

Abstract
Central core disease (CCD) is a human myopathy that involves a dysregulation in muscle Ca2+ homeostasis caused by mutations in the gene encoding the skeletal muscle ryanodine receptor (RyR1), the protein that comprises the calcium release channel of the SR. Although genetic studies have clearly demonstrated linkage between mutations in RyR1 and CCD, the impact of these mutations on release channel function and excitation-contraction coupling in skeletal muscle is unknown. Toward this goal, we have engineered the different CCD mutations found in the NH2-terminal region of RyR1 into a rabbit RyR1 cDNA (R164C, I404M, Y523S, R2163H, and R2435H) and characterized the functional effects of these mutations after expression in myotubes derived from RyR1-knockout (dyspedic) mice. Resting Ca2+ levels were elevated in dyspedic myotubes expressing four of these mutants (Y523S > R2163H > R2435H R164C > I404M RyR1). A similar rank order was also found for the degree of SR Ca2+ depletion assessed using maximal concentrations of caffeine (10 mM) or cyclopiazonic acid (CPA, 30 μM). Although all of the CCD mutants fully restored L-current density, voltage-gated SR Ca2+ release was smaller and activated at more negative potentials for myotubes expressing the NH2-terminal CCD mutations. The shift in the voltage dependence of SR Ca2+ release correlated strongly with changes in resting Ca2+, SR Ca2+ store depletion, and peak voltage–gated release, indicating that increased release channel activity at negative membrane potentials promotes SR Ca2+ leak. Coexpression of wild-type and Y523S RyR1 proteins in dyspedic myotubes resulted in release channels that exhibited an intermediate degree of SR Ca2+ leak. These results demonstrate that the NH2-terminal CCD mutants enhance release channel sensitivity to activation by voltage in a manner that leads to increased SR Ca2+ leak, store depletion, and a reduction in voltage-gated Ca2+ release. Two fundamentally distinct cellular mechanisms (leaky channels and EC uncoupling) are proposed to explain how altered release channel function caused by different mutations in RyR1 could result in muscle weakness in CCD.
Keywords: excitation-contraction coupling, calcium channels, muscle disease, calcium homeostasis
INTRODUCTION
Rapid elevations of intracellular Ca2+ in skeletal muscle are controlled by a unique, bidirectional signaling interaction between L-type Ca2+ channels (L-channels or DHPRs) in the sarcolemma and Ca2+ release channels (ryanodine receptors or RyR1s) of the SR. During skeletal muscle excitation-contraction (EC) coupling, action potentials in the sarcolemma induce conformational changes in dihydropyridine receptors (DHPRs) that rapidly trigger the opening of nearby SR Ca2+ release channels (orthograde coupling; Melzer et al. 1995). Interestingly, DHPR channel activity is strongly dependent on the presence of RyR1 (retrograde coupling), thus demonstrating the reciprocal nature of the DHPR–RyR1 interaction (Nakai et al. 1996; Grabner et al. 1999; Avila and Dirksen 2000). The central roles of DHPR and RyR1 proteins in skeletal muscle Ca2+ homeostasis is emphasized by the fact that point mutations in both of these proteins account for several different congenital human muscle diseases (e.g., hypokalemic periodic paralysis, malignant hyperthermia, and central core disease [CCD]; Lorenzon and Beam 2000).
CCD, first described by Shy and Magee 1956, is a human autosomal dominant, nonprogressive myopathy characterized by hypotonia and proximal muscle weakness in infancy. Muscle weakness of the lower extremities leading to delayed attainment of motor skill milestones is the most common complication. However, the symptoms can vary from mild to severe with a marked diversity in skeletal muscle involvement. Diagnosis of CCD is confirmed by histological examination of muscle biopsy tissue in which type 1 fibers exhibit amorphous central areas (cores) that lack both mitochondria and oxidative enzyme activity (Dubowitz and Pearse 1960). Electron microscopic analysis of central cores shows disintegration of the contractile apparatus and alterations in the structure and amount of SR and transverse tubule (t-tubule) membranes (Hayashi et al. 1989). Less dramatic abnormalities are observed in the noncore or peripheral regions of the muscle fibers. Finally, CCD patients are typically susceptible to episodes of malignant hyperthermia (MH), a pharmacogenetic syndrome in which individuals respond to potent inhalation anesthetics (e.g., halothane) and depolarizing skeletal muscle relaxants (e.g., succinylcholine) with high fever, skeletal muscle rigidity, hypermetabolism, lactic acidosis, hypoxia, and tachycardia (Jurkat-Rott et al. 2000)
CCD and MH are localized to human chromosome 19q13.1, which includes the locus of RyR1 (MacKenzie et al. 1990; Kausch et al. 1991). So far, a total of 26 different point mutations in RyR1 have been shown to account for MH, nine of which are also associated with CCD (McCarthy et al. 2000). A single point mutation in RyR1 (R615C) accounts for all cases of porcine MH, suggesting its origin in a founder animal (Fujii et al. 1991). All of the identified MH and CCD point mutations in RyR1 occur within three relatively restricted regions of the RyR1 protein: MH/CCD region 1 (residues 35–614), MH/CCD region 2 (residues 2,129–2,458), and MH/CCD region 3 (COOH-terminal region, including residues 4,637–4,898). The MH and CCD mutations in RyR1 are hypothesized to lead to the formation of SR Ca2+ release channels that exhibit varying degrees of Ca2+ leak (MacLennan 1992; Zhang et al. 1993; MacLennan and Phillips 1995). According to this hypothesis, MH mutations result in release channels that exhibit lesser degrees of Ca2+ leak, whereas the CCD mutations result in release channels that are severely leaky to Ca2+, which leads to Ca2+-induced alterations and damage to the core of the muscle fiber. Support for this hypothesis comes from the observation that resting Ca2+ is elevated and intracellular Ca2+ stores are depleted in HEK-293 cells transfected with MH and CCD mutant RyR1 proteins (Tong et al. 1997, Tong et al. 1999; Lynch et al. 1999; Monnier et al. 2000).
However, the physiological effects of the CCD mutations in RyR1 on EC coupling and intracellular Ca2+ homeostasis in intact skeletal muscle have yet to be systematically studied. As a step toward this goal, we recently evaluated the functional properties of a CCD mutation in MH/CCD region 3 (I4897T) after expression in skeletal myotubes derived from “RyR1-knockout” (dyspedic) mice (Nakai et al. 1996). Contrary to the results obtained with the same mutation expressed in HEK 293 cells (Lynch et al. 1999), we found that I4897T-containing release channels reconstituted in dyspedic myotubes are not abnormally leaky to Ca2+, but rather, release SR Ca2+ ineffectively in response to sarcolemmal depolarization (termed “EC uncoupling”; Avila et al. 2001a). Thus, functional analysis of each of the CCD mutants operating within dyspedic myotubes is advantageous since: dyspedic myotubes express other proteins (Buck et al. 1997) critical for the modulation of release channel function (e.g., calmodulin, FK-506 binding protein, triadin, calsequestrin, and DHPR), several of which are relatively muscle specific; the effects of the mutations on the orthograde and retrograde signals of EC coupling can only be studied in the context of muscle expression systems; and the influence of endogenous compensatory mechanisms recruited in response to the introduction of mutant release channels should be more accurately reflected after reconstitution in dyspedic myotubes.
Five amino acid residues (R163, I403, Y522, R2163, and R2435) account for the six different CCD mutations in the NH2-terminal or cytoplasmic “foot” region of the human RyR1 protein (MH/CCD regions 1 and 2; Quane et al. 1993, Quane et al. 1994; Zhang et al. 1993; Manning et al. 1998; Barone et al. 1999). The effects of these cytoplasmic CCD mutations on release channel activity in skeletal muscle have not been determined. Therefore, we have evaluated the functional consequences (on resting Ca2+, SR Ca2+ content, and EC coupling) of the NH2-terminal CCD mutations engineered into a rabbit RyR1 cDNA after expression in dyspedic myotubes.
MATERIALS AND METHODS
Preparation and cDNA Injections of Dyspedic Myotubes
Myotubes were prepared from primary cultures of dyspedic muscle as described previously (Nakai et al. 1996; Avila and Dirksen 2000). All animal protocols were reviewed and approved by the University Committee on Animal Resources at the University of Rochester School of Medicine and Dentistry. Each of the different CCD point mutations in RyR1 used in this study (R164C, I404M, Y523S, R2163H, R2435H, and I4897T) was introduced into a rabbit RyR1 cDNA using standard two-step site-directed mutagenesis strategies. Expression of wild-type RyR1 and the different NH2-terminal CCD mutations in RyR1 was achieved by nuclear microinjection of cDNAs encoding CD8 (0.2 μg/μl) and the appropriate expression plasmid (0.5 μg/μl) 4–6 d after initial planting of myoblasts (Avila et al. 2001b). A 1:1 cDNA mixture (0.25 μg/μl each) was used in experiments of dyspedic myotubes coexpressing RyR1 and Y523S (see Fig. 5). Expressing myotubes were identified 2–4 d after nuclear microinjection by incubation with CD8 antibody beads.
Figure 5.
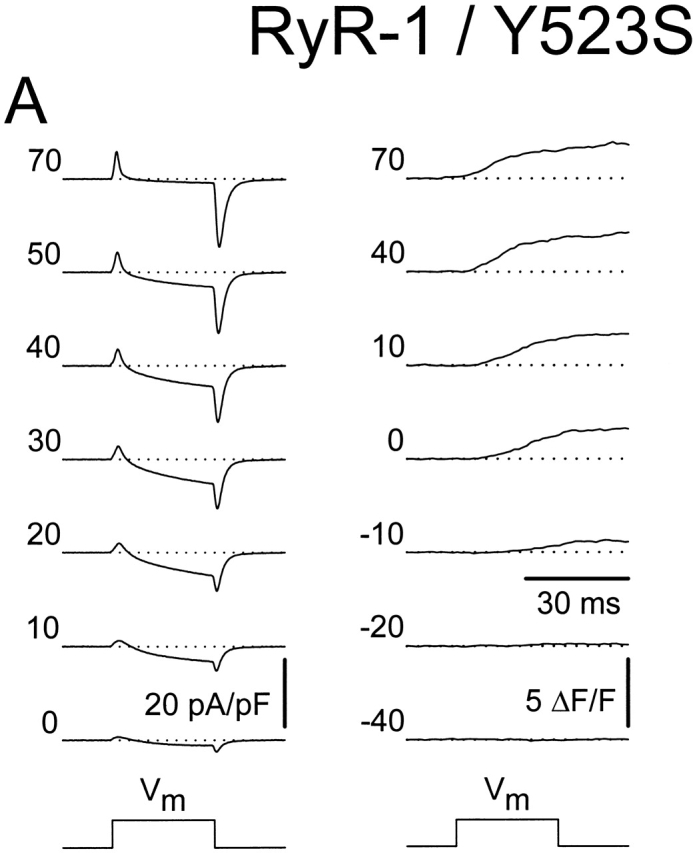
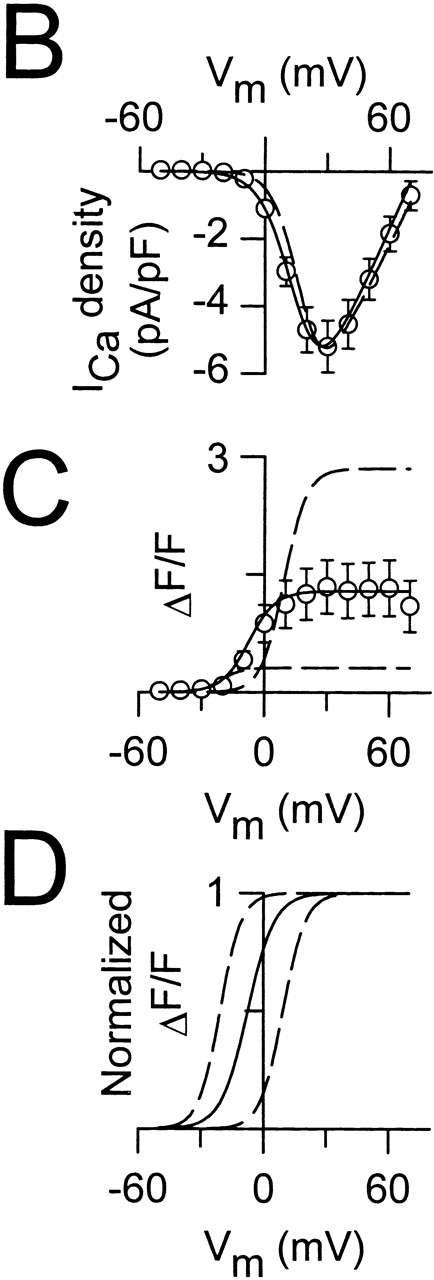
Coexpression of the wild-type RyR1 and Y523S causes an intermediate shift in 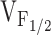 and a moderate reduction in voltage-gated SR Ca2+ release. (A) L-currents (left) and intracellular Ca2+ transients (right) obtained from a representative RyR1/Y523S-expressing dyspedic myotube. Average voltage dependence of maximal L-currents (B) and Ca2+ transients (C) in RyR-1/Y523S-expressing myotubes (n = 13). The continuous lines were calculated using (B) and (C) from the values reported in Table . The average I-V and F-V curves for RyR1- and Y523S-expressing myotubes are illustrated for comparison (dashed lines). (D) The ΔF/F-V curves in C were normalized to their respective maximal value (ΔF/F)max.
and a moderate reduction in voltage-gated SR Ca2+ release. (A) L-currents (left) and intracellular Ca2+ transients (right) obtained from a representative RyR1/Y523S-expressing dyspedic myotube. Average voltage dependence of maximal L-currents (B) and Ca2+ transients (C) in RyR-1/Y523S-expressing myotubes (n = 13). The continuous lines were calculated using (B) and (C) from the values reported in Table . The average I-V and F-V curves for RyR1- and Y523S-expressing myotubes are illustrated for comparison (dashed lines). (D) The ΔF/F-V curves in C were normalized to their respective maximal value (ΔF/F)max.
Intracellular Ca2+ Measurements in Intact Myotubes
Measurements of resting Ca2+ were obtained in intact, Indo-1 AM–loaded myotubes (Molecular Probes) as described previously (Avila et al. 2001b). The ratio of fluorescence emission at 405 and 485 (F405/F485) was converted to cytosolic Ca2+ levels using an in situ calibration approach (Avila et al. 2001b). Relative changes in global Ca2+ levels were subsequently monitored after application of rodent Ringer (see results) containing either 10 mM caffeine (see Fig. 1) or 30 μM cyclopiazonic acid (CPA; see Fig. 2). Caffeine- and CPA-induced Ca2+ transients were not converted to free Ca2+ since Ca2+ levels above ∼150 nM resulted in contractions that could introduce movement artifacts into the calibration (Avila et al. 2001b). To resolve rapid elevations in intracellular Ca2+ levels, caffeine was applied through a local, rapid (response time <5s) perfusion system (Warner Instrument Corporation). Several myotubes within a single dish could be monitored separately since the perfusion system limits caffeine accumulation in the bath, and responses to multiple applications of caffeine separated by ∼10-min wash intervals were similar. Simple gravity perfusion (<30s for complete solution exchange) was used to monitor the slower changes in intracellular Ca2+ induced by application of 30 μM CPA (Avila et al. 2001a).
Figure 1.

Intact dyspedic myotubes expressing NH2-terminal CCD mutants exhibit higher resting Ca2+ levels and a reduced maximal caffeine-induced Ca2+ release. (A) Representative caffeine-induced Ca2+ responses (10 mM caffeine, black bars) in Indo-1 AM–loaded dyspedic myotubes expressing wild-type RyR1, I404M, R164C, R2435H, R2163H, or Y523S. The abscissa for each panel corresponds to 5 min. (B) Average resting Ca2+ levels in dyspedic myotubes expressing RyR1 and the different CCD mutations. Unstimulated Indo-1 fluorescence ratios were converted to resting Ca2+ levels using an in situ calibration approach (Avila et al. 2001a). (C) Average maximal caffeine-induced Ca2+ release (Δ Ratio = Rcaffeine − Rbaseline). Asterisks indicate significant differences (P < 0.05) compared with RyR1.
Figure 2.
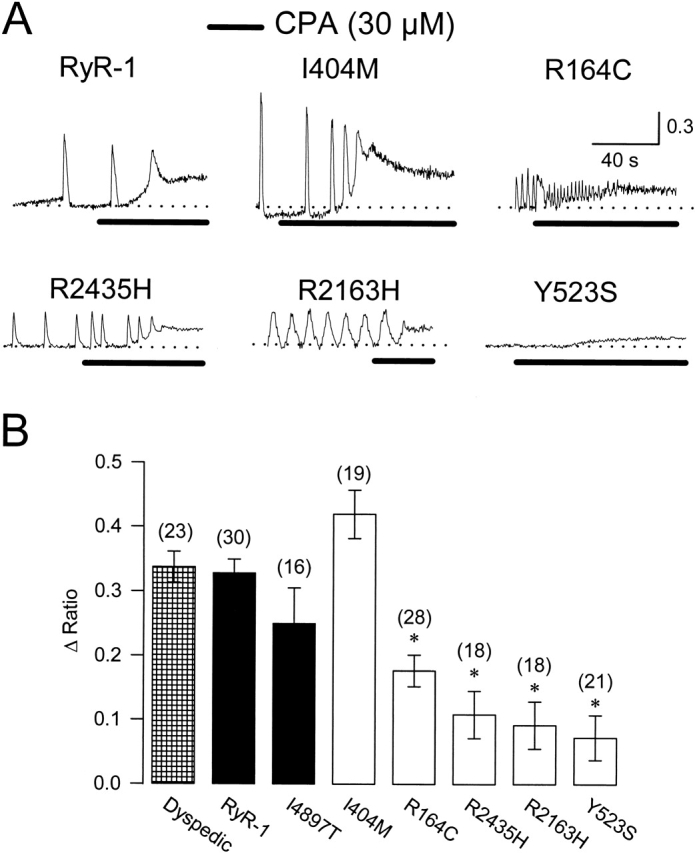
Intact dyspedic myotubes expressing NH2-terminal CCD mutants exhibit reduced CPA-induced Ca2+ release. (A) Representative CPA-induced Ca2+ responses (30 μM, black bars) in Indo-1 AM–loaded dyspedic myotubes expressing wild-type RyR1, I404M, R164C, R2435H, R2163H, or Y523S. For comparison, fluorescence ratios are aligned to their respective baseline levels (dotted lines). (B) Average steady-state CPA-induced Ca2+ release (Δ Ratio = RCPA − Rbaseline) measured 2–3 min after the initial application of CPA. Asterisks indicate significant differences (P < 0.05) compared with RyR-1.
Simultaneous Measurements of Macroscopic Ca2+ Currents and Ca2+ Transients
The whole-cell patch-clamp technique was used to simultaneously measure voltage-gated L-currents and Ca2+ transients in expressing myotubes (Avila and Dirksen 2000; Avila et al. 2001a,Avila et al. 2001b). Sodium and T-type Ca2+ currents were eliminated using a standard prepulse protocol consisting of a 1-s depolarization usually to −30 mV followed by a 25-ms repolarization to −50 mV before each test pulse (Avila and Dirksen 2000). L-currents were leak subtracted (−P/3) and normalized to cell capacitance (pA/pF). Peak L-currents were plotted as a function of test potential and fitted according to:
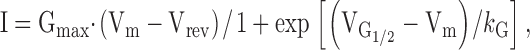 |
1 |
where Vrev is the extrapolated reversal potential of the Ca2+ current, Vm is the membrane potential during the test pulse, Gmax is the maximum L-channel conductance, 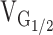 is the voltage for half activation of Gmax, and k
G is a slope factor. Relative changes in cytosolic Ca2+ in patch-clamp experiments were recorded in Fluo-3-dialyzed myotubes. Fluorescence traces were analogue-filtered (τ = 0.5 ms) before digitization (10 kHz), and subsequently expressed as ΔF/F. Amplitudes at the end of each test pulse were plotted as a function of the membrane potential, and fitted according to:
is the voltage for half activation of Gmax, and k
G is a slope factor. Relative changes in cytosolic Ca2+ in patch-clamp experiments were recorded in Fluo-3-dialyzed myotubes. Fluorescence traces were analogue-filtered (τ = 0.5 ms) before digitization (10 kHz), and subsequently expressed as ΔF/F. Amplitudes at the end of each test pulse were plotted as a function of the membrane potential, and fitted according to:
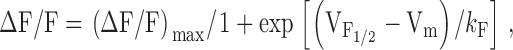 |
2 |
where (ΔF/F)max is the calculated maximal fluorescence change during the test pulse, 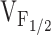 is the midpoint potential, and k
F is a slope factor.
is the midpoint potential, and k
F is a slope factor.
Recording Solutions
Cytosolic Ca2+ levels were monitored in myotubes bathed in normal rodent Ringer containing the following (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 10 HEPES, pH 7.4. For measurements of macroscopic Ca2+ currents and intracellular Ca2+ transients, the pipette solution contained the following (in mM): 145 cesium aspartate, 10 CsCl, 0.1 Cs2-EGTA, 1.2 MgCl2, 5 Mg-ATP, 0.2 K5-Fluo-3 (Molecular Probes), and 10 HEPES, pH 7.4. In these experiments, the external solution contained the following (in mM): 145 TEA-Cl, 10 CaCl2, 0.003 TTX, and 10 HEPES, pH 7.4. Except where noted, all chemical reagents were obtained from Sigma-Aldrich.
RESULTS
Effects of CCD Mutations on Resting Ca2+ and SR Ca2+ Content
Compared with wild-type RyR1, HEK-293 cells expressing NH2-terminal CCD mutations in RyR1 exhibited elevated resting Ca2+ levels and a reduction in the Ca2+ content of the ER (Tong et al. 1999). To evaluate the functional impact of the CCD mutations on Ca2+ dynamics in skeletal muscle, we expressed wild-type RyR1 and five of the different NH2-terminal CCD mutations in RyR1 (I404M, R164C, R2435H, R2163H, and Y523S) in dyspedic myotubes (which lack RyR1 proteins). Resting Ca2+ and caffeine responses were evaluated in intact Indo-1 AM–loaded myotubes (Fig. 1). RyR1- and I4897T-expressing dyspedic myotubes exhibited similar resting Ca2+ levels, which were higher than that of uninjected myotubes (Fig. 1 B; Avila et al. 2001a). Interestingly, dyspedic myotubes expressing four of the five different NH2-terminal CCD mutations in RyR1 (R164C, R2435H, R2163H, and Y523S, but not I404M) exhibited significant (P < 0.05) elevations in resting Ca2+ (Fig. 1A and Fig. B) according to the following rank order: Y523S > R2163H > R2435H > R164C > I404M ∼ RyR1. For clarity, the data for each of the different constructs in Fig. 1 Fig. 2 Fig. 3 Fig. 4 are presented using this rank order. Under control conditions, some of the myotubes expressing either wild-type RyR1 or the different NH2-terminal CCD mutants exhibited spontaneous intracellular Ca2+ transients that varied in frequency, amplitude, and duration (Fig. 1 A). The percentages of myotubes exhibiting spontaneous Ca2+ oscillations during the first ∼60 s of recording were the following: 33% (14/43), 25% (5/20), 54% (15/28), 65% (13/20), 54% (15/28), and 10% (2/20), for RyR1-, I404M-, R164C-, R2435H-, R2163H-, and Y523S-expressing myotubes, respectively. In contrast, none of the I4897T-expressing myotubes (0/16) or uninjected dyspedic myotubes (0/33) studied exhibited spontaneous Ca2+ oscillations. In general, resting Ca2+ levels were elevated and Ca2+ oscillations were smaller and of higher frequency in the NH2-terminal CCD-expressing myotubes (Fig. 1 A and 2 A). However, since the frequency and magnitude of Ca2+ oscillations could be influenced by differences in spontaneous electrical activity, the patterns of these Ca2+ oscillations for the different constructs were not characterized in greater detail. Interestingly, a similar pattern of Ca2+ oscillations were also observed at the holding potential (−80 mV) in whole-cell patch-clamp experiments (data not shown), suggesting that increased spontaneous release does not solely arise from elevated intracellular Ca2+ levels or membrane depolarization.
Figure 3.

The NH2-terminal CCD mutants fully restore retrograde coupling. (A) Representative whole-cell L-currents recorded in response to 30-ms depolarizing pulses to the indicated membrane potentials (left). (B) Average peak I-V curves for dyspedic myotubes expressing wild-type RyR1 (n = 20), I404M (n = 7), R164C (n = 10), R2435H (n = 8), R2163H (n = 6), or Y523S (n = 11). The average values (±SEM) for the parameters obtained by fitting each myotube within a group separately to are given in Table (I–V data). The solid lines through the data were generated using and the corresponding parameters given in Table ([dashed line] RyR1 and [continuous line] CCD mutants).
Figure 4.

The NH2-terminal CCD mutants increase SR Ca2+ release channel sensitivity to activation by voltage and reduce maximal voltage-gated SR Ca2+ release. (A) Intracellular Ca2+ transients (ΔF/F) elicited by 30-ms test pulses to the indicated potentials (left). (B) Average voltage dependence of peak intracellular Ca2+ transients as recorded in A. The average values (±SEM) for the parameters obtained by fitting each myotube within a group separately to are given in Table (ΔF/F -V data). The solid lines through the data were generated using and the corresponding parameters given in Table ([dashed line] RyR1 and [continuous lines] CCD mutants). (C) The ΔF/F-V data and curves in B were normalized to their respective maximal value ((ΔF/F)max) ([dashed line] RyR-1 and [continuous lines] CCD mutants). The decline in the magnitude of the Y523S Ca2+ transients at potentials greater than +30 mV arises from a reduction in L-current magnitude at these potentials.
The trend for the different CCD mutations in RyR1 to cause an elevation in resting Ca2+ as well as smaller amplitude Ca2+ oscillations is consistent with the notion that the CCD mutations lead to the expression of leaky SR Ca2+ release channels. Since leaky SR Ca2+ release channels may result in SR Ca2+ store depletion we evaluated SR Ca2+ content after application of a maximal concentration of caffeine (10 mM; Fig. 1 A). Dyspedic myotubes expressing four of the five different NH2-terminal CCD mutant RyR1 proteins (R164C, R2435H, R2163H, and Y523S) exhibited a significantly smaller response to caffeine compared with that of wild-type RyR1 (P < 0.05; Fig. 1). Interestingly, repetitive Ca2+ oscillations were typically observed throughout the caffeine application in 90% (17/19) of RyR1-expressing myotubes. However, repetitive caffeine-induced Ca2+ oscillations were less frequent: 43% (6/14), 31% (4/13), 43% (3/7), 0% (0/11), and 0% (0/8) for I404M, R164C, R2435H, R2163H, and Y523S, respectively. A reduction in repetitive Ca2+ oscillations could arise from differences in the caffeine-induced plateau Ca2+ level or reductions in either SR Ca2+ content and/or release channel inhibition by high Ca2+ (Fill et al. 1990).
The data presented in Fig. 1 suggest that expression of NH2-terminal CCD mutant RyR1 proteins in dyspedic myotubes results in a significant depletion of SR Ca2+ stores. This idea was further tested by evaluating SR Ca2+ content using a method to release Ca2+ by a mechanism that is independent of RyR1 activation (Fig. 2). For these experiments, luminal SR Ca2+ stores were assessed after treatment with CPA, an agent that increases cytosolic Ca2+ by reversibly inhibiting SR Ca2+-ATPase pumps, and thereby prevents the reuptake of Ca2+ lost through passive leak pathways. Application of CPA (Fig. 2, 30 μM, black bars) induced similar increases in cytosolic Ca2+ in intact dyspedic myotubes either expressing RyR1 or I404M. However, dyspedic myotubes expressing the other NH2-terminal CCD mutants exhibited significantly smaller elevations in cytosolic Ca2+ after application of CPA, which is consistent with these mutants reducing luminal SR Ca2+ levels. Although caffeine fails to activate Ca2+ release in I4897T-expressing myotubes (Fig. 1 C), these myotubes exhibit a resting Ca2+ level and CPA-sensitive Ca2+ store comparable to that of RyR1-expressing myotubes (Fig. 1 A and 2 B; Avila et al. 2001a). These results indicate that I4897T-containing release channels do not result in the formation of overactive, or leaky Ca2+ release channels, but rather release Ca2+ inefficiently after activation (by caffeine or voltage).
Effects on Retrograde and Orthograde Signals of EC Coupling
EC coupling in skeletal muscle involves a unique bidirectional signaling interaction between RyR1s and DHPRs. Accordingly, while DHPRs trigger the activation of RyR1 proteins in response to a sarcolemmal depolarization (orthograde coupling), the functional activity of the DHPR (retrograde coupling) is in turn strongly influenced by the presence of RyR1 (Nakai et al. 1996; Grabner et al. 1999; Avila and Dirksen 2000). We used the whole-cell patch-clamp technique in conjunction with a Ca2+-sensitive dye to evaluate the effects of the different NH2-terminal CCD mutations in RyR1 on the retrograde (Fig. 3) and orthograde (Fig. 4) signals of skeletal muscle EC coupling. Uninjected dyspedic myotubes exhibit a very low L-current density and completely lack voltage-gated SR Ca2+ release. Reintroduction of RyR1 proteins restores both robust L-currents and voltage-gated SR Ca2+ release (Nakai et al. 1996). Dyspedic myotubes expressing each of the different NH2-terminal CCD mutant RyR1s exhibited L-currents of similar magnitude, kinetics, and voltage dependence as those recorded from wild-type RyR1-expressing dyspedic myotubes (Fig. 3). These results indicate that each of the mutants are synthesized and targeted to sarcolemmal–SR junctions and are able to functionally interact with DHPRs present within the junctions. Moreover, the results indicate that none of the NH2-terminal CCD mutations in RyR1 markedly influence retrograde coupling between DHPR and RyR1 proteins.
Although the NH2-terminal CCD mutations in RyR1 did not alter retrograde coupling, significant effects on orthograde coupling were observed (Fig. 4). Fig. 4 A illustrates representative voltage-gated Ca2+ transients recorded under whole-cell voltage clamp for dyspedic myotubes expressing wild-type RyR1 and each of the different CCD mutant RyR1 proteins. In general, voltage-gated Ca2+ transients attributable to the NH2-terminal CCD mutant release channels were smaller and activated at more negative potentials than Ca2+ transients arising from wild-type RyR1. This can best be appreciated by comparing voltage-gated Ca2+ transients arising from RyR1 (Fig. 4 A, upper left) and Y523S (Fig. 4 A, lower right). Even though the Y523S transients illustrated in Fig. 4 A were the largest we recorded (n = 11), the maximal transient (at +70 mV) for this construct was still more than three times smaller than that of wild-type RyR1. In addition, the threshold for activation of Y523S-expressing myotubes was ∼40 mV more hyperpolarized than that of RyR1-expressing myotubes. The other NH2-terminal CCD mutations in RyR1 also resulted in reductions in maximal voltage-gated Ca2+ release and a negative shift in the voltage dependence of release, although to a lesser extent that Y523S. Thus, although the largest maximal voltage-gated Ca2+ transients were recorded from wild-type RyR1-expressing myotubes, the NH2-terminal CCD mutants actually exhibited larger Ca2+ transients at threshold potentials. For example, the Ca2+ transient amplitudes at −10 mV were the following (in ΔF/F): 0.04 ± 0.02, 0.11 ± 0.05, 0.72 ± 0.19, 0.33 ± 0.10, 1.18 ± 0.45, and 0.27 ± 0.15, for RyR1, I404M, R164C, R2435H, R2163H, and Y523S, respectively. To provide a more quantitative description of this observation, Boltzmann fits to the voltage dependence of SR Ca2+ release for each construct were used to determine the maximal change in fluorescence (ΔF/Fmax), the half-maximal activation voltage (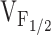 ), and voltage sensitivity (k
F; Fig. 4 B). This analysis revealed that each of the NH2-terminal CCD
), and voltage sensitivity (k
F; Fig. 4 B). This analysis revealed that each of the NH2-terminal CCD 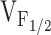 mutations in RyR1 caused a significant negative shift in without changing k
F (Table ). The various degrees of hyperpolarizing shift in the activation of voltage-gated SR Ca2+ release caused by the different NH2-terminal CCD mutations in RyR1 are best appreciated by comparing the normalized voltage dependence of release for each mutant to that of wild-type RyR1 (Fig. 4 C).
mutations in RyR1 caused a significant negative shift in without changing k
F (Table ). The various degrees of hyperpolarizing shift in the activation of voltage-gated SR Ca2+ release caused by the different NH2-terminal CCD mutations in RyR1 are best appreciated by comparing the normalized voltage dependence of release for each mutant to that of wild-type RyR1 (Fig. 4 C).
Table 1.
Parameters of Fitted I-V and ΔF/F-V Curves
| I-V Data | ΔF/F-V Data | |||||||
|---|---|---|---|---|---|---|---|---|
| n | Gmax |
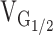
|
k G | vrev | (ΔF/F)max |
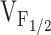
|
k F | |
| nS/nF | mV | mV | mV | mV | mV | |||
| RyR-1 | 20 | 126 ± 9.1 | 17.7 ± 1.0 | 6.1 ± 0.1 | 77 ± 1.4 | 2.8 ± 0.3 | 9.5 ± 1.1 | 5.4 ± 0.4 |
| 1404M | 7 | 126 ± 20 | 19.8 ± 2.8 | 6.8 ± 0.3 | 80 ± 1.8 | 2.7 ± 0.6 | 5.0 ± 2.4 | 5.2 ± 0.8 |
| R164C | 10 | 125 ± 11 | 20.5 ± 1.7 | 7.2 ± 0.3 | 75 ± 3.0 | 2.0 ± 0.4 | −5.6 ± 2.8 | 6.0 ± 0.5 |
| R2435H | 8 | 123 ± 12 | 23.2 ± 1.5 | 8.1 ± 0.2 | 80 ± 4.1 | 1.5 ± 0.3 | −1.0 ± 2.6 | 6.0 ± 0.8 |
| R2163H | 6 | 153 ± 16 | 22.1 ± 2.2 | 7.5 ± 0.4 | 74 ± 6.9 | 2.3 ± 0.5 | −9.4 ± 2.2 | 5.0 ± 0.3 |
| Y523S | 11 | 119 ± 16 | 18.1 ± 1.4 | 8.5 ± 0.2 | 85 ± 2.2 | 0.3 ± 0.1 | −21 ± 6.1 | 5.1 ± 1.1 |
| RYR-1/Y523S | 13 | 139 ± 14 | 15.4 ± 1.0 | 7.4 ± 0.2 | 72 ± 2.8 | 1.3 ± 0.3 | −7.5 ± 2.6 | 6.4 ± 0.7 |
Since individuals with CCD are typically heterozygous for the causative RyR1 mutation, we also investigated the effects of coexpression of RyR1 and Y523S in dyspedic myotubes (Fig. 5). We evaluated the impact of RyR1/Y523S coexpression since this mutation resulted in the most severe disruption in Ca2+ homeostasis (Fig. 1 and Fig. 2) and voltage-gated SR Ca2+ release (Fig. 4). Not surprisingly, RyR1/Y523S-expressing dyspedic myotubes displayed L-currents with a similar magnitude, kinetics, and voltage dependence as those recorded from homozygous RyR1-expressing myotubes (Fig. 5a and Fig. B, and Table ). However, RyR1/Y523S-expressing myotubes exhibited the following: only a moderate elevation in resting Ca2+ (for RyR1, 49 ± 7.8 nM, n = 43; for RyR1/Y523S, 94 ± 11 nM, n = 23); and voltage-gated Ca2+ transients that were intermediate in magnitude and voltage dependence to those arising from homozygous expression of either construct (Fig. 5C and Fig. D). The effects on voltage-gated Ca2+ release are qualitatively similar to those observed after coexpression of RyR1/I4897T (Avila et al. 2001a). Thus, although voltage-gated SR Ca2+ release is reduced after coexpression of both Y523S and I4897T, the mutations differ with regard to their effects on resting 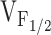 Ca2+ and SR Ca2+ content (Avila et al. 2001a).
Ca2+ and SR Ca2+ content (Avila et al. 2001a).
Correlation between Effects on Ca2+ Dynamics and Release Channel Sensitivity to Activation by Voltage (V F12)
To test whether changes in release channel sensitivity to voltage (i.e., 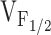 ) might account for the observed changes in resting Ca2+, SR Ca2+ content, and maximal voltage-gated Ca2+ release, a linear correlation analysis was conducted for the data obtained from dyspedic myotubes expressing RyR1 and each of the NH2-terminal CCD mutations (Fig. 6). The elevation in resting Ca2+ correlated strongly with the degree of shift in
) might account for the observed changes in resting Ca2+, SR Ca2+ content, and maximal voltage-gated Ca2+ release, a linear correlation analysis was conducted for the data obtained from dyspedic myotubes expressing RyR1 and each of the NH2-terminal CCD mutations (Fig. 6). The elevation in resting Ca2+ correlated strongly with the degree of shift in 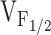 (Fig. 6 A, r = 0.97, P < 0.05). In addition, myotube responsiveness to maximal concentrations of both caffeine (Fig. 6 B, r = 0.97, P < 0.05) and CPA (Fig. 6 C, r = 0.80, P = 0.05), two different means of assessing SR Ca2+ content, also correlated well with
(Fig. 6 A, r = 0.97, P < 0.05). In addition, myotube responsiveness to maximal concentrations of both caffeine (Fig. 6 B, r = 0.97, P < 0.05) and CPA (Fig. 6 C, r = 0.80, P = 0.05), two different means of assessing SR Ca2+ content, also correlated well with 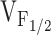 . Finally, the degree of reduction in maximal voltage-gated SR Ca2+ release also correlated with release channel sensitivity to activation by voltage (Fig. 6 D, r = 0.86, P < 0.05) across the different RyR1 constructs. Although these correlational analyses fall short of establishing causation, the results suggest that increased release channel sensitivity to activation by voltage contributes to the enhanced SR Ca2+ leak, Ca2+ store depletion, and a reduction in Ca2+ released during EC coupling in dyspedic myotubes expressing the different NH2-terminal CCD mutations in RyR1.
. Finally, the degree of reduction in maximal voltage-gated SR Ca2+ release also correlated with release channel sensitivity to activation by voltage (Fig. 6 D, r = 0.86, P < 0.05) across the different RyR1 constructs. Although these correlational analyses fall short of establishing causation, the results suggest that increased release channel sensitivity to activation by voltage contributes to the enhanced SR Ca2+ leak, Ca2+ store depletion, and a reduction in Ca2+ released during EC coupling in dyspedic myotubes expressing the different NH2-terminal CCD mutations in RyR1.
Figure 6.

The degree of altered release channel activity for the different NH2-terminal CCD mutations in RyR1 correlates with a hyperpolarizing shift in 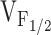 . Average resting Ca2+ levels (A), maximal caffeine response (B), maximal CPA response (C), and maximal voltage-gated Ca2+ release (D) are plotted as a function of
. Average resting Ca2+ levels (A), maximal caffeine response (B), maximal CPA response (C), and maximal voltage-gated Ca2+ release (D) are plotted as a function of 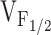 for RyR1 and the different NH2-terminal CCD mutants. Symbols represent RyR1 (closed circle), R164C (open diamond), I404M (open triangle), Y523S (open circle), R2163H (open square), R2435H (inverted open triangle), and RyR1/Y523S (dotted closed circle).
for RyR1 and the different NH2-terminal CCD mutants. Symbols represent RyR1 (closed circle), R164C (open diamond), I404M (open triangle), Y523S (open circle), R2163H (open square), R2435H (inverted open triangle), and RyR1/Y523S (dotted closed circle).
DISCUSSION
Functional Impact of NH2-terminal CCD Mutations on RyR1 Activity in Dyspedic Myotubes
Our experiments are the first to characterize the relative effects of the different NH2-terminal CCD mutations in RyR1 on release channel activity operating within a skeletal muscle environment. The results indicate that dyspedic myotubes expressing the different NH2-terminal CCD mutations in RyR1 exhibit different degrees of SR Ca2+ leak as judged by the presence of parallel, but opposing, changes in resting Ca2+ and SR Ca2+ content. None of the NH2-terminal CCD mutants markedly altered L-current magnitude, kinetics, or voltage dependence (Fig. 3). However, the NH2-terminal CCD mutations reduced maximal voltage-gated Ca2+ release and caused a negative shift in 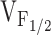 (Fig. 4), indicating that the CCD mutations in RyR1 selectively alter the orthograde signal of skeletal muscle EC coupling. The degree of reduction in voltage-gated release correlated well with the shift in
(Fig. 4), indicating that the CCD mutations in RyR1 selectively alter the orthograde signal of skeletal muscle EC coupling. The degree of reduction in voltage-gated release correlated well with the shift in 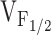 across the different NH2-terminal CCD mutations in RyR1. These data are the first to suggest that increased release channel activity at negative membrane potentials results in an elevation in resting Ca2+ and a partial reduction in SR Ca2+ content. In spite of these correlations, we should point out that our experiments have not definitively demonstrated that the observed increased sensitivity to activation by voltage (i.e., negative shift in
across the different NH2-terminal CCD mutations in RyR1. These data are the first to suggest that increased release channel activity at negative membrane potentials results in an elevation in resting Ca2+ and a partial reduction in SR Ca2+ content. In spite of these correlations, we should point out that our experiments have not definitively demonstrated that the observed increased sensitivity to activation by voltage (i.e., negative shift in 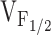 ) observed for the NH2-terminal CCD mutants is directly responsible for an increase in SR Ca2+ leak.
) observed for the NH2-terminal CCD mutants is directly responsible for an increase in SR Ca2+ leak.
Two Distinct Cellular Mechanisms for Muscle Weakness in CCD
Since our results obtained with the NH2-terminal CCD mutants are in contrast to those obtained after expression of I4897T (Avila et al. 2001a), we propose two distinct cellular mechanisms to account for muscle weakness in CCD (Fig. 7). The models presented in Fig. 7 are based on the 5-state kinetic model that was recently used to account for the voltage dependence of both L-currents and SR Ca2+ release in porcine skeletal myotubes homozygously expressing the R615C MH mutation (Dietze et al. 2000). In this model, the equilibrium constant (Fig. 7, “K”) governing a voltage-independent transition of the release channel is linked to ligand-modulated gating of RyR1, such that triggering agents (e.g., caffeine, halothane, and voltage sensor) promote release channel opening by reducing the value of K (i.e., stabilizing the open state of the release channel). Accordingly, an eightfold reduction in K quantitatively accounts for the selective hyperpolarizing shift in 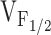 observed in porcine myotubes (Dietze et al. 2000). The beauty of this model is that alterations in a single voltage-independent equilibrium constant occurs independent of the precise molecular mechanism by which the DHPR and RyR1 are coupled. Thus, mutations in vastly different regions of RyR1 that exert different degrees of influence on K would result in variable, but selective, alterations in
observed in porcine myotubes (Dietze et al. 2000). The beauty of this model is that alterations in a single voltage-independent equilibrium constant occurs independent of the precise molecular mechanism by which the DHPR and RyR1 are coupled. Thus, mutations in vastly different regions of RyR1 that exert different degrees of influence on K would result in variable, but selective, alterations in 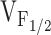 . Our finding of variable shifts in
. Our finding of variable shifts in 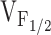 (ranging from 5 to 31 mV) caused by the NH2-terminal CCD mutations in RyR1 could, therefore, arise from variable reductions in the value of K.
(ranging from 5 to 31 mV) caused by the NH2-terminal CCD mutations in RyR1 could, therefore, arise from variable reductions in the value of K.
Figure 7.

Schematic representation for alterations in Ca2+ homeostasis and EC coupling in dyspedic myotubes expressing normal (left, RyR1), leaky (middle, Y523S), and EC uncoupled (right, I4897T) SR Ca2+ release channels. Each model is based on the 5-state reaction scheme for coupled DHPRs and RyR1s (Dietze et al. 2000). A thin black line connecting DHPR (black channels) and RyR1 proteins (white and gray channels) is used to represent mechanical coupling. The number of Ca2+ symbols represents relative myoplasmic and luminal SR Ca2+ levels. In each scheme, L-channels are assumed to exist in one of three distinct states governed by two voltage-dependent transitions (V12 and V23): (1) resting (closed), (2) preactive (closed), and (3) open (open). In each of these states, coupled SR Ca2+ release channels can exist in either a closed (states 1–3) or open configuration (states 4 and 5). A reduction in the voltage-independent equilibrium constant (K) for the release channel open-closed reaction causes a selective negative shift in 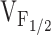 without an alteration in
without an alteration in 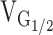 . The selective, but variable shifts in
. The selective, but variable shifts in 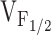 produced by the different NH2-terminal CCD mutations in RyR1, could arise from different degrees of reductions in K in the model for Y523S. Increased release channel activity at negative potentials could result in SR Ca2+ leak and the subsequent depletion of SR Ca2+ stores. Our previous results demonstrated that I4897T-expressing myotubes lack voltage-gated SR Ca2+ release in the absence of a change in either resting Ca2+ levels or SR Ca2+ content (Avila et al. 2001a). However, the I4897T mutation apparently does not alter K, since the value of
produced by the different NH2-terminal CCD mutations in RyR1, could arise from different degrees of reductions in K in the model for Y523S. Increased release channel activity at negative potentials could result in SR Ca2+ leak and the subsequent depletion of SR Ca2+ stores. Our previous results demonstrated that I4897T-expressing myotubes lack voltage-gated SR Ca2+ release in the absence of a change in either resting Ca2+ levels or SR Ca2+ content (Avila et al. 2001a). However, the I4897T mutation apparently does not alter K, since the value of 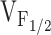 was similar for dyspedic myotubes expressing RyR1 alone and both RyR1 and I4897T. Thus, the I4897T mutation may result in release channels that conduct Ca2+ poorly after activation (depicted as gray release channels). Both cellular mechanisms (“leaky” and “uncoupled” SR Ca2+ release channels) would be expected to result in muscle weakness as a consequence of reduced voltage-gated SR Ca2+ release. Disorganization of the contractile proteins and a reduction in energy supply within the core regions are also likely to contribute to muscle weakness in CCD (Loke and MacLennan 1998; MacLennan and Phillips 1995).
was similar for dyspedic myotubes expressing RyR1 alone and both RyR1 and I4897T. Thus, the I4897T mutation may result in release channels that conduct Ca2+ poorly after activation (depicted as gray release channels). Both cellular mechanisms (“leaky” and “uncoupled” SR Ca2+ release channels) would be expected to result in muscle weakness as a consequence of reduced voltage-gated SR Ca2+ release. Disorganization of the contractile proteins and a reduction in energy supply within the core regions are also likely to contribute to muscle weakness in CCD (Loke and MacLennan 1998; MacLennan and Phillips 1995).
We propose two fundamentally distinct cellular mechanisms (leaky channels and EC uncoupling) to explain how altered SR Ca2+ release channel activity caused by different mutations in RyR1 may result in muscle weakness in CCD (Fig. 7). The three basic schemes presented in Fig. 7 are based on our results obtained from expression of RyR1, Y523S, and I4898T in dyspedic myotubes and are based on the 5-state kinetic model of DHPR-RyR1 coupled gating (Dietze et al. 2000). Because RyR1 channel opening is coupled to the voltage dependence of L-channel opening in this model, changes in the value of the voltage-independent equilibrium constant K result in a selective shift of 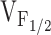 for Ca2+ release without significantly altering the voltage dependence of L-channel opening. Thus, our finding of a selective shift in
for Ca2+ release without significantly altering the voltage dependence of L-channel opening. Thus, our finding of a selective shift in 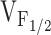 with expression of Y523S mutation in RyR1 (as well as R2163H, RyR1/Y523S, R164C, R2435H, and I404M) in dyspedic myotubes can also be explained by a reduction in K. The increased sensitivity of Y523S-containing release channels to activation at negative voltages should result in a partial depletion of the SR Ca2+ store. If store content is reduced enough, SR Ca2+ release elicited by a single action potential would be smaller than usual, thus, resulting in muscle weakness. Coexpression of Y523S with RyR1 resulted in an intermediate shift in
with expression of Y523S mutation in RyR1 (as well as R2163H, RyR1/Y523S, R164C, R2435H, and I404M) in dyspedic myotubes can also be explained by a reduction in K. The increased sensitivity of Y523S-containing release channels to activation at negative voltages should result in a partial depletion of the SR Ca2+ store. If store content is reduced enough, SR Ca2+ release elicited by a single action potential would be smaller than usual, thus, resulting in muscle weakness. Coexpression of Y523S with RyR1 resulted in an intermediate shift in 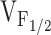 , a more moderate reduction in peak voltage-gated SR Ca2+ release, and a resting Ca2+ level that was more than RyR1 but less than Y523S. Thus, similar to our previous findings regarding I4897T, the Y523S mutation in RyR1 exerts a dominant negative action on the activity of the resulting SR Ca2+ release channels, which is consistent with the autosomal dominant pattern of inheritance of this human myopathy (Isaacs et al. 1975).
, a more moderate reduction in peak voltage-gated SR Ca2+ release, and a resting Ca2+ level that was more than RyR1 but less than Y523S. Thus, similar to our previous findings regarding I4897T, the Y523S mutation in RyR1 exerts a dominant negative action on the activity of the resulting SR Ca2+ release channels, which is consistent with the autosomal dominant pattern of inheritance of this human myopathy (Isaacs et al. 1975).
We have previously reported that expression in dyspedic myotubes of release channels harboring the I4898T CCD mutation do not function as leaky SR Ca2+ release channels, but rather reflect a functional uncoupling of sarcolemmal excitation from SR Ca2+ release (EC uncoupling) (Avila et al. 2001a). This assertion was based on the observation that I4897T-expressing dyspedic myotubes lack voltage-gated SR Ca2+ release (orthograde coupling) in spite of the presence of normal resting intracellular Ca2+ levels, CPA-responsiveness, and DHPR channel activity (retrograde coupling). Although coexpression of I4897T and RyR1 only partially restored orthograde coupling, the 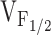 values of RyR1- and RyR1/I4897T-expressing dyspedic myotubes did not differ significantly (Avila et al. 2001a). Taken together, these data indicate that the effects of the I4897T CCD mutation cannot be described by a simple reduction in
values of RyR1- and RyR1/I4897T-expressing dyspedic myotubes did not differ significantly (Avila et al. 2001a). Taken together, these data indicate that the effects of the I4897T CCD mutation cannot be described by a simple reduction in 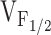 , and thus, likely involves a fundamentally distinct mechanism than that of the NH2-terminal CCD mutations. One possibility is that the lack of voltage-gated Ca2+ release after homozygous expression of I4897T could arise from a selective disruption in orthograde coupling, such that the transition governed by K becomes essentially nonpermissible (i.e., K approaches infinity; not depicted in Fig. 7). A reduction in the maximal voltage-gated Ca2+ transient occurring in the absence of a change in
, and thus, likely involves a fundamentally distinct mechanism than that of the NH2-terminal CCD mutations. One possibility is that the lack of voltage-gated Ca2+ release after homozygous expression of I4897T could arise from a selective disruption in orthograde coupling, such that the transition governed by K becomes essentially nonpermissible (i.e., K approaches infinity; not depicted in Fig. 7). A reduction in the maximal voltage-gated Ca2+ transient occurring in the absence of a change in 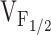 after RyR1/I4897T coexpression could arise from certain heteromeric release channel combinations (e.g., consisting predominantly of RyR1) that undergo the transition governed by K normally, whereas other combinations (e.g., consisting predominantly of I4897T) almost never undergo this transition. Alternatively, the I4897T mutation may drastically alter release channel permeation and/or gating in such a way that Ca2+ flux through activated channels is severely compromised (Fig. 7 C, shaded release channels). Thus, release channels comprised of I4897T subunits may undergo the voltage-independent activating transition governed by K normally, but Ca2+ may only conduct poorly through activated I4897T release channels. Recent findings that the I4897T residue contributes to a conserved RyR pore-forming sequence (Balshaw et al. 1999; Zhao et al. 1999; Gao et al. 2000) and that conservative mutations I4897 result in marked reductions in single-channel conductance, activation by Ca2+, and Ca2+ permeation (Gao et al. 2000) supports the latter interpretation. In this case, heteromeric release channels that permit intermediate degrees of overall Ca2+ permeation could explain the partial restoration of release channel activity after coexpression of RyR1 and I4897T in dyspedic myotubes. Thus, we propose two parallel but converging mechanisms to account for muscle weakness caused by NH2- and COOH-terminal (I4897T) CCD mutations in RyR1 (Fig. 7). Clearly, it will be important for future work to determine whether newly identified CCD mutations in RyR1 expressed in dyspedic myotubes also result in “leaky” or “EC uncoupled” SR Ca2+ release channels.
after RyR1/I4897T coexpression could arise from certain heteromeric release channel combinations (e.g., consisting predominantly of RyR1) that undergo the transition governed by K normally, whereas other combinations (e.g., consisting predominantly of I4897T) almost never undergo this transition. Alternatively, the I4897T mutation may drastically alter release channel permeation and/or gating in such a way that Ca2+ flux through activated channels is severely compromised (Fig. 7 C, shaded release channels). Thus, release channels comprised of I4897T subunits may undergo the voltage-independent activating transition governed by K normally, but Ca2+ may only conduct poorly through activated I4897T release channels. Recent findings that the I4897T residue contributes to a conserved RyR pore-forming sequence (Balshaw et al. 1999; Zhao et al. 1999; Gao et al. 2000) and that conservative mutations I4897 result in marked reductions in single-channel conductance, activation by Ca2+, and Ca2+ permeation (Gao et al. 2000) supports the latter interpretation. In this case, heteromeric release channels that permit intermediate degrees of overall Ca2+ permeation could explain the partial restoration of release channel activity after coexpression of RyR1 and I4897T in dyspedic myotubes. Thus, we propose two parallel but converging mechanisms to account for muscle weakness caused by NH2- and COOH-terminal (I4897T) CCD mutations in RyR1 (Fig. 7). Clearly, it will be important for future work to determine whether newly identified CCD mutations in RyR1 expressed in dyspedic myotubes also result in “leaky” or “EC uncoupled” SR Ca2+ release channels.
Comparison of Results with Previous Reports
Due to the presence of the porcine model for MH, the functional effects of the R615C MH mutation in RyR1 has been the most thoroughly characterized of the different disease mutations in RyR1 (for reviews see Mickelson and Louis 1996; Loke and MacLennan 1998; Jurkat-Rott et al. 2000; McCarthy et al. 2000). In skeletal muscle bundles and myotubes obtained from MH pigs, contractions exhibit enhanced sensitivity to activation via both caffeine and sarcolemmal depolarization (Gallant and Lentz 1992; Gallant and Jordan 1996). The increased contractile sensitivity to depolarization arises from a hyperpolarizing shift in the voltage dependence of SR Ca2+ release (Dietze et al. 2000) that occurs in the absence of an effect on the magnitude or voltage dependence of L-currents (Dietze et al. 2000; Gallant et al. 1996; Gallant and Jordan 1996). These abnormalities, which may be potentiated by inhalation anesthetics and depolarizing skeletal muscle relaxants, would be anticipated to result in supersensitive or overactive SR Ca2+ release channels. Our results are the first to demonstrate that the NH2-terminal CCD mutations in RyR1 also shift 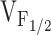 without markedly altering the voltage dependence of L-channel activation.
without markedly altering the voltage dependence of L-channel activation.
Heterologous expression of MH and CCD mutations in nonmuscle cells has been an alternate approach used to compare the functional properties of mutant release channels (Treves et al. 1994; Lynch et al. 1999; Tong et al. 1997, Tong et al. 1999; Monnier et al. 2000). The studies of Tong et al. 1997, Tong et al. 1999 represent the most thorough comparison of the functional impact of the different MH and CCD mutations in RyR1 evaluated in the context of a single genetic background. In these studies, resting Ca2+ levels, Ca2+ content of the ER, and release channel sensitivity to caffeine and halothane were monitored in HEK-293 cells expressing 15 of the different MH and CCD mutations in RyR1. These studies found that the MH and CCD mutant RyR1 proteins exhibited enhanced sensitivity to caffeine and halothane (Tong et al. 1997), as well as variable degrees of both store depletion and elevations in resting Ca2+ (Tong et al. 1999). These results indicate that the MH/CCD mutations in RyR1 result in “leaky” intracellular Ca2+ release channels. In particular, the Y523S and R2163H CCD mutants represented the most leaky channels, since they exhibited the largest degree of store depletion and corresponding elevations in resting Ca2+. Interestingly, our results in dyspedic myotubes also indicate that the Y523S and R2163H mutations result in the most severe disruption of SR Ca2+ release channel function of all the NH2-terminal CCD mutations in RyR1. Nevertheless, differences in the conclusions obtained from expression of the I4897T mutation in HEK-293 cells (Lynch et al. 1999) and dyspedic myotubes (Avila et al. 2001a) underscores the importance of confirming data obtained in heterologous expression systems with those observed within a skeletal muscle context.
Effects of CCD Mutations in RyR1 on Resting Ca2+
The influence of the MH/CCD mutations in RyR1 on resting intracellular Ca2+ levels is still one of the most controversial and hotly debated issues in the field (for review see Mickelson and Louis 1996). Compared with wild-type RyR1, HEK-293 cells transfected with either Y523S or R2163H exhibited significantly higher resting Ca2+ levels as monitored using Fura-2 (Tong et al. 1999). Our results indicate that dyspedic myotubes expressing certain CCD mutations in RyR1 (R164C, R2435H, R2163H, and Y523S, but not I4898T or I404M) exhibit significant elevations in resting Ca2+ compared with that of wild-type RyR1 (Fig. 1 B). Clearly, it will be important for future studies to determine whether or not resting intracellular Ca2+ levels are altered after expression of the different MH-selective mutations in RyR1 in dyspedic myotubes.
The precise mechanism(s) underlying how leaky SR Ca2+ release channels lead to long-term elevations in resting Ca2+ is still unclear. If SR Ca2+ leak exceeds calcium uptake and extrusion, a new dynamic equilibrium of calcium mobilization could be established that leads to a higher resting calcium level. However, since the predominant Ca2+ extrusion processes of the plasma membrane (Ca2+ pumps and Na/Ca2+ exchangers) are stimulated by elevations in resting Ca2+, then over longer periods of time these transport mechanisms may be sufficient to compensate for the Ca2+ leak, ultimately resulting in a return of Ca2+ toward normal levels. Under such a scenario, the long-term effect of SR leak would manifest as a similar resting Ca2+ level in the face of SR Ca2+ store depletion. The caveat to this would be the presence of restricted intracellular domains in which Ca2+ diffusion to the plasma membrane is limited via some currently unknown mechanism (e.g., a central core; MacLennan and Phillips 1995; Loke and MacLennan 1998). Within such a diffusion-limited domain, increased SR Ca2+ leak could result in localized changes in resting Ca2+. However, elevations in global Ca2+ levels are found after transient expression of CCD mutant RyR1 proteins in both dyspedic myotubes (Fig. 1 B) and HEK-293 cells (Tong et al. 1999). Such changes in global Ca2+ levels may involve alterations in the activity of sarcolemmal Ca2+ transport mechanisms after introduction of mutant SR Ca2+ release channels. For example, changes in steady-state Ca2+ influx through L-type Ca2+ channels (due to differences in L-channel window current), Ca2+ release activated Ca2+ channels (after store depletion), and/or the plasma membrane Ca2+ ATPases (subsequent to mitochondrial dysfunction) represent a few mechanisms by which altered RyR1 function could potentially influence sarcolemmal Ca2+ transport processes. Clearly, future investigations are required to delineate the precise cellular mechanism(s) that underlies the elevation in resting Ca2+ induced by certain CCD mutations in RyR1.
Implications for the Pathophysiology of CCD
Although diagnosis of CCD is determined through identification of amorphous central areas in type 1 muscle fibers that lack mitochondria and oxidative enzyme activity, the underlying processes involved in the formation of central cores and their biochemical composition is still a mystery. Global resting Ca2+ levels are elevated after transient expression of CCD mutations in RyR1 in HEK-293 cells (Tong et al. 1999) and dyspedic myotubes (Fig. 1 B). Consequently, long-term changes in Ca2+-regulated gene transcription (Ginty 1997; Avila et al. 2001b) may lead to alterations in muscle fiber structure and function. However, our observation that the I4897T CCD mutation does not lead to a change in resting Ca2+ levels argues against the notion that an increase in resting Ca2+ is an absolute requirement for core formation. Rather, our data indicate that a reduction in the magnitude of voltage-gated Ca2+ transients, which is a common feature for both NH2-terminal and COOH-terminal mutants, may contribute to core formation. However, a reduction in Ca2+ transient magnitude alone may not be sufficient, since central cores are not observed for other conditions in which skeletal muscle activity is markedly compromised (e.g., other debilitating muscular dystrophies, after paralysis, etc.). Thus, the CCD mutations are likely to alter other factors that also contribute to the pathogenesis of central cores. A recent study has identified a functional mitochondrial ryanodine receptor that participates in dynamic changes in mitochondrial Ca2+ levels in cardiac ventricular myocytes (Beutner et al. 2001). If ryanodine receptors are also found to be present in skeletal muscle mitochondria, then the expression of mutant RyR1s with altered Ca2+ permeability properties may result in dire consequences for the proper control of intramitochondrial Ca2+ levels. Subsequent alterations in mitochondrial Ca2+ homeostasis and Ca2+-dependent energy metabolism that leads to an increased generation of reactive oxygen species and mitochondrial dysfunction (Gunter et al. 1994) could explain the loss of oxidative enzyme activity within the core regions. Clearly, it will be important for future work to determine the downstream mechanisms and processes that underlie core formation. Since it is unlikely that transient expression approaches will recapitulate the process of core formation, the answer to this question must likely await the development of appropriate animal models for CCD.
Acknowledgments
We would like to thank Drs. Kurt G. Beam and Paul D. Allen for providing us access to the dyspedic mice used in this study, as well as for their advice and continued support. We would also like to thank Drs. W. Melzer, S.-S. Sheu, and Ms. K.M.S. O'Connell for helpful discussions and comments on the manuscript, and Linda Groom for excellent technical assistance.
This work was supported by a grant from the National Institutes of Health (AR44657 to R.T. Dirksen), a Neuromuscular Disease Research grant from the Muscular Dystrophy Association (to R.T. Dirksen), and a Consejo Nacional de Ciencia y Tecnologia postdoctoral fellowship (to G. Avila).
Footnotes
Abbreviations used in this paper: CCD, central core disease; CPA, cyclopiazonic acid; DHPR, dihydropyridine receptor; EC, excitation-contraction; L-channel, L-type Ca2+ channel; MH, malignant hyperthermia.
References
- Avila G., Dirksen R.T. Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca2+ channel. J. Gen. Physiol. 2000;4:467–480. doi: 10.1085/jgp.115.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila G., O'Brien J.J., Dirksen R.T. Excitation-contraction uncoupling by a human central core disease mutation in the ryanodine receptor Proc. Natl. Acad. Sci. USA. 7 2001. 4215 4220a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila G., O'Connell K.M.S., Groom L., Dirksen R.T. Ca2+ release through ryanodine receptors regulates skeletal muscle L-type Ca2+ channel expression J. Biol. Chem. 276 2001. 17732 17738b [DOI] [PubMed] [Google Scholar]
- Balshaw D., Gao L., Meissner G. Luminal loop of the ryanodine receptora pore-forming segment? Proc. Natl. Acad. Sci. USA. 1999;7:3345–3347. doi: 10.1073/pnas.96.7.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone V., Massa O., Intravaia E., Bracco A., Di Martino A., Tegazzin V., Cozzolino S., Sorrentino V. Mutation screening of the RYR1 gene and identification of two novel mutations in Italian malignant hyperthermia families. J. Med. Genet. 1999;2:115–118. [PMC free article] [PubMed] [Google Scholar]
- Beutner G., Sharma V.K., Giovannucci D.R., Yule D.I., Sheu S.-S. Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 2001;276:21482–21488. doi: 10.1074/jbc.M101486200. [DOI] [PubMed] [Google Scholar]
- Buck E.D., Nguyen H.T., Pessah I.N., Allen P.D. Dyspedic mouse skeletal muscle expresses major elements of the triadic junction but lacks detectable ryanodine receptor protein and function. J. Biol. Chem. 1997;272:7360–7367. doi: 10.1074/jbc.272.11.7360. [DOI] [PubMed] [Google Scholar]
- Dietze B., Henke J., Eichinger H.M., Lehmann-Horn F., Melzer W. Malignant hyperthermia mutation Arg615Cys in the porcine ryanodine receptor alters voltage dependence of Ca2+ release. J. Physiol. 2000;526:507–514. doi: 10.1111/j.1469-7793.2000.t01-1-00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowitz V., Pearse A.G.E. Oxidative enzymes and phosphorylase in central-core disease of muscle. Lancet. 1960;2:23–24. doi: 10.1016/s0140-6736(60)92665-9. [DOI] [PubMed] [Google Scholar]
- Fill M., Coronado R., Mickelson J.R., Vilven J., Ma J.J., Jacobson B.A., Louis C.F. Abnormal ryanodine receptor channels in malignant hyperthermia. Biophys. J. 1990;3:471–475. doi: 10.1016/S0006-3495(90)82563-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii J., Otsu K., Zorzato F., de Leon S., Khanna V.K., Weiler J.E., O'Brien P.J., MacLennan D.H. Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science. 1991;5018:448–451. doi: 10.1126/science.1862346. [DOI] [PubMed] [Google Scholar]
- Gallant E.M., Lentz L.R. Excitation-contraction coupling in pigs heterozygous for malignant hyperthermia. Am. J. Physiol. 1992;262:C422–C426. doi: 10.1152/ajpcell.1992.262.2.C422. [DOI] [PubMed] [Google Scholar]
- Gallant E.M., Jordan R.C. Porcine malignant hyperthermiagenotype and contractile threshold of immature muscles. Muscle Nerve. 1996;1:68–73. doi: 10.1002/(SICI)1097-4598(199601)19:1<68::AID-MUS9>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Gallant E.M., Balog E.M., Beam K.G. Slow calcium current is not reduced in malignant hyperthermic porcine myotubes. Muscle Nerve. 1996;4:450–455. doi: 10.1002/(SICI)1097-4598(199604)19:4<450::AID-MUS4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Gao L., Balshaw D., Xu L., Tripathy A., Xin C., Meissner G. Evidence for a role of the lumenal M3-M4 loop in skeletal muscle Ca2+ release channel (ryanodine receptor) activity and conductance. Biophys. J. 2000;2:828–840. doi: 10.1016/S0006-3495(00)76339-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginty D.D. Calcium regulation of gene expressionisn't that spatial? Neuron. 1997;2:183–186. doi: 10.1016/s0896-6273(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Grabner M., Dirksen R.T., Suda N., Beam K.G. The II-III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 1999;31:21913–21919. doi: 10.1074/jbc.274.31.21913. [DOI] [PubMed] [Google Scholar]
- Gunter T.E., Gunter K.K., Sheu S.-S., Gavin C.E. Mitochondrial calcium transportphysiological and pathological relevance. Am. J. Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Hayashi K., Miller R.G., Brownell A.K. Central core diseaseultrastructure of the sarcoplasmic reticulum and t-tubules. Muscle Nerve. 1989;2:95–102. doi: 10.1002/mus.880120203. [DOI] [PubMed] [Google Scholar]
- Isaacs H., Heffron J.J., Badenhorst M. Central core disease. A correlated genetic, histochemical, ultramicroscopic, and biochemical study. J. Neurol. Neurosurg. Psychiatry. 1975;12:1177–1186. doi: 10.1136/jnnp.38.12.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat-Rott K., McCarthy T., Lehmann-Horn F. Genetics and pathogenesis of malignant hyperthermia. Muscle Nerve. 2000;1:4–17. doi: 10.1002/(sici)1097-4598(200001)23:1<4::aid-mus3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Kausch K., Lehmann-Horn F., Janka M., Wieringa B., Grimm T., Muller C.R. Evidence for linkage of the central core disease locus to the proximal long arm of human chromosome 19. Genomics. 1991;3:765–769. doi: 10.1016/0888-7543(91)90461-m. [DOI] [PubMed] [Google Scholar]
- Loke J., MacLennan D.H. Malignant hyperthermia and central core diseasedisorders of Ca2+ release channels. Am. J. Med. 1998;5:470–486. doi: 10.1016/s0002-9343(98)00108-9. [DOI] [PubMed] [Google Scholar]
- Lorenzon N.M., Beam K.G. Calcium channelopathies. Kidney Intl. 2000;3:794–802. doi: 10.1046/j.1523-1755.2000.00917.x. [DOI] [PubMed] [Google Scholar]
- Lynch P.J., Tong J., Lehane M., Mallet A., Giblin L., Heffron J.J., Vaughan P., Zafra G., MacLennan D.H., McCarthy T.V. A mutation in the transmembrane/luminal domain of the ryanodine receptor is associated with abnormal Ca2+ release channel function and severe central core disease. Proc. Natl. Acad. Sci. USA. 1999;7:4164–4169. doi: 10.1073/pnas.96.7.4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie A.E., Korneluk R.G., Zorzato F., Fujii J., Phillips M., Iles D., Wieringa B., Leblond S., Bailly J., Willard H.F. The human ryanodine receptor geneits mapping to 19q13.1, placement in a chromosome 19 linkage group, and exclusion as the gene causing myotonic dystrophy. Am. J. Hum. Gen. 1990;6:1082–1089. [PMC free article] [PubMed] [Google Scholar]
- MacLennan D.H. The genetic basis of malignant hyperthermia. Trends Pharmacol. Sci. 1992;8:330–334. doi: 10.1016/0165-6147(92)90101-b. [DOI] [PubMed] [Google Scholar]
- MacLennan D.H., Phillips M.S. The role of the skeletal muscle ryanodine receptor (RYR1) gene in malignant hyperthermia and central core disease. Soc. Gen. Physiol. Series. 1995;50:89–100. [PubMed] [Google Scholar]
- Manning B.M., Quane K.A., Ording H., Urwyler A., Tegazzin V., Lehane M., O'Halloran J., Hartung E., Giblin L.M., Lynch P.J. Identification of novel mutations in the ryanodine-receptor gene (RYR1) in malignant hyperthermiagenotype-phenotype correlation. Am. J. Hum. Genet. 1998;3:599–609. doi: 10.1086/301748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy T.V., Quane K.A., Lynch P.J. Ryanodine receptor mutations in malignant hyperthermia and central core disease. Hum. Mutat. 2000;5:410–417. doi: 10.1002/(SICI)1098-1004(200005)15:5<410::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Melzer W., Herrmann-Frank A., Luttgau H.C. The role of Ca2+ ions in excitation-contraction coupling of skeletal muscle fibres. Biochim. Biophys. Acta. 1995;1:59–116. doi: 10.1016/0304-4157(94)00014-5. [DOI] [PubMed] [Google Scholar]
- Mickelson J.R., Louis C.F. Malignant hyperthermiaexcitation-contraction coupling, Ca2+ release channel, and cell Ca2+ regulation defects. Physiol. Rev. 1996;2:537–592. doi: 10.1152/physrev.1996.76.2.537. [DOI] [PubMed] [Google Scholar]
- Monnier N., Romero N.B., Lerale J., Nivoche Y., Qi D., MacLennan D.H., Fardeau M., Lunardi J. An autosomal dominant congenital myopathy with cores and rods is associated with a neomutation in the RYR1 gene encoding the skeletal muscle ryanodine receptor. Hum. Mol. Genet. 2000;18:2599–2608. doi: 10.1093/hmg/9.18.2599. [DOI] [PubMed] [Google Scholar]
- Nakai J., Dirksen R.T., Nguyen H.T., Pessah I.N., Beam K.G., Allen P.D. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 1996;6569:72–75. doi: 10.1038/380072a0. [DOI] [PubMed] [Google Scholar]
- Quane K.A., Healy J.M., Keating K.E., Manning B.M., Couch F.J., Palmucci L.M., Doriguzzi C., Fagerlund T.H., Berg K., Ording H. Mutations in the ryanodine receptor gene in central core disease and malignant hyperthermia. Nat. Genet. 1993;1:51–55. doi: 10.1038/ng0993-51. [DOI] [PubMed] [Google Scholar]
- Quane K.A., Keating K.E., Healy J.M., Manning B.M., Krivosic-Horber R., Krivosic I., Monnier N., Lunardi J., McCarthy T.V. Mutation screening of the RYR1 gene in malignant hyperthermiadetection of a novel Tyr to Ser mutation in a pedigree with associated central cores. Genomics. 1994;1:236–239. doi: 10.1006/geno.1994.1483. [DOI] [PubMed] [Google Scholar]
- Shy G.M., Magee K.R. A new congenital non-progressive myopathy. Brain. 1956;79:610–621. doi: 10.1093/brain/79.4.610. [DOI] [PubMed] [Google Scholar]
- Tong J., Oyamada H., Demaurex N., Grinstein S., McCarthy T.V., MacLennan D.H. Caffeine and halothane sensitivity of intracellular Ca2+ release is altered by 15 calcium release channel (ryanodine receptor) mutations associated with malignant hyperthermia and/or central core disease. J. Biol. Chem. 1997;42:26332–26339. doi: 10.1074/jbc.272.42.26332. [DOI] [PubMed] [Google Scholar]
- Tong J., McCarthy T.V., MacLennan D.H. Measurement of resting cytosolic Ca2+ concentrations and Ca2+ store size in HEK-293 cells transfected with malignant hyperthermia or central core disease mutant Ca2+ release channels. J. Biol. Chem. 1999;2:693–702. doi: 10.1074/jbc.274.2.693. [DOI] [PubMed] [Google Scholar]
- Treves S., Larini F., Menegazzi P., Steinberg T.H., Koval M., Vilsen B., Andersen J.P., Zorzato F. Alteration of intracellular Ca2+ transients in COS-7 cells transfected with the cDNA encoding skeletal-muscle ryanodine receptor carrying a mutation associated with malignant hyperthermia. Biochem. J. 1994;3:661–665. doi: 10.1042/bj3010661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Chen H.S., Khanna V.K., de Leon S., Phillips M.S., Schappert K., Britt B.A., Browell A.K., MacLennan D.H. A mutation in the human ryanodine receptor gene associated with central core disease. Nat. Genet. 1993;1:46–50. doi: 10.1038/ng0993-46. [DOI] [PubMed] [Google Scholar]
- Zhao M., Li P., Li X., Zhang L., Winkfein R.J., Chen S.R. Molecular identification of the ryanodine receptor pore-forming segment. J. Biol. Chem. 1999;37:25971–25974. doi: 10.1074/jbc.274.37.25971. [DOI] [PubMed] [Google Scholar]