281. doi: 10.1074/jbc.M403413200. [DOI] [PubMed] [Google Scholar]
- 32.Lopes CM, Zhang H, Rohacs T, Jin T, Yang J, Logothetis DE. Alterations in conserved Kir channel-PIP2 interactions underlie channelopathies. Neuron. 2002;34:933–944. doi: 10.1016/s0896-6273(02)00725-0. [DOI] [PubMed] [Google Scholar]
- 33.Lin D, Sterling H, Lerea KM, Giebisch G, Wang WH. Protein kinase C (PKC)-induced phosphorylation of ROMK1 is essential for the surface expression of ROMK1 channels. J Biol Chem. 2002;277:44278–44284. doi: 10.1074/jbc.M203702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sterling H, Lin DH, Gu RM, Dong K, Hebert SC, Wang WH. Inhibition of protein-tyrosine phosphatase stimulates the dynamin-dependent endocytosis of ROMK1. J Biol Chem. 2002;277:4317–4323. doi: 10.1074/jbc.M109739200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang WH, Lin DH, Sterling H. Regulation of ROMK channels by protein tyrosine kinase and tyrosine phosphatase. Trends Cardiovasc Med. 2002;12:138–142. doi: 10.1016/s1050-1738(02)00155-x. [DOI] [PubMed] [Google Scholar]
- 36.Wang WH. Regulation of ROMK (Kir1.1) channels: new mechanisms and aspects. Am J Physiol Renal Physiol. 2006;290:F14–F19. doi: 10.1152/ajprenal.00093.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeng WZ, Babich V, Ortega B, Quigley R, White SJ, Welling PA, Huang CL. Evidence for endocytosis of ROMK potassium channel via clathrin-coated vesicles. Am J Physiol Renal Physiol. 2002;283:F630–F639. doi: 10.1152/ajprenal.00378.2001. [DOI] [PubMed] [Google Scholar]
- 38.Beguin P, Nagashima K, Nishimura M, Gonoi T, Seino S. PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 1999;18:4722–4732. doi: 10.1093/emboj/18.17.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hibino H, Horio Y, Fujita A, Inanobe A, Doi K, Gotow T, Uchiyama Y, Kubo T, Kurachi Y. Expression of an inwardly rectifying K(+) channel, Kir4.1, in satellite cells of rat cochlear ganglia. Am J Physiol. 1999;277:C638–C644. doi: 10.1152/ajpcell.1999.277.4.C638. [DOI] [PubMed] [Google Scholar]
- 40.Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. 2004;279:44065–44073. doi: 10.1074/jbc.M405985200. [DOI] [PubMed] [Google Scholar]
- 41.Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K(+) channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- 42.Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, Tomoike H, Kurachi Y. Immunolocalization of an inwardly rectifying K+ channel, K(AB)-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett. 1996;388:11–15. doi: 10.1016/0014-5793(96)00502-9. [DOI] [PubMed] [Google Scholar]
- 43.Kusaka S, Horio Y, Fujita A, Matsushita K, Inanobe A, Gotow T, Uchiyama Y, Tano Y, Kurachi Y. Expression and polarized distribution of an inwardly rectifying K+ channel, Kir4.1, in rat retinal pigment epithelium. J Physiol. 1999;520(Pt 2):373–381. doi: 10.1111/j.1469-7793.1999.00373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu H, Yang Z, Cui N, Giwa LR, Abdulkadir L, Patel M, Sharma P, Shan G, Shen W, Jiang C. Molecular determinants for the distinct pH sensitivity of Kir1.1 and Kir4.1 channels. Am J Physiol Cell Physiol. 2000;279:C1464–C1471. doi: 10.1152/ajpcell.2000.279.5.C1464. [DOI] [PubMed] [Google Scholar]
- 45.Xu H, Yang Z, Cui N, Chanchevalap S, Valesky WW, Jiang C. A single residue contributes to the difference between Kir4.1 and Kir1.1 channels in pH sensitivity, rectification and single channel conductance. J Physiol. 2000;528(Pt 2):267–277. doi: 10.1111/j.1469-7793.2000.00267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Whole-cell currents were recorded from HEK293 cells 3 days post-transfection of the Kir4.1-Kir5.1 cDNA. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −120 mV to 80 mV was applied to the cell and the corresponding currents were recorded. Following stabilization 100 nM thymeleatoxin was applied to the bath solution and inhibited the Kir4.1-Kir5.1 currents by 51.4±2.6% (n=4). Application of DMSO alone barely inhibited the Kir4.1-Kir5.1 currents. During the experiments with thymeleatoxin and DMSO, the whole cell capacitance and series resistance were monitored. Neither capacitance nor series resistance changed significantly upon application of 100 nM thymeleatoxin or DMSO to the bath solution.
Immunocytochemistry was performed on HEK293 cells 48 hours post-transfection of the Kir4.1-Kir5.1 dimer cDNA together with the GFP cDNA. A. Prior to PMA exposure the channel was mainly expressed on the surface of the HEK293 cells, suggesting membrane expression. B. Following application of 15 nM PMA for 25 minutes the channel maintained expression on the cell surface. ∧ Denotes GFP fluorescence imaging. * Denotes Kir5.1 antibody was used as the primary antibody. GFP was used as an indicator of transfection efficiency. Note that all HEK293 cells expressing GFP also expressed the Kir4.1-Kir5.1 channel although the GFP intensity was compromised by the fixation process. Bar=10 μm.
A. Using TEVC whole-cell Kir4.1-Kir5.1 currents were recorded from an oocyte 3 days post-injection of the Kir4.1-Kir5.1 dimer cDNA. With 90 mM K+ in the extracellular solution inward rectifying currents were recorded at baseline. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −160 mV to 140 mV with a 20-mV increment was applied to the cell. Exposure to 100 nM PMA strongly inhibited the Kir4.1-Kir5.1 currents. B. The same experiment was carried out using the 4α-PDD (an inactive analogue of PMA) instead of PMA. Exposure to 20 nM 4α-PDD failed to inhibit the Kir4.1-Kir5.1 channel. C, D. Shows the I-V curve relationships for the experiments carried out in A and B. E. The time profile shows that the Kir4.1-Kir5.1 current amplitude decreased rapidly when PMA was present in the bath solution at a high concentration (100 nM), and reached maximum inhibition in ∼25 min.
A. Using TEVC whole-cell currents were recorded from an oocyte 3 days post-injection of the Kir4.1-Kir5.1T174D mutant. With 90 mM K+ in the extracellular solution inward rectifying currents were recorded at baseline. Membrane potential (Vm) was held at 0 mV. A series of command pulse potentials from −160 mV to 140 mV with a 20-mV increment was applied to the cell. Exposure to 15 nM PMA failed to inhibit the T174D currents. B. The same experiment was carried out using 1μM PMA. Exposure to 1μM PMA also failed to inhibit the Kir4.1-Kir5.1T174D channel. C. Unlike the wild-type Kir4.1-Kir5.1 channel that is strongly inhibited by 15% CO2, the T174D currents were unaffected by such an exposure. D, E. Single channel recordings and analyses shows that the T174D mutant has a higher basal Popen than the wild-type Kir4.1-Kir5.1 although the conductances are the very similar (n≥5). F. Shows the I-V curve relationships for the experiments carried out in A, B and C. G. Bargraph showing the effects of 15nM PMA on the T174 mutants. Note that the T174D mutant had a much larger basal current and failed to be inhibited by PMA in comparison to the wild-type and the other T174 mutants (n≥5).