

Abstract
Tumor cells gene-modified to produce GM-CSF potently stimulate antitumor immune responses, in part, by causing the growth and differentiation of dendritic cells (DC). However, GM-CSF-modified tumor cells must be γ-irradiated or they will grow progressively, killing the host. We observed that 23 of 75 (31%) human tumor lines and two commonly used mouse tumor lines spontaneously produced GM-CSF. In mice, chronic GM-CSF production by tumors suppressed Ag-specific CD8+ T cell responses. Interestingly, an inhibitory population of adherent CD11b(Mac-1)/Gr-1 double-positive cells caused the observed impairment of CD8+ T cell function upon direct cell-to-cell contact. The inhibitory cells were positive for some markers associated with Ag presenting cells, like F4/80, but were negative for markers associated with fully mature DC like DEC205, B7.2, and MHC class II. We have previously reported that a similar or identical population of inhibitory “immature” APC was elicited after immunization with powerful recombinant immunogens. We show here that these inhibitory cells can be elicited by the administration of recombinant GM-CSF alone, and, furthermore, that they can be differentiated ex vivo into “mature” APC by the addition of IL-4 and GM-CSF. Thus, tumors may be able to escape from immune detection by producing “unopposed” GM-CSF, thereby disrupting the balance of cytokines needed for the maturation of fully functional DC. Further, CD11b/Gr-1 double-positive cells may function as “inhibitory” APC under the influence of GM-CSF alone.
Every tumor that results in the death of its host is a tumor that was not destroyed by the immune system. Workers once speculated that tumors were not recognized by immune cells because they failed to express “recognizable” Ags, but the molecular identification of tumor-associated Ags (TAA)3 has largely disproved this hypothesis. Tumor cells indeed can express TAA that are specifically recognized by immune cells, and under some circumstances that have not yet been fully elucidated, the immune system can mediate the destruction of tumor cells in both mouse and man (1–8).
It now seems likely that tumors are not destroyed by immune cells because the tumor microenvironment is a poor site for the initiation of immune responses. Tumor cells generally do not express the requisite costimulatory and cytokine molecules that signal an inflammatory state. In this regard, tumors are like most normal tissues in the body. Activation of the cellular immune response appears to be largely effected by “professional” APC, including dendritic cells (DC) (9–12). Professional APC originate from granulocyte-monocyte lineage progenitors that have the ability to capture Ag but lack the requisite accessory signals for T cell activation (13, 14). Under the influence of cytokines, such as GM-CSF, IL-4, and TNF-α, progenitor cells undergo terminal differentiation to professional APC capable of presenting Ags in the context of immunostimulatory molecules that result in the activation T cell reactivities (9, 13, 15).
Local production of GM-CSF by γ-irradiated cDNA-transduced tumors is thought to recruit and expand professional APCs that present Ags released by apoptotic tumor cells (16–18). In the B16 mouse model, the presentation of tumor Ags by professional APCs to naive lymphocytes can result in memory T cell responses that prevent subsequent challenge with the wild-type tumor (18). Gene modification of tumor cells with GM-CSF in the absence of γ-irradiation, however, does not generally change the growth rate or lethality of tumor cells in their nonirradiated state. Thus, the induction of apoptosis by γ-irradiation in GM-CSF gene-modified tumor cells might be required to enhance the processing and presentation of TAA by DC (19, 20). Indeed, the production of GM-CSF in the absence of an apoptotic “danger signal” could have very different effects on DC recruitment and maturation. It is perplexing that some murine tumor cells spontaneously produce GM-CSF (21). Furthermore, the production of GM-CSF has been correlated with the ability of mouse tumor cells to spontaneously metastasize (22). Human prostate cancers (23) and human melanoma cells (our unpublished data) have been found to spontaneously produce GM-CSF.
In the present article, we attempt to delineate the role of GM-CSF production by tumors on the recruitment and maturation of APC. We studied a panel of human tumors for the production of GM-CSF and found that 31% of human tumors spontaneously secreted this cytokine. To characterize the immunological significance of this finding, GM-CSF-producing and -nonproducing murine tumors were grown in vivo. The production of GM-CSF by tumors was found to induce a population of cells that morphologically resembled granulocyte-monocyte progenitor cells and phenotypically expressed the granulocyte-monocyte markers C11b (Mac-1) and Gr-1 (Ly-6G). We have previously reported that these cells can cause apoptotic death of CD8+ T cells after immunization with powerful vaccinia virus-based immunogens. (24). We show here that these CD11b+/Gr-1+ cells could mature in vitro under the influence of GM-CSF and IL-4 to produce functional APCs that are capable of enhancing T cell responses in a mixed lymphocyte reaction. We explore the hypothesis that tumors may evade immune destruction by dysregulating the maturation of APC through the unopposed secretion of GM-CSF.
Materials and Methods
Tumor cell lines and GM-CSF determination
CT26.WT, the β-galactosidase (β-gal)-expressing CT26.CL25 clone, EL4 thymoma, and its lacZ transfectants E22 have been described (25, 26). The mouse mammary adenocarcinoma tumor, TS/A, was kindly provided by Dr. Guido Forni (University of Turin, Orbassano, Italy). MBL-2 and LSTRA are Moloney virus-induced lymphomas. Cell lines were maintained in culture media (CM) consisting of RPMI 1640, 10% heat-inactivated FBS (Biofluids, Rockville, MD), 0.03% L-glutamine, 100 μg/ml streptomycin, 100 μg/ml penicillin, and 50 μg/ml gentamicin sulfate (National Institutes of Health Media Center, Bethesda, MD). The following human tumor cell lines were obtained from American Type Culture Collection (Manassas, VA): BT474, MCF-7, SKOV3, MB 361, BT549, MDA 231, MS 751, SIHA, CASKI, IGROV-1, OVCAR-3, OV-1, WIDR, H508, COLO 205, SW480, LOVO, and SK23. The following human cells were provided by Suzanne Topalian (National Cancer Institute (NCI; Bethesda, MD): LNCaP, DU 145, PC-3, and 1542 CP3TX. The following human cell lines were generated in the Surgery Branch, NCI: 1312, 1338, 501A, 1143, 537, 1353, 553B, 677 II, 1173, 1402, 1290−2, 1199, 888, 1287, 1011, 624, 1359, 1363, 1495, 1280−1, 697, 1362, 526, 1308, 1335, 586, 1182, 1479, 1102, 836, 938, 1379, 624, 883, 1286, 397, 1123, 1390, 1448 Y, and 1195. The following human cell lines were generated in the Urologic Oncology Branch, NCI: Ce-RCC, M-RCC, Gr-RCC, R-RCC, Ca-RCC, Gi-RCC, N-RCC, P-RCC, Ba-RCC, Bu-RCC, and W-RCC. Cells were plated in duplicate in 96-well plates (Costar, Cambridge, MA) at a concentration of 105 cells/well in 250 μl of CM. Values are expressed as pg/ml produced in 16 h by 105 cells, unless otherwise stated. Genetic modification of B16 tumor cells to secrete GM-CSF was performed as follows. The E86 packaging cell line carrying the LmGM-CSFSN retroviral vector expressing the mouse GM-CSF was a gift of Mario Colombo (Istituto Nazionale Tumori, Milan, Italy). B16 melanoma was trans-infected with the supernatant of the packaging cell line in the presence of Polybrene (8 μg/ml) and then subcloned. Stable transfectants were screened for cytokine production after 2 wk of continuous in vitro passages in the presence of G418 (0.2 mg/ml). The subclones of the B16 tumor engineered with GM-CSF were plated at 106 cells in 1 ml of CM. ELISA kits for human IFN-γ and mouse GM-CSF were purchased from Endogen (Cambridge, MA), and kits for human GM-CSF were purchased from R&D Systems (Minneapolis, MN). All values refer to duplicate samples.
Peptides, Abs, and cytokines
The following synthetic peptides were synthesized by Peptide Technologies (Washington, DC) to a purity of >99% as determined by HPLC and amino acid analyses: TPHPARIGL (amino acids 876−884 of β-gal, H-2Ld-restricted (27)), DAPIYTNV (amino acids 96−103 of β-gal, H-2Kb-restricted (28)). Rat mAb recognizing NLDC-145 (DEC-205) and F4/80 were purchased from BMA Biomedicals (Augst, Switzerland). FITC- or PE-labeled mAb recognizing mouse CD3, CD8, CD4, CD11b (Mac-1), CD45R/B220, CD11c, CD80 (B7−1), CD86 (B7−2), Ly-6G (Gr-1), anti-H-2 I-Ed/I-Ad, Kd, and the isotype-matched controls were purchased from PharMingen (San Diego, CA). The mAb 24G.2 (CD16/CD23; PharMingen), which recognizes the extracellular domain of the mouse Fcγ-RIII and RII, was used to block the nonspecific mAb binding. Recombinant mouse GM-CSF and IL-4 were purchased from PreproTech (Rocky Hill, NJ) and resuspended in HBSS containing 1% mouse serum (Sigma, St. Louis, MO).
Isolation and depletion of Gr-1+ splenocytes
A panning technique employing flasks coated with mouse anti-rat Abs (T-25 AIS MicroCell; Applied Immune Sciences, Santa Clara, CA) was used to enrich or eliminate Gr-1+ splenocytes from either tumor-bearing mice or mice treated with recombinant GM-CSF. Briefly, spleens were depleted of RBC through ACK lysis buffer (Biofluids) and resuspended in HBSS containing 1 mM EDTA and 10% mouse serum (HBSS-EDTA-MS). Cells were transferred on ice, incubated for 30 min with anti-Gr-1 Ab at a concentration of 10 μg/107 cells, and washed with cold HBSS-EDTA. Cells were resuspended in ice-cold HBSS-EDTA-MS, transferred to flasks coated with the secondary Ab (anti-rat), and incubated for 1 h at 4°C. The nonadherent cells were then dislodged, and fluorescence labeling confirmed a >95% depletion. Cells that remained attached to the flasks were provided with CM and incubated for further studies. Adherent cells were usually recovered by gentle scraping in Versene solution (1:5000; Biofluids).
Maturation of CD11b+/Gr-1+ cells in vitro
Gr-1+ splenocytes were isolated as described above. Isolated cells were grown for 6 days in culture with 100 ng/ml GM-CSF, 100 ng/ml IL-4, a combination of both GM-CSF and IL-4, or in media alone. After 6 days of culture, the adherent cells were analyzed for phenotypic markers or added to a 6-day mixed lymphocyte stimulation. In the mixed lymphocyte reaction, BALB/c splenocytes (H-2d) at 3 × 106 were incubated with 3 × 106 irradiated C57BL/6n splenocytes (H-2b) in the presence or absence of 0.18 × 106 Gr-1+ splenocytes (H-2d). After 6 days of stimulation, the BALB/c lymphocytes were tested in a standard chromium release assay against syngeneic (CT26 or LSTRA) or allogeneic (MBL-2) targets.
Enrichment of spleen-derived DC
Spleens were collected aseptically, minced, and incubated for 30 min at room temperature in HBSS containing collagenase (1 mg/ml; Sigma). Spleens were depleted of RBC through ACK lysis buffer and washed. The single cell suspension was resuspended in CM and plated in a 150 × 25-mm tissue culture dish (Falcon, Cockeysville, MD). Splenocytes were incubated for 2 h at 37°C, and the nonadherent cells were removed by gentle washing. Adherent cells were further incubated for 18 h in CM containing 3 ng/ml mouse GM-CSF. At the end of the incubation, the supernatant was collected, and DC were finally enriched by centrifugation (1900 × g) over a 45% Percoll cushion. At this point, >80% of the cells were B7−2+, B220−, class II MHC +, CD11c+.
Evaluation of CD8+ T cell responses
Eight- to twelve-week-old female BALB/c or C57BL/6 mice (Animal Production Colonies, Frederick Cancer Research Facility, National Institutes of Health, Frederick, MD) were immunized with 5 × 106 PFU/mouse of the construct VJS6, a recombinant vaccinia virus (rVV) expressing β-gal (β-gal-rVV) (25). Three weeks after immunization, two to three mice per group were injected s.c. with 0.1 ml of HBSS containing 106 TS/A tumor cells. Two to five weeks after tumor challenge, spleens were collected, separated into a single cell suspension, and cultured in CM containing 1 mg/ml of the immunodominant β-gal peptides (27, 28). After 6 days, cultures were tested for their ability to lyse β-gal-positive targets in a 6-h 51Cr release assay using target cells previously incubated with 200 μCi Na51CrO4 (51Cr) for 90 min (together with 1 μg/ml of peptide) (25).The amount of 51Cr released was determined by γ-counting, and the percentage of specific lysis was calculated from triplicate samples using the formula: [(experimental cpm – spontaneous cpm)/(maximal cpm – spontaneous cpm)] × 100. In some experiments, mice received a 3-day cycle of GM-CSF (5 μg/mouse, i.p. twice daily) 3 wk following immunization with β-gal rVV.
The calculation of LU was performed because it conveys the activity of the culture more effectively than simply showing a single E:T ratio. Calculations of LU per 106 effector cells were performed as follows: divide 106 by the number of cells giving 30% lysis of specific target cells (we used 2 × 103 β-gal peptide-pulsed EL4 cells). There was no lysis of unpulsed target cells in this assay.
In cell separation experiments, splenocytes were cultured at the same cell concentration in 24-well plates (Costar) containing a culture chamber insert with 0.4-μm pores (Millipore, Bedford, MA).
Results
Human tumor cell lines spontaneously produce GM-CSF
GM-CSF secretion by tumor lines has been sporadically described in the literature (29, 30), but to our knowledge, the prevalence of GM-CSF production by tumors has not been systematically studied. Thus, we evaluated a panel of 75 human tumor cell lines (Table I) and found that 23 produced levels of GM-CSF higher than the background (>10 pg/ml/105 cells), especially those lines originating from kidney and prostate cancers. Moreover, IFN-γ release during the same assay was not above the detection limit (12 pg/ml), indicating the specific release of GM-CSF. Thus, our finding that human tumors spontaneously produce GM-CSF is consistent with previous reports.
Table I.
Production of GM-CSF and IFN-γ by human tumor cell linesa
| Tumor Cell Line | GM-CSF | IFN-γ | Tumor Cell Line | GM-CSF | IFN-γ |
|---|---|---|---|---|---|
| Breast cancer | Melanoma | ||||
| BT474 | 0 | 0 | 1312 | 0 | 0 |
| MCF-7 | 0 | 0 | 1338 | 0 | 0 |
| SKOV3 | 0 | 0 | 501A | 0 | 0 |
| MB361 | 4 | 0 | 1143 | 0 | 0 |
| BT549 | 5 | 0 | 537 | 0 | 0 |
| MDA231 | 668 | 0 | 1353 | 0 | 0 |
| 553B | 0 | 0 | |||
| Cervical cancer | 677II | 0 | 0 | ||
| MS751 | 0 | 0 | 1173 | 0 | 0 |
| SIHA | 16 | 0 | 1402 | 0 | 0 |
| CASKI | 488 | 12 | 1290−2 | 0 | 0 |
| 1199 | 0 | 0 | |||
| Ovarian cancer | 888 | 0 | 0 | ||
| IGROV-1 | 0 | 0 | 1287 | 0 | 0 |
| OVCAR-3 | 0 | 0 | 1011 | 0 | 0 |
| OV-1 | 17 | 0 | 624 | 0 | 0 |
| 1359 | 0 | 0 | |||
| Prostate cancer | 1363 | 0 | 0 | ||
| LNCaP | 0 | 0 | 1495 | 0 | 0 |
| Du 145 | 45 | 0 | 1280−1 | 0 | 0 |
| PC 3 270.4 | 270 | 0 | 697 | 0 | 0 |
| 1542 CP3 | 1,035 | 0 | 1362 | 0 | 0 |
| Tx | |||||
| 526 | 0 | 0 | |||
| Colon cancer | 1308 | 0 | 0 | ||
| WIDR | 0 | 0 | 1335 | 0 | 0 |
| H508 | 3 | 0 | 586 | 0 | 0 |
| COLO 205 | 7 | 0 | 1182 | 0 | 0 |
| SW480 | 34 | 16 | 1479 | 0 | 0 |
| LOVO | 36 | 0 | 1102 | 0 | 0 |
| 836 | 0 | 0 | |||
| Renal cancer | 938 | 0 | 0 | ||
| Ce-RCC | 0 | 0 | 1379 | 0 | 0 |
| M-RCC | 25 | 0 | SK23 | 0 | 0 |
| Gr-RCC | 29 | 0 | 624 | 0 | 0 |
| R-RCC | 98 | 0 | 883 | 0 | 0 |
| Ca-RCC | 125 | 0 | 1286 | 0 | 0 |
| Gi-RCC | 675 | 0 | 397 | 4 | 0 |
| N-RCC | 1,125 | 120 | 1123 | 30 | 0 |
| P-RCC | 1,134 | 0 | 1390 | 43 | 0 |
| Ba-RCC | 1,723 | 0 | 1448Y | 277 | 0 |
| Bu-RCC | 1,707 | 0 | 1195 | 280 | 0 |
| W-RCC | 39,390 | 0 |
Values are expressed as picograms/ml produced in 16 h by 105 cells. Values for culture media alone were <12 pg/ml for IFN-γ and 2 pg/ml for GM-CSF. All values were obtained from duplicate samples.
GM-CSF-producing tumors induce CD11b+/Gr-1+ splenocytes that inhibit CD8+ T cells
To test the immune consequences of spontaneous GM-CSF production by tumors on the maturation of APC, two experimental tumor lines were grown in vivo. The colon adenocarcinoma, CT26.WT, and the mammary adenocarcinoma, TS/A, spontaneously produced significant levels of GM-CSF (average 50 and 250 pg/ml/106 cells/day, respectively). Mice bearing unmanipulated s.c. TS/A tumors ∼1 cm in diameter were splenectomized. A representative cytofluorometric analysis of spleen cells (shown in Fig. 1A) demonstrates an increase in the numbers of CD11b+/Gr-1+ cells. These cells comprised a mean of 22.3 ± 1.83% of all splenocytes in five independent experiments, compared with an average of 2.4 ± 0.43% in mice with a small but palpable tumor. Evaluation of splenocytes from tumor-bearing mice did not reveal substantial changes in the percentages of lymphocytes with T or B cell markers (31).
FIGURE 1.

Tumor growth results in the induction of CD11b+/Gr-1+ cells that inhibit CD8+ T cells. A, Spleens of tumor-bearing mice contain large numbers of the CD11b+/Gr-1+ cells. Freshly harvested splenocytes were stained with FITC anti-Gr-1 and PE anti-CD11b mAbs. Only mice bearing palpable tumors 2−3 mm in diameter (left) or tumors with diameters >1 cm (right) are shown, as results in non-tumor-bearing mice and mice bearing a small but palpable tumor were identical. B, Inhibition of the anamnestic response in tumor-bearing mice is abrogated by the depletion of Gr-1+ cells. Three weeks after immunization with β-gal-rVV, BALB/c mice were injected s.c. with TS/A tumor cells. Duration of the tumor-bearing state was 35 days for mice with tumors >1 cm in diameter, and 15 days for tumors that were ∼2−3 mm in diameter (open triangles). Splenocytes were collected, pooled, then cultured with β-gal peptide. Splenocytes originating from mice bearing tumors >1 cm2 were depleted with anti-Gr-1 (filled circles) or with a control, IgG2b, Ab (open squares) before the culture was established. Cytotoxicity shown was measured in a single assay after 6 days of incubation with the target peptide. Target cells were CT26.WT (H-2d), CT26.WT pulsed with the Ld-restricted β-gal peptide (CT26. WT+pep), the lacZ-transfected, β-gal-expressing clone CT26.CL25, and the negative control E22 (lacZ-transfected β-gal-expressing EL4 cells, H-2b haplotype). E:T cell ratios were 100:1, then 1:3 dilutions. No difference in β-gal-specific cytolytic activity between immune mice bearing a small tumor (control) vs normal immune mice was observed (not shown). These findings were obtained reproducibly in five independently performed experiments.
We sought to explore whether or not the growth of a GM-CSF-producing tumor had any impact on CD8+ T cell responses. Therefore, we immunized mice with an rVV encoding lacZ under the control of a strong promoter (25). This immunization has been previously shown to induce strong CD8+ T cell responses to the model Ag, β-gal (β-gal rVV). Mice were then allowed to develop a “memory” immune response. After 3 wk, mice were randomized to receive TS/A tumor cells that were allowed to grow s.c. for either 35 days, which resulted in tumor nodules >1 cm2, or for only 15 days, which resulted in small but palpable tumor nodules. Mice with small but palpable tumors mounted a vigorous Ag-specific immune response to targets expressing the Ld-restricted, β-gal epitope (Fig. 1B, control). However, mice bearing large tumors failed to mount an anamnestic, β-gal-specific CD8+ T cell response.
To determine whether or not the Gr-1+ population that was so prevalent in mice bearing large tumors was responsible for the observed inhibition of Ag-specific CD8+ T cell responses, a fraction of the splenocytes from mice bearing large tumors was depleted with a Gr-1-specific Ab, or an isotype-controlled (IgG2b) Ab before restimulation. The simple depletion of Gr-1+ cells normalized CD8+ T cell function. Indeed, the lytic levels observed were nearly as great as those seen in β-gal-immune mice bearing small, but palpable, tumors (Fig. 1B, compare anti-Gr-1 and control). Isotype-matched mAb did not normalize T cell function (Fig. 1B, IgG2b). Experiments with the CT26 tumor cell line, which also naturally produces GM-CSF, yielded similar results (data not shown). Our results with mice bearing CT26 are consistent with those previously published by G. Nabel and colleagues (32) in which mice bearing this tumor were found to have large numbers of CD11b+ cells in their spleens and inhibition of allospecific responses. Therefore, the newly induced population of CD11b+/Gr-1+ cells appeared to be responsible for the abrogation of CD8+ T cell function.
We have previously shown that purified CD8+ T lymphocytes obtained from splenic preparations of mice with high levels of CD11b (Mac-1)+/Gr-1+ suppressor cells are normally responsive (24). Thus, the inhibition observed is not a quality of the CD8+ T cells, but is solely a quality of the CD11b/Gr-1 double-positive population.
GM-CSF, not the tumor-bearing state, drives the production of CD11b+/Gr-1+ cells
To determine whether or not the CD11b+/Gr-1+ cells were induced by GM-CSF or some other stimulus from the tumor, the murine melanoma, B16, which does not normally produce GM-CSF, was retrovirally transduced with the cDNA for mouse GM-CSF. Immediately following transduction, transduced B16 were cloned. To minimize clonotypic variability, several clones were isolated that released comparable amounts of the cytokine (600−900 pg/ml; Table II).
Table II.
Tumor cells engineered to secrete GM-CSF suppress anamnestic CTL response
| Tumor | Tumor Size (mm)b | GM-CSF in vitro (pg/ml)a | GM-CSF in Vivo (pg/ml)b | CD11b+ Splenocytesb | Anti-β-Gal Response (LU30/106 Cells)c |
|---|---|---|---|---|---|
| No tumor | Negative | — | ND | 1.8 | 213 |
| B16 wt | 17 × 16 | 0 | 0 | 3.0 | 196 |
| GM42 | 17 × 16 | 671 | 84.5 | 12.2 | <10 |
| GM32 | 13 × 13 | 719 | 301.2 | 12.0 | 15 |
| GM15 | 17 × 14 | 894 | 800.7 | 17.4 | <10 |
| GM19 | 11 × 10 | 868 | 115.2 | 9.3 | <10 |
| GM37 | 17 × 13 | 690 | 158.5 | 13.1 | <10 |
| GM34 | 10 × 10 | 877 | 146.0 | 7.5 | 14 |
| GM12 | 18 × 12 | 702 | 11.7 | 8.3 | 10 |
| GM47 | 18 × 15 | 626 | 172.1 | 11.2 | 10 |
Levels of GM-CSF released by 106 tumor cells after 18 h were evaluated on aliquots of the same cellular preparation injected s.c. to establish the tumor nodules.
Major tumor diameters were measured in a blind fashion immediately before the mice were sacrificed to remove the spleens and set up the peptide-stimulated cultures. On the same day, blood samples were drawn from each mouse to determine the serum levels of GM-CSF (GM-CSF in vivo), and the spleens were analyzed for the presence of CD11b+ splenocytes.
LU30 defined as the number of lymphocytes necessary to achieve 30% lysis of 2 × 103 β-gal peptide-pulsed target cells in a 4-h assay, were calculated from recovered viable cells.
Mice were vaccinated with β-gal rVV to establish β-gal-specific memory T cells (33, 34). Three weeks later, they were inoculated s.c. with either parental B16 (B16 wt) or one of the GM-CSF-producing clones. Tumors were allowed to grow for 14 days. Interestingly, no significant difference in the growth rate was observed between any of the GM-CSF-producing clones and the parental tumor, indicating that the levels of GM-CSF secretion were not sufficient to cause tumor rejection (Table II). Two weeks after tumor inoculation, we were able to detect increased serum levels of GM-CSF in mice bearing gene-modified tumors but not in mice bearing parental tumor, indicating that cytokine production had not been silenced in vivo (Table II). As seen with CT26 and TS/A, cytofluorometric analysis of spleens from mice bearing GM-CSF-transduced tumors demonstrated an increase in the percentage of the CD11b+ cells (Table II).
The lytic activity against β-gal after in vitro stimulation of the spleens with the Kb-restricted β-gal peptide in control mice were similar to those found in mice bearing the B16 wild-type tumor (B16 wt). In sharp contrast, growth of all the subclones producing GM-CSF (GM15−47) resulted in the inhibition of the anamnestic CD8+ T cell responses, as indicated by the reduction of the lytic units in the peptide-stimulated cultures. These data suggest that GM-CSF production by tumors cells can inhibit CD8+ T cell responses.
Recombinant GM-CSF administered alone induces CD11b+/Gr-1+ cells and inhibits CD8+ T cell function
To further test the hypothesis that GM-CSF was directly responsible for the increase in CD11b+/Gr-1+ cells, a 3-day cycle of recombinant mouse GM-CSF was administered twice daily to β-gal immune mice and naive controls. The administration of GM-CSF caused an increase in CD11b+/Gr-1+ cells in the spleens of treated mice (Fig. 2A) and inhibited the immune responses observed after stimulation with β-gal peptide (Fig. 2, B and C). These data demonstrated that a exposure to GM-CSF was sufficient to induce both the appearance of a distinct population of CD11b+/Gr-1+ cells and to induce CD8+ T cell inhibition.
FIGURE 2.
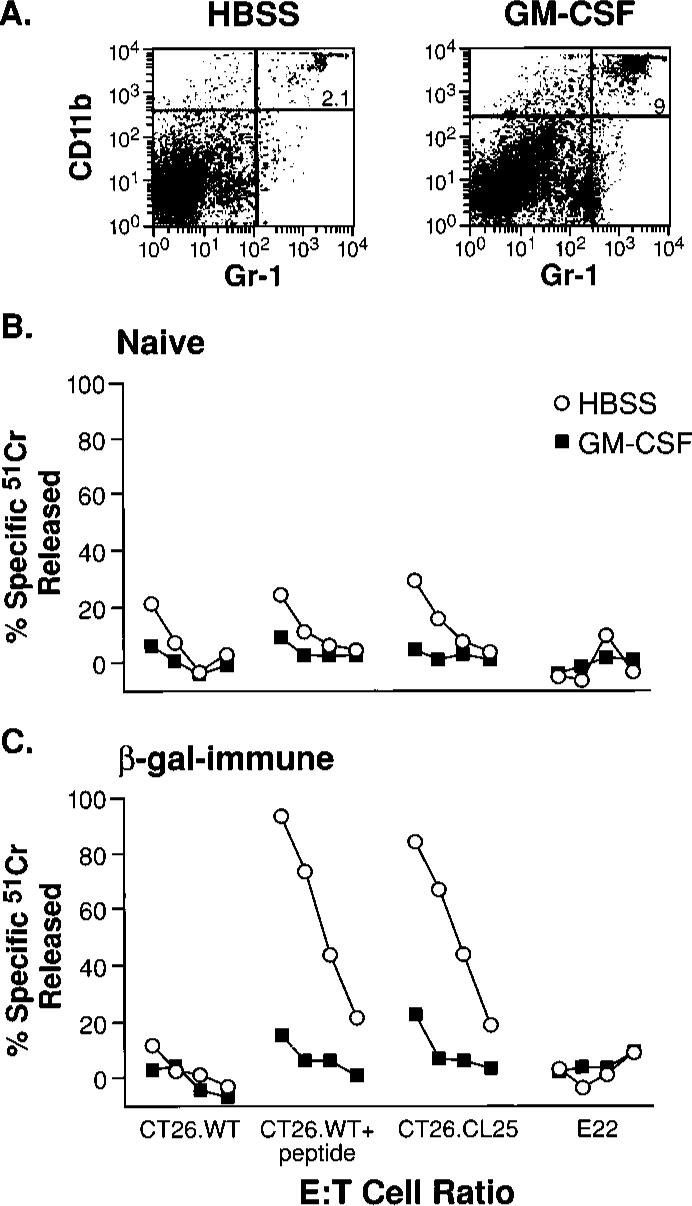
Induction of inhibitory cells in vivo using recombinant GM-CSF alone. Administration of GM-CSF causes an increase in CD11b/Gr-1 double-positive splenocytes. BALB/c mice were injected either with saline alone (“Naive”), or saline containing β-gal-rVV (“β-gal immune”). After 3 wk, the two groups of mice were randomly separated into smaller groups of three animals each that were inoculated i.p. with HBSS alone (HBSS), or HBSS containing 5 μg of mouse GM-CSF (GM-CSF), twice daily for a total of 3 days. At the end of this cycle of inoculations, spleens were removed and cultured with β-gal peptide for 6 days. A, Cytofluorometric staining with FITC anti-Gr-1 and PE anti-CD11b mAbs of splenocytes from immune mice inoculated with GM-CSF (right panel) or with vehicle alone (left panel). Similar percentages of CD11b+/Gr-1+ cells were found in spleens of nonimmunized mice (data not shown). B and C, Functional consequence of CD11b+/Gr-1+ induction. The cytolytic response against a panel of β-gal-positive and -negative targets is shown in naive (B) and β-gal-immune (C) mice. E:T cell ratio was 100:1, then 3-fold dilutions (100:1, 33:1, 11:1, 4:1). Repeat experiments gave nearly identical results.
Inhibitory cells express some immature APC markers such as CD11b and F4/80 and are CD11clow but lack the expression of costimulatory molecules and MHC class II
To characterize the inhibitory population of CD11b+ cells in greater detail, splenocytes were positively selected from the spleens of TS/A-bearing mice with anti-Gr-1 mAb and were cultured in vitro in standard medium. After 7 days of culture, a homogenous population of weakly adherent cells was isolated that retained its ability to inhibit T lymphocyte function (see Fig. 4A, “no cytokine”). We sought to characterize the phenotypes of these cells in more detail. For comparison, we simultaneously prepared a population of “classical” DC from naive mice using an established protocol (compare Fig. 3A with granulocyte-monocyte progenitor cells, Fig. 3B).
FIGURE 4.
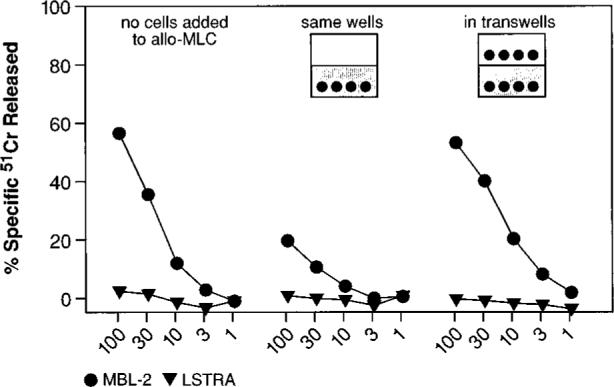
A cell-cell contact is required for suppression of CD8+ T lymphocyte response. Gr-1+ cells were enriched through panning with a specific mAb from the spleens of mice that had received TS/A tumor inoculation. Following culture in medium without cytokines for 7 days, adherent cells were harvested and added at a final concentration of 0.18 × 106 cells (3%) to a MLC consisting of BALB/c splenocytes and γ-irradiated C57BL/6n splenocytes. The Gr-1+-derived cells were either admixed in the same well (cell contact) or separated by a semipermeable membrane (no cell contact). Control cultures did not receive the third part population of suppressor cells (shown in the leftmost panel). After 5 days, cytolytic activity in the cultures was assayed against either a syngeneic (H-2d) target, LSTRA, or an allogeneic (H-2b) target, MBL-2. This experiment was repeated twice with similar results.
FIGURE 3.

Phenotypic characterization of inhibitory cells and their comparison with “classical” DC. Comparison between mature DC (A) and the CD11b+/Gr-1+ cells from a tumor-bearing animal (B). Gr-1+ cells from the spleens of mice s.c. inoculated 35 days earlier with TS/A cells were enriched through panning and cultured for 7 days in complete medium. Before staining with the different mAb, nonadherent cells were removed, and adherent cells were collected in PBS, 1 mM EDTA by gentle scraping, then blocked for nonspecific staining with unlabelled anti-Fc Ab. The phenotypic pattern shown in this figure was also observed in repeat experiments, despite some variability in the percentage and intensity of staining with anti-CD11c. Isotype-matched control Abs are shaded.
Immature markers for APC in the granulocyte-monocyte lineage stained positive including F4/80 and CD11b (also known as CD11b, integrin αM chain and Ly-40) (Fig. 3B). The expression of CD11c (integrin αχ chain) in the purified inhibitory cell was low, and cytofluorometric quantification of these cells varied somewhat from experiment to experiment with CD11cdim cells comprising between 25 and 60% (compare with Fig. 4B, “no cytokine”). Interestingly, expression of Gr-1 (Ly-6G), another granulocyte-monocyte lineage marker, was present early but was consistently lost both when the cells were prepared from tumor-bearing mice or from mice that received a short course of GM-CSF.
Clearly, these inhibitory cells differed from “classical” mature DC and activated monocytes and macrophages most significantly by their lack of uniform expression of the costimulatory molecule B7−2 (CD86/B70/Ly-58). They also lacked the expression of DEC 205 (NLDC-145), a protein involved in carbohydrate endocytosis and Ag presentation (Fig. 3A vs 3B) (35). “Classical” DC, and activated macrophages for that matter, express high levels of MHC class II, whereas the inhibitory cells were consistently negative. While the cell surface marker CD16 (FcγRIII) is found in low quantities on DC and is a developmental marker on monocytes and macrophages, the inhibitory cells were negative. Normal levels of MHC class I were present, and there was no expression of T and B lymphocyte markers (data not shown), excluding the possibility that these CD11b+/Gr-1+ were CD8a+ as recently described for a subpopulation of DC with a suppressive function (36, 37). Cells with similar granulocyte-monocyte progenitor markers were found in C57BL/6n and BALB/c after induction, in vivo, with twice daily regimen of GM-CSF alone given for 3 days (data not shown).
Inhibitory cells prevent CD8+ T cell generation through a contact-dependent mechanism
We next investigated the mechanism used by CD11b+/Gr-1+ cells to inhibit CD8+ cell function. To determine whether soluble factors were responsible for the suppression, we did experiments in which inhibitory APC were cultured either together with, or in close proximity to, an allogeneic MLC. The anti-H-2b MLC was generated by admixing BALB/c (H-2d) splenocytes with irradiated C57BL/6n (H-2b) splenocytes. Inhibitory cells, which were enriched from mice bearing TS/A tumors, were allowed to differentiate in vitro in medium devoid of exogenously added cytokines. These inhibitory cells were then added to MLC, either in the same well, or in a transwell. The effectors were then tested in a micro-cytotoxicity assay against syngeneic (LSTRA) or allogeneic (MBL-2) targets.
The secretion of soluble factors, such as inhibitory cytokines, by the CD11b+/Gr-1+ population may be expected to inhibit the allo-MLC reaction when the inhibitory cells were placed in diffusion chambers. However, as shown in Fig. 4, no such inhibition was observed, and the MLC functioned normally when the inhibitory cells were in transwells. Indeed, it appeared from this experiment that cell-cell contact was required for the immunosuppressive activity of the inhibitory cells, since suppression of allospecific cytolysis was observed only when the Gr-1+-derived population was not separated by a membrane from the population responsive to stimulation with allogeneic cells.
Inhibitory cells can be differentiated into fully functional APC when GM-CSF is given in combination with IL-4
Based on their phenotype, we hypothesized that these cells were immature APC in the granulocyte-monocyte lineage, developmentally blocked in their ability to mature into function APC capable of activating T lymphocytes. One way of measuring the T cell stimulatory capacity of APC is the use of a MLR (9, 38).
To test the hypothesis that these inhibitory cells were APC progenitors, arrested in their development, we did an experiment in which we isolated cells previously shown to be inhibitory (see Fig. 2), as described above, from TS/A-bearing mice. These inhibitory cells were then exposed to either no cytokines, GM-CSF alone, IL-4 alone, or to a combination of GM-CSF and IL-4. After 6 days of differentiation, these cells were tested for their function and phenotype. The cells were cytofluorometrically analyzed for three markers (MHC class II, B7−2, and CD11c) of “mature” APC and simultaneously added to a MLC to test their function. Specifically, these cytokine treated cells comprised 3% of the total cells in a MLC in which BALB/c splenocytes (H-2d) were mixed with γ-irradiated C57BL/6n splenocytes (H-2b).
Phenotypically, cells partially differentiated under the influence of GM-CSF alone had an up-regulation of B7−2 expression (Fig. 5B). Functionally, these cells retained their ability to inhibit T cells, although some variability was observed (Fig. 5A). Addition of IL-4 alone resulted in a high level of CD11c expression but a low level of B7−2. Again, these cells remained inhibitory. But when the cells were differentiated with a combination of GM-CSF and IL-4, we observed a significant up-regulation of B7−2 and CD11c, but no significant MHC class II was induced (I-Ad/I-Ed). More importantly, cells differentiated under the combined effects of GM-CSF and IL-4 resulted in an enhanced anti-H-2b response.
FIGURE 5.

Inhibitory cells can be differentiated into functionally mature, activating APC under the influence of combined GM-CSF and IL-4. To assess the influences of GM-CSF and IL-4 alone or in combination on the differentiation of inhibitory cells, we isolated Gr-1+ splenocytes from mice that had received TS/A tumor inoculation. Inhibitory cells were then exposed to either no cytokines, GM-CSF alone, IL-4 alone, or a combination of GM-CSF and IL-4. After 6 days of culture, these cells were tested for their function (A) and phenotype (B). A, Cytokine-treated inhibitory cells were added at a final concentration of 0.18 × 106 cells (3%) to a MLC consisting of 3 × 106 BALB/c splenocytes (H-2d) together with an equal number of γ-irradiated C57BL/6n splenocytes (H-2b). The MLC was assessed in a standard 51Cr release assay for activity after an additional 6 days of culture using an H-2b target, MBL-2, and a control H-2d target, CT26, at E:T ratios starting at 100:1, followed by 3-fold dilutions (100:1, 33:1, 11:1, 4:1, 1:1, 0.4:1). B, Phenotypic characterization of cells after the 6-day cytokine regimen. Isotype matched controls are shaded.
These data indicated that the CD11b+/Gr-1+ cells could be differentiated into functional APC when placed in the appropriate cytokine environment. Nevertheless, coinoculation of 5 μg of mouse IL-4, twice daily for 3 days, together with GM-CSF, was not sufficient to revert suppression of CD8+ T lymphocyte anamnestic response to β-gal, despite a 40−50% reduction in the number of CD11b+/Gr-1+ splenocytes (V.B. and N.P.R., unpublished data). Apparently, the in vivo effect of GM-CSF on recruitment and maturation of suppressor cell precursors was dominant.
Discussion
Our results indicate that some tumor cells can inhibit CD8+ T cell function through their production of GM-CSF and its effects on the development of cells in the granulocyte-monocyte lineage. Initially, this finding appears to conflict with the previously described adjuvant effects of tumor cells gene-modified with GM-CSF (18). Like many other investigators, we have independently confirmed that γ-irradiated B16, gene-modified to produce GM-CSF, can induce long-lasting CD8+ T cell responses against the parental tumor (data not shown).
It is puzzling that the growth of B16 clones secreting GM-CSF inhibited CD8+ T cell function while the same tumor cells, irradiated and administered s.c., protected from a challenge with the parental B16. One of several possibilities is that when tumor cells are lethally irradiated, their GM-CSF production may be limited in duration compared with nonirradiated tumors that grow progressively. Short-term, local expression of GM-CSF might allow for the terminal differentiation of dermal DC (Langerhans cells) (39), whereas, the more sustained or systemic presence of GM-CSF alone may cause the egress of inhibitory granulocyte-monocyte progenitors from the bone marrow. Indeed, our data demonstrate that a GM-CSF-producing tumor must grow for 2−5 wk in vivo before CD11b+/Gr-1+ cells appear in the spleen (see Fig. 1A). There is also likely to be a threshold serum level at which GM-CSF acts in this fashion. Furthermore, γ-irradiation of cells has been linked with the induction of caspase-dependent apoptotic death (40–42). Apoptotic death of tumor cells may elicit the production of inflammatory cytokines by resident macrophages. IL-1β, in particular, is activated by caspase-1, also known as ICE (IL-1β converting enzyme), the homologue of CED-3 (43). Together with other inflammatory cytokines, the microenvironment of the dying GM-CSF gene-modified tumor may stimulate the maturation of APC (15). In addition, apoptotic death may result in the selective uptake of fragmented cells by mature DC and other activating APC (44).
The main reason why we should be concerned about the ectopic release of GM-CSF by tumors relates to our current efforts to develop effective therapeutic cancer vaccines (45). Although these efforts have met with some real success in the clinic, most patients do not respond to our attempts at intervention (3).
GM-CSF can act effectively in concert with other cytokines to induce the maturation of APC (9). However, we found that GM-CSF given alone appears to elicit granulocyte-monocyte progenitor cells from the bone marrow that fail to develop into functional APC in the periphery. Other reports are consistent with these observations. The daily administration of GM-CSF (at doses similar to those used in this study) had virtually no effect on the splenocytes with a DC phenotype (46). In another study, transgenic mice overproducing GM-CSF exhibited only a modest increase in the number of DC in the thymus and spleen (47). Bone marrow cell cultures stimulated with GM-CSF alone do not induce mature DC, but induce a population of what Starzl and colleagues (48) call “DC-progenitors.” These bone marrow cells, phenotypically characterized as class IIlow, B7−1dim, B7−2 negative, functionally induce hyporesponsiveness in allogeneic T cells (48). Further, these cells were able to significantly prolong cardiac allograft survival when transferred to graft recipients (49). Finally, the addition of IL-4 to the GM-CSF regimen corrected the defects in class II and costimulatory molecule expression and eliminated the anergizing capability of these “dendritic precursor” cells (48). Thus, GM-CSF appears to have an important, but not singular influence on APC homeostasis and differentiation.
Other cytokines produced by tumors may act in concert with GM-CSF to dysregulate the maturation of APCs and inhibit T cell activity. TGF-β interferes with the development and function of DC (9). IL-10 can suppress DC function and differentiation in addition to shifting the balance of a Th1 response toward the Th2 type response (50–53). Vascular endothelial growth factor (VEGF) has also been shown to impair the generation of mature DC from granulocyte-monocyte progenitor cells (53, 54). TS/A tumor cells used in this study have been shown to produce VEGF and TGF-β, in addition to various CSF (55, 56). In addition, tumor cells have been shown to evade immune recognition by a variety of mechanisms that include poor Ag processing, loss of β2-micro-globulin, loss of Ag, and production of immunoinhibitory cytokines (57–59). The expression of Fas ligand by tumor cells is still controversial (60, 61). In any case, tumor cells may use one or more of these mechanisms to subvert immune responses, although the administration GM-CSF by itself was sufficient to abrogate the development of mature APC.
The mechanism used by the granulocyte-monocyte progenitor cells to inhibit the effector function of CD8+ T cells is unknown. However, we are currently investigating several potential mechanisms. It has been reported that inhibition of T lymphocyte reactivity could be induced by a subclass of CD8a+ lymphoid-derived DC. This inhibition could be reversed with exogenous IL-2 (36). Recently, these authors went on to demonstrate that the apoptotic death of CD4+ T cells induced by this subpopulation of DC was mediated by Fas (CD95/Apo-1) ligand (37). The cells described in this report are phenotypically quite different from the CD8a+ DC described above, but they have a striking resemblance to the bone marrow-derived “DC-progenitors” that develop under the influence of GM-CSF (48, 49). The T cell defects induced by these previously reported “DC-progenitors” could be reversed by the addition of an anti-CD28 mAb or with exogenous IL-2 (48). Thus, the unresponsiveness of the CD8+ T lymphocytes upon stimulation through their clonotypic receptors may result from a deficit in costimulation and cytokine signaling.
The beneficial effect of GM-CSF administration in immunocompromised patients after bone marrow or stem cell transplantation has been documented since the late 1980s (62). This effect may be in apparent conflict with our results. However, GM-CSF is thought to accelerate the recovery of mature granulocytes, a component of the innate immune system. Granulocytes and other phagocytes are the first line of defense in the nonspecific neutralization of infectious agents. Our results suggest that GM-CSF may inhibit the adaptive immune system, including CD8+ T cell function, while favoring the function of the innate immune system.
On the basis of our findings, we propose that the alterations in the immune response induced by some malignancies are a consequence of the induction of granulocyte-monocyte progenitor cells that are unable to mature into functional APC due to the unopposed secretion of GM-CSF. In the absence of costimulation and the appropriate cytokine mileu provided by mature APC, it may not be possible to initiate or sustain immune reactivity at the tumor site. Additional experiments are needed to evaluate whether or not these CD11b+/Gr-1+ cells are recruited when GM-CSF is employed as immune or hemopoietic adjuvant, especially when large doses of this cytokine are administered systemically. The induction and isolation of granulocyte-monocyte progenitor cells may represent a powerful therapeutic tool to control the unrestrained immune reactivity associated with autoimmune diseases and transplant rejections.
Acknowledgments
We thank P. Segato for editing the manuscript, D. R. Surman for help and advice with many aspects of the work presented here, P. Spiess and D. Jones for help with the animal experiments, M. Blalock for expert graphics, E. P. Shulman and K. R. Irvine for tissue culture preparations, and A. Mixon for the flow cytometry experiments.
Footnotes
This work was supported in part by the Strong Children's Research Center (Rochester, NY) (to M.W.) and by the Istituto Superiore Sanità (ISS) Italy-US cooperation program for the therapy of cancer, Grant 981/A.14 (to V.B., E.A., and A.C.).
Abbreviations used in this paper: TAA, tumor-associated Ags; DC, dendritic cell; β-gal, β-galactosidase; CM, culture medium; rVV, recombinant vaccinia virus.
References
- 1.Overwijk WW, Tsung A, Irvine KR, Parkhurst MR, Goletz TJ, Tsung K, Carroll MW, Liu C, Moss B, Rosenberg SA, Restifo NP. gp100/pmel 17 is a murine tumor rejection antigen: induction of “self”-reactive, tumoricidal T cells using high-affinity, altered peptide ligand. J. Exp. Med. 1998;188:277. doi: 10.1084/jem.188.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Overwijk WW, Lee DS, Surman DR, Irvine KR, Touloukian CE, Chan C-C, Carroll MW, Moss B, Rosenberg SA, Restifo NP. Vaccination with a recombinant vaccinia virus encoding a “self” antigen induces autoimmune vitiligo and tumor destruction in mice: Requirement for CD4+ T lymphocytes. Proc. Natl. Acad. Sci. USA. 1999 doi: 10.1073/pnas.96.6.2982. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang, JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL, Restifo NP, Dudley ME, Schwarz SL, Spiess PJ, et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998;4:321. doi: 10.1038/nm0398-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pan ZK, Ikonomidis G, Lazenby A, Pardoll D, Paterson Y. A recombinant Listeria monocytogenes vaccine expressing a model tumour antigen protects mice against lethal tumour cell challenge and causes regression of established tumours. Nat. Med. 1995;1:471. doi: 10.1038/nm0595-471. [DOI] [PubMed] [Google Scholar]
- 5.Dyall R, Bowne WB, Weber LW, LeMaoult J, Szabo P, Moroi Y, Piskun G, Lewis JJ, Houghton AN, Nikolic-Zugic J. Heteroclitic immunization induces tumor immunity. J. Exp. Med. 1998;188:1553. doi: 10.1084/jem.188.9.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pardoll DM. Cancer vaccines. Nat. Med. 1998;4:525. doi: 10.1038/nm0598supp-525. [DOI] [PubMed] [Google Scholar]
- 7.Restifo NP, Surman DR, Zheng H, Palese P, Rosenberg SA, Garcia-Sastre A. Transfectant influenza A viruses are effective recombinant immunogens in the treatment of experimental cancer. Virology. 1998;249:89. doi: 10.1006/viro.1998.9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Restifo NP. Cancer vaccines '98: a reductionistic approach. Mol. Med. Today. 1998;4:327. doi: 10.1016/s1357-4310(98)01304-5. [DOI] [PubMed] [Google Scholar]
- 9.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 10.Bronte V, Carroll MW, Goletz TJ, Wang M, Overwijk WW, Marincola F, Rosenberg SA, Moss B, Restifo NP. Antigen expression by dendritic cells correlates with the therapeutic effectiveness of a model recombinant poxvirus tumor vaccine. Proc. Natl. Acad. Sci. USA. 1997;94:3183. doi: 10.1073/pnas.94.7.3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J. Exp. Med. 1998;188:1075. doi: 10.1084/jem.188.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ashley DM, Faiola B, Nair S, Hale LP, Bigner DD, Gilboa E. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J. Exp. Med. 1997;186:1177. doi: 10.1084/jem.186.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oehler L, Majdic O, Pickl WF, Stockl J, Riedl E, Drach J, Rappersberger K, Geissler K, Knapp W. Neutrophil granulocyte-committed cells can be driven to acquire dendritic cell characteristics. J. Exp. Med. 1998;187:1019. doi: 10.1084/jem.187.7.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inaba K, Inaba M, Deguchi M, Hagi K, Yasumizu R, Ikehara S, Muramatsu S, Steinman RM. Granulocytes, macrophages, and dendritic cells arise from a common major histocompatibility complex class II-negative progenitor in mouse bone marrow. Proc. Natl. Acad. Sci. USA. 1993;90:3038. doi: 10.1073/pnas.90.7.3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cella M, Engering A, Pinet V, Pieters J, Lanzavecchia A. Inflammatory stimuli induce accumulation of MHC class II complexes on dendritic cells. Nature. 1997;388:782. doi: 10.1038/42030. [DOI] [PubMed] [Google Scholar]
- 16.Levitsky HI, Montgomery J, Ahmadzadeh M, Staveley-O'Carroll K, Guarnieri F, Longo DL, Kwak LW. Immunization with granulocyte-macrophage colony-stimulating factor-transduced, but not B7−1-transduced, lymphoma cells primes idiotype-specific T cells and generates potent systemic anti-tumor immunity. J. Immunol. 1996;156:3858. [PubMed] [Google Scholar]
- 17.Thomas MC, Greten TF, Pardoll DM, Jaffee EM. Enhanced tumor protection by granulocyte-macrophage colony-stimulating factor expression at the site of an allogeneic vaccine. Hum. Gene Ther. 1998;9:835. doi: 10.1089/hum.1998.9.6-835. [DOI] [PubMed] [Google Scholar]
- 18.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, Jackson V, Hamada H, Pardoll D, Mulligan RC. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA. 1993;90:3539. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 20.Albert ML, Pearce SA, Francisco LM, Sauter B, Roy P, Silverstein RL, Bhardwaj N. Immature dendritic cells phagocytose apoptotic cells via αvβ5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J. Exp. Med. 1998;188:1359. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsuchiya Y, Igarashi M, Suzuki R, Kumagai K. Production of colony-stimulating factor by tumor cells and the factor- mediated induction of suppressor cells. J. Immunol. 1988;141:699. [PubMed] [Google Scholar]
- 22.Takeda K, Hatakeyama K, Tsuchiya Y, Rikiishi H, Kumagai K. A correlation between GM-CSF gene expression and metastases in murine tumors. Int. J. Cancer. 1991;47:413. doi: 10.1002/ijc.2910470318. [DOI] [PubMed] [Google Scholar]
- 23.Rokhlin OW, Griebling TL, Karassina NV, Raines MA, Cohen MB. Human prostate carcinoma cell lines secrete GM-CSF and express GM-CSF-receptor on their cell surface. Anticancer Res. 1996;16:557. [PubMed] [Google Scholar]
- 24.Bronte V, Wang M, Overwijk WW, Surman DR, Pericle F, Rosenberg SA, Restifo NP. Apoptotic death of CD8+ T lymphocytes after immunization: induction of a suppressive population of Mac-1+/Gr-1+ cells. J. Immunol. 1998;161:5313. [PMC free article] [PubMed] [Google Scholar]
- 25.Bronte V, Tsung K, Rao JB, Chen PW, Wang M, Rosenberg SA, Restifo NP. IL-2 enhances the function of recombinant poxvirus-based vaccines in the treatment of established pulmonary metastases. J. Immunol. 1995;154:5282. [PMC free article] [PubMed] [Google Scholar]
- 26.Wang M, Bronte V, Chen PW, Gritz L, Panicali D, Rosenberg SA, Restifo NP. Active immunotherapy of cancer with a nonreplicating recombinant fowlpox virus encoding a model tumor-associated antigen. J. Immunol. 1995;154:4685. [PMC free article] [PubMed] [Google Scholar]
- 27.Gavin MA, Gilbert MJ, Riddell SR, Greenberg PD, Bevan MJ. Alkali hydrolysis of recombinant proteins allows for the rapid identification of class I MHC-restricted CTL epitopes. J. Immunol. 1993;151:3971. [PubMed] [Google Scholar]
- 28.Overwijk WW, Surman DR, Tsung K, Restifo NP. Identification of a Kb-restricted CTL epitope of β-galactosidase: potential use in development of immunization protocols for “self” antigens. Methods. 1997;12:117. doi: 10.1006/meth.1997.0461. [DOI] [PubMed] [Google Scholar]
- 29.Stephens ND. GM-CSF secretion in primary cultures of normal and cancerous human renal cells. Kidney Int. 1996;50:1044. doi: 10.1038/ki.1996.407. [DOI] [PubMed] [Google Scholar]
- 30.Trutmann M. GM-CSF gene expression and protein production in human colorectal cancer cell lines and clinical tumor specimens. Int. J. Cancer. 1998;77:378. doi: 10.1002/(sici)1097-0215(19980729)77:3<378::aid-ijc12>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 31.Pericle F, Kirken RA, Bronte V, Sconocchia G, DaSilva L, Segal DM. Immunocompromised tumor-bearing mice show a selective loss of STAT5a/b expression in T and B lymphocytes. J. Immunol. 1997;159:2580. [PubMed] [Google Scholar]
- 32.Jaffe ML, Arai H, Nabel GJ. Mechanisms of tumor-induced immunosuppression: evidence for contact-dependent T cell suppression by monocytes. Mol. Med. 1996;2:692. [PMC free article] [PubMed] [Google Scholar]
- 33.Irvine KR, Chamberlain RS, Shulman EP, Surman DR, Rosenberg SA, Restifo NP. Enhancing efficacy of recombinant anticancer vaccines with prime/boost regimens that use two different vectors. J. Natl. Cancer Inst. 1997;89:1595. doi: 10.1093/jnci/89.21.1595. [DOI] [PubMed] [Google Scholar]
- 34.Irvine KR, Chamberlain RS, Shulman EP, Rosenberg SA, Restifo NP. Route of immunization and the therapeutic impact of recombinant anticancer vaccines. J. Natl. Cancer Inst. 1997;89:390. doi: 10.1093/jnci/89.5.390. [DOI] [PubMed] [Google Scholar]
- 35.Jiang W, Swiggard WJ, Heufler C, Peng M, Mirza A, Steinman RM, Nussenzweig MC. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 1995;375:151. doi: 10.1038/375151a0. [DOI] [PubMed] [Google Scholar]
- 36.Kronin V, Winkel K, Suss G, Kelso A, Heath W, Kirberg J, von Boehmer H, Shortman K. A subclass of dendritic cells regulates the response of naive CD8 T cells by limiting their IL-2 production. J. Immunol. 1996;157:3819. [PubMed] [Google Scholar]
- 37.Suss G, Shortman K. A subclass of dendritic cells kills CD4 T cells via Fas/Fas-ligand-induced apoptosis. J. Exp. Med. 1996;183:1789. doi: 10.1084/jem.183.4.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steinman RM, Witmer MD. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc. Natl. Acad. Sci. USA. 1978;75:5132. doi: 10.1073/pnas.75.10.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witmer-Pack MD, Olivier W, Valinsky J, Schuler G, Steinman RM. Granulocyte/macrophage colony-stimulating factor is essential for the viability and function of cultured murine epidermal Langerhans cells. J. Exp. Med. 1987;166:1484. doi: 10.1084/jem.166.5.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, et al. Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998;94:339. doi: 10.1016/s0092-8674(00)81477-4. [DOI] [PubMed] [Google Scholar]
- 41.Sarin A, Wu ML, Henkart PA. Different interleukin-1β converting enzyme (ICE) family protease requirements for the apoptotic death of T lymphocytes triggered by diverse stimuli. J. Exp. Med. 1996;184:2445. doi: 10.1084/jem.184.6.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reap EA, Roof K, Maynor K, Borrero M, Booker J, Cohen PL. Radiation and stress-induced apoptosis: a role for Fas/Fas ligand interactions. Proc. Natl. Acad. Sci. USA. 1997;94:5750. doi: 10.1073/pnas.94.11.5750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chinnaiyan AM, O'Rourke K, Lane BR, Dixit VM. Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science. 1997;275:1122. doi: 10.1126/science.275.5303.1122. [DOI] [PubMed] [Google Scholar]
- 44.Bender A, Albert M, Reddy A, Feldman M, Sauter B, Kaplan G, Hellman W, Bhardwaj N. The distinctive features of influenza virus infection of dendritic cells. Immunobiology. 1998;198:552. doi: 10.1016/S0171-2985(98)80078-8. [DOI] [PubMed] [Google Scholar]
- 45.Restifo NP, Rosenberg SA. Developing recombinant and synthetic vaccines for the treatment of melanoma. Curr. Opin. Oncol. 1999;11:50. doi: 10.1097/00001622-199901000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maraskovsky E, Brasel K, Teepe M, Roux ER, Lyman SD, Shortman K, McKenna HJ. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J. Exp. Med. 1996;184:1953. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vremec D, Lieschke GJ, Dunn AR, Robb L, Metcalf D, Shortman K. The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur. J. Immunol. 1997;27:40. doi: 10.1002/eji.1830270107. [DOI] [PubMed] [Google Scholar]
- 48.Lu L, McCaslin D, Starzl TE, Thomson AW. Bone marrow-derived dendritic cell progenitors (NLDC 145+, MHC class II+, B7−1dim, B7−2) induce alloantigen-specific hyporesponsiveness in murine T lymphocytes. Transplantation. 1995;60:1539. doi: 10.1097/00007890-199560120-00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu F, Li Y, Qian S, Lu L, Chambers F, Starzl TE, Fung JJ, Thomson AW. Costimulatory molecule-deficient dendritic cell progenitors (MHC class II+, CD80dim, CD86−) prolong cardiac allograft survival in nonimmunosuppressed recipients. Transplantation. 1996;62:659. doi: 10.1097/00007890-199609150-00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qin Z, Noffz G, Mohaupt M, Blankenstein T. Interleukin-10 prevents dendritic cell accumulation and vaccination with granulocyte-macrophage colony-stimulating factor gene-modified tumor cells. J. Immunol. 1997;159:770. [PubMed] [Google Scholar]
- 51.Dummer W, Becker JC, Schwaaf A, Leverkus M, Moll T, Brocker EB. Elevated serum levels of interleukin-10 in patients with metastatic malignant melanoma. Melanoma Res. 1995;5:67. doi: 10.1097/00008390-199502000-00008. [DOI] [PubMed] [Google Scholar]
- 52.Chen Q, Daniel V, Maher DW, Hersey P. Production of IL-10 by melanoma cells: examination of its role in immunosuppression mediated by melanoma. Int. J. Cancer. 1994;56:755. doi: 10.1002/ijc.2910560524. [DOI] [PubMed] [Google Scholar]
- 53.Schuler G, Steinman RM. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J. Exp. Med. 1997;186:1183. doi: 10.1084/jem.186.8.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996;2:1096. doi: 10.1038/nm1096-1096. [Published erratum appears in 1996 Nat. Med. 2:1267.] [DOI] [PubMed] [Google Scholar]
- 55.Nicoletti G. Colony-stimulating activity from the new metastatic TS/A cell line and its high- and low-metastatic clonal derivatives. Br. J. Cancer. 1985;52:215. doi: 10.1038/bjc.1985.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giovarelli M. Local release of IL-10 by transfected mouse mammary adenocarcinoma cells does not suppress but enhances antitumor reaction and elicits a strong cytotoxic lymphocyte and antibody-dependent immune memory. J. Immunol. 1995;155:3112. [PubMed] [Google Scholar]
- 57.Restifo NP, Esquivel F, Kawakami Y, Yewdell JW, Mule JJ, Rosenberg SA, Bennink JR. Identification of human cancers deficient in antigen processing. J. Exp. Med. 1993;177:265. doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional β2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J. Natl. Cancer Inst. 1996;88:100. doi: 10.1093/jnci/88.2.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chouaib S, Asselin-Paturel C, Mami-Chouaib F, Caignard A, Blay JY. The host-tumor immune conflict: from immunosuppression to resistance and destruction. Immunol. Today. 1997;18:493. doi: 10.1016/s0167-5699(97)01115-8. [DOI] [PubMed] [Google Scholar]
- 60.Chappell DB, Restifo NP. T cell-tumor cell: a fatal interaction? Cancer Immunol. Immunother. 1998;47:65. doi: 10.1007/s002620050505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chappell DB, Zaks TZ, Rosenberg SA, Restifo NP. Human melanoma cells do not express Fas (Apo-1/CD95) ligand. Cancer Res. 1999;59:59. [PMC free article] [PubMed] [Google Scholar]
- 62.Brandt SJ. Effect of recombinant human granulocyte-macrophage colony-stimulating factor on hematopoietic reconstitution after high-dose chemotherapy and autologous bone marrow transplantation. N. Engl. J. Med. 1988;318:869. doi: 10.1056/NEJM198804073181401. [DOI] [PubMed] [Google Scholar]