

Abstract
Members of the hyperpolarization-activated cation (HCN) channel family generate HCN currents (Ih) that are directly regulated by cAMP and contribute to pacemaking activity in heart and brain. The four different HCN isoforms show distinct biophysical properties. In cell-free patches from Xenopus oocytes, the steady-state activation curve of HCN2 channels is 20 mV more hyperpolarized compared with HCN1. Whereas the binding of cAMP to a COOH-terminal cyclic nucleotide binding domain (CNBD) markedly shifts the activation curve of HCN2 by 17 mV to more positive potentials, the response of HCN1 is much less pronounced (4 mV shift). A previous deletion mutant study suggested that the CNBD inhibits hyperpolarization-gating in the absence of cAMP; the binding of cAMP shifts gating to more positive voltages by relieving this inhibition. The differences in basal gating and cAMP responsiveness between HCN1 and HCN2 were proposed to result from a greater inhibitory effect of the CNBD in HCN2 compared with HCN1. Here, we use a series of chimeras between HCN1 and HCN2, in which we exchange the NH2 terminus, the transmembrane domain, or distinct domains of the COOH terminus, to investigate further the molecular bases for the modulatory action of cAMP and for the differences in the functional properties of the two channels. Differences in cAMP regulation between HCN1 and HCN2 are localized to sequence differences within the COOH terminus of the two channels. Surprisingly, exchange of the CNBDs between HCN1 and HCN2 has little effect on basal gating and has only a modest one on cAMP modulation. Rather, differences in cAMP modulation depend on the interaction between the CNBD and the C-linker, a conserved 80–amino acid region that connects the last (S6) transmembrane segment to the CNBD. Differences in basal gating depend on both the core transmembrane domain and the COOH terminus. These data, taken in the context of the previous data on deletion mutants, suggest that the inhibitory effect of the CNBD on basal gating depends on its interactions with both the C-linker and core transmembrane domain of the channel. The extent to which cAMP binding is able to relieve this inhibition is dependent on the interaction between the C-linker and the CNBD.
Keywords: pacemaker channel, Ih • potassium channel, cyclic nucleotide, chimera
INTRODUCTION
The recent cloning of a family of four mammalian genes encoding hyperpolarization-activated cation (HCN) channels (Santoro et al. 1997, Santoro et al. 1998; Ludwig et al. 1998) provides a potential molecular basis for the pacemaker current, Ih or If, present in neuronal and cardiac cells (DiFrancesco 1993; Santoro and Tibbs 1999; Kaupp and Seifert 2001). The four genes (HCN1–4) encode highly similar proteins that belong to the voltage-gated K+ channel superfamily (Jan and Jan 1997): they contain six transmembrane segments, a pore region, and cytosolic NH2 and COOH termini. The COOH terminus of the HCN channels contains additionally a cyclic nucleotide binding domain (CNBD) homologous to those of other cyclic nucleotide binding proteins, including the CNG channels of photoreceptors and olfactory neurons (Zagotta and Siegelbaum 1996). Unlike most voltage-gated K+ channels, HCN channels activate in response to hyperpolarization, opening slowly over hundreds of milliseconds. Intracellular cAMP, by directly binding to the CNBD, shifts the voltage dependence of these channels to less hyperpolarized potentials, hence, facilitating channel opening. These biophysical characteristics contribute to the crucial pacemaking functions these channels are believed to serve (for reviews see DiFrancesco 1993; Pape 1996; Santoro and Tibbs 1999; Kaupp and Seifert 2001).
A comparison of the functional properties of two members of the HCN family, HCN1 and HCN2, reveals some intriguing differences. HCN1 activates faster and requires less hyperpolarization to open compared with HCN2, which activates at potentials that are 20 mV more negative (Santoro et al. 1998, Santoro et al. 2000). Moreover, HCN2 channels are more strongly modulated by cAMP (17-mV shift) than are HCN1 channels (4-mV shift; Ludwig et al. 1998, Ludwig et al. 1999; Santoro et al. 1998; Chen et al. 2001). The molecular bases for some of these differences were previously investigated through a series of deletion mutants (Wainger et al. 2001). This earlier study showed that deletion of the CNBD mimicked the effect of cAMP by shifting the activation curves of HCN1 and HCN2 in the depolarizing direction, by an amount that was similar to the respective voltage shifts in gating produced by saturating concentrations of cAMP. Thus, deleting the CNBD in HCN2 produced a much greater positive voltage shift compared with the effect of a similar deletion in HCN1. As a result of this differential shift, the HCN1 and HCN2 CNBD deletion mutants activated with nearly identical voltage dependence, even though the full-length channels showed a 20-mV difference in their voltage dependence.
These results suggested a simple model in which the CNBD inhibits the gating of HCN channels in the absence of cAMP, shifting activation to more hyperpolarized voltages. The binding of cAMP acts to relieve this inhibition. According to this model, both the more hyperpolarized gating and greater response to cAMP of HCN2 relative to HCN1 are explained by a greater inhibitory effect of the CNBD on HCN2 than on HCN1. Although the deletion mutant experiments indicate that the CNBD is necessary for the inhibition of channel gating, they do not show whether this domain is sufficient, nor do they show how the CNBD exerts its inhibition. Is the inhibition due to an autonomous action of the CNBD, or does it depend on an interaction of the CNBD with other regions of the channel? Is the greater inhibition of gating in HCN2 compared with HCN1 due to sequence differences solely contained in the CNBDs of the two channels, or are there important sequence differences in other regions of the channel that influence the inhibitory action of the CNBD?
To examine further the molecular mechanism of cAMP modulation and the basis for gating differences between HCN1 and HCN2, we constructed a series of chimeras between these channels, exchanging the NH2 terminus, the transmembrane region, and three distinct domains within the COOH terminus. As expected, the difference in the extent of modulation by cAMP between HCN1 and HCN2 is largely accounted for by sequence differences within their COOH terminus. As predicted by the model of Wainger et al. 2001, sequence differences in the COOH terminus also partly account for differences in basal gating between HCN1 and HCN2. However, sequence differences in the core transmembrane domain were equally important. Finally, by subdividing the COOH terminus, we found that the effect of the CNBD on basal gating and cAMP modulation depends on its interaction with the core transmembrane domain and the C-linker, an 80–amino acid region that couples the transmembrane domain to the CNBD and that plays an important role in the gating by ligand of the CNG channels (Gordon and Zagotta 1995a,Gordon and Zagotta 1995b; Broillet and Firestein 1996; Broillet et al. 1997; Gordon et al. 1997; Paoletti et al. 1999).
MATERIALS AND METHODS
Molecular Biology
Mouse HCN1 (Santoro et al. 1998) and HCN2 (Ludwig et al. 1998) were subcloned into the pGH19 and pGHE expression vectors, respectively (Santoro et al. 2000; Chen et al. 2001). Deletion and chimeric mutants were made by a PCR/subcloning strategy, and the resulting mutant HCN channels were verified by dideoxy chain termination sequencing.
In one series of chimeras, we exchanged the entire cytoplasmic NH2 terminus, the S1–S6 core transmembrane domain, or the cytoplasmic COOH terminus between HCN1 and HCN2. We identify such chimeras using the nomenclature HCNXYZ, where X, Y, or Z is a number (either 1 or 2) that refers to the identity of the NH2 terminus, core transmembrane domain, or COOH terminus, respectively. Thus, in HCN112, the COOH-terminal amino acids, D390–L910, of HCN1 were substituted by the COOH-terminal amino acids, D443–L863, of HCN2. Conversely, in HCN221, amino acids D443–L863 of HCN2 were substituted by D390–L910 of HCN1. In HCN211, the NH2-terminal amino acids, M1–S128, of HCN1 were substituted by the NH2-terminal amino acids, M1–S181, of HCN2. Conversely, in HCN122, amino acids M1–S181 of HCN2 were substituted by M1–S128 of HCN1. In HCN121, the S1–S6 transmembrane domain amino acids, D129–L389, of HCN1 were substituted by the transmembrane domain amino acids, D182–L442, of HCN2. Conversely, in HCN212, amino acids D182–L442 of HCN2 were substituted by D129–L389 of HCN1.
In a second series of chimeras, we subdivided the cytoplasmic COOH terminus into three subregions: the 82–amino acid C-linker (L) that couples the transmembrane domain to the CNBD, the 119–amino acid CNBD (B), and the final extreme COOH-terminal 200–300 amino acids (X). We refer to these chimeras as HCNQ-ST, where Q refers to the parent channel (HCN1 or HCN2), S refers to the identity of the donor channel from which the subregion was derived, and T refers to the subregion of the COOH terminus that has been replaced (that is, the L, B and/or X subregions). Moreover, in this series of experiments, we deleted amino acids G3–E74 at the extreme NH2 terminus of the HCN1 parent channel to improve the level of current expression. Deletion of these nonconserved residues had no effect on the function of the parent channel (see results). We refer to this HCN1 deletion mutant as HCN1′. Thus, in HCN1′-2L, the C-linker amino acids, D390–N471, of HCN1′ (numbered according to position in HCN1) were replaced by the C-linker amino acids, D443–N524, of HCN2. Conversely, in HCN2–1L, amino acids D443–N524 of HCN2 were replaced by D390–N471 of HCN1. In HCN1′-2B, the CNBD amino acids, F472–L590, of HCN1′ were replaced by the CNBD amino acids, F525–L643, of HCN2. Conversely, in HCN2–1B, amino acids F525–L643 of HCN2 were replaced by F472–L590 of HCN1. In HCN1′-2X, all amino acids COOH-terminal to the CNBD of HCN1′, L591–L910, were replaced by the corresponding HCN2 amino acids, L644–L863. Conversely, in HCN2–1X, amino acids L644–L863 of HCN2 were replaced by L591–L910 of HCN1. In HCN1′-2LB, both the C-linker and CNBD amino acids of HCN1′, D390–L590, were replaced by the corresponding region of HCN2, D443– L643. Conversely, in HCN2–1LB, amino acids D443– L643 of HCN2 were replaced by D390–L590 of HCN1.
In HCN1ΔC-term, all cytoplasmic residues, starting three residues COOH-terminal to the last (S6) transmembrane segment (S391–L910), were deleted (Wainger et al. 2001). In HCN1–2LΔCNBD, amino acids S444–F525 of HCN2 (the C-linker) were added to the COOH terminus of HCN1ΔC-term.
Expression in Xenopus Oocytes
cRNA was transcribed from NheI-linearized DNA (for HCN1 and mutants based on the HCN1 background) or SphI-linearized DNA (for HCN2 and mutants based on the HCN2 background) using a T7 RNA polymerase (Message Machine; Ambion). 50 ng of cRNA was injected into Xenopus oocytes as described previously (Goulding et al. 1992).
Electrophysiological Recordings
Cell-free inside-out patches were obtained 3–6 d after cRNA injection, and data were acquired using a patch-clamp amplifier (model Axopatch 200A; Axon Instruments). A symmetrical recording solution was used on both sides of the membrane, containing the following (in mM): 107 KCl, 5 NaCl, 10 HEPES, 1 MgCl2, and 1 EGTA, pH 7.3. Patch pipettes had resistances of 1–3 MΩ and were coated with Sylgard to minimize capacitance. The holding potential was −40 mV. A Ag-AgCl ground wire was connected to the bath solution by a 3-M KCl agar bridge, and junction potential was compensated before the formation of each patch. Hyperpolarizing voltage pulses (3 s long) in 10-mV step increments were applied to inside-out patches from the holding potential. All recordings were obtained at room temperature (22–26°C). Linear leak current was not subtracted. Data were filtered at 1 kHz with the Axopatch 200A built-in 4-pole low-pass Bessel filter and sampled at 2 kHz with an ITC-18 interface. Analysis was done using PulseFit, IgorPro, and Sigma Plot.
Data Analysis
Activation curves were determined from the amplitudes of tail currents measured on return to −40 mV after hyperpolarizing steps to different test voltages. Tail current amplitudes were measured after the decay of the capacitive transient by averaging the current during the plateau of the tail. Current values were plotted as a function of the step voltages and fit with the Boltzmann equation: I = A1 + A2/{1 + exp[(V − V1/2)/s]}, where A1 is an offset caused by a nonzero holding current, A2 is the maximal tail current amplitude, V is voltage during the hyperpolarizing test pulse (in millivolts), V1/2 is the midpoint activation voltage, and s is the slope of the relation (in millivolts). To obtain average activation curves, the tail current amplitudes from individual patches first were normalized by subtracting the fitted value of A1, and then dividing by the fitted value of A2. These normalized data were then averaged among the different experiments. The averaged, normalized data were then fitted by the Boltzmann equation with A1 set to 0 and A2 set to 1. These normalized curves are plotted in Fig. 2. In Fig. 3 Fig. 4 Fig. 5 Fig. 6 Fig. 7 and Table and Table , the mean V1/2 and slope were obtained by averaging the individual values determined for each patch for the different constructs. The Hill equation was fitted to the cAMP dose–response data: ΔV1/2 = ΔV1/2 max/{1 + (K1/2/[cAMP])h}, where ΔV1/2 is the shift in V1/2 produced by a given [cAMP], ΔV1/2 max is the maximal shift produced at saturating cAMP, K1/2 is the concentration of cAMP that produces half of the maximal effect, and h is the Hill coefficient.
Figure 2.

Tail current activation curves for HCN1, HCN2, and COOH-terminal chimeras in the absence and presence of cAMP. Mean, normalized tail current amplitudes (materials and methods) are plotted on the y-axis as a function of test voltage. Solid curves show fitted Boltzmann relation. (closed symbols) In the presence of cAMP; (open symbols) in the absence of cAMP. Bars indicate SEM. (A) HCN1. V1/2 and s values from fitted Boltzmann relations are, respectively, −115.3 and 6.2 mV in the absence of cAMP and −111.2 and 5.0 mV in the presence of cAMP (n = 7 patches). (B) HCN2. V1/2 and s equal −135.7 and 5.1 mV, respectively, in the absence of cAMP, and −118.8 and 5.2 mV in the presence of cAMP, respectively (n = 10). (C) HCN221. V1/2 and s values from fitted Boltzmann relations are, respectively, −121.8 and 5.5 mV in the absence of cAMP and −116.2 and 5.1 mV in the presence of cAMP (n = 6). (D) HCN112. V1/2 and s values from fitted Boltzmann relations are, respectively, −124.4 and 4.8 mV in the absence of cAMP and −105.7 and 5.8 mV in the presence of cAMP (n = 10 patches).
Figure 3.

cAMP dose–response curves for HCN1, HCN2, and COOH-terminal chimeras. Shifts in V1/2 (ΔV1/2) are plotted as a function of [cAMP]. Solid lines show fits of Hill equation (materials and methods). Bars indicate SEM. (A) HCN1. Fit of the Hill equation yields: maximal shift = 4.1 mV, K1/2 = 0.06 μM, and h = 1.0 (n = 26 patches). (B) HCN2. Maximal shift = 17.4 mV, K1/2 = 0.10 μM, and h = 1.1 (n = 16). (C) HCN221. Maximal shift = 5.4 mV, K1/2 = 0.02 μM, and h = 1.4 (n = 14). (D) HCN112. Maximal shift = 20.4 mV, K1/2 = 0.07 μM, and h = 1.1 (n = 17).
Figure 4.

Basal voltage dependence and cAMP modulation of wild-type HCN1, HCN2, and their NH2-terminal, transmembrane domain, and COOH-terminal chimeras. Icons at left represent HCN1 and HCN2 channels sequences as bold and light lines, respectively. Data plotted next to each channel show steady-state V1/2 in the absence of cAMP (left graph) and magnitude of shift in V1/2 in response to 10 μM cAMP (right graph); bars indicate SEM. Mean V1/2 values, slope values, and number of patches are in Table .
Figure 5.
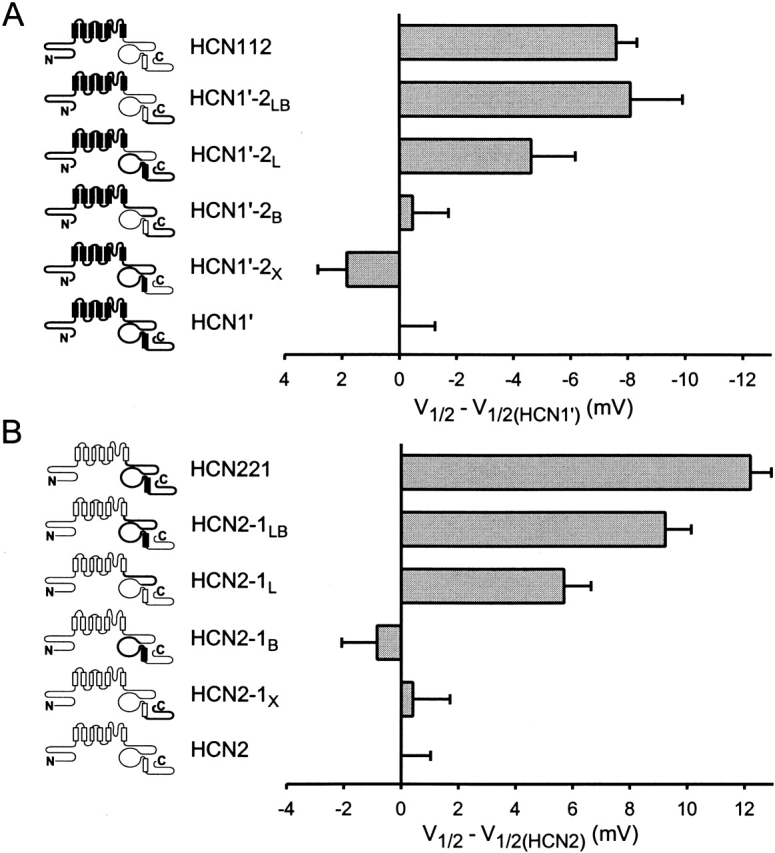
Contribution of COOH-terminal subdomains to differences in basal voltage dependence of activation between HCN1 and HCN2. (A). Effect of replacing different regions of the COOH terminus of HCN1′ with corresponding regions of HCN2. V1/2 of chimeras are plotted relative to the V1/2 of HCN1′ (i.e., V1/2 of a given channel minus V1/2 of HCN1′). Note the negative voltage scale (indicating negative shift in V1/2 for a given chimera relative to HCN1′). (B) Effect of replacing different regions of the COOH terminus of HCN2 with corresponding regions of HCN1. Note the positive voltage scale (indicating positive shift in V1/2 for a given chimera relative to HCN2). Mean values for V1/2, slope of Boltzmann fit, and number of patches for HCN1′ and the COOH-terminal chimeras are in Table . Data for HCN2, HCN221, and HCN112 are in Table .
Figure 6.
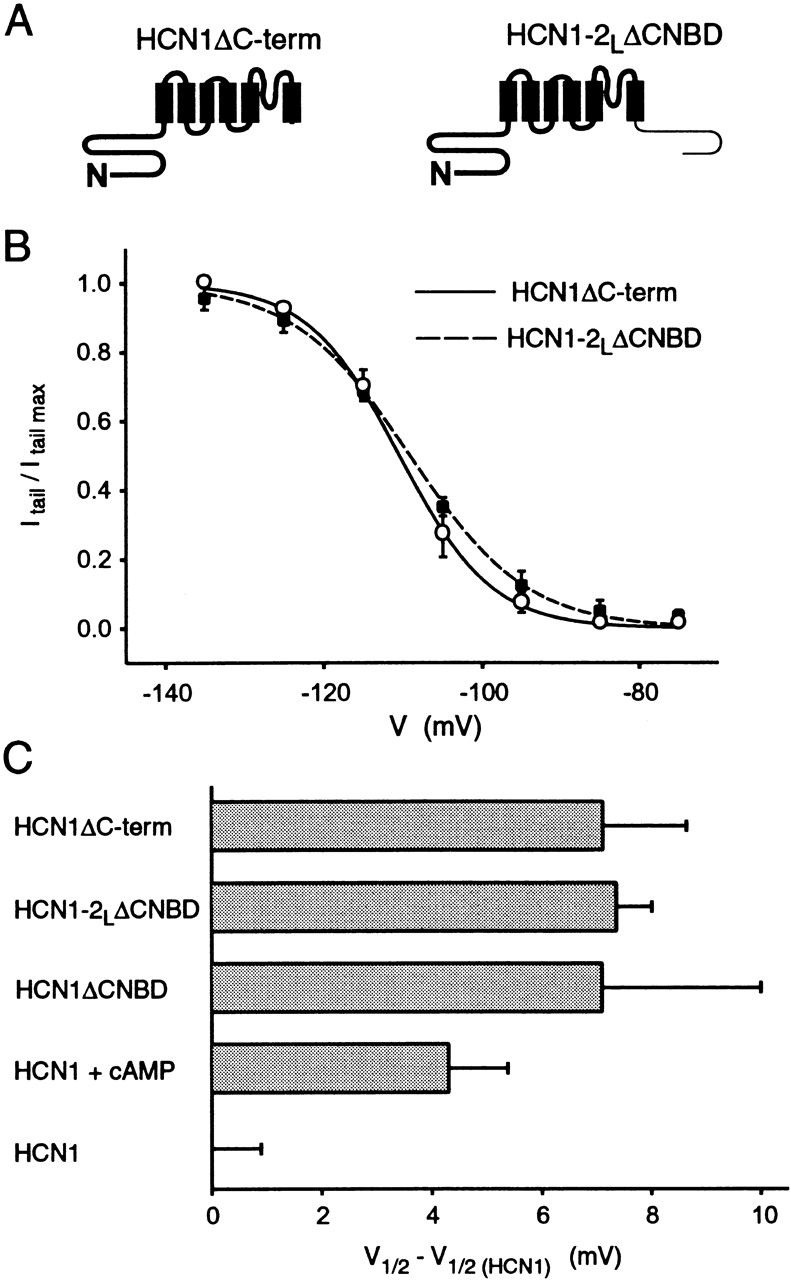
The C-linker has no independent effect on voltage dependence of gating. (A) Schematic drawings of HCN1ΔC-term and HCN1–2LΔCNBD (bold line indicates HCN1 and light line HCN2). (B) Steady-state tail current activation curves for HCN1ΔC-term (solid curve, open circles) and HCN1–2LΔCNBD (dashed curve, closed squares). The curves show the fits of Boltzmann relations that yield the following parameters: for HCN1ΔC-term, V1/2 = −110.5 mV and s = 5.8 mV (11 patches); and for HCN1–2LΔCNBD, V1/2 = −109.1 mV and s = 7.4 mV (4 patches). (C) Difference between the V1/2 of HCN1ΔC-term, HCN1–2LΔCNBD, or HCN1ΔCNBD (data from Wainger et al. 2001) and the V1/2 of wild-type HCN1 (i.e., V1/2 of a given deletion mutant minus the V1/2 of HCN1). Also shown is the shift in V1/2 of HCN1 by saturating [cAMP] (data in Table ). Error bars show SEM.
Figure 7.

Contribution of COOH-terminal subdomains to differences in cAMP modulation between HCN1 and HCN2. (A) Change in response to cAMP upon replacement of COOH-terminal regions of HCN1′ with corresponding region of HCN2 (terminology as in Fig. 5). Maximal shift in V1/2 in response to 10 μM cAMP for a given channel is plotted relative to the maximal shift for HCN1′ (i.e., shift in V1/2 in response to cAMP for a given chimera minus the shift for HCN1′). Positive values indicate that a given construct shows a larger positive shift in response to cAMP compared with HCN1′. (B) Change in response to cAMP upon replacement of COOH-terminal regions of HCN2 with corresponding region of HCN1. Maximal shift in V1/2 in response to 10 μM cAMP for a given channel is plotted relative to the maximal shift for HCN2 (i.e., shift in V1/2 for a chimeric channel minus the shift for HCN2). Negative values indicate that a given construct shows a smaller positive shift in response to cAMP compared with HCN2. Mean values for the voltage shifts and the number of patches for HCN1′ and the COOH-terminal chimeras are in Table . Data for HCN2, HCN221, and HCN112 are in Table .
Table 1.
Parameters of Steady-state Voltage Dependence and cAMP Modulation for HCN Wild-type and Chimeric Channels
| Steady-state voltage dependence | cAMP modulation | ||||
|---|---|---|---|---|---|
| Clones | V1/2 | Slope | n | ΔV1/2 | n |
| mV | mV | mV | |||
| HCN2 | −137.2 ± 1.0 | 4.7 ± 0.3 | 19 | 17.1 ± 0.3 | 8 |
| HCN122 | −136.5 ± 0.8 | 3.5 ± 0.4 | 6 | 15.8 ± 0.5 | 6 |
| HCN112 | −126.2 ± 0.7 | 4.8 ± 0.2 | 20 | 20.4 ± 0.7 | 4 |
| HCN212 | −127.3 ± 0.7 | 4.9 ± 0.4 | 6 | 19.4 ± 0.5 | 7 |
| HCN121 | −123.4 ± 0.6 | 5.5 ± 0.3 | 10 | 6.8 ± 0.7 | 9 |
| HCN221 | −125.0 ± 0.7 | 5.2 ± 0.3 | 22 | 5.4 ± 0.7 | 5 |
| HCN211 | −114.8 ± 1.3 | 5.2 ± 0.2 | 7 | 3.5 ± 0.4 | 6 |
| HCN1 | −117.1 ± 0.9 | 6.2 ± 0.2 | 23 | 4.3 ± 0.4 | 6 |
Values ± SEM for V1/2 and slope (s) from Boltzmann relations. ΔV1/2: difference in V1/2 in the presence and absence of 10 μM cAMP.
Table 2.
Parameters of Steady-state Voltage Dependence and cAMP Modulation for HCN1′ and the COOH-terminal Chimeras
| Steady-State voltage dependence | cAMP modulation | ||||
|---|---|---|---|---|---|
| Clones | V1/2 | Slope | n | ΔV1/2 | n |
| mV | mV | mV | |||
| HCN1′ | −118.5 ± 1.2 | 5.8 ± 0.3 | 13 | 4.4 ± 0.7 | 5 |
| HCN1′-2L | −123.2 ± 1.6 | 4.0 ± 0.3 | 6 | 8.9 ± 0.4 | 5 |
| HCN1′-2B | −119.0 ± 1.3 | 4.9 ± 0.6 | 6 | 8.5 ± 0.6 | 5 |
| HCN1′-2X | −116.4 ± 1.0 | 5.7 ± 0.3 | 14 | 5.0 ± 0.7 | 12 |
| HCN1′-2LB | −126.7 ± 1.8 | 5.1 ± 0.4 | 10 | 19.0 ± 0.8 | 7 |
| HCN2-1L | −131.5 ± 0.9 | 4.2 ± 0.2 | 11 | 11.7 ± 0.8 | 4 |
| HCN2-1B | −138.0 ± 1.2 | 4.1 ± 0.2 | 7 | 13.5 ± 0.7 | 5 |
| HCN2-1X | −136.8 ± 1.3 | 4.2 ± 0.3 | 7 | 16.3 ± 0.6 | 6 |
| HCN2-1LB | −127.9 ± 0.9 | 5.3 ± 0.3 | 10 | 5.5 ± 0.7 | 10 |
Values ± SEM for V1/2 and slope (s) from Boltzmann relations. n is number of patches. ΔV1/2: difference between V1/2 in the presence of cAMP minus V1/2 in the absence of cAMP.
Some of the more slowly activating constructs, such as HCN2, did not reach steady-state levels of activation by the end of the 3-s pulses, especially for pulses to voltages at or positive to the V1/2. In whole-cell recordings, this can lead to erroneous estimates of V1/2 values that are negative to the true, steady-state values (Seifert et al. 1999; Santoro et al. 2000). To estimate the potential error, we compared activation curves for HCN2, the slowest-activating of our constructs, using 3- or 10-s pulses (the latter of which permits the channels to approach steady-state activation). Under the cell-free conditions of our recordings, we found nearly identical V1/2 (<2 mV difference) and slope values from Boltzmann fits to the two activation curves. Thus, we have routinely used for these experiments 3-s pulses, since they are less likely to cause patch breakdown during hyperpolarization.
Upon formation of the cell-free patches, there is a fairly rapid negative shift in the position of the V1/2 for both HCN1 and HCN2 channels (Chen et al. 2001). However, this shift generally reached completion after ∼9 min. All steady-state activation data included in this paper were obtained after the V1/2 measurements had stabilized.
RESULTS
The COOH Terminus Has a Profound Effect on cAMP Modulation and Contributes to the Steady-state Voltage Dependence of HCN Channels
To explore the contribution of different channel domains to HCN gating and cAMP modulation, we have taken advantage of the functional differences between HCN1 and HCN2. In inside-out patches, HCN1 has a threshold of activation in response to hyperpolarization of around −95 mV, whereas HCN2 does not begin to activate until steps to −125 mV (Fig. 1 A). Moreover, the activation and deactivation kinetics of HCN1 are more rapid than those of HCN2, measured during steps to the same voltages (Fig. 1A and Fig. C). Finally, cAMP has a minimal effect on either the threshold or kinetics of activation of HCN1, but markedly accelerates the kinetics of HCN2 and shifts its threshold for activation by ∼20 mV (Fig. 1 B).
Figure 1.

Comparison of hyperpolarization-activated currents and response to cAMP for HCN1, HCN2, and their COOH-terminal chimeras. Icons in the top row represent wild-type HCN1 (sequences represented by bold lines, leftmost icon), HCN2 (sequences represented by light lines, rightmost icon), and their COOH-terminal chimeras HCN112 and HCN221. (A) Currents elicited by 3-s hyperpolarizations to indicated potentials from inside-out patches obtained from oocytes injected with cRNA of (from left to right) HCN1, HCN112, HCN221, or HCN2. Patches were stepped to voltages ranging from −85 to −165 mV in 10-mV steps from a holding potential of −40 mV. (B) Currents shown for same patches in the presence of 10 μM cAMP in internal (bath) solution. (C) Comparison of activation kinetics during a step to −135 mV in the presence and absence of 10 μM cAMP. Superimposed traces from records shown in A and B were scaled so that the current amplitudes appear equal to show the differences in activation kinetics.
We next studied a series of chimeras to probe the regions of these channels that are responsible for these differences. We first replaced the COOH terminus of HCN1 by that of HCN2, to generate the chimera HCN112 (where the three numbers refer to the identity of the cytoplasmic NH2 terminus, transmembrane domain, and cytoplasmic COOH terminus, respectively; as described in materials and methods and icons of Fig. 1). We also examined the converse chimera, HCN221. The voltage dependence and activation kinetics of these two chimeras in the absence of cAMP are intermediate between those of HCN1 and HCN2 (Fig. 1A and Fig. C). This suggests that the COOH terminus contributes to basal voltage gating properties, similar to the conclusions of the deletion mutant study (Wainger et al. 2001).
We then compared the effect of cAMP on the gating of the wild-type channels and the chimeras. Whereas cAMP shifted the threshold of activation of HCN112 in the positive direction by ∼20 mV, similar to its effect on HCN2, the agonist had little effect on the threshold of HCN221, similar to its action on HCN1 (Fig. 1 B). In addition, cAMP significantly increased the rate of activation of HCN112 (Fig. 1 C), but caused minimal change in HCN221. These results suggest that differences in the effect of cAMP on HCN1 and HCN2 are principally due to sequence differences in the COOH terminus.
To quantify the role of different regions of the channel in gating, we measured tail current activation curves for the different constructs, in the absence and presence of cAMP (Fig. 2). Fits of the Boltzmann equation to tail current data from several patches (see materials and methods) provided estimates of the voltage at which the channels were half-activated (V1/2), the slope (s) of the activation curves, and the extent to which cAMP shifted the midpoint voltage of activation to more positive values (ΔV1/2). These data confirm that HCN1 activates at voltages 20 mV positive to those required to activate HCN2 and that cAMP causes a much smaller voltage shift in steady-state activation for HCN1 (4 mV) compared with its larger shift in HCN2 (17 mV; Fig. 2 and Table ).
According to the model of Wainger et al. 2001, the CNBD of HCN2 produces a larger inhibitory effect on gating compared with the effect of the CNBD of HCN1. Consistent with this view, we find that HCN112, which contains the CNBD of HCN2, activates at voltages 9 mV hyperpolarized to those needed to activate HCN1. Conversely, HCN221, which contains the CNBD of HCN1, activates at voltages that are >10 mV depolarized to those required to activate HCN2. However, these shifts in V1/2 values upon exchange of the COOH terminus account for only about half of the full 20-mV difference between the activation curves of HCN1 and HCN2, indicating an additional role for either the core transmembrane domain or NH2 terminus (which we investigate below).
In contrast to the partial shift in basal V1/2 values upon exchange of the COOH terminus, the response to cAMP of the chimeras is fully determined by the identity of the COOH terminus (Fig. 2 and Table ). Thus, HCN221 shows a 5-mV shift with 10 μM cAMP, similar to the 4-mV shift observed for HCN1. Conversely, HCN112 shows a 20-mV shift with cAMP, similar to the 17-mV shift observed for HCN2. The different responses to cAMP could be due either to differences in sensitivity to ligand (i.e., the EC50 or K1/2) or to differences in efficacy (i.e., the maximal shift at saturating ligand concentration). To distinguish between these possibilities, we compared dose–response curves for the wild-type channels and the chimeras, in which the shift in V1/2 is plotted as a function of cAMP concentration (Fig. 3). The data were fit by the Hill equation (materials and methods). This analysis shows that all four channels have a very high sensitivity to cAMP (with K1/2 values ranging from 0.02 to 0.1 μM, although HCN221 appears to be more sensitive than either parent channel). Thus, the difference in the extent to which cAMP shifts gating to more positive voltages is due to a difference in efficacy. Furthermore, the COOH terminus determines that efficacy.
The Transmembrane Domains and the COOH Terminus Interact to Inhibit HCN2 Channel Gating
Because the COOH terminus only partially determines the difference in the basal voltage dependence of gating between HCN1 and HCN2, we constructed four additional chimeras to study the role of the NH2 terminus and the transmembrane domain (Fig. 4 and Table ). Exchange of the NH2 terminus between HCN1 and HCN2 has little or no effect on cAMP modulation or basal gating (Fig. 4; ≤2.5-mV shift). In contrast, exchange of the core transmembrane domain does significantly alter basal voltage dependence (Fig. 4). Thus, replacing the transmembrane region of HCN2 with that of HCN1, yielding the chimera HCN212, shifts the V1/2 by 10 mV to more positive potentials (relative to HCN2). Conversely, replacing the transmembrane domain of HCN1 with that of HCN2, yielding the chimera HCN121, shifts the V1/2 by 6 mV to more negative potentials (relative to HCN1). In contrast to its effect on basal gating, exchange of the transmembrane domain has little effect on the response to cAMP (≤2.5-mV change). Thus, the difference in basal gating between HCN1 and HCN2 depends on sequence differences located in both the core transmembrane domain and the COOH terminus. For both regions, residues in HCN2 channels exert a larger inhibitory shift on gating compared with the corresponding residues from HCN1 channels. In contrast, sequence differences responsible for differences in the extent of cAMP modulation between HCN1 and HCN2 are largely confined to the COOH terminus of the channels.
The COOH Terminus Inhibits Gating through an Interaction of the C-linker and the Cyclic Nucleotide Binding Domain
Which regions of the COOH terminus are important for determining its effects on basal gating and cAMP modulation? Does the CNBD uniquely determine the influence of the COOH terminus? Or does the C-linker region, important for coupling binding to gating in CNG channels (Paoletti et al. 1999), also play an important role? What is the function of the nonconserved 200–300–amino acid domain at the extreme COOH terminus of the channel? To address such questions, we constructed a series of COOH-terminal chimeras in which we exchanged the C-linker (L), CNBD (B), or extreme COOH-terminal region (X) between HCN1 and HCN2 channels. Because of low expression of HCN1, we used a construct (HCN1′) in which amino acids G3–E74 at the poorly conserved, extreme NH2-terminal region of the channel were deleted as the background for HCN1. This deletion greatly increased the hyperpolarization-activated current magnitude with little or no effect on steady-state V1/2, activation kinetics, or cAMP modulation (see Fig. 5 and Fig. 7 and Table ).
We first examined the effect of exchanging between HCN1 and HCN2 each of the three regions of the COOH terminus, individually, on the basal voltage dependence of gating (Fig. 5). We expressed the effect of the various substitutions on gating of the chimeras relative to the V1/2 of the parent channel (i.e., by subtracting the V1/2 of the parent channel, HCN1′ in Fig. 5 A or HCN2 in Fig. 5 B, from the V1/2 for each chimera). Substitution of the extreme COOH-terminal nonconserved sequences of HCN1′ with those of HCN2, yielding the chimera HCN1′-2X (see Fig. 5 and materials and methods for nomenclature), has little effect on gating (≤2-mV shift; Fig. 5 A and Table ). Similarly, substitution of the extreme COOH-terminal sequences of HCN2 with those of HCN1, generating the chimera HCN2–1X, also has little effect on gating (<1-mV shift; Fig. 5 B and Table ). The lack of effect of exchanging the extreme COOH-terminal region is in agreement with the lack of effect on gating upon its deletion (Wainger et al. 2001).
Based on the previous finding that deletion of the CNBD of HCN2 causes a much larger positive shift in gating compared with the effect of deletion of the CNBD of HCN1 (25- vs. 7-mV shift; Wainger et al. 2001), we expected that the exchange of the CNBDs between HCN1 and HCN2 would significantly alter basal gating. To our surprise, exchange of the CNBD between HCN1 and HCN2 has no effect on the V1/2 in the absence of cAMP (Fig. 5 and Table ). Thus, the gating of the HCN1′-2B chimera (HCN1′ with the CNBD of HCN2) is similar to that of HCN1′. Similarly, the gating of the converse chimera, HCN2–1B, is similar to that of HCN2. The lack of effect of exchanging the CNBDs is also unexpected given the large shifts in gating (∼10 mV) we observed upon exchange of the entire COOH terminus between HCN1 and HCN2 (Fig. 2 and Fig. 4).
How can we reconcile the deletion mutant results (Wainger et al. 2001), showing a markedly greater voltage shift upon deletion of the CNBD of HCN2 compared with deletion of the CNBD of HCN1, with our findings above that exchange of the HCN1 and HCN2 CNBDs has little effect on basal gating? And why does exchange of the entire COOH terminus significantly alter gating, whereas exchange of the CNBD alone does not? One simple idea is that the inhibitory effect of the CNBD requires an interaction between the CNBD and some other region in the COOH terminus; it is the strength of this interaction that differs in HCN1 and HCN2. Indeed, we show below that the inhibitory effect of the CNBD depends on its interaction with the C-linker.
Thus, in contrast to the lack of effect of exchange of the CNBD on gating, replacement of the C-linker of HCN1 by that of HCN2 produces a marked shift in the activation curve. Replacement of the C-linker of HCN1′ with that of HCN2, yielding the chimera HCN1′-2L (HCN1′ with the C-linker of HCN2), produces a 5-mV hyperpolarizing shift in gating relative to HCN1′. Conversely, replacement of the C-linker of HCN2 by that of HCN1, yielding the chimera HCN2–1L, produces a 6-mV depolarizing shift relative to HCN2. These shifts in gating observed upon substitution of the C-linker account for 50–60% of the total voltage shift (8–12 mV) observed upon exchange of the entire COOH terminus (Fig. 2, Fig. 4, and Fig. 5 and Table and Table ).
Although the C-linker clearly influences basal gating, our finding that exchange of the C-linker only partially reproduces the effect of exchange of the entire COOH terminus indicates that the C-linker must interact with some other COOH-terminal region to produce the full gating phenotype. Therefore, we reexamined the role of the binding domain by exchanging it in the background of the C-linker chimeras. Replacing the HCN1 CNBD of HCN1′-2L with the CNBD of HCN2, yielding the chimera HCN1′-2LB, shifts gating by 3.5 mV to more negative voltages, relative to the gating of HCN1′-2L. As a result, the V1/2 of HCN1′-2LB is now identical to that of the COOH-terminal chimera, HCN112. Conversely, replacing the CNBD of HCN2–1L with that of HCN1, yielding the chimera, HCN2–1LB, shifts gating by 3.5 mV to more positive potentials relative to the gating of HCN2–1L. As a result, the V1/2 value of HCN2–1LB is similar (although not identical) to that of HCN221 (Fig. 5 and Table ). Thus, the identity of the binding domain does influence the voltage dependence of gating of HCN1 and HCN2. The effect of exchanging the HCN1 and HCN2 binding domain, however, depends on the identity of the C-linker and core transmembrane domain of the channel. Exchange of the CNBD alters the voltage dependence of gating only if the C-linker and transmembrane domain are derived from different channels. This implies that the CNBD, C-linker, and transmembrane domain must interact to regulate gating.
The C-linker Does Not Directly Alter Gating Independently of the CNBD
The above finding that exchange of the C-linker alters basal gating is consistent with one of two models: either the C-linker passively couples an inhibitory action exerted by the CNBD on the gating of the core transmembrane domain, similar to the role of the C-linker in CNG channels (Paoletti et al. 1999), or the C-linker itself exerts an intrinsic, regulatory effect. The results of Wainger et al. 2001 suggest that the HCN1 C-linker has no independent inhibitory effect on gating, because the gating of an HCN1 deletion mutant that contains the C-linker, but lacks the CNBD (HCN1ΔCNBD), is identical to the gating of an HCN1 deletion mutant that lacks the C-linker and the CNBD (HCN1ΔC-term, entire COOH terminus deletion). However, since the HCN2 COOH-terminal deletion mutant (HCN2ΔC-term) does not express functional channels, the results of Wainger et al. 2001 do not exclude the possibility that the HCN2 C-linker has an extra inhibitory effect on gating that is not seen with the HCN1 C-linker.
The intrinsic action of the HCN2 C-linker could not be examined in the background of HCN2 because of the lack of expression of HCN2ΔC-term. However, we were able to explore the effect of the HCN2 C-linker by attaching it to the COOH terminus of HCN1ΔC-term, generating the deletion chimera HCN1–2LΔCNBD (Fig. 6 A). If the HCN2 C-linker has an extra intrinsic inhibitory action then its presence should inhibit the gating of HCN1–2LΔCNBD relative to that of HCN1ΔC-term (similar to its observed effect to inhibit the gating of HCN1′-2L relative to HCN1′). However, we find that the steady-state activation curves for these two constructs are nearly identical (Fig. 6 B). The V1/2 value of both HCN1ΔC-term and HCN1–2LΔCNBD are shifted by similar amounts (+7 mV) relative to wild-type HCN1, which is similar to the shift in V1/2 produced by cAMP for wild-type HCN1 (Fig. 6 C).
These results confirm the finding of Wainger et al. 2001 that the CNBD exerts an inhibitory influence on gating that is relieved by cAMP (Wainger et al. 2001). Because the C-linker does not exert an intrinsic inhibitory effect on gating in the absence of the CNBD, the effect of exchanging the C-linker on gating in the chimeras between HCN1 and HCN2 is likely to result from an interaction of the C-linker with the CNBD. The simplest interpretation of these results is that the C-linker of HCN2 more efficiently couples an inhibitory action of the CNBD to the gating of the transmembrane domain compared with the effect of the C-linker of HCN1. Finally, the X region appears to have a fairly minor role in gating, as the exchange of this region on any background gives only a 0.5–3-mV voltage shift in gating (Fig. 5 and Table ).
The Extent to which cAMP Binding Relieves Inhibition Also Depends on an Interaction of the C-linker and the Cyclic Nucleotide Binding Domain
If the C-linker and CNBD do indeed interact to inhibit basal gating then the extent to which cAMP binding is able to relieve inhibition and, thus, shift the activation curve should depend on the identity of both the CNBD and the C-linker. To test this idea, we examined the effect of cAMP on the activation curves of the various COOH-terminal chimeras relative to their parent channels, HCN1′ or HCN2 (Fig. 7 and Table ). Not surprisingly, exchange of the nonconserved sequences COOH-terminal to the CNBD (HCN1′-2X and HCN2–1X), has only a minimal effect on the extent to which cAMP shifts voltage-dependent gating. However, exchange of either the CNBD or the C-linker between HCN1 and HCN2 (the B and L chimeras) does significantly alter the extent of the shift produced by cAMP. Chimeras that contain either the HCN2 C-linker or the HCN2 CNBD show a greater response to cAMP compared with chimeras that contain the corresponding regions from HCN1 (Fig. 7 and Table ). However, exchange of either the C-linker or the CNBD alone generates channels with intermediate phenotypes that do not fully reproduce the cAMP response characteristic of either the wild-type channels or the full COOH-terminal chimeras. Simultaneous exchange of both the C-linker and CNBD are required to fully reproduce the appropriate cAMP response (HCN1′-2LB and HCN2–1LB in Fig. 7 and Table ). Thus, similar to the effects of the COOH terminus on basal gating, the extent of cAMP modulation appears to depend on the identity of both the C-linker and the CNBD.
DISCUSSION
Using a series of chimeras between HCN1 and HCN2 channels, we have identified domains important for differences in both the basal voltage dependence of gating and the extent to which cAMP shifts this gating to more positive potentials. Differences in voltage dependence of gating are due to differences in both the core transmembrane domain (S1–S6) and COOH terminus of the two channels. Channels containing either the core transmembrane domain or COOH terminus of HCN2 channels activate at more negative voltages compared with channels that contain the corresponding regions of HCN1. In contrast, differences in cAMP modulation are largely localized to the COOH terminus, with channels containing the HCN2 COOH terminus showing a larger shift in response to cAMP compared with channels containing the HCN1 COOH terminus.
In a previous study using HCN1 and HCN2 deletion mutants, Wainger et al. 2001 showed that deletion of the CNBD shifted the gating of HCN1 and HCN2 to more positive potentials, similar to the effect of cAMP. This suggested a simple model in which the CNBD inhibits basal gating, shifting it to more negative voltages. cAMP binding to the CNBD would shift gating to more positive voltages by relieving this inhibition. According to this model, the more negative basal voltage dependence and larger positive shift with cAMP in HCN2 compared with HCN1 are explained by a greater basal inhibitory effect of the CNBD in HCN2. Our chimera results both confirm and extend certain key features of this model. Moreover, our results allow us to determine whether the inhibitory effect of the CNBD on gating is an autonomous property of the CNBD or whether it depends on interactions with other regions of the channel.
Interactions of the COOH Terminus and Core Transmembrane Domain Control Basal Gating
One surprising finding, given the previous deletion mutant results of Wainger et al. 2001, is that exchange of the CNBD between HCN1 and HCN2 has no effect on basal gating of the chimeras. This indicates that the differential effect on gating of deleting the CNBD of HCN1 and HCN2 is not solely due to sequence differences within the two CNBDs, but must depend on differences in the interaction of the CNBD with other regions of the channel. Moreover, this interaction must inhibit gating to a larger extent in HCN2 than in HCN1. We identified two regions of the channel that are important in this interaction: one is the core transmembrane domain and the other is the C-linker.
Evidence implicating the transmembrane domain comes from experiments showing that replacement of the transmembrane domain of HCN1 with the homologous region of HCN2 shifts the gating of the chimera to more negative potentials (Fig. 4 and Table ). Moreover, this effect is not due to an intrinsic difference in gating of the two core transmembrane domains because HCN1 and HCN2 deletion mutants that lack the CNBD show an identical voltage dependence of gating (Wainger et al. 2001). Thus, the shift in gating observed upon exchange of the core transmembrane domain must result from a differential inhibitory interaction with the COOH terminus of the channel.
Interaction of the CNBD with the C-linker Inhibits Basal Gating and Mediates cAMP Modulation
Our data further show that the inhibitory effect of the CNBD depends on its interaction with the C-linker. Thus, whereas exchange of the CNBD alone does not alter basal gating, exchange of the C-linker has a significant effect on both basal gating and cAMP modulation. Moreover, these chimeras reveal that the HCN2 C-linker exerts a larger inhibitory effect on gating than does the HCN1 C-linker. This is not an autonomous effect of the C-linker on gating, however, because we observed no change in basal gating when we attached the C-linker of either HCN1 or HCN2 to an HCN1 deletion mutant that lacks its entire COOH terminus (Wainger et al. 2001; Fig. 6). Additionally, reconstitution of the phenotype of the entire COOH terminus chimeras requires the simultaneous exchange of both the C-linker and the CNBD between HCN1 and HCN2. Therefore, the effect of the C-linker requires an interaction with the CNBD.
In CNG channels, the C-linker has an important influence on gating and is believed to act to couple ligand binding to the channel opening (Paoletti et al. 1999). In CNG channels, the NH2 terminus also has been shown to interact with the CNBD and C-linker to modulate gating (Gordon et al. 1997; Varnum and Zagotta 1997). Although we do not observe a prominent role for the NH2 terminus here, our experiments only can pinpoint regions of the HCN1 and HCN2 channels that are responsible for differences in channel function.
cAMP Modulation of Gating Also Depends on the Interaction of the CNBD and C-linker with the Transmembrane Domain
According to the simple model proposed by Wainger et al. 2001, differences in the magnitude of the response to cAMP among different channels are largely due to differences in the extent to which the CNBD inhibits basal gating. Channels that show a larger inhibition of basal gating, reflected in a more negative V1/2, should show a larger response to cAMP due to the relief of this larger inhibition. This simple relation is obeyed for most chimeras on a qualitative level. However, in general, we observe a quantitative discrepancy between the change in basal gating and the change in the response to cAMP between a given chimera and its parent channel. For example, HCN112 shows a −9-mV shift in basal gating relative to HCN1, but a 16-mV greater response to cAMP than seen with HCN1, nearly twice as great as predicted by the increased basal inhibition. Conversely, HCN121 shows a −6-mV shift in basal gating relative to HCN1 (i.e., greater inhibition) but only a 2.5-mV increase in the response to cAMP over that seen with HCN1.
These discrepancies can be explained if the chimeric mutations not only alter the extent of basal inhibition but also alter the efficacy with which cAMP binding is able to relieve this inhibition. Even for wild-type channels, this efficacy must be less than one, because the voltage shifts produced by the CNBD deletions are greater than the voltage shifts produced by saturating concentrations of cAMP (Wainger et al. 2001). Thus, for HCN2, deletion of the binding domain shifts gating by +25 mV, whereas saturating cAMP shifts gating by +17 mV, suggesting an efficacy of cAMP of 0.7. Likewise in HCN1, deletion of the binding domain shifts gating by +7 mV, whereas saturating cAMP shifts gating by +5, also yielding an efficacy of 0.7. Changes in efficacy can account for much of our chimera data if we postulate that cAMP is able to relieve inhibition with a higher than normal efficacy when the core transmembrane domain of HCN1 interacts with the COOH terminus of HCN2. This hypothesis explains the larger than expected shift by cAMP in HCN112 and HCN212. Conversely, efficacy would be reduced when the core transmembrane domain of HCN2 interacts with the COOH terminus of HCN1, which explains the smaller than expected shift by cAMP in HCN121 and HCN221.
In contrast to the relatively simple results of the deletion mutant studies, our chimera studies show that the inhibitory actions of the CNBD are mediated by a complex series of interactions among two COOH-terminal regions and the core transmembrane domain. Such interactions are, in retrospect, not surprising. The hyperpolarization-activation of the channel is likely to be mediated by movements of the S4 voltage sensor in the core transmembrane domain (Chen et al. 2000; Vaca et al. 2000). Movements of S4 must, in turn, be allosterically coupled to a conformational change that opens the activation gate, perhaps involving a movement of the S6 inner helix (Shin et al. 2001), similar to conformational changes proposed for KcsA (Perozo et al. 1999) and depolarization-activated Shaker K channels (del Camino et al. 2000). Studies on CNG channels have previously shown the importance of the C-linker in coupling cyclic nucleotide binding to conformational changes that activate the channel (Gordon and Zagotta 1995a,Gordon and Zagotta 1995b; Broillet and Firestein 1996; Broillet et al. 1997; Gordon et al. 1997; Paoletti et al. 1999). Based on structural prediction models, the C-linker in both CNG channels and HCN channels is likely to adopt a rigid, helical structure, allowing it to couple movements of the S6 helix to movements of the CNBD. Thus, although the HCN channels function in a modular manner, with a distinct core transmembrane domain that controls voltage gating and a modulatory COOH-terminal CNBD, this study demonstrates that these domains must interact through the interposed C-linker region to couple ligand binding to alterations in the energetics of channel gating.
Acknowledgments
We thank Edgar Young, Brian Wainger, and Damian Bell for helpful discussions and assistance, Eric Odell for help in preparing the manuscript and Huan Yao and John Riley for their technical assistance. Clones HCN1ΔC-term and HCN1-2LΔCNBD were prepared by Matthew DeGennaro and Brian Wainger, respectively.
This work was partially supported by grant RO1 NS-36658 from the National Institutes of Health to S.A. Siegelbaum. J. Wang was supported by the Medical Scientist Training Program.
Footnotes
Abbreviations used in this paper: CNBD, cyclic nucleotide binding domain; HCN, hyperpolarization-activated cation.
References
- Broillet M.C., Firestein S. Direct activation of the olfactory cyclic nucleotide-gated channel through modification of sulfhydryl groups by NO compounds. Neuron. 1996;16:377–385. doi: 10.1016/s0896-6273(00)80055-0. [DOI] [PubMed] [Google Scholar]
- Broillet M.C., Huang Y., Kamawura D.H., Firestein S. Mechanisms of nitric oxide activation of olfactory cyclic nucleotide-fated channels Soc. Neurosci. Abs. 23 1997. 289(Abstr.) [Google Scholar]
- Chen J., Mitcheson J.S., Lin M., Sanguinetti M.C. Functional roles of charged residues in the putative voltage sensor of the HCN2 pacemaker channel. J. Biol. Chem. 2000;275:36465–36471. doi: 10.1074/jbc.M007034200. [DOI] [PubMed] [Google Scholar]
- Chen S., Wang J., Siegelbaum S.A. Properties of hyperpolarization-activated pacemaker current defined by co-assembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J. Gen. Physiol. 2001;117:491–503. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino D., Holmgren M., Liu Y., Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 2000;403:321–325. doi: 10.1038/35002099. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D. Pacemaker mechanisms in cardiac tissue. Annu. Rev. Physiol. 1993;55:455–472. doi: 10.1146/annurev.ph.55.030193.002323. [DOI] [PubMed] [Google Scholar]
- Gordon S.E., Zagotta W.N. A histidine residue associated with the gate of the cyclic nucleotide-activated channels in rod photoreceptors Neuron. 14 1995. 177 183a [DOI] [PubMed] [Google Scholar]
- Gordon S.E., Zagotta W.N. Localization of regions affecting an allosteric transition in cyclic nucleotide-activated channels Neuron. 14 1995. 857 864b [DOI] [PubMed] [Google Scholar]
- Gordon S.E., Varnum M.D., Zagotta W.N. Direct interaction between amino- and carboxyl-terminal domains of cyclic nucleotide-gated channels. Neuron. 1997;19:431–441. doi: 10.1016/s0896-6273(00)80951-4. [DOI] [PubMed] [Google Scholar]
- Goulding E.H., Ngai J., Kramer R.H., Colicos S., Axel R., Siegelbaum S.A., Chess A. Molecular cloning and single-channel properties of the cyclic nucleotide-gated channel from catfish olfactory neurons. Neuron. 1992;8:45–58. doi: 10.1016/0896-6273(92)90107-o. [DOI] [PubMed] [Google Scholar]
- Jan L.Y., Jan Y.N. Cloned potassium channels from eukaryotes and prokaryotes. Annu. Rev. Neurosci. 1997;20:91–123. doi: 10.1146/annurev.neuro.20.1.91. [DOI] [PubMed] [Google Scholar]
- Kaupp U.B., Seifert R. Molecular diversity of pacemaker ion channels. Annu. Rev. Physiol. 2001;63:235–257. doi: 10.1146/annurev.physiol.63.1.235. [DOI] [PubMed] [Google Scholar]
- Ludwig A., Zong X., Jeglitsch M., Hofmann F., Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Ludwig A., Zong X., Stieber J., Hullin R., Hofmann F., Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO. J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pape H.C. Queer current and pacemakerthe hyperpolarization-activated cation current in neurons. Annu. Rev. Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Paoletti P., Young E., Siegelbaum S.A. C-linker of cyclic nucleotide-gated channels controls coupling of ligand binding to channel gating. J. Gen. Physiol. 1999;113:17–33. doi: 10.1085/jgp.113.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E., Cortes D.M., Cuello L.G. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- Santoro B., Tibbs G.R. The HCN gene familymolecular basis of the hyperpolarization-activated pacemaker channels. Ann. NY Acad. Sci. 1999;868:741–764. doi: 10.1111/j.1749-6632.1999.tb11353.x. [DOI] [PubMed] [Google Scholar]
- Santoro B., Grant S.G., Bartsch D., Kandel E.R. Interactive cloning with the SH3 domain of N-src identifies a new brain specific ion channel protein, with homology to eag and cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. USA. 1997;94:14815–14820. doi: 10.1073/pnas.94.26.14815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B., Liu D.T., Yao H., Bartsch D., Kandel E.R., Siegelbaum S.A., Tibbs G.R. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- Santoro B., Chen A., Luthi P., Pavlidis G.P., Shumyatsky G.R., Tibbs, Siegelbaum S.A. Molecular and functional heterogeneity of hyperpolarization-activated pacemaker channels in the mouse CNS. J. Neurosci. 2000;20:5264–5275. doi: 10.1523/JNEUROSCI.20-14-05264.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert R., Scholten A., Gauss R., Mincheva A., Lichter P., Kaupp U.B. Molecular characterization of a slowly gating human hyperpolarization-activated channel predominantly expressed in thalamus, heart, and testis. Proc. Natl. Acad. Sci. USA. 1999;96:9391–9396. doi: 10.1073/pnas.96.16.9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K., Rothberg B., Yellen G. Blocker state dependence and trapping in hyperpolarization-activated cation channels. Evidence for an intracellular activation gate. J. Gen. Physiol. 2001;117:91–102. doi: 10.1085/jgp.117.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaca L., Stieber J., Zong X., Ludwig A., Hofmann F., Biel M. Mutations in the S4 domain of a pacemaker channel alter its voltage dependence. FEBS Lett. 2000;479:35–40. doi: 10.1016/s0014-5793(00)01837-8. [DOI] [PubMed] [Google Scholar]
- Varnum M.D., Zagotta W.N. Interdomain interactions underlying activation of cyclic nucleotide-gated channels. Science. 1997;278:110–113. doi: 10.1126/science.278.5335.110. [DOI] [PubMed] [Google Scholar]
- Wainger B.J., DeGennaro M., Santoro B., Siegelbaum S.A., Tibbs G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Zagotta W.N., Siegelbaum S.A. Structure and function of cyclic nucleotide- gated channels. Annu. Rev. Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]