

Abstract
Calmodulin is a ubiquitous Ca2+ binding protein that modulates the in vitro activity of the skeletal muscle ryanodine receptor (RyR1). Residues 3614–3643 of RyR1 comprise the CaM binding domain and mutations within this region result in a loss of both high-affinity Ca2+-bound calmodulin (CaCaM) and Ca2+-free CaM (apoCaM) binding (L3624D) or only CaCaM binding (W3620A). To investigate the functional role of CaM binding to this region of RyR1 in intact skeletal muscle, we compared the ability of RyR1, L3624D, and W3620A to restore excitation–contraction (EC) coupling after expression in RyR1-deficient (dyspedic) myotubes. W3620A-expressing cells responded normally to 10 mM caffeine and 500 μM 4-chloro-m-cresol (4-cmc). Interestingly, L3624D-expressing cells displayed a bimodal response to caffeine, with a large proportion of cells (∼44%) showing a greatly attenuated response to caffeine. However, high and low caffeine-responsive L3624D-expressing myotubes exhibited Ca2+ transients of similar magnitude after activation by 4-cmc (500 μM) and electrical stimulation. Expression of either L3624D or W3620A in dyspedic myotubes restored both L-type Ca2+ currents (retrograde coupling) and voltage-gated SR Ca2+ release (orthograde coupling) to a similar degree as that observed for wild-type RyR1, although L-current density was somewhat larger and activated at more hyperpolarized potentials in W3620A-expressing myotubes. The results indicate that CaM binding to the 3614–3643 region of RyR1 is not essential for voltage sensor activation of RyR1.
Keywords: ryanodine receptor, muscle, calcium channel, caffeine, sarcoplasmic reticulum
INTRODUCTION
Excitation–contraction (EC)* coupling is the process by which membrane depolarization triggers Ca2+ release from the sarcoplasmic reticulum (SR), thereby resulting in muscle contraction. In contrast to cardiac EC coupling where the influx of extracellular Ca2+ triggers SR Ca2+ release through RyRs (Nabauer et al., 1989), EC coupling in skeletal muscle does not depend on Ca2+ influx (Armstrong et al., 1972), but rather relies on a unique bidirectional mechanical interaction between dihydropyridine receptors (DHPRs) in the surface membrane and RyRs in the SR (Nakai et al., 1996; Grabner et al., 1999; Avila and Dirksen, 2000). The skeletal muscle DHPR is a voltage-gated L-type Ca2+ channel (L-channel) that also functions as the voltage sensor for skeletal-type EC coupling. The skeletal muscle isoform of the ryanodine receptor (RyR1) functions as a large Ca2+-conducting intracellular Ca2+ release channel located in the terminal cisternae of the SR. Depolarization-induced conformational changes in the skeletal muscle DHPR are transmitted to the release channel via interactions between the II-III loop of the α1S subunit of the DHPR (Tanabe et al., 1990) and multiple cytoplasmic region(s) of RyR1 (Nakai et al., 1998), which ultimately result in the activation of RyR1 and the subsequent release of Ca2+ from the SR.
Both the DHPR α1S subunit and RyR1 are required for the EC-coupling process in skeletal muscle; the absence of either results in a complete loss of both voltage-gated SR Ca2+ release and EC coupling. In α1S-null (dysgenic) mice, the absence of the voltage sensor precludes voltage-gated activation of RyR1. Similarly, in RyR1-knockout (dyspedic) mice, the absence of the SR Ca2+ release channel eliminates the pathway for Ca2+ release from the SR. Interestingly, although dyspedic myotubes express sarcolemmal voltage sensors, there is a significant reduction in L-type Ca2+ channel function that is restored after reintroduction of RyR1 (Nakai et al., 1996). This observation provides strong evidence for a bidirectional interaction between the DHPR and RyR1, in which DHPRs not only send an orthograde signal to RyR1 to trigger SR Ca2+ release, but also receive a retrograde signal from RyR1 that enhances the Ca2+-conducting activity of the DHPR (Nakai et al., 1996; Grabner et al., 1999; Avila and Dirksen, 2000).
Although activation of RyR1 during EC coupling depends on a reciprocal gating interaction with the skeletal muscle DHPR, RyR1 proteins also interact with a diverse group of other regulatory proteins and small molecules. The RyR1 homotetramer binds four FKBP12 proteins, one per monomer, and binding of FKBP12 regulates the proper gating of the release channel (Jayaraman et al., 1992; Brillantes et al., 1994). RyR1 also interacts with other proteins such as triadin, junctin, calsequestrin, and calmodulin (CaM) as well as small molecules such as Ca2+, Mg2+, and ATP (for review see MacKrill, 1999). In addition, the β-subunit of the DHPR also strongly influences voltage sensor activation of RyR1 (Beurg et al., 1999). All of these accessory proteins of EC coupling are present within sarcolemmal–SR junctions, suggesting that the functional unit of EC coupling in skeletal muscle may actually represent a macromolecular complex composed of the DHPR, RyR1, and various other modulatory proteins/molecules.
One of these modulatory proteins is the ubiquitous Ca2+-sensing protein CaM. CaM is a small, highly conserved calcium-sensing protein that binds to a diverse group of targets and plays an important role in many cellular signaling pathways. The protein has a high degree of internal symmetry, containing four EF-hand Ca2+ binding motifs, two in the NH2-terminal lobe and two in the COOH-terminal lobe. This structural division between the NH2-terminal and COOH-terminal regions reflects a functional division between the two lobes (Jurado et al., 1999; Saimi and Kung, 2002). The protein undergoes a conformational change upon binding Ca2+, exposing hydrophobic pockets in either lobe that permit effector binding (Jurado et al., 1999). Most CaM binding proteins bind CaCaM, whereas a few, including both α1S (Pate et al., 2000; Sencer et al., 2001) and RyR1 (Tripathy et al., 1995; Moore et al., 1999; Yamaguchi et al., 2001; Samso and Wagenknecht, 2002), are capable of binding both apoCaM and CaCaM. Although the functional importance of CaM binding to the skeletal muscle DHPR remains to be resolved, there has been considerable progress toward understanding the role of CaM in the modulation of RyR1 activity.
Each RyR1 tetramer binds four CaM molecules (1 CaM per RyR1 subunit) in both the absence and presence of Ca2+. Interestingly, apoCaM and CaCaM bind to overlapping sites between residues 3614–3643 of RyR1 (Moore et al., 1999; Yamaguchi et al., 2001; Samso and Wagenknecht, 2002). In the absence of CaM, activation of RyR1 exhibits a bell-shaped dependence on Ca2+, with micromolar cytosolic concentrations causing activation and millimolar concentrations of Ca2+ inhibiting RyR1 activity (Tripathy et al., 1995). Addition of CaM in the presence of submicromolar Ca2+ causes further activation of RyR1, whereas at micromolar to millimolar Ca2+, CaM inhibits RyR1 activity (Tripathy et al., 1995; Rodney et al., 2000). Thus, in in vitro assays, addition of CaM shifts the Ca2+ sensitivity of RyR1 to lower concentrations of Ca2+. Although there is considerable evidence to suggest that CaM exerts a profound modulatory effect on isolated RyR1 activity, the prominent role of CaM in multiple cellular signaling pathways presents an obstacle to isolating the effect of CaM on EC coupling in intact muscle cells. However, the recent identification of two residues within the 3614–3643 region that disrupt specific RyR1–CaM interactions provides a valuable tool for examining the role of CaM in EC coupling. Substitution of an alanine residue for W3620 selectively disrupts the interaction of CaCaM with RyR1, whereas an aspartate substitution at L3624 disrupts the binding and regulation by both apoCaM and CaCaM (Yamaguchi et al., 2001).
To investigate the impact of apoCaM and CaCaM binding to RyR1 on EC coupling, we have functionally characterized the activity of dyspedic myotubes expressing either wild-type RyR1, W3620A, or L3624D. Our results indicate that neither apoCaM nor CaCaM binding to the 3614–3643 region of RyR1 is required for bidirectional signaling with the skeletal muscle DHPR. Although neither L3624D nor W3620A eliminated voltage-dependent Ca2+ release, the W3620A mutation resulted in a significant increase in L-channel conductance and a slight hyperpolarizing shift in the voltage dependence of activation of L-channel conductance and SR Ca2+ release. Nevertheless, the response of W3620A-expressing myotubes to both caffeine and 4-chloro-m-cresol (4-cmc) was indistinguishable from that of wild-type RyR1. Surprisingly, a significant population of myotubes expressing L3624D (∼44%) showed a markedly attenuated response to caffeine, whereas normal responsiveness to activation by both 4-cmc and depolarization was retained. The results indicate that CaM binding to RyR1 is not essential for voltage-dependent Ca2+ release by RyR1, but does impact selective ligand activation of the in situ SR Ca2+ release channel.
MATERIALS AND METHODS
Preparation and Microinjection of Dyspedic Myotubes
Myotubes were prepared from the skeletal muscle of newborn normal and dyspedic mice as described previously (Nakai et al., 1996; Avila and Dirksen, 2000, 2001). L3624D and W3620A were constructed as described previously (Yamaguchi et al., 2001). Expression of RyR1, L3624D, and W3620A in individual dyspedic myotubes was accomplished by microinjection of nuclei with cDNA encoding CD8 (0.2 μg/μl) and either RyR1, L3624D, or W3620A (0.5 μg/μl). Expressing myotubes were subsequently identified 48–72 h postinjection by incubation with CD8 antibody-coated beads (Dynabeads; Dynal ASA).
Intracellular Ca2+ Measurements in Intact Myotubes
Intracellular Ca2+ was measured in intact (nonpatched) myotubes loaded with the fluorescent Ca2+ indicator Indo-1 AM (Molecular Probes) as described previously (Avila et al., 2001). Indo-1–loaded myotubes were excited at 350 nm using a DeltaRam illumination system (Photon Technology, Inc.). Fluorescence emission at 405 and 485 nm was monitored using a 40× (1.35 NA) oil immersion objective, collected using a photomultiplier detection system, and the results presented as the ratio (R) of F405/F485. Caffeine (10 mM) and 4-chloro-m-cresol (4-cmc, 500 μM) were prepared in normal rodent Ringer's solution (see below) and applied using a rapid perfusion system (Warner Instruments, Inc.) that permits fast, local application of agonist as well as rapid washout with control solution (Avila et al., 2001). During each experiment, myotubes were continuously perfused with either control or agonist-containing Ringer's solution. Multiple myotubes were measured per dish and bulk gravity perfusion (∼10 ml) with control Ringer's was used to wash the dish between each measurement. Peak intracellular Ca2+ changes in response to agonist application are expressed as ΔRatio (Ragonist − Rbaseline). For measurements of electrically evoked Ca2+ transients in intact myotubes, Indo-1 AM–loaded cells were stimulated (8 V for 20 ms at 0.5 Hz for 30 s) with an extracellular pipette (Avila et al., 2001). Data were analyzed using FeliX (Photon Technology, Inc.) and SigmaPlot 2000 (SPSS, Inc.) software packages. Caffeine response histograms were fitted according a standard Gaussian equation of the following general form:
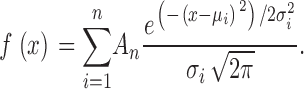 |
(1) |
Simultaneous Measurement of Voltage-gated L-Channel Activity and SR Ca2+ Release
The whole-cell patch clamp technique was used to simultaneously measure voltage-gated L-type Ca2+ currents (L-currents) and Ca2+ transients in RyR1-, L3624D-, and W3620A-expressing myotubes. A 1-s prepulse to −30 mV was used to inactivate Na+ channels and T-type Ca2+ channels. L-currents were online leak-subtracted using −P/3 delivered from the holding potential (−80 mV) before each test pulse. Peak currents were normalized to total cell capacitance (pA/pF), plotted as a function of test potential, and fitted according to:
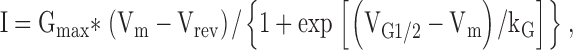 |
(2) |
where Gmax is the maximal L-channel conductance, Vm is the test potential, Vrev is the extrapolated reversal potential, VG1/2 is the potential for half-maximal activation of Gmax, and kG is a slope factor.
To measure relative changes in voltage-gated SR Ca2+ release in patch-clamped myotubes, the Ca2+ indicator K5-Fluo-3 (Molecular Probes) was included in the patch pipette internal solution and allowed to dialyze ∼5 min before all recordings. Myotubes were excited at 488 nm and fluorescence emission at 535 nm was collected using a photomultiplier system (Photon Technology, Inc.). Fluorescence traces were analogue filtered (τ = 0.5 ms) before digitization at 10 kHz and are expressed as ΔF/F ([Fpeak − FBase]/FBase). Peak fluorescence during each test pulse was plotted as a function of test potential (Vm) and fitted according to:
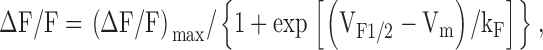 |
(3) |
where (ΔF/F)max is the maximal fluorescence change, VF1/2 is the potential for half-maximal activation, and kF is a slope factor.
Recording Solutions
Intracellular Ca2+ changes in Indo-1–loaded myotubes were measured from myotubes bathed in normal rodent Ringer's (in mM): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, and 10 Hepes, pH 7.4 with NaOH. The internal solution for macroscopic L-current recordings contained (in mM): 145 Cs-aspartate, 0.1 Cs2-EGTA, 1.2 MgCl2, 5 MgATP, 0.2 K5-Fluo-3 (Molecular Probes), and 10 Hepes, pH 7.4 with CsOH. The external solution contained (in mM): 145 TEACl, 10 CaCl2, 0.003 TTX (Molecular Probes), and 10 HEPES, pH 7.4 with TEA-OH. Unless otherwise indicated, all chemicals were from Sigma-Aldrich.
RESULTS
Effects of Mutations within the CaM Binding Region of RyR1 on Agonist-induced SR Ca2+ Release
In normal myotubes (derived from skeletal muscle of phenotypically normal mice and expressing normal amounts of RyR1 protein), application of 10 mM caffeine typically elicited a rapid peak Ca2+ release that is followed by a maintained plateau phase (Fig. 1 A, left). Less frequently, application of 10 mM caffeine produced repetitive Ca2+ oscillations (Fig. 1 A, right). RyR1-expressing dyspedic myotubes also displayed both of these type of responses (Fig. 1 B); however, a larger proportion of RyR1-expressing dyspedic myotubes exhibited Ca2+ oscillations (76%; 98/129). By contrast, uninjected dyspedic myotubes respond only very weakly, if at all, to 10 mM caffeine (Fig. 1 C), which likely arises from variable expression of RyR3 in dyspedic myotubes, as suggested previously (Takeshima et al., 1994).
Figure 1.

Caffeine response of Indo-1 AM–loaded dyspedic myotubes expressing CaM binding–deficient RyR1 mutants. Caffeine-induced Ca2+ release (10 mM, black bar) for normal myotubes (A), dyspedic myotubes (C), RyR1- (B), W3620A- (D), and L3624D-expressing (E) myotubes. Bars, 60 s. (F) Average peak caffeine-induced Ca2+ release (ΔRatio = Rcaffeine − Rbaseline; where Rcaffeine is the peak ratio following caffeine application; *, P < 0.001). (G) Percentage of RyR1-, L3624D-, and W3620A-expressing myotubes exhibiting a peak caffeine–induced Ca2+ release with ΔRatio >0.4.
[3H]Ryanodine binding and single channel measurements have shown that CaM shifts the Ca2+ sensitivity of RyR1 toward lower concentrations of Ca2+ (Tripathy et al., 1995; Fruen et al., 2000; Rodney et al., 2000). Application of 10 mM caffeine to dyspedic myotubes expressing W3620A, which binds apoCaM but not CaCaM (Yamaguchi et al., 2001), resulted in a response identical in amplitude and profile to that of RyR1, with 83% (88/106) of W3620A-expressing myotubes exhibiting Ca2+ oscillations (Fig. 1 D, W3620A). Expression of L3624D, which almost completely abolishes both apoCaM and CaCaM binding (Yamaguchi et al., 2001), resulted in an average peak caffeine response that is significantly lower than that observed for RyR1-expressing myotubes (Fig. 1 F). However, underlying this decrease in the average response are two distinct populations of L3624D-expressing myotubes: one that responds to caffeine much like RyR1 (Fig. 1 E, left) and another that displays a very weak response to 10 mM caffeine (Fig. 1 E, right). For subsequent analysis, we have defined a weak caffeine response as any myotube in which ΔRatio was ≤0.4; ∼44% (68/153) of all L3624D-expressing myotubes responded with a ΔRatio <0.4 (Fig. 1 G). In an attempt to more rigorously quantify this observation, we plotted the ΔRatio values recorded from a large number of RyR1-, L3624D-, and W3620A-expressing myotubes as a frequency distribution (Fig. 2 A). Not surprisingly, data for both RyR1 (n = 129) and W3620A (n = 106) are reasonably well described by a single Gaussian distribution. However, data obtained from L3624D-expressing myotubes (n = 153) clearly fall into two distinct populations, one centered around ΔRatio ∼0.2 and another centered around ΔRatio ∼0.8 (Fig. 2 A), consistent with the observation that some L3624D-expressing cells respond similarly to RyR1, whereas the rest respond only weakly to 10 mM caffeine. It is unlikely that this difference arises from an increase in the threshold for caffeine activation, as application of 30 mM caffeine also failed to elicit a normal release in L3624D-expressing myotubes that showed an attenuated response to 10 mM caffeine (unpublished data).
Figure 2.

The caffeine response of L3624D-expressing myotubes exhibits a caffeine response distribution. (A) Caffeine response histograms. Data are binned at 0.05 ΔRatio units and fit according to Eq.1 as described in materials and methods. RyR1- and W3620A-expressing myotubes exhibit a single Gaussian distribution (A = 5.19, μ = 0.96, σ = 0.38 for RyR1, n = 129 and A = 6.0, μ = 0.95, σ = 0.48 for W3620A, n = 106). The caffeine response for L3624D-expressing myotubes exhibits a bimodal distribution (A1 = 2.7, μ1 = 0.16, σ1 = 0.14 and A2 = 4.4, μ2 = 0.68, σ2 = 0.45 for L3624D, n = 153). (B) Resting F405/F485 ratios (*, P < 0.001 with respect to dyspedic). (C) RyR1, W3620A, and both populations of L3624D-expressing myotubes exhibit similar, rapidly activating global intracellular Ca2+ transients.
One possible explanation for this observation is that the L3624D mutation significantly impairs expression and/or assembly of functional RyR1 channels, since the mean ΔRatio of the weakly responsive population of L3624D-expressing cells is only slightly higher than that observed for uninjected dyspedic myotubes. We have previously documented that dyspedic myotubes exhibit a significantly lower resting Ca2+ level than RyR1-expressing myotubes (Avila et al., 2001). Therefore, resting Ca2+ measurements provide a means of verifying expression of functional SR Ca2+ release channels. Fig. 2 B shows resting F405/F485 ratios for dyspedic, RyR1-, L3624D-, and W3620A-expressing myotubes. In accordance with previous results, reintroduction of RyR1 increases cytosolic Ca2+ levels above that found in uninjected dyspedic myotubes (resting F405/F485 ratios were 0.46 ± 0.01 and 0.65 ± 0.01 for uninjected [or CD8 sham-injected; Avila et al., 2001] and RyR1-expressing dyspedic myotubes, respectively). W3620A-expressing myotubes and both low and high caffeine-responsive L3624D-expressing myotubes exhibited resting ratios that were not significantly different from that of RyR1. However, for each expression condition (RyR1-, W3620A-, and both low and high caffeine-responsive L3624D-expressing myotubes), resting ratios were significantly higher than that of uninjected dyspedic myotubes (Fig. 2 B). These results indicate that functional Ca2+ release channels are expressed in myotubes microinjected with cDNA encoding L3624D, regardless of their response to caffeine.
Another indication of functional release channel expression in L3624D cDNA-injected myotubes that responded poorly to caffeine was the presence of spontaneous global Ca2+ transients (Fig. 2 C) in these cells. Spontaneous, action potential–evoked global Ca2+ transients such as those depicted in Fig. 2 C are characterized by a rapid rise (∼10 ms) and decay (∼200 ms) and are generally smaller than caffeine-induced Ca2+ transients. Thus, these rapid global Ca2+ release events likely arise from spontaneous electrical activity that result in DHPR activation of SR Ca2+ release channels. Additionally, there was no difference in the population of cells that displayed spontaneous fast transients among RyR1, L3624D, and W3620A-expressing myotubes (with 11.8%, 8.5%, and 12.4% of RyR1-, W3620A-, and L3624D-expressing myotubes exhibiting spontaneous transients, respectively). Moreover, similar percentages were observed for L3624D-expressing myotubes that displayed a robust response to caffeine (ΔRatio >0.4; 12.9%) and those that only weakly responded to caffeine (ΔRatio <0.4; 11.8%). By comparison, due to the absence of RyR1, dyspedic myotubes are incapable of exhibiting rapid global changes in [Ca2+]i that is activated by spontaneous electrical activity (EC coupling). Thus, the presence of spontaneous global Ca2+ transients in L3624D-expressing myotubes that exhibited a weak caffeine response (i.e., ΔRatio <0.4) provides further evidence for the expression of functional release channels in these cells and suggests that the L3624D mutation may selectively alter caffeine activation of the SR Ca2+ release channel.
To determine whether the decrease in activation by caffeine in L3624D-expressing cells was specific for caffeine or reflects a general loss of pharmacological activation of the release channel, we also examined the sensitivity to activation by 4-chloro-m-cresol (4-cmc). 4-cmc is a potent RyR1-selective agonist that, unlike caffeine, has no effect on RyR3, presumably because the site of action for 4-cmc is distinct from that of caffeine (Fessenden et al., 2000). Consistent with this observation, application of 10 mM caffeine elicits a small response in dyspedic myotubes while 500 μM 4-cmc generates almost no response (Fig. 3 A, top left). Application of 500 μM 4-cmc to both RyR1- (Fig. 3 A, top right) and W3620A-expressing dyspedic myotubes produces a response that is similar in amplitude to that of 10 mM caffeine (Fig. 3 B). Not surprisingly, application of 4-cmc to L3624D-expressing myotubes that exhibit a robust caffeine response also displayed a large response to 500 μM 4-cmc (Fig. 3 A, bottom left). However, 4-cmc also elicited large Ca2+ transients even in L3624D-expressing myotubes that only weakly responded to 10 mM caffeine (Fig. 3 A, bottom right, and B). Furthermore, repetitive trains of electrical stimulation (8 V for 20 ms at 0.5Hz for 30 s initiated at the arrows in Fig. 3) evoked Ca2+ transients of similar magnitude and kinetics (see insets to Fig. 3 A) in both caffeine-responsive and nonresponsive L3624D-expressing myotubes. Similar trains of electrically evoked Ca2+ transients were also observed in RyR1-expressing (Fig. 3 A) and W3620A-expressing myotubes (unpublished data).
Figure 3.

CaM binding–deficient RyR1 mutants respond normally to both electrical stimulation and 4-cmc. (A) Representative traces for dyspedic, RyR1-, and L3624D-expressing myotubes. Arrows indicate the beginning of a train of electrical stimuli (30 s, 20-ms pulses of 8 V at 0.5 Hz). The light gray bars (60 s) define periods of 10 mM caffeine application and black bars (60 s) define periods of 500 μM 4-cmc application. (Insets) The final electrically evoked Ca2+ transient from each train is shown on an expanded scale (scale bars: 0.2 ΔRatio and 0.2 s shown for RyR1 applies to each inset). (B) Average peak response to 4-cmc (ΔRatio = R4-cmc − Rbaseline, *, P < 0.001) for uninjected dyspedic myotubes, RyR1-expressing myotubes, low and high caffeine–responsive L3624D-expressing myotubes, and W3620A-expressing myotubes. Number of experiments is given in parentheses above each bar.
Effects of the L3624D and W3620A Mutations on the Orthograde and Retrograde Signals of Skeletal Muscle EC Coupling
EC coupling in skeletal muscle involves a unique bidirectional signaling interaction between RyR1 and DHPR proteins, with the DHPR functioning as an activator of SR Ca2+ release (orthograde coupling) and RyR1 enhancing the Ca2+ conducting activity of the DHPR (retrograde coupling) (Nakai et al., 1996; Grabner et al., 1999; Avila and Dirksen, 2000). To examine the role of CaM binding to RyR1 on these two coupling mechanisms, we used the whole-cell patch clamp technique in conjunction with intracellular Ca2+ measurements to simultaneously measure voltage-gated L-type Ca2+ currents and SR Ca2+ release from dyspedic myotubes expressing either wild-type RyR1, L3624D, or W3620A. In agreement with previous reports (Nakai et al., 1996; Avila et al., 2001), dyspedic myotubes did not release Ca2+ upon membrane depolarization and displayed a very low L-current density (unpublished data). Reintroduction of RyR1 into dyspedic myotubes restored both robust L-channel activity (retrograde coupling) (Fig. 4 A) and voltage-gated SR Ca2+ release (orthograde coupling) (Fig. 5 A). Expression of L3624D and W3620A also resulted in restoration of both retrograde and orthograde coupling. L-channel conductance of L3624D-expressing myotubes was nearly identical in amplitude and voltage dependence to that of RyR1-expressing myotubes (RyR1: Gmax = 202 ± 11 nS/nF, VG1/2 = 9.7 ± 1.0 mV; L3624D: Gmax = 203 ± 11 nS/nF, VG1/2 = 10.4 ± 1.4 mV) (Fig. 4 B, Table I) . Importantly, all cells positively identified as expressing L3624D displayed robust L-currents and Ca2+ transients, suggesting that the failure of some L3624D-expressing myotubes to fully respond to caffeine does not extend to activation by the voltage sensor. In contrast to L3624D, myotubes expressing W3620A exhibited an L-channel current density that was significantly larger (and slightly shifted to more negative potentials) than that of RyR1-expressing myotubes (W3620A: Gmax= 270 ± 16 nS/nF, VF1/2 = 3.8 ± 2.2 mV).
Figure 4.

CaM binding–deficient RyR1 mutants restore voltage-gated L-channel activity (retrograde coupling) after expression in dyspedic myotubes. (A) L-currents elicited in response to 200-ms depolarizations to the indicated potentials for representative RyR1- (left), L3624D- (middle), and W3620A-expressing (right) myotubes. (B) Average peak I-V relationships for RyR1-, L3624D-, and W3620A-expressing myotubes. The lines through the data represent Boltzmann fits of the average data using Eq. 2. For L3624D and W3620A, a dashed line representing the fit of the RyR1 data is also shown for comparison.
Figure 5.

CaM binding–deficient RyR1 mutants restore voltage-gated Ca2+ release (orthograde coupling) after expression in dyspedic myotubes. (A) Ca2+ transients elicited by 200 ms depolarization to the indicated potentials for representative RyR1- (left), L3624D- (middle), and W3620A-expressing (right) myotubes. (B) Average peak F-V relationship for RyR1-, L3624D-, and W3620A-expressing myotubes. The lines through the data represent Boltzmann fits to the average data using Eq. 3. For L3624D and W3620A, a dashed line representing the fit of the RyR1 data is also shown for comparison.
TABLE I.
Parameters of I-V and F-V Curves
| I-V | F-V | |||||||
|---|---|---|---|---|---|---|---|---|
| n | Gmax | k | V1/2 | Vrev | ΔF/Fmax | k | V1/2 | |
| nS/nF | mV | mV | mV | mV | mV | |||
| RyR1 | 27 | 202 ± 11 | 5.1 ± 0.2 | 9.7 ± 1.0 | 71.8 ± 1.4 | 4.0 ± 0.5 | 3.7 ± 0.3 | 0.1 ± 1.1 |
| L3624D | 28 | 203 ± 11 | 4.8 ± 0.3 | 10.4 ± 1.4 | 74.5 ± 1.5 | 3.6 ± 0.4 | 3.7 ± 0.3 | 1.6 ± 1.4 |
| W3620A | 18 | 270 ± 16a | 4.1 ± 0.4 | 3.8 ± 2.2a | 70.4 ± 1.8 | 3.9 ± 0.6 | 3.0 ± 0.3 | −5.9 ± 1.7a |
I-V and F-V data are from the number of experiments indicated in column “n”. Parameters are given as mean ± SEM and were obtained by fitting each myotube within a group to the appropriate equation (I-V, Eq. 2 and F-V, Eq. 3). The average cell capacitance for each group was (in pF): 487 ± 29, 397 ± 22, and 439 ± 42 for RyR1, L3624D, and W3620A, respectively. The average series resistance (Rs, MΩ) was: 0.9 ± 0.1, 0.9 ± 0.1, and 0.8 ± 0.1 for RyR1, L3624D, and W3620A, respectively. The average Verror (Rs × Ipeak; mV) was: 3.2 ± 0.4, 3.1 ± 0.3, and 4.0 ± 0.4 for RyR1, L3624D, and W3620A, respectively.
P < 0.01 compared to RyR1.
Fig. 5 shows that disruption of CaM binding to RyR1 does not alter the magnitude or voltage dependence of orthograde coupling. Myotubes expressing L3624D displayed Ca2+ transients that had a peak fluorescence and voltage dependence not significantly different from that of RyR1 (RyR1: ΔF/Fmax = 4.0 ± 0.5, VF1/2 = 0.1 ± 1.1; L3624D: ΔF/Fmax = 3.6 ± 0.4, VF1/2 = 1.6 ± 1.4). W3620A-expressing myotubes also had Ca2+ transients of similar magnitude as RyR1-expressing myotubes (ΔF/Fmax = 3.9 ± 0.6, VF1/2 = −5.9 ± 1.7), although the voltage dependence of these transients was shifted to approximately the same degree as that observed for L-channel conductance (∼6 mV). Together, the results of Figs. 4 and 5 indicate that CaM binding to RyR1 is not required for the targeting or assembly of functional RyR1 ion channels, their activation by the DHPR/voltage sensor, or RyR1-mediated enhancement of DHPR Ca2+ channel activity.
DISCUSSION
Evidence has accumulated over the past two decades that calmodulin is a potent modulator of RyR1 activity as assessed using a variety of in vitro approaches. Early studies indicated that the Ca2+-bound form of calmodulin (CaCaM) inhibits RyR1 in the absence of ATP, which suggested inhibition via a direct interaction rather than through phosphorylation (Meissner, 1986; Smith et al., 1989). More recent [3H]ryanodine and single channel measurements indicate that at low free Ca2+ concentrations (<1 μM), apoCaM has a stimulatory effect on RyR1 channel activity (Tripathy et al., 1995; Fruen et al., 2000; Rodney et al., 2000). Binding studies show that apoCaM and CaCaM exert their opposite effects on RyR1 activity by binding to and dissociating from RyR1 on a time scale of seconds to minutes (Moore et al., 1999; Yamaguchi et al., 2001). It is therefore likely that CaM is constitutively bound to the receptor.
In the present study, the role of CaM binding to a specific domain of RyR1 (3614–3643) in modulating EC coupling in intact skeletal myotubes was examined. Given the extensive biochemical evidence that CaM is a potent modulator of RyR1 activity, a surprising finding was that expression of L3624D or W3620A in dyspedic myotubes restored EC coupling to an extent nearly indistinguishable from that of wild-type RyR1. Both L3624D and W3620A appeared to form functional release channels that are correctly targeted to the junction, based on their ability to restore spontaneous action potential-evoked Ca2+ release, 4-cmc responsiveness, and both orthograde and retrograde coupling with the DHPR.
Effect of L3624D and W3620A on Ligand Activation of RyR1
The most prominent effect of disrupting the RyR1–CaM interaction in our experiments was an altered caffeine response of a subpopulation of L3624D-expressing myotubes. There are several possible explanations for the attenuated caffeine response. First, those cells that showed an attenuated response to caffeine may have failed to express a sufficient number of functional mutant SR Ca2+ release channels. However, our data argue strongly against this explanation. First, low-resting Ca2+ levels of dyspedic myotubes were significantly elevated in L3624D-expressing myotubes that showed an attenuated response to caffeine, indicating that these myotubes express functional release channels. Second, spontaneous and electrically evoked global Ca2+ release transients were observed in weakly caffeine-responsive L3624D-expressing myotubes. Due to their extremely rapid activation (<10 ms), these spontaneous and evoked transients clearly represent functional DHPR-RyR1 orthograde coupling. It is unlikely that low endogenous RyR3 levels play a significant role in initiating these Ca2+ release events since RyR3 cannot substitute for RyR1 in EC coupling (Fessenden et al., 2000). Third, if >40% of myotubes injected with L3624D cDNA resulted in the expression of nonfunctional channels, we would have expected this to have also been reflected in patch-clamp experiments. However, all L3624D-expressing myotubes exhibited large voltage-gated L-currents and SR Ca2+ release in our patch clamp experiments. Finally, SR Ca2+ release triggered by 4-chloro-m-cresol was similar for both RyR1-expressing myotubes and weakly caffeine-responsive L3624D-expressing myotubes.
Caffeine and 4-cmc act at distinct sites in RyR1 (Fessenden et al., 2002), which may explain the discordance between caffeine and 4-cmc responsiveness of L3624D-expressing myotubes. Although its precise mechanism of action in intact cells is unclear, caffeine augments activation of RyR1 by apoCaM (Balshaw et al., 2001). Therefore, caffeine may be unable to sufficiently activate L3624D mutant release channels in myotubes since the L3624D mutation abolishes apoCaM binding to RyR1 (Yamaguchi et al., 2001). Caffeine activation of SR Ca2+ release in a subpopulation of L3624D-expressing myotubes could be explained by the presence of higher levels of RyR3 in these myotubes, and that Ca2+ release from RyR3 may serve to facilitate activation of L3624D channels even in the absence of apoCaM binding. On the other hand, activation by 4-cmc may not be dependent on the binding of apoCaM to RyR1, as it likely acts via a different mechanism (Fessenden et al., 2000). This hypothesis could be tested in future experiments by evaluating the ability of caffeine to activate SR Ca2+ release after expression L3624D mutant release channels in RyR1/RyR3 double knockout myotubes.
CaM Binding to the 3614–3643 Region of RyR1 Is not Required for Bidirectional DHPR/RyR1 Signaling
Given the postulated role of apoCaM to activate and CaCaM to inhibit Ca2+ release through RyR1, it might have been expected that disruption of both apoCaM and CaCaM (L3624D) or only CaCaM (W3620A) binding to RyR1 would significantly affect voltage-gated Ca2+ release during EC coupling. Morphological evidence from freeze-fracture studies has suggested that in junctional SR, only every other release channel is associated with a tetrad of DHPRs (Block et al., 1988), raising the question of how “uncoupled” release channels are activated and inactivated. One hypothesis is that Ca2+ released from the mechanically coupled ryanodine receptors activates and inactivates adjacent, uncoupled release channels. In such a scenario, CaM may act to fine-tune the activation of uncoupled release channels by increasing (apoCaM) and lowering (CaCaM) their intrinsic Ca2+ sensitivity. However, maximal SR Ca2+ release in response to depolarization was unaffected by either mutation and in L3624D-expressing myotubes the voltage dependence of Ca2+ release was identical to that of RyR1. Additionally, no significant difference was seen in either the t 1/2 (time to 50%) of Ca2+ transient activation in voltage-clamp experiments (t 1/2 at +50 mV was 24.1 ± 1.9 ms, 22.1 ± 1.8 ms, and 26.8 ± 2.4 ms for RyR1-, W3620A-, and L3624D-expressing myotubes, respectively) or the t 1/2 of decay of spontaneous Ca2+ transients measured in intact Indo-1–loaded myotubes (t 1/2 was 0.11 ± 0.06 s, 0.11 ± 0.02 s, and 0.09 ± 0.01 s for RyR1-, W3620A-, and L3624D-expressing myotubes, respectively). Thus, apoCaM and CaCaM appear to impart little, if any, impact on SR Ca2+ release triggered by the voltage sensor.
Interestingly, expression of W3620A caused a significant increase in maximal L-channel conductance (see Table I). In fact, this increase in L-channel conductance likely represents an underestimation of the actual effect of the W3620A mutation because many of the W3620A-expressing myotubes patch clamped during the course of our experiments had to be excluded from the final analysis due to the presence of excessively large L-currents that resulted in significant compromise of the voltage clamp. Since the estimated voltage error due to series resistance, after compensation, for the myotubes used in Fig. 5 was only slightly larger for W3620A-expressing myotubes (see legend to Table I), the increase in L-channel conductance of W3620A-expressing myotubes may only partially account for the small (∼6 mV) hyperpolarizing shift in VG1/2 and VF1/2 values observed in these experiments. There are several possible mechanisms to explain the increased conductance through L-channels in W3620A-expressing dyspedic myotubes. The W3620A mutation in RyR1 may promote DHPR subunit synthesis and/or membrane insertion, thus resulting in an increase in L-channel surface expression. Alternatively, a conformational change in RyR1 caused by the W3620A mutation could alter the efficiency of retrograde coupling between RyR1 and DHPR. In any event, the increase in L-channel activity is apparently specific for the W3620A mutation, since L-currents from L3624D-expressing myotubes were identical to that of RyR1.
The results presented here do not entirely exclude a role for CaM in SR Ca2+ release during skeletal muscle EC coupling. First of all, CaM regulation of RyR1 may have long-term or more subtle effects that are not detected under the experimental conditions used here. Furthermore, we used single amino acid mutations to disrupt RyR1–CaM interaction that eliminate high-affinity [35S]CaM binding in in vitro assays (Yamaguchi et al., 2001). While these mutations were effective in those assays, it is possible that because of interactions with the DHPR and/or other accessory proteins, the mutant release channels adopt a conformation that permits CaM binding when expressed within junctions of intact muscle cells. Another, however unlikely, possibility is that native RyR1 interactions with other cellular protein(s) unmask a CaM binding site that is not detected in in vitro assays. In addition, although EC coupling in cultured myotubes is widely used as a model for skeletal muscle EC coupling, it is possible that CaM–RyR1 interactions are developmentally regulated such that CaM–RyR1 binding is more prevalent in mature skeletal muscle. Finally, although the experiments in Fig. 3 A indicate that voltage-gated SR Ca2+ release elicited by low frequency (0.5 Hz) repetitive stimulation is similar for RyR1 and the CaM binding–deficient mutants, it will be important for future experiments to determine whether CaM modulates Ca2+ release during rapid or tetanic stimulation.
In conclusion, our results illustrate the importance of studying the function of EC coupling proteins operating within an intact muscle cell environment. Voltage-dependent conformational changes in the DHPR/L-type Ca2+ channel, and not Ca2+ or ligand activation paradigms, are the physiologically relevant trigger for skeletal muscle EC coupling. Although biochemical studies provide valuable insights into the function of proteins of EC coupling, their impact on bidirectional DHPR–RyR1 functional coupling have yet to be readily reconstituted in any heterologous expression system. Our results indicate that under the experimental conditions used here, the control of RyR1 gating by the skeletal muscle voltage sensor is stronger than any possible modulatory effects of CaM.
Acknowledgments
We would like to thank Drs. Kurt G. Beam and Paul D. Allen for providing access to the dyspedic mice used in this study and to Linda Groom for excellent technical assistance.
This work was supported by grants from the National Institutes of Health (AR44657 to R.T. Dirksen, AR18687 to G. Meissner, and DA07232 to K.M.S. O'Connell), and an American Heart Association Grant-in-Aid (to R.T. Dirksen).
Footnotes
Abbreviations used in this paper: CaM, calmodulin; DHPR, dihydropyridine receptor; EC, excitation–contraction; 4-cmc, 4-chloro-m-cresol; L-channel, L-type Ca2+ channel; RyRs, ryanodine receptors; SR, sarcoplasmic reticulum.
References
- Armstrong, C.M., F.M. Bezanilla, and P. Horowicz. 1972. Twitches in the presence of ethylene glycol bis(-aminoethyl ether)-N,N'-tetracetic acid. Biochim. Biophys. Acta. 267:605–608. [DOI] [PubMed] [Google Scholar]
- Avila, G., and R.T. Dirksen. 2000. Functional impact of the ryanodine receptor on the skeletal muscle L-type Ca2+ channel. J. Gen. Physiol. 115:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila, G., and R.T. Dirksen. 2001. Functional effects of central core disease mutations in the cytoplasmic region of the skeletal muscle ryanodine receptor. J. Gen. Physiol. 118:277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila, G., J.J. O'Brien, and R.T. Dirksen. 2001. Excitation-contraction uncoupling by a human central core disease mutation in the ryanodine receptor. Proc. Natl. Acad. Sci. USA. 98:4215–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balshaw, D.M., L. Xu, N. Yamaguchi, D.A. Pasek, and G. Meissner. 2001. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J. Biol. Chem. 276:20144–20153. [DOI] [PubMed] [Google Scholar]
- Beurg, M., C.A. Ahern, P. Vallejo, M.W. Conklin, P.A. Powers, R.G. Gregg, and R. Coronado. 1999. Involvement of the carboxy-terminus region of the dihydropyridine receptor beta1a subunit in excitation-contraction coupling of skeletal muscle. Biophys. J. 77:2953–2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block, B.A., T. Imagawa, K.P. Campbell, and C. Franzini-Armstrong. 1988. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 107:2587–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brillantes, A.B., K. Ondrias, A. Scott, E. Kobrinsky, E. Ondriasova, M.C. Moschella, T. Jayaraman, M. Landers, B.E. Ehrlich, and A.R. Marks. 1994. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 77:513–523. [DOI] [PubMed] [Google Scholar]
- Fessenden, J.D., C.F. Perez, I.N. Pessah, and P.D. Allen. 2002. Localization of key determinants on the ryanodine receptor type 1 (RyR1) required for activation by 4-chloro-m-cresol. Biophys. J. 82:83A. [Google Scholar]
- Fessenden, J.D., Y. Wang, R.A. Moore, S.R. Chen, P.D. Allen, and I.N. Pessah. 2000. Divergent functional properties of ryanodine receptor types 1 and 3 expressed in a myogenic cell line. Biophys. J. 79:2509–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruen, B.R., J.M. Bardy, T.M. Byrem, G.M. Strasburg, and C.F. Louis. 2000. Differential Ca2+ sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am. J. Physiol. Cell Physiol. 279:C724–C733. [DOI] [PubMed] [Google Scholar]
- Grabner, M., R.T. Dirksen, N. Suda, and K.G. Beam. 1999. The II-III loop of the skeletal muscle dihydropyridine receptor is responsible for the Bi-directional coupling with the ryanodine receptor. J. Biol. Chem. 274:21913–21919. [DOI] [PubMed] [Google Scholar]
- Jayaraman, T., A.M. Brillantes, A.P. Timerman, S. Fleischer, H. Erdjument-Bromage, P. Tempst, and A.R. Marks. 1992. FK506 binding protein associated with the calcium release channel (ryanodine receptor). J. Biol. Chem. 267:9474–9477. [PubMed] [Google Scholar]
- Jurado, L.A., P.S. Chockalingam, and H.W. Jarrett. 1999. Apocalmodulin. Physiol. Rev. 79:661–682. [DOI] [PubMed] [Google Scholar]
- MacKrill, J.J. 1999. Protein-protein interactions in intracellular Ca2+-release channel function. Biochem. J. 337:345–361. [PMC free article] [PubMed] [Google Scholar]
- Meissner, G. 1986. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J. Biol. Chem. 261:6300–6306. [PubMed] [Google Scholar]
- Moore, C.P., G. Rodney, J.Z. Zhang, L. Santacruz-Toloza, G. Strasburg, and S.L. Hamilton. 1999. Apocalmodulin and Ca2+ calmodulin bind to the same region on the skeletal muscle Ca2+ release channel. Biochemistry. 38:8532–8537. [DOI] [PubMed] [Google Scholar]
- Nabauer, M., G. Callewaert, L. Cleemann, and M. Morad. 1989. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 244:800–803. [DOI] [PubMed] [Google Scholar]
- Nakai, J., R.T. Dirksen, H.T. Nguyen, I.N. Pessah, K.G. Beam, and P.D. Allen. 1996. Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature. 380:72–75. [DOI] [PubMed] [Google Scholar]
- Nakai, J., N. Sekiguchi, T.A. Rando, P.D. Allen, and K.G. Beam. 1998. Two regions of the ryanodine receptor involved in coupling with L-type Ca2+ channels. J. Biol. Chem. 273:13403–13406. [DOI] [PubMed] [Google Scholar]
- Pate, P., J. Mochca-Morales, Y. Wu, J.Z. Zhang, G.G. Rodney, I.I. Serysheva, B.Y. Williams, M.E. Anderson, and S.L. Hamilton. 2000. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J. Biol. Chem. 275:39786–39792. [DOI] [PubMed] [Google Scholar]
- Rodney, G.G., B.Y. Williams, G.M. Strasburg, K. Beckingham, and S.L. Hamilton. 2000. Regulation of RYR1 activity by Ca2+ and calmodulin. Biochemistry. 39:7807–7812. [DOI] [PubMed] [Google Scholar]
- Saimi, Y., and C. Kung. 2002. Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 64:289–311. [DOI] [PubMed] [Google Scholar]
- Samso, M., and T. Wagenknecht. 2002. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. 277:1349–1353. [DOI] [PubMed] [Google Scholar]
- Sencer, S., R.V. Papineni, D.B. Halling, P. Pate, J. Krol, J.Z. Zhang, and S.L. Hamilton. 2001. Coupling of RYR1 and L-type calcium channels via calmodulin binding domains. J. Biol. Chem. 276:38237–38241. [DOI] [PubMed] [Google Scholar]
- Smith, J.S., E. Rousseau, and G. Meissner. 1989. Calmodulin modulation of single sarcoplasmic reticulum Ca2+-release channels from cardiac and skeletal muscle. Circ. Res. 64:352–359. [DOI] [PubMed] [Google Scholar]
- Takeshima, H., M. Iino, H. Takekura, M. Nishi, J. Kuno, O. Minowa, H. Takano, and T. Noda. 1994. Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature. 369:556–559. [DOI] [PubMed] [Google Scholar]
- Tanabe, T., K.G. Beam, B.A. Adams, T. Niidome, and S. Numa. 1990. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 346:567–569. [DOI] [PubMed] [Google Scholar]
- Tripathy, A., L. Xu, G. Mann, and G. Meissner. 1995. Calmodulin activation and inhibition of skeletal muscle Ca2+ release channel (ryanodine receptor). Biophys. J. 69:106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, N., C. Xin, and G. Meissner. 2001. Identification of apocalmodulin and Ca2+-calmodulin regulatory domain in skeletal muscle Ca2+ release channel, ryanodine receptor. J. Biol. Chem. 276:22579–22585. [DOI] [PubMed] [Google Scholar]