

Abstract
ATP-sensitive potassium (KATP) channels are formed by the coassembly of four Kir6.2 subunits and four sulfonylurea receptor subunits (SUR). The cytoplasmic domains of Kir6.2 mediate channel gating by ATP, which closes the channel, and membrane phosphoinositides, which stabilize the open channel. Little is known, however, about the tertiary or quaternary structures of the domains that are responsible for these interactions. Here, we report that an ion pair between glutamate 229 and arginine 314 in the intracellular COOH terminus of Kir6.2 is critical for maintaining channel activity. Mutation of either residue to alanine induces inactivation, whereas charge reversal at positions 229 and 314 (E229R/R314E) abolishes inactivation and restores the wild-type channel phenotype. The close proximity of these two residues is demonstrated by disulfide bond formation between cysteine residues introduced at the two positions (E229C/R314C); disulfide bond formation abolishes inactivation and stabilizes the current. Using Kir6.2 tandem dimer constructs, we provide evidence that the ion pair likely forms by residues from two adjacent Kir6.2 subunits. We propose that the E229/R314 intersubunit ion pairs may contribute to a structural framework that facilitates the ability of other positively charged residues to interact with membrane phosphoinositides. Glutamate and arginine residues are found at homologous positions in many inward rectifier subunits, including the G-protein–activated inwardly rectifying potassium channel (GIRK), whose cytoplasmic domain structure has recently been solved. In the GIRK structure, the E229- and R314-corresponding residues are oriented in opposite directions in a single subunit such that in the tetramer model, the E229 equivalent residue from one subunit is in close proximity of the R314 equivalent residue from the adjacent subunit. The structure lends support to our findings in Kir6.2, and raises the possibility that a homologous ion pair may be involved in the gating of GIRKs.
Keywords: inward rectifiers, salt bridge, KATP, PIP2, gating
INTRODUCTION
Currents through inwardly rectifying potassium (Kir) channels are important determinants of the resting membrane potential in many cell types (Nichols and Lopatin, 1997; Hille, 2001). The Kir family has many members comprising seven subfamilies, and functional channels are tetrameric assemblies that may be homo- or heteromeric. All Kir subunits share the two transmembrane domain topology and distinct primary sequence homology, yet among the different subtypes, channel gating is controlled by a variety of stimuli (Nichols and Lopatin, 1997; Hille, 2001). Among them are ATP-sensitive potassium (KATP) channels that are gated by intracellular ATP and ADP, reflecting the energetic state of a cell (Bryan and Aguilar-Bryan, 1997; Ashcroft and Gribble, 1998; Aguilar-Bryan and Bryan, 1999). KATP channels underlie numerous important physiological processes, including regulation of insulin secretion, control of vascular tone, and electrophysiological changes during ischemia in cardiac and neuronal cells (Aguilar-Bryan and Bryan, 1999).
KATP channels are assembled from four Kir6.2 subunits and in addition require four sulfonylurea receptor subunits (SUR)* (Inagaki et al., 1995, 1997; Clement et al., 1997; Shyng and Nichols, 1997). The activity of KATP channels is determined by orchestrated interactions between the channel subunits and intracellular ATP and ADP. ATP inhibits channel opening by binding to the pore-forming Kir6.2 subunits, an effect that does not involve ATP hydrolysis (Tucker et al., 1997, 1998; Drain et al., 1998; Tanabe et al., 1999). On the other hand, ATP stimulates channel opening by interacting with SUR1, an effect that requires Mg2+ and ATP hydrolysis at the second nucleotide binding fold, or NBF2 (Shyng et al., 1997b; Ashcroft and Gribble, 1998; Gribble et al., 1998b). ADP, accompanied by Mg2+, also interacts with NBF2 of SUR1 and stabilizes the protein in a postnucleotide hydrolysis state to stimulate channel activity (Nichols et al., 1996; Gribble et al., 1997; Shyng et al., 1997b; Zingman et al., 2001). In addition to intracellular nucleotides, membrane phosphoinositides (PIPs), such as phosphatidylinositol bisphosphate (PIP2), and phosphatidylinositol trisphosphate (PIP3), have a profound effect on channel activity (Fan and Makielski, 1997; Baukrowitz et al., 1998; Shyng and Nichols, 1998). PIP2 and PIP3 increase the open probability of the channel and antagonize the inhibitory effect of ATP. Although many reports support a model in which ATP and phosphoinositides bind directly to the cytoplasmic domains of Kir6.2 to promote channel closure and opening, respectively (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Tucker et al., 1998; Tanabe et al., 1999; MacGregor et al., 2002; Wang et al., 2002), little is known about the tertiary or quaternary structural elements in these domains that are involved in ligand binding and channel gating (Cukras et al., 2002a,b; MacGregor et al., 2002).
In an earlier study, we systematically mutated each positively charged amino acid in the cytoplasmic domain of Kir6.2 to alanine to identify residues that are involved in interactions between the KATP channel and the negatively charged phosphate groups in PIPs (Shyng et al., 2000b). Three of the arginine-to-alanine mutations (R192A, R301A, and R314A) that exhibited reduced response to exogenously applied PIP2 also displayed rapid spontaneous decay of currents after membrane excision into ATP-free solutions, a behavior which we have termed “inactivation” (Shyng et al., 2000b). Channel inactivation appears different from the “rundown” phenomenon frequently observed with wild-type (WT) channels in that it is faster and can be recovered by exposure and then washout of ATP even in the absence of Mg2+ (Ribalet et al., 2000; Shyng et al., 2000b).
In this study, the mechanisms underlying KATP channel inactivation were investigated. Although it is possible that the three arginine residues (192, 301, and 314) are directly responsible for binding to the negatively charged phosphate groups in PIP2, an alternative hypothesis is that the arginine residues are involved in maintaining structural conformations necessary for channel activity. Specifically, we tested whether the R192A, R301A, and R314A mutations cause channel inactivation by disrupting electrostatic interactions within the channel complex. Through systematic mutation of each of the negatively charged residues in the cytoplasmic regions of Kir6.2 to alanine, we found that mutation E229A also induces fast channel inactivation similar to that seen in the R192A, R301A, and R314A mutants. Double charge reversal at positions 229 and 314 (E229R/R314E) abolished inactivation and restored the WT channel phenotype, indicating that these residues form an ion pair. The close proximity of these two residues was supported by disulfide bond formation between cysteine residues introduced at the two positions (E229C/R314C); disulfide bond formation abolished inactivation and stabilized the current. Experiments using a series of Kir6.2 tandem dimer constructs containing mutations at position 229 and/or 314 in one or both subunits showed that the E229/R314 ion pair is likely formed between two adjacent Kir6.2 subunits.
MATERIALS AND METHODS
Molecular Biology
Mouse Kir6.2 cDNA in the pCMV6b vector was used as a template for introducing point mutations in Kir6.2. Site-directed mutagenesis was performed by overlap extension at the junctions of the relevant residues by sequential polymerase chain reaction (PCR) or by the QuickChange site-directed mutagenesis kit (Stratagene). All constructs were sequenced to verify the correct mutations. Mutant clones from multiple PCR reactions were analyzed to avoid false results caused by undesired mutations introduced by PCR.
The Kir6.2 tandem dimer was constructed as follows. Two Kir6.2 PCR fragments with unique restriction sites and linkers were generated. The first fragment has a ClaI site at the 5′ end and a five glutamine linker with an NheI site replacing the stop codon at the 3′ end. The second fragment has an NheI site followed by a five glutamine linker at the 5′ end and an EcoRI site at the 3′ end. The two fragments were ligated into the pCMV6b vector. The resulting construct has a QQQQQASQQQQQ linker between the two Kir6.2 subunits.
Inside-out Patch-clamp Recordings
COSm6 cells were tranfected using Fugene 6 (Roche) and plated onto coverslips. The cDNA for the green fluorescent protein (GFP) was cotransfected with SUR1 and Kir6.2 to facilitate identification of positively transfected cells. Patch-clamp recordings were performed 36–72 h posttransfection at room temperature, in a chamber which allowed rapid exchange of bathing solution. Micropipettes were pulled from nonheparinized Kimble glass (Fisher Scientific) on a horizontal puller (Sutter Instrument Co.). Electrode resistance was typically 0.5–1 MΩ when filled with K-INT (below). Inside-out patches were voltage-clamped with an Axopatch 1B amplifier (Axon Instruments, Inc.). The standard bath (intracellular) and pipette (extracellular) solution (K-INT) had the following composition: 140 mM KCl, 10 mM K-HEPES, 1 mM K-EGTA, pH 7.3. ATP and AMP-PNP (Sigma-Aldrich) were added as the potassium salt. In the majority of the experiments, 1 mM EDTA was included in the K-INT solution to chelate residual Mg2+. PIP2 (Roche) was prepared as described previously (Shyng and Nichols, 1998). Dithiothreotol (DTT) at 1 mM was used to reduce disulfide bonds. All currents were measured at a membrane potential of −50 mV (pipette voltage = 50 mV) and inward currents were shown as upward deflections. Data were filtered at 2 kHz and analyzed using pCLAMP software. All data are expressed as means ± SEM.
Immunofluorescent Staining
For cell surface staining of FLAG-tagged SUR1, living COSm6 cells transiently expressing FLAG-tagged SUR1 and various Kir6.2 mutants were incubated with 10 μg/ml anti-FLAG M2 monoclonal antibody (in 0.1 mg BSA/ml OptiMEM) for 1 h at 4°C. They were washed with ice-cold PBS and incubated with Cy-3–conjugated donkey anti–mouse secondary antibodies (diluted in 0.1 mg BSA/ml OptiMEM) for 30 min at 4°C. After extensive wash in ice cold PBS, cells were fixed with 4% paraformaldehyde and viewed using a Leica fluorescent microscope.
RESULTS
Mutation of Glutamate 229 in Kir6.2 to Alanine Induces KATP Channel Inactivation
KATP currents generated by coexpression of SUR1 and Kir6.2 mutants R192A, R301A, or R314A have been shown previously to inactivate after inside-out patch excision into ATP-free solutions (Shyng et al., 2000b). We hypothesized that the three arginines may form ion pairs with negatively charged residues in Kir6.2 and that these ion pairs are important for maintaining channel activities; if true, neutralization of the negatively charged residues should induce similar channel inactivation. There are 30 negatively charged residues in the cytoplasmic regions of Kir6.2, each of which was individually replaced with an alanine and screened for inactivation. Mutation of glutamate 229 to alanine (E229A) resulted in robust current inactivation (Figs. 1 B, and 2, A and B), with characteristics similar to those seen with the R192A, R301A, and R314A channels (Shyng et al., 2000b).
Figure 1.
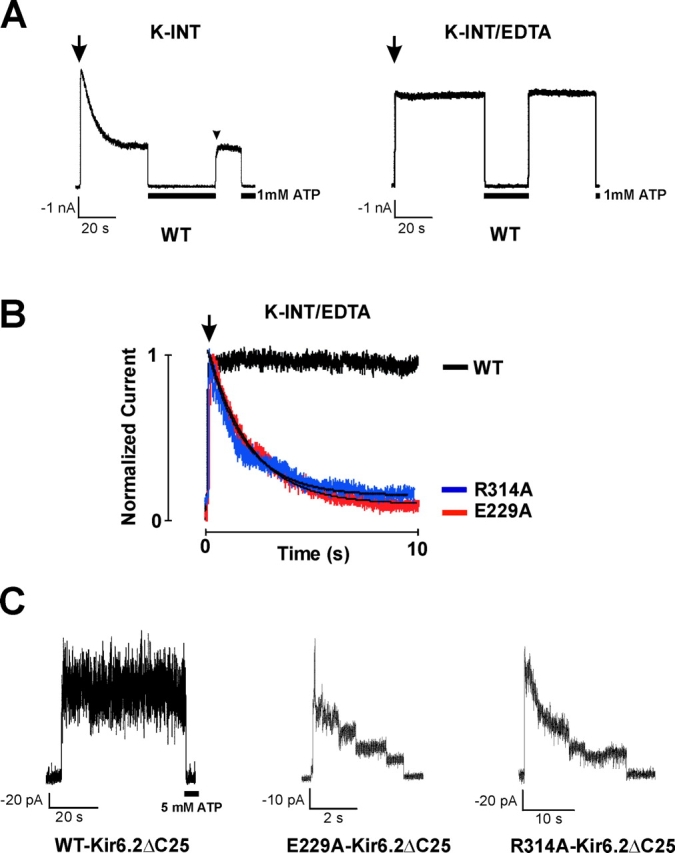
Identification of E229A as an inactivation mutant. (A) WT channel activities recorded in inside-out membrane patches using K-INT (left) or K-INT + 1 mM EDTA (right) bath solution. Arrows indicate patch excision. Channel rundown is dramatically reduced by the addition of 1 mM EDTA to the bath solution. (B) Using K-INT/EDTA solution, inactivation is observed in the E229A mutant channels. Representative currents recorded from inside-out membrane patches containing WT, the E229A, or the R314A mutant channels are superimposed for comparison. The arrow indicates patch excision into K-INT/EDTA. The kinetics of inactivation of the E229A channels are similar to the R314A channels. The inactivation time course of both E229A and R314A channels was fitted by a single exponential (indicated by the black curves). (C) Channels formed by WT-Kir6.2ΔC25 in the absence of SUR1 exhibited stable activities after patch excision into K-INT/EDTA (left). In contrast, channels formed by E229A-Kir6.2ΔC25 (middle) or R314A-Kir6.2ΔC25 (right) in the absence of SUR1 showed rapid current decay. Currents in this and subsequent figures were recorded at −50 mV membrane potential. Inward currents are shown as upward deflections.
Quantitation of Channel Inactivation: Comparison with Channel Rundown
KATP currents run down after patch excision into K-INT solution (140 mM KCl, 10 mM HEPES, 1 mM EGTA, pH 7.3; Fig. 1 A, left). The rate of rundown varies and can be rapid in some patches, making it difficult to distinguish from inactivation. Ribalet et al. (2000) have reported previously that channel rundown involves multiple factors including loss of MgADP stimulation, loss of membrane PIP2, and protein dephosphorylation. We reasoned that since many protein phosphatases, as well as phospholipases and lipid phosphatases that break down membrane PIPs require Mg2+ or Ca2+ (Cohen, 1989; Matzaris et al., 1994; Palmer et al., 1994; Hilgemann and Ball, 1996; Singer et al., 1997), removal of residual Mg2+ and other metal ions in the bath solution might prevent rundown. We found that addition of 1 mM EDTA to the K-INT solution (K-INT/EDTA) dramatically reduced channel rundown (Fig. 1 A, right). Using K-INT/EDTA bath solution, the WT current decayed only by 10.72 ± 3.66% over a 20-s period after patch excision (n = 10; Table I) , in contrast to 57.06 ± 3.37% in K-INT solution without EDTA (n = 38). The significant reduction of channel rundown made it possible to quantitatively compare the current stability of WT to that of inactivation mutants (Fig. 1 B). In K-INT/EDTA solution, the current from the E229A mutant still inactivated rapidly; the current decay was 81.19 ± 5.39% at 20 s after patch excision (n = 7; Fig. 1 B, Table I). The time course of inactivation of E229A channels was fit by a single exponential, yielding a time constant (τi) of 6.64 ± 1.80 s. Under the same recording condition, the inactivation time constants were 4.79 ± 1.34, 0.32 ± 0.03, and 3.08 ± 0.43 s for R192A, R301A, and R314A channels, respectively (n = 7–9 patches in each case as shown in Table I; the slow current decay in WT could not be fit by a single exponential, therefore no value was given).
TABLE I.
Current Stability of Channels Formed by SUR1 and Kir6.2 Monomers
| Mutation | Percentage current decay in 20 s |
Inactivation time constanta τi |
Number of patches (n) |
|---|---|---|---|
| s | |||
| WT | 10.72 ± 3.66 | N/A | 10 |
| E229A | 81.19 ± 5.39 | 6.64 ± 1.80 | 7 |
| R192A | 78.24 ± 5.92 | 4.79 ± 1.34 | 9 |
| R301A | >99.00 | 0.32 ± 0.03 | 8 |
| R314A | 94.98 ± 1.89 | 3.08 ± 0.43 | 8 |
| E229R | 92.94 ± 2.22 | 3.02 ± 0.31 | 3 |
| R192E | 84.83 ± 1.10 | 5.19 ± 0.67 | 9 |
| R301E | No currents | N/A | 16 |
| R314E | 78.15 ± 3.44 | 1.28 ± 0.13 | 3 |
| E229R/R192E | 66.06 ± 3.98 | 7.70 ± 0.06 | 11 |
| E229R/R301E | No currents | N/A | 9 |
| E229R/R314E | 6.95 ± 3.27 | N/A | 5 |
| E229C | 75.14 ± 7.65 | 4.83 ± 0.96 | 7 |
| R314C | No currents | N/A | 20 |
| E229C/R314C | 46.98 ± 5.96 | N/A | 6 |
Values were given only for those mutants whose time course of inactivation could be fit well by a single exponential.
The fast phase of channel rundown has been largely attributed to loss of MgADP-dependent functional coupling between SUR1 and Kir6.2 (Ribalet et al., 2000). We asked whether the current decay seen in the inactivation mutants could be due to uncoupling between SUR1 and Kir6.2. Kir6.2, when its distal COOH-terminal 25 or 35 amino acids are deleted, can form functional channels in the absence of SUR1 (Tucker et al., 1997). We constructed Kir6.2ΔC25 containing either the E229A or the R314A mutation and tested whether the resulting channels also exhibited rapid current decay. Fig. 1 C shows that, in contrast to WT-Kir6.2ΔC25 channels whose activities were stable over the course of the recording, the E229A- and R314A-Kir6.2ΔC25 channels inactivated similar to E229A and R314A channels formed in the presence of SUR1. The extent of current decay measured at 20 s after patch excision and the estimated inactivation time constant are 100 ± 0% (n = 3) and 2.5 s (n = 2) for E229A-Kir6.2ΔC25, and 90.3 ± 11.2% (n = 4) and 5.2 s (n = 2) for R314A-Kir6.2ΔC25. The fact that inactivation occurs even in the absence of SUR1 indicates that a structural change in the Kir6.2 tetramer itself is a major mechanism underlying inactivation.
Channel Inactivation Caused by the E229A Mutation Can Be Recovered by Exposure to ATP and Subsequent Washout of ATP, and Is Reversed by PIP2
A defining feature of channel inactivation is that after exposure to ATP and subsequent washout of ATP, the inactivated channels become reactivated (Shyng et al., 2000b); this is also seen in the E229A mutant channels (Fig. 2 A). The “reactivation” effect by ATP is dose and time dependent: the higher the concentration and the longer the exposure time, the more complete the reactivation effect. A nonhydrolysable ATP analogue, AMP-PNP, mimics the effect of ATP in reactivating the E229A and R314A mutant channels (Fig. 2 B). Therefore, the reactivation effect of ATP is not mediated by nucleotide hydrolysis at the NBF2 of SUR1 (Zingman et al., 2001), or by phosphorylation of the channel proteins (Beguin et al., 1999; Light et al., 2000; Lin et al., 2000). Interestingly, in channels formed by E229A-Kir6.2ΔC25 alone, current decay could not be recovered by ATP exposure and its subsequent removal (Fig. 2 C, left). However, when coexpressed with SUR1, inactivated E229A-Kir6.2ΔC25 channels could be reactivated by ATP and subsequent washout of ATP (Fig. 2 C, right). Similar observations were made with the R314A mutation (unpublished data). These results suggest that SUR1 plays a role in mediating the reactivation effect of ATP.
Figure 2.

Channel inactivation can be recovered by exposure to ATP and subsequent washout of ATP. (A) Inactivated R314A (left) and E229A (right) channels were reactivated by exposure to ATP (5 mM) and subsequent washout of ATP. (B) A nonhydrolyzable ATP analogue, AMP-PNP mimicked the reactivation effect of ATP in both R314A (left) and E229A (right) channels. (C) The reactivation effect of ATP was not observed in channels formed by E229A-Kir6.2ΔC25 alone (left), but was retained in channels formed by E229A-Kir6.2ΔC25 in the presence of SUR1 (right).
Aside from exposure to ATP and subsequent removal of ATP, PIP2 can also reverse channel inactivation caused by the E229A mutation, similar to that found in the R192A, R301A, or R314A mutant channels (Shyng et al., 2000b). After inactivation of E229A channels, PIP2 gradually increased the current to a stable maximum that exceeded the amplitude observed at the time of patch excision; PIP2 also rendered the channel less sensitive to ATP inhibition (Fig. 3) .
Figure 3.

Inactivation of E229A mutant channels is reversed by PIP2. Application of PIP2 (10 μM) reversed inactivation and stimulated channel activity; it also decreased the ATP sensitivity of the channel, as was previously reported for WT channels (Baukrowitz et al., 1998; Shyng and Nichols, 1998).
The Double Mutation E229R/R314E Rescues Channel Inactivation Induced by E229R or R314E
The similar inactivation behavior caused by charge neutralization of E229 and one of the R192, R301, and R314 residues prompted us to examine the possibility that E229 may form a salt bridge with one of the three arginine residues to maintain channel activities. If true, disruption of the ion pair should cause inactivation, whereas exchanging the residues at the two positions should preserve the electrostatic interaction and retain WT channel behavior. Currents from patches containing channels harboring the individual charge reversal mutations E229R, R192E, or R314E all inactivated like the charge neutralization mutants at those positions, with similar kinetics (Fig. 4 A, Table I), while no macroscopic currents were detected in patches from cells transfected with the R301E Kir6.2 mutant. Immunostaining and Western blot experiments detected only very low levels of the R301E Kir6.2 mutant protein (unpublished data), suggesting that the R301E mutation may have adversely affected the stability of the protein.
Figure 4.
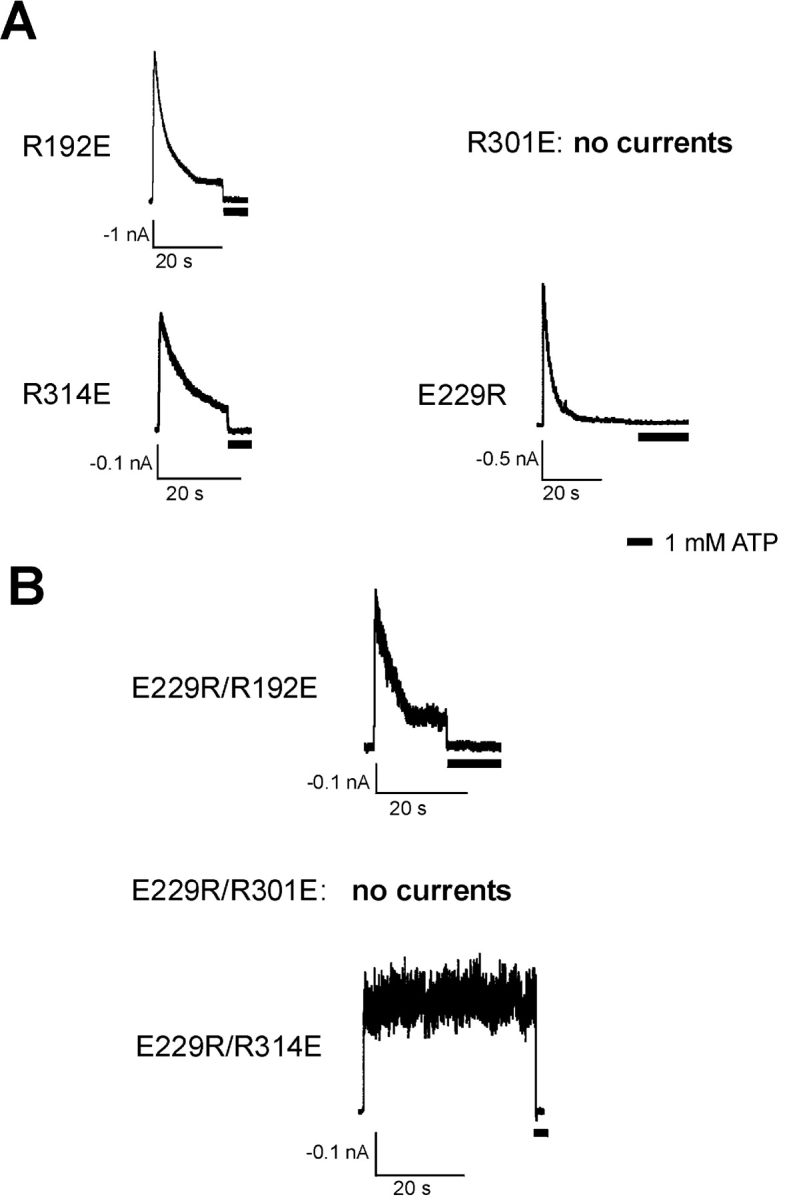
Open state stability is retained in the E229R/R314E double reverse mutant channels. (A) Representative currents recorded from inside-out membrane patches containing R192E, R301E, R314E, or E229R mutant channels. R301E channels did not give rise to measurable currents. All the other three mutant channels exhibited inactivation. (B) Currents recorded from membrane patches isolated from cells expressing the double charge reversal mutant E229R/R192E, E229R/R301E, or E229R/R314E. No currents were detected for the E229R/R301E channels. The E229R/R192E mutant channels still showed inactivation. In contrast, the E229R/R314E double mutant did not inactivate and behaved like WT channels.
Next, the three double mutants, E229R/R192E, E229R/R301E, and E229R/R314E, were constructed in which the residues at the two positions in each potential ion pair were exchanged. E229R/R301E failed to generate measurable currents (Fig. 4 B), whereas E229R/R192E displayed inactivation similar to the E229R and the R192E single mutations (66.06 ± 3.98% current decay over 20 s in K-INT/EDTA, with τi = 7.70 ± 0.06 s; Table I). In contrast, the E229R/R314E double mutant exhibited WT-like behavior, lacking the rapid current decay induced by either the E229R or the R314E single charge reversal mutations (Fig. 4 B). For E229R/R314E channels, the extent of current decay measured at 20 s after patch excision into K-INT/EDTA is only 6.95 ± 3.27% (n = 5), comparable to that of WT channels (10.72 ± 3.66%; see Table I). These results are consistent with the idea that an ion pair formed between E229 and R314 stabilizes KATP channel activity.
E229 and R314 of Kir6.2 Form an Ion Pair that Increases KATP Channel Activity
Although the data presented above suggest electrostatic interactions between E229 and R314, they do not demonstrate definitively that the two residues interact directly with each other. One approach to assess the proximity of protein domains is to exploit the ability of cysteine residues to form disulfide bonds. This approach has been successfully used to study the structure-function relationships for a number of ion channels, including shaker potassium channels, cyclic nucleotide–gated (CNG) channels, and GABAA receptors (Gordon and Zagotta, 1995; Papazian et al., 1995; Gordon et al., 1997; Larsson and Elinder, 2000; Horenstein et al., 2001). Accordingly, we further probed the interaction between E229 and R314 by substituting them with two cysteine residues. If the two residues are in close proximity, it should be possible to form a disulfide bond, and this covalent bonding should stabilize channel opening.
In channels carrying a single cysteine mutation at position 229 of Kir6.2, rapid inactivation was observed, similar to that of E229A or E229R mutant channels (Fig. 5 A, Table I). However, mutation of R314 to cysteine resulted in channels that did not give rise to detectable currents, although Western blots confirmed expression of the mutant protein (unpublished data). To see whether the R314C mutant Kir6.2 was indeed incorporated into the channel complex, we performed surface immunostaining for the FLAG-epitope in cells cotransfected with R314C Kir6.2 and FLAG-tagged SUR1 (Cartier et al., 2001). Surface expression of FLAG-SUR1 requires that the protein coassemble with Kir6.2 to form an octameric channel complex, which shields the RKR tripeptide ER retention/retrieval signals in the individual subunits (Zerangue et al., 1999). Fig. 5 B shows that the surface FLAG staining in cells coexpressing FLAG-SUR1 and R314C Kir6.2 is equivalent to cells coexpressing FLAG-SUR1 and WT Kir6.2, indicating that the R314C Kir6.2 does form octameric channel complexes with SUR1 (Zerangue et al., 1999), but that the open probability of the resulting channels may be too low to be detected. These results are consistent with those of John et al. (2001) obtained with R314C-Kir6.2 linked to GFP.
Figure 5.

Characterization of the E229C and the R314C mutant channels. (A) A representative current trace recorded from an inside-out membrane patch containing the E229C mutant channels. The channels displayed inactivation similar to that observed in the E229A and E229R channels upon patch excision into the K-INT/EDTA solution. Exposure to 1 mM ATP (indicated by black bars below the recordings) partially reactivated the channels, which again underwent rapid inactivation. (B) The R314C Kir6.2 mutant protein is expressed and incorporated into the KATP channel complex. Surface immunofluorescent staining of cells coexpressing FLAG-tagged SUR1 and either the WT or the R314C mutant Kir6.2. Immunostaining was performed in living cells at 4°C using the M2 anti-FLAG monoclonal antibody followed by Cy3-conjugated secondary antibody. Surface labeling of the FLAG-SUR1 was observed in both cells coexpressing the WT or the R314C mutant Kir6.2, demonstrating that the R314C Kir6.2 is properly incorporated into the KATP channel complex.
The E229C/R314C double cysteine mutant channels opened, upon patch isolation into ATP-free K-INT/EDTA solution. The initial current amplitude after patch excision was low, and the rate of current decay was faster than WT channels, but slower than the E229A and the R314A channels (the percentage of current decay in 20 s is 46.98 ± 5.96, compared with 10.72 ± 3.66, 81.19 ± 5.39, and 94.98 ± 1.89 for WT, E229A, and E314A channels, respectively; see Table I). We found that given enough time, nearly all patches (21 out of a total of 23 patches) exhibited gradual increases and stabilization of the currents, after repeated exposure to 5 mM ATP (fold increase = 6.02 ± 1.01, n = 21; two examples shown in Fig. 6) . The rate at which this current increase occurred varied. In general, the more frequent and longer the patch was exposed to ATP, the faster the current increase was observed upon subsequent washout of ATP. Since the cytoplasmic environment is reducing and does not favor disulfide-bond formation, it is likely that immediately after patch excision most channels are already in the “inactivated” state, explaining the low initial current amplitude. After patch excision and exposure to ATP to close the channels, subsequent washout of ATP permits the channels to become “reactivated” (see discussion). The patch now is exposed to an oxidizing environment and spontaneous disulfide bond formation between 229C and 314C stabilizes the reactivated channels and increases current amplitudes. Consistent with this interpretation, subsequent exposure of these patches to the reducing reagent DTT (1 mM) caused rapid inactivation of the reactivated channels in a reversible manner (Fig. 6, top trace). Interestingly, the open probability of the E229C/R314C channel after its current has reached a maximum is ∼0.5, comparable to that observed in WT channels under normal conditions (estimated by noise analysis; Shyng et al., 1997a). Application of 5 μM PIP2 further stimulated the current by ∼2-fold (Fig. 6, bottom trace).
Figure 6.

Disulfide bond formation between C229 and C314 in the E229C/R314C mutant channel stabilizes channel opening. Currents recorded from two separate membrane patches containing the E229C/R314C mutant channels. Upon patch excision into K-INT/EDTA, the channel exhibited low open probability and mild inactivation. However, the current gradually increased in amplitude and stability. Exposure of the patch to 5 mM ATP (indicated by the black bars underneath the current traces) accelerated the current reactivation process. (Top trace) Application of 1 mM DTT (in K-INT/EDTA, indicated by the gray bar) after current stabilization induced rapid inactivation of the channel in a reversible manner, indicating that the current stabilization was due to spontaneous disulfide bond formation. (Bottom trace) After the current has reached the maximum, application of 5 μM PIP2 (see materials and methods) further stimulated channel activity by ∼2-fold.
It is conceivable that disulfide bond formation is mediated by other endogenous cysteine residues in the channel, or by single engineered cysteine from two different Kir6.2 subunits. However, neither WT channels nor the single cysteine mutant channels, E229C and R314C, exhibited the time-dependent current increase as that seen in the E229C/R314C double cysteine mutant channels (Fig. 7 , n = 4–5 in each case; no recording for R314C is shown since there were no measurable currents). Further, DTT had little effects on the kinetics of either WT or E229C channels (Fig. 7). Together, these results strongly support disulfide bond formation between C229 and C314, and that the disulfide bond increases the stability of channel activities.
Figure 7.

Disulfide bond formation that promotes channel opening in the E229C/R314C mutant does not involve endogenous cysteines. Representative recordings of WT and E229C channels showing the lack of time-dependent current increase in K-INT/EDTA and the lack of response to DTT.
The E229-R314 Ion Pair Is Formed between Two Kir6.2 Subunits
To determine whether the ionic interaction between E229 and R314 occurs between two Kir6.2 subunits, or within a single Kir6.2 subunit, we conducted experiments using Kir6.2 tandem dimers. Tandem dimers were constructed such that the COOH terminus of subunit one is joined to the NH2 terminus of subunit two by a linker of 12 amino acids (see materials and methods). Work on other potassium and CNG channels has suggested that assembly of such dimers likely follow the head-to-tail direction, such that the leading subunits of the two dimers are positioned diagonally to each other (Isacoff et al., 1990; Gordon and Zagotta, 1995; Pascual et al., 1995; Liu et al., 1998; He et al., 2000; Shapiro and Zagotta, 2000). Such head-to-tail cyclic symmetry was also observed in the crystal structure of G-protein–activated inwardly rectifying potassium channel (GIRK1) (Nishida and MacKinnon, 2002). In principle, with tandem dimers, one can constrain the ability of the channel to form intra- or intersubunit ion pairing, by introducing mutations at the E229 and R314 positions in designated subunits. For example, only intersubunit ion pairing is possible, but no intrasubunit ion pairing is possible, in a construct that has R314 mutated to E in the first subunit, and E229 mutated to R in the second subunit (we name this the EE-RR dimer, where the amino acids at position 229 and 314 shown in single letter codes in the first subunit are joined by a hyphen with the amino acids at position 229 and 314 of the second subunit; Fig. 8 B). On the other hand, only intrasubunit ion pairing is possible, but no intersubunit ion pairing is possible, in a construct in which the first subunit carries the E229R/R314E double charge reversal mutations and the second subunit is WT (the RE-ER dimer; Fig. 8 D).
Figure 8.
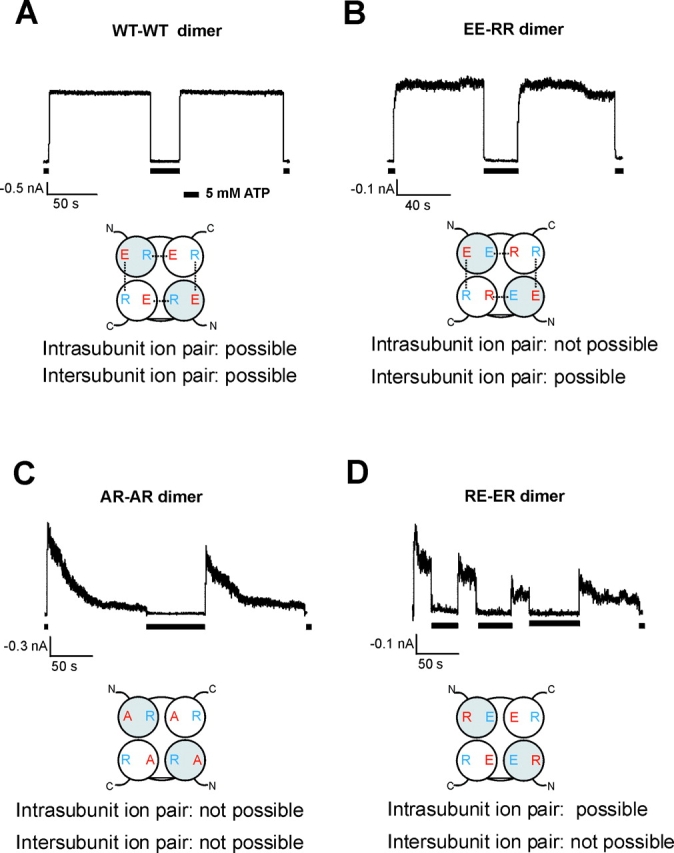
The E229/R314 ion pair is formed between two adjacent Kir6.2 subunits. (A) Currents recorded from a membrane patch containing Kir6.2 WT-WT (or ER-ER) dimer channels in the K-INT/EDTA bath solution. The dimer channel currents were stable and were inhibited by 5 mM ATP. Below the current trace is a cartoon illustrating the expected tetramer formed by the tandem dimer. The 229 residue in each subunit is shown in red and the 314 residue shown in blue. For each tandem dimer, the leading subunit is in gray and the trailing subunit in white. The expected ion pairs are connected by dotted lines. (B) In the EE-RR dimer channels, the possibility of ion pair formation in a single subunit is excluded. The channels behaved similarly to the WT-WT dimer channels; the rate of current decay is only slightly faster (see Table II). (C) In the AR-AR dimer channels, neither intra- nor intersubunit ion pair formation is possible. Currents from these channels decayed much faster than the WT-WT dimer channels, as expected. (D) In the RE-ER dimer channels, the possibility of ion pair formation between two adjacent subunits is excluded. Currents from these channels decayed much faster than the WT-WT, or the EE-RR dimer channels (see Table II).
When coexpressed with SUR1, the WT-WT Kir6.2 tandem dimer (or the ER-ER dimer) gave rise to channels with stable currents that were inhibited by ATP (Fig. 8 A). Mutation of E229 to alanine in both subunits (the AR-AR dimer) induced marked current inactivation, as expected (Fig. 8 C). The time course and the extent of current decay seen from AR-AR dimer channels were not as pronounced as those seen from the E229A monomer channels. A potential explanation for this difference is that the dimer channels exhibit a slightly higher open probability (Po = 0.88 ± 0.02; n = 6) than channels formed by Kir6.2 monomers (Po=0.67 ± 0.05; n = 8); this higher Po likely exerts an antagonizing effect on channel inactivation induced by the E229A mutation. Because of the slower inactivation rate seen in the dimer mutant channels, we measured the extent of current decay at 40 s after patch excision into K-INT/EDTA, as shown in Table II . Currents from the WT-WT and the AR-AR dimer channels decayed by 6.08 ± 2.19% and 48.35 ± 6.54%, respectively, at 40 s after patch excision. The EE-RR dimer, in which intrasubunit ion pair formation is prohibited but intersubunit ion pair formation is allowed, generated channels that had relatively stable currents in ATP-free solution, resembling WT and WT-WT dimer channels (Fig. 8 B). The percentage of current decay is 14.94 ± 6.73% at 40 s after patch excision into K-INT/EDTA solution (Table II). By contrast, the ER-RE dimer, in which only intrasubunit ion pair formation is allowed, generated currents that decayed much faster than the WT-WT and the EE-RR dimers (Fig. 8 D). The percentage of current decay is 38.05 ± 6.07% at 40 s after patch excision (Table II). Collectively, these results are consistent with the E229/R314 ion pair being formed between two Kir6.2 subunits.
TABLE II.
Current Stability of Channels Formed by SUR1 and Kir6.2 Tandem Dimers
| Mutation | Percentage current decay in 40 s |
Number of patches (n) |
|---|---|---|
| WT-WT (ER-ER) dimer | 6.08 ± 2.19 | 17 |
| AR-AR dimer | 48.35 ± 6.54 | 15 |
| EE-RR dimer | 14.94 ± 6.73 | 8 |
| ER-RE dimer | 38.05 ± 6.07 | 14 |
DISCUSSION
Electrostatic and hydrogen bonding interactions between two oppositely charged amino acids are important determinants of ion channels structures and their disruption can often lead to altered or lost functions (Papazian et al., 1995; Yang et al., 1997; Doyle et al., 1998). The data presented here demonstrate that electrostatic interactions between glutamate 229 and arginine 314 in Kir6.2 are critical for maintaining KATP channel activity. First, mutation of either charged residue to a neutral alanine, or to an oppositely charged amino acid, induces rapid inactivation of KATP currents. Second, exchanging the amino acids at positions 229 and 314 restores current stability comparable to the WT channel. Third, currents are stabilized by disulfide bond formation between two engineered cysteine residues at positions 229 and 314 in Kir6.2. Disulfide bond formation between the two cysteine residues indicates that the distance between the α-carbons of the two cysteines is ∼5.6 Å; (Careaga and Falke, 1992; Krovetz et al., 1997).
Relationship between Channel Inactivation and Channel Rundown: the Role of SUR1 and Kir6.2
Channel inactivation and the fast-phase of channel rundown both result in rapid current decay. What is the relationship between these two phenomena? One major mechanism accounting for the fast-phase rundown in WT channels is uncoupling between Kir6.2 and SUR1 due to loss of MgADP at NBF2; channels formed by Kir6.2 alone do not show fast-phase rundown (Ribalet et al., 2000). Our result that channels formed by E229A- or R314A-Kir6.2ΔC25 still inactivate at rates comparable to channels formed by SUR1 and E229A- or R314A-Kir6.2, demonstrates that the rapid current decay induced by the E229A or R314A mutation is due, to a large extent, to an effect on the Kir6.2 tetramer structure itself. However, one cannot exclude the possibility that disrupting the ion pair may also affect Kir6.2-SUR1 interactions, which may in turn contribute to channel inactivation.
Mechanisms of Inactivation
Channels in which the ion pair between complementary charges at positions 229 and 314 has been disrupted open upon exposure to ATP-free internal solution, but the current decays rapidly. This current decay can be recovered by three manipulations: (a) reestablishment of the physical interaction between residue 229 and 314 through disulfide bonds; (b) application of PIP2; or (c) reapplication of ATP, and subsequent washout of ATP. What is the molecular mechanism of inactivation and recovery from inactivation in KATP channels, and how do ATP and PIP2 affect these processes?
Membrane phospholipids such as PIP2 and PIP3 stimulate KATP channel activity by increasing the open probability of the channel (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Baukrowitz and Fakler, 2000). Depleting membrane phospholipids or interfering with the interactions between the channels and membrane phospholipids leads to loss of channel activity (Baukrowitz et al., 1998; Gribble et al., 1998a; Shyng and Nichols, 1998; Xie et al., 1999; Shyng et al., 2000a,b; Liu et al., 2001). Structure-functional studies suggest that the effect of PIP2 is mediated largely by direct electrostatic interactions between the cytoplasmic domain of Kir6.2 and phospholipids in the membrane (Baukrowitz et al., 1998; Shyng and Nichols, 1998; Enkvetchakul et al., 2000). Recent biochemical studies that demonstrate binding of phospholipids to recombinant proteins containing the COOH terminus of Kir6.2 lend further support to this hypothesis (MacGregor et al., 2002; Wang et al., 2002). However, a role of SUR in channel response to PIP2 is also evident (Baukrowitz et al., 1998; Shyng and Nichols, 1998; John et al., 2001; Song and Ashcroft, 2001). Channels formed by SUR and Kir6.2 have a much higher open probability and respond to PIP2 better than channels formed by Kir6.2 alone, suggesting that physical association between SUR and Kir6.2 may facilitate channel interaction with membrane PIP2. One model consistent with our data is that, in the WT SUR1–Kir6.2 channel complex, E229 and R314 are in close proximity and form an ion pair. This ion pair provides structural stability to a channel conformation that facilitates interactions between membrane PIP2 and channel protein residues that are directly involved in PIP2 binding (Baukrowitz et al., 1998; Huang et al., 1998; Shyng and Nichols, 1998; Shyng et al., 2000b). Mutations which disrupt the E229/R314 ion pair destabilize channel interaction with PIP2, effectively lowering the apparent affinity of the channel for PIP2, which in turn results in channel inactivation and decreased channel activity (Fig. 9 A). Application of exogenous PIP2 overcomes the reduced affinity between inactivated channels and PIP2, and reverses channel inactivation. In the E229C/R314C mutant, by covalently linking the two residues through a disulfide bond, channel interaction with PIP2 is restabilized and channel inactivation reversed. It is worth noting that disulfide bond formation between 229C and 314C in the E229C/R314C mutant does not increase channel activity beyond that observed for WT channels under normal conditions (see Fig. 6, bottom trace). This suggests that although close interactions between the 229 and 314 residues are necessary to stabilize the active states of the channel, other factors, including the concentration of phospholipids in the membrane and the intrinsic affinity of the channel to phospholipids, determine the overall activity of the channel (Baukrowitz et al., 1998; Huang et al., 1998; Shyng and Nichols, 1998; Xie et al., 1999; Zhang et al., 1999; Shyng et al., 2000a).
Figure 9.
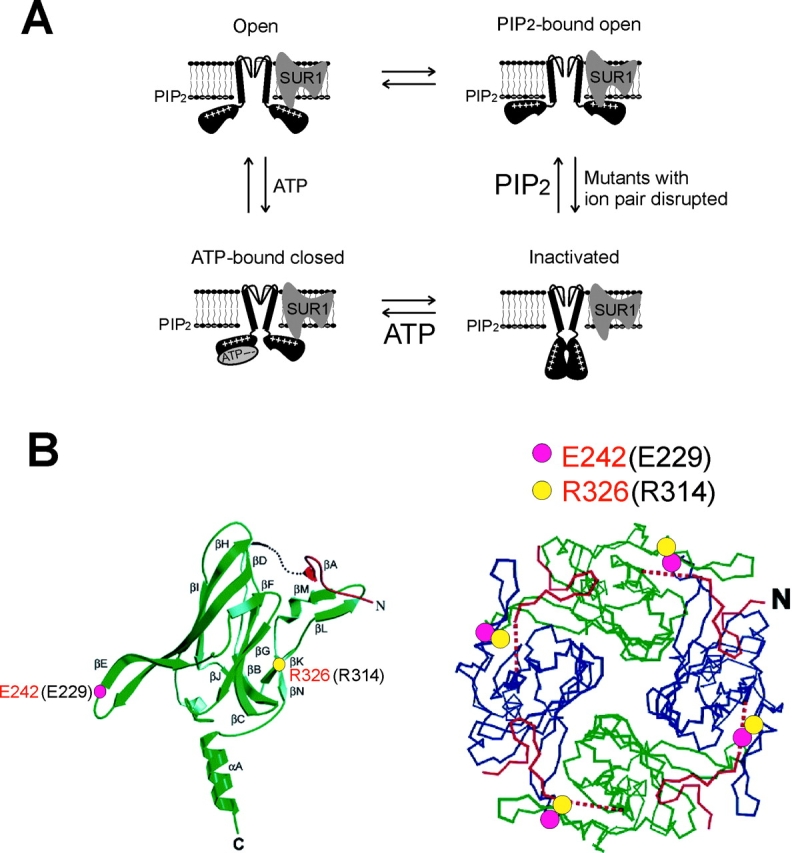
Proposed model for KATP channel inactivation. (A) Cartoon illustrating proposed physical relationships between the channel, ATP, and PIP2 in the inactivation mutants. Disruption of the ion pair causes a structural change in the Kir6.2 tetramer, and possibly also a change in Kir6.2/SUR1 interactions, leading to channel inactivation. These structural changes can be overcome by increasing membrane PIP2, or by ATP binding to the channel in a SUR1-dependent manner. Note that the four states presented should not be taken as detailed kinetic gating states of the channel, and that the transitions between the states are indicated only to illustrate the recovery effects of PIP2 and ATP on inactivated channels. (B) Positions of the two ion pair-forming corresponding residues in the single (left) and tetramer (right) GIRK1 channel structures (Nishida and MacKinnon, 2002). Residue numbering in Kir6.2 is in black, and in GIRK1, red. The Kir6.2-E229 corresponding residue is shown as pink circles, and R314 corresponding residues as yellow circles.
Exposure to ATP and subsequent removal of ATP reopen inactivated channels, which undergo another round of inactivation (Fig. 2 A). The ATP recovery effect is unlikely to involve nucleotide hydrolysis at SUR1, protein phosphorylation, or replenishment of membrane PIPs since the effect can be mimicked by the nonhydrolyzable ATP analogue, AMP-PNP (Fig. 2 B). Though speculative, ATP binding to the inactivated channel may induce a conformational change that brings 229 and 314 residues to close proximity resembling that seen in the WT channel, such that channels can again open upon subsequent removal of ATP (Fig. 9 A). Consistent with this idea, in the E229C/R314C channels, covalent bonding between 229 and 314 does not prevent the channel from entering the ATP-bound closed state or from retransitioning back to the open state (Fig. 6), implying that in WT channels the salt bridge is maintained in the ATP-bound closed state. Further, simultaneous application of DTT (1 mM) and ATP (5 mM) to the E229C/R314C channels abolishes the ability of ATP to recover channels from inactivation (unpublished data), implicating that the two residues form a disulfide bond in the ATP-bound state. Of note, the recovery effect of ATP appears to require SUR1; inactivation of channels formed by E229A- or R314A-Kir6.2ΔC25 alone cannot be recovered by exposure to ATP and subsequent removal of ATP (Fig. 2 C, left). Future studies will determine the mechanism by which SUR1 mediates the reactivation effect of ATP.
Structural Implications
The KATP channel pore is formed by assembly of four Kir6.2 subunits. The electrostatic interaction between E229 and R314 could occur within a single Kir6.2 subunit or between two subunits. The results from experiments using Kir6.2 tandem dimers with mutations introduced into specific positions indicate that the E229-R314 ion pair is formed between two adjacent Kir6.2 subunits rather than within a single subunit. In the EE-RR dimer channels, in which intrasubunit ion pair formation is prohibited but intersubunit ion pair formation is allowed, K+ currents remain relatively stable in ATP-free solutions. On the other hand, the RE-ER dimer, which prohibits intersubunit ion pair formation, but allows intrasubunit ion pair formation, forms channels that display current decay similar to inactivation. The conclusion that the E229/R314 ion pair is formed between two adjacent Kir6.2 subunits is consistent with the recently published cytoplasmic domain structure of GIRK1 (Nishida and MacKinnon, 2002), which shares significant sequence homology (∼40%) with Kir6.2. In the structure of a single GIRK1 subunit, the E229 and R314 corresponding residues, E242 and R326, are located immediately before the βE strand and at the beginning of the βK strand, respectively (Fig. 8 B, left). Such opposite orientations make it highly unlikely for the two residues to form an intrasubunit salt bridge. Rather, the two residues likely form an intersubunit ion pair, as one would predict from the GIRK1 tetramer model, in which E242 from one subunit lies in close proximity of R326 from the adjacent subunit (Fig. 8 B, right). In the AMPA-type glutamate receptor, alterations in the strength of subunit–subunit interfaces have been shown to underlie receptor desensitization (Sun et al., 2002). We speculate that the E229/R314 ion pair may be important in maintaining the quaternary structure of the Kir6.2 tetramer and/or its interaction with SUR1, and that disruption of this ion pair may cause inactivation of KATP channels by destabilizing subunit-subunit interactions. In this regard, it would be important to determine in the future whether R192 and R301 are also involved in subunit–subunit interactions by forming noncovalent chemical bonds, such as hydrogen bonds, with residues from adjacent Kir6.2 or SUR1 subunits.
In summary, our study demonstrates the critical role of an intersubunit ion pair formed by two residues in the COOH terminus of Kir6.2 in maintaining KATP channel activity. The study provides important insight into the structure of the COOH-terminal region of Kir6.2. The two residues at positions homologous to E229 and R314 of Kir6.2 are conserved in Kir1, 2, and 3. And like KATP, the activities of these channels are modulated by membrane PIPs (Huang et al., 1998; Zhang et al., 1999; Lopes et al., 2002). Future studies will determine whether an ion pair similar to the one we reported here plays a role in the gating of other Kir channels.
Acknowledgments
We thank Dr. Anthony Auerbach for helpful suggestions on the experiments. We are grateful to Drs. John Adelman, Peter H. Larsson, and Colin Nichols for comments on the manuscript, and to Drs. Joe Bryan and S. Seino for providing the hamster SUR1 and mouse Kir6.2 cDNA.
This work was supported by National Institutes of Health Grant DK57699 (to S.-L. Shyng), a research grant from the March of Dimes Birth Defects Foundation (to S.-L. Shyng), and a research grant from the Juvenile Diabetes Foundation (to S.-L. Shyng).
David C. Gadsby served as editor.
Footnotes
Abbreviations used in this paper: GIRK, G-protein–activated inwardly rectifying potassium channel; PCR, polymerase chain reaction; SUR, sulfonylurea receptor subunits; WT, wild-type.
References
- Aguilar-Bryan, L., and J. Bryan. 1999. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 20:101–135. [DOI] [PubMed] [Google Scholar]
- Ashcroft, F.M., and F.M. Gribble. 1998. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 21:288–294. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., and B. Fakler. 2000. K(ATP) channels: linker between phospholipid metabolism and excitability. Biochem. Pharmacol. 60:735–740. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., U. Schulte, D. Oliver, S. Herlitze, T. Krauter, S.J. Tucker, J.P. Ruppersberg, and B. Fakler. 1998. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 282:1141–1144. [DOI] [PubMed] [Google Scholar]
- Beguin, P., K. Nagashima, M. Nishimura, T. Gonoi, and S. Seino. 1999. PKA-mediated phosphorylation of the human K(ATP) channel: separate roles of Kir6.2 and SUR1 subunit phosphorylation. EMBO J. 18:4722–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan, J., and L. Aguilar-Bryan. 1997. The ABCs of ATP-sensitive potassium channels: more pieces of the puzzle. Curr. Opin. Cell Biol. 9:553–559. [DOI] [PubMed] [Google Scholar]
- Careaga, C.L., and J.J. Falke. 1992. Thermal motions of surface alpha-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. J. Mol. Biol. 226:1219–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier, E.A., L.R. Conti, C.A. Vandenberg, and S.L. Shyng. 2001. Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc. Natl. Acad. Sci. USA. 98:2882–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement, J.P.T., K. Kunjilwar, G. Gonzalez, M. Schwanstecher, U. Panten, L. Aguilar-Bryan, and J. Bryan. 1997. Association and stoichiometry of K(ATP) channel subunits. Neuron. 18:827–838. [DOI] [PubMed] [Google Scholar]
- Cohen, P. 1989. The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 58:453–508. [DOI] [PubMed] [Google Scholar]
- Cukras, C.A., I. Jeliazkova, and C.G. Nichols. 2002. a. The role of NH(2)-terminal positive charges in the activity of inward rectifier K(ATP) channels. J. Gen. Physiol. 120:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukras, C.A., I. Jeliazkova, and C.G. Nichols. 2002. b. Structural and functional determinants of conserved lipid interaction domains of inward rectifying kir6.2 channels. J. Gen. Physiol. 119:581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle, D.A., J. Morais Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Drain, P., L. Li, and J. Wang. 1998. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 95:13953–13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul, D., G. Loussouarn, E. Makhina, S.L. Shyng, and C.G. Nichols. 2000. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys. J. 78:2334–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, Z., and J.C. Makielski. 1997. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 272:5388–5395. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., M.D. Varnum, and W.N. Zagotta. 1997. Direct interaction between amino- and carboxyl-terminal domains of cyclic nucleotide-gated channels. Neuron. 19:431–441. [DOI] [PubMed] [Google Scholar]
- Gordon, S.E., and W.N. Zagotta. 1995. Subunit interactions in coordination of Ni2+ in cyclic nucleotide-gated channels. Proc. Natl. Acad. Sci. USA. 92:10222–10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble, F.M., P. Proks, B.E. Corkey, and F.M. Ashcroft. 1998. a. Mechanism of cloned ATP-sensitive potassium channel activation by oleoyl-CoA. J. Biol. Chem. 273:26383–26387. [DOI] [PubMed] [Google Scholar]
- Gribble, F.M., S.J. Tucker, and F.M. Ashcroft. 1997. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 16:1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble, F.M., S.J. Tucker, T. Haug, and F.M. Ashcroft. 1998. b. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. USA. 95:7185–7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, Y., M. Ruiz, and J.W. Karpen. 2000. Constraining the subunit order of rod cyclic nucleotide-gated channels reveals a diagonal arrangement of like subunits. Proc. Natl. Acad. Sci. USA. 97:895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann, D.W., and R. Ball. 1996. Regulation of cardiac Na +,Ca2 + exchange and KATP potassium channels by PIP2. Science. 273:956–959. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Sinauer, Sunderland, MA.
- Horenstein, J., D.A. Wagner, C. Czajkowski, and M.H. Akabas. 2001. Protein mobility and GABA-induced conformational changes in GABA(A) receptor pore-lining M2 segment. Nat. Neurosci. 4:477–485. [DOI] [PubMed] [Google Scholar]
- Huang, C.L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 391:803–806. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., T. Gonoi, J.P.T. Clement, N. Namba, J. Inazawa, G. Gonzalez, L. Aguilar-Bryan, S. Seino, and J. Bryan. 1995. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 270:1166–1170. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., T. Gonoi, and S. Seino. 1997. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 409:232–236. [DOI] [PubMed] [Google Scholar]
- Isacoff, E.Y., Y.N. Jan, and L.Y. Jan. 1990. Evidence for the formation of heteromultimeric potassium channels in Xenopus oocytes. Nature. 345:530–534. [DOI] [PubMed] [Google Scholar]
- John, S.A., J.N. Weiss, and B. Ribalet. 2001. Regulation of cloned ATP-sensitive K channels by adenine nucleotides and sulfonylureas: interactions between SUR1 and positively charged domains on Kir6.2. J. Gen. Physiol. 118:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krovetz, H.S., H.M. VanDongen, and A.M. VanDongen. 1997. Atomic distance estimates from disulfides and high-affinity metal- binding sites in a K+ channel pore. Biophys. J. 72:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson, H.P., and F. Elinder. 2000. A conserved glutamate is important for slow inactivation in K+ channels. Neuron. 27:573–583. [DOI] [PubMed] [Google Scholar]
- Light, P.E., C. Bladen, R.J. Winkfein, M.P. Walsh, and R.J. French. 2000. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc. Natl. Acad. Sci. USA. 97:9058–9063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y.F., Y.N. Jan, and L.Y. Jan. 2000. Regulation of ATP-sensitive potassium channel function by protein kinase A-mediated phosphorylation in transfected HEK293 cells. EMBO J. 19:942–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D.T., G.R. Tibbs, P. Paoletti, and S.A. Siegelbaum. 1998. Constraining ligand-binding site stoichiometry suggests that a cyclic nucleotide-gated channel is composed of two functional dimers. Neuron. 21:235–248. [DOI] [PubMed] [Google Scholar]
- Liu, G.X., P.J. Hanley, J. Ray, and J. Daut. 2001. Long-chain acyl-coenzyme A esters and fatty acids directly link metabolism to K(ATP) channels in the heart. Circ. Res. 88:918–924. [DOI] [PubMed] [Google Scholar]
- Lopes, C.M., H. Zhang, T. Rohacs, T. Jin, J. Yang, and D.E. Logothetis. 2002. Alterations in conserved Kir channel-PIP(2) interactions underlie channelopathies. Neuron. 34:933–944. [DOI] [PubMed] [Google Scholar]
- MacGregor, G.G., K. Dong, C.G. Vanoye, L. Tang, G. Giebisch, and S.C. Hebert. 2002. Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc. Natl. Acad. Sci. USA. 99:2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzaris, M., S.P. Jackson, K.M. Laxminarayan, C.J. Speed, and C.A. Mitchell. 1994. Identification and characterization of the phosphatidylinositol-(4,5)-bisphosphate 5-phosphatase in human platelets. J. Biol. Chem. 269:3397–3402. [PubMed] [Google Scholar]
- Nichols, C.G., and A.N. Lopatin. 1997. Inward rectifier potassium channels. Annu. Rev. Physiol. 59:171–191. [DOI] [PubMed] [Google Scholar]
- Nichols, C.G., S.L. Shyng, A. Nestorowicz, B. Glaser, J.P.T. Clement, G. Gonzalez, L. Aguilar-Bryan, M.A. Permutt, and J. Bryan. 1996. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 272:1785–1787. [DOI] [PubMed] [Google Scholar]
- Nishida, M., and R. MacKinnon. 2002. Structural basis of inward rectification. Cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell. 111:957–965. [DOI] [PubMed] [Google Scholar]
- Palmer, F.B., R. Theolis, Jr., H.W. Cook, and D.M. Byers. 1994. Purification of two immunologically related phosphatidylinositol-(4,5)-bisphosphate phosphatases from bovine brain cytosol. J. Biol. Chem. 269:3403–3410. [PubMed] [Google Scholar]
- Papazian, D.M., X.M. Shao, S.A. Seoh, A.F. Mock, Y. Huang, and D.H. Wainstock. 1995. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 14:1293–1301. [DOI] [PubMed] [Google Scholar]
- Pascual, J.M., C.C. Shieh, G.E. Kirsch, and A.M. Brown. 1995. K+ pore structure revealed by reporter cysteines at inner and outer surfaces. Neuron. 14:1055–1063. [DOI] [PubMed] [Google Scholar]
- Ribalet, B., S.A. John, and J.N. Weiss. 2000. Regulation of cloned ATP-sensitive K channels by phosphorylation, MgADP, and phosphatidylinositol bisphosphate (PIP(2)): a study of channel rundown and reactivation. J. Gen. Physiol. 116:391–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, M.S., and W.N. Zagotta. 2000. Structural basis for ligand selectivity of heteromeric olfactory cyclic nucleotide-gated channels. Biophys. J. 78:2307–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., T. Ferrigni, and C.G. Nichols. 1997. a. Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J. Gen. Physiol. 110:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., T. Ferrigni, and C.G. Nichols. 1997. b. Regulation of KATP channel activity by diazoxide and MgADP. Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. J. Gen. Physiol. 110:643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S., and C.G. Nichols. 1997. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 110:655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., A. Barbieri, A. Gumusboga, C. Cukras, L. Pike, J.N. Davis, P.D. Stahl, and C.G. Nichols. 2000. a. Modulation of nucleotide sensitivity of ATP-sensitive potassium channels by phosphatidylinositol-4-phosphate 5-kinase. Proc. Natl. Acad. Sci. USA. 97:937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., C.A. Cukras, J. Harwood, and C.G. Nichols. 2000. b. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J. Gen. Physiol. 116:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., and C.G. Nichols. 1998. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 282:1138–1141. [DOI] [PubMed] [Google Scholar]
- Singer, W.D., H.A. Brown, and P.C. Sternweis. 1997. Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu. Rev. Biochem. 66:475–509. [DOI] [PubMed] [Google Scholar]
- Song, D.K., and F.M. Ashcroft. 2001. ATP modulation of ATP-sensitive potassium channel ATP sensitivity varies with the type of SUR subunit. J. Biol. Chem. 276:7143–7149. [DOI] [PubMed] [Google Scholar]
- Sun, Y., R. Olson, M. Horning, N. Armstrong, M. Mayer, and E. Gouaux. 2002. Mechanism of glutamate receptor desensitization. Nature. 417:245–253. [DOI] [PubMed] [Google Scholar]
- Tanabe, K., S.J. Tucker, M. Matsuo, P. Proks, F.M. Ashcroft, S. Seino, T. Amachi, and K. Ueda. 1999. Direct photoaffinity labeling of the Kir6.2 subunit of the ATP- sensitive K+ channel by 8-azido-ATP. J. Biol. Chem. 274:3931–3933. [DOI] [PubMed] [Google Scholar]
- Tucker, S.J., F.M. Gribble, P. Proks, S. Trapp, T.J. Ryder, T. Haug, F. Reimann, and F.M. Ashcroft. 1998. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17:3290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, S.J., F.M. Gribble, C. Zhao, S. Trapp, and F.M. Ashcroft. 1997. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 387:179–183. [DOI] [PubMed] [Google Scholar]
- Wang, C., K. Wang, W. Wang, Y. Cui, and Z. Fan. 2002. Compromised ATP binding as a mechanism of phosphoinositide modulation of ATP-sensitive K(+) channels. FEBS Lett. 532:177–182. [DOI] [PubMed] [Google Scholar]
- Xie, L.H., M. Horie, and M. Takano. 1999. Phospholipase C-linked receptors regulate the ATP-sensitive potassium channel by means of phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. USA. 96:15292–15297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., M. Yu, Y.N. Jan, and L.Y. Jan. 1997. Stabilization of ion selectivity filter by pore loop ion pairs in an inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. USA. 94:1568–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue, N., B. Schwappach, Y.N. Jan, and L.Y. Jan. 1999. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 22:537–548. [DOI] [PubMed] [Google Scholar]
- Zhang, H., C. He, X. Yan, T. Mirshahi, and D.E. Logothetis. 1999. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat. Cell Biol. 1:183–188. [DOI] [PubMed] [Google Scholar]
- Zingman, L.V., A.E. Alekseev, M. Bienengraeber, D. Hodgson, A.B. Karger, P.P. Dzeja, and A. Terzic. 2001. Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 31:233–245. [DOI] [PubMed] [Google Scholar]