

Abstract
The nematode Caenorhabditis elegans offers significant experimental advantages for defining the genetic basis of diverse biological processes. Genetic and physiological analyses have demonstrated that inositol-1,4,5-trisphosphate (IP3)–dependent Ca2+ oscillations in intestinal epithelial cells play a central role in regulating the nematode defecation cycle, an ultradian rhythm with a periodicity of 45–50 s. Patch clamp studies combined with behavioral assays and forward and reverse genetic screening would provide a powerful approach for defining the molecular details of oscillatory Ca2+ signaling. However, electrophysiological characterization of the intestinal epithelium has not been possible because of its relative inaccessibility. We developed primary intestinal epithelial cell cultures that circumvent this problem. Intestinal cells express two highly Ca2+-selective, voltage-independent conductances. One conductance, IORCa, is constitutively active, exhibits strong outward rectification, is 60–70-fold more selective for Ca2+ than Na+, is inhibited by intracellular Mg2+ with a K1/2 of 692 μM, and is insensitive to Ca2+ store depletion. Inhibition of IORCa with high intracellular Mg2+ concentrations revealed the presence of a small amplitude conductance that was activated by passive depletion of intracellular Ca2+ stores. Active depletion of Ca2+ stores with IP3 or ionomycin increased the rate of current activation ∼8- and ∼22-fold compared with passive store depletion. The store-operated conductance, ISOC, exhibits strong inward rectification, and the channel is highly selective for Ca2+ over monovalent cations with a divalent cation selectivity sequence of Ca2+ > Ba2+ ≈ Sr2+. Reversal potentials for ISOC could not be detected accurately between 0 and +80 mV, suggesting that PCa/PNa of the channel may exceed 1,000:1. Lanthanum, SKF 96365, and 2-APB inhibit both IORCa and ISOC reversibly. Our studies provide the first detailed electrophysiological characterization of voltage-independent Ca2+ conductances in C. elegans and form the foundation for ongoing genetic and molecular studies aimed at identifying the genes that encode the intestinal cell channels, for defining mechanisms of channel regulation and for defining their roles in oscillatory Ca2+ signaling.
Keywords: calcium oscillations; biorhythm; calcium channel; inositol-1,4,5-trisphosphate; MIC; CRAC
INTRODUCTION
Fluctuating intracellular Ca2+ concentration is a ubiquitous signaling mechanism that controls numerous cellular processes, including gene expression, exocytosis and secretion, motility, cell proliferation, programmed cell death, and differentiation (Berridge et al., 2000). Elevation of cytoplasmic Ca2+ levels is brought about by Ca2+ release from intracellular stores and by influx across the plasma membrane. In excitable cells, Ca2+ influx is mediated to a large extent by voltage- and ligand-gated cation channels. Calcium influx into nonexcitable cells, such as blood cells and endothelial and epithelial cells, occurs primarily via second messenger– and store-operated Ca2+ channels (SMOCCs and SOCCs)* (Elliott, 2001; Zitt et al., 2002).
The ER is the principal Ca2+ store in nonexcitable cells. Agonist binding to plasma membrane tyrosine kinase– or G protein–coupled receptors activates phospholipase C, leading to the production of inositol 1,4,5-trisphosphate (IP3). IP3, in turn, activates IP3 receptors in the ER membrane, inducing Ca2+ release that leads to either a sustained elevation of cytoplasmic Ca2+ concentration or Ca2+ oscillations (Shuttleworth, 1999; Berridge et al., 2000). Sustained Ca2+ elevation is often observed with high agonist concentrations and occurs in a biphasic manner. The first phase involves ER Ca2+ release. As the stores are depleted of Ca2+, SOCCs are activated, allowing Ca2+ to enter from the extracellular medium (Parekh and Penner, 1997; Taylor and Thorn, 2001). Cytoplasmic Ca2+ levels and Ca2+ influx remain elevated as long as the stimulus is maintained (Putney and McKay, 1999; Shuttleworth, 1999).
Lower concentrations of agonists typically trigger Ca2+ oscillations (Shuttleworth, 1999). The role of plasma membrane Ca2+ entry in generating and maintaining the oscillations is unclear (Shuttleworth, 1999). Calcium oscillations in some cell types continue for long periods in the absence of extracellular Ca2+ (Lechleiter and Clapham, 1992). In contrast, oscillatory Ca2+ signals in other cell types are strictly dependent on Ca2+ influx (Torihashi et al., 2002; Wu et al., 2002).
The molecular identity of both SOCCs and SMOCCs, the mechanisms by which they are regulated, and their precise functional roles in local and global Ca2+ signaling are unclear. Genetic model organisms provide a number of powerful experimental advantages for defining the genes and genetic pathways involved in biological processes such as Ca2+ signaling. The nematode Caenorhabditis elegans is a particularly attractive model system for such studies (Barr, 2003; Strange, 2003). C. elegans has a short life cycle, is genetically tractable, and has a fully sequenced and well-annotated genome. It is also relatively easy and economical to manipulate and hence characterize gene function in this organism.
C. elegans exhibits a number of relatively simple stereotyped behaviors that have formed the bases for powerful forward genetic screens. The defecation cycle is one such behavior. Defecation is an ultradian rhythm that occurs once every 45–50 s when nematodes are feeding and is mediated by sequential contraction of the posterior body wall muscles, anterior body wall muscles, and enteric muscles (Iwasaki and Thomas, 1997). Loss-of-function mutations in the IP3 receptor gene itr-1 slow or eliminate the defecation cycle, whereas overexpression of the gene increases the rate of defecation (Dal Santo et al., 1999). Oscillatory changes in intestinal epithelial cell Ca2+ levels track the defecation cycle, with Ca2+ levels peaking just before the initiation of posterior body wall muscle contraction. Calcium oscillations are slowed or absent in animals with loss-of-function mutations in itr-1 (Dal Santo et al., 1999). Dal Santo et al. (1999) have suggested that IP3-dependent Ca2+ signals may control the secretion of a factor from the intestinal epithelium that regulates contraction of surrounding body wall muscles.
The ability to combine physiological tools, such as patch clamp analysis and Ca2+ imaging, with behavioral assays and forward and reverse genetic screening would provide a powerful approach for defining the molecular details of intestinal cell IP3-dependent Ca2+ signaling. However, electrophysiological characterization of somatic cells in C. elegans is difficult due to the small size of the animal and the presence of a tough, pressurized cuticle that limits access. To circumvent this problem, we recently developed methods that allow the primary culture and terminal differentiation of nematode embryo cells (Christensen et al., 2002).
We report here the electrophysiological characterization of cultured C. elegans intestinal epithelial cells. Intestinal cells express two highly Ca2+-selective whole-cell cation conductances. One conductance is insensitive to store depletion, shows strong outward rectification, and is inhibited by intracellular Mg2+. Intracellular Ca2+ store depletion activates an inwardly rectifying SOCC current. These studies provide the first detailed electrophysiological characterization of voltage-independent Ca2+-selective cation conductances in C. elegans and form the foundation for ongoing genetic and molecular studies aimed at identifying the genes that encode the channels, for defining mechanisms of channel regulation and for defining their roles in oscillatory Ca2+ signaling.
MATERIALS AND METHODS
C. elegans Strains
All strains were derived from the wild-type N2 line and maintained at 20–25°C using standard methods (Brenner, 1974). The elt-2–GFP strains used in these studies were JR1838 (wIs84) and JM63 (caIs13). GFP strains contain integrated transgenes.
C. elegans Embryonic Cell Culture
Embryonic cells were prepared by treating synchronized adult nematodes with an alkaline hypochlorite solution (0.5 M NaOH and 1% NaOCl) for 5 min (Lewis and Fleming, 1995). Eggs released by this treatment were pelleted by centrifugation and then washed three times with egg buffer containing 118 mM NaCl, 48 mM KCl, 2 mM CaCl2, 2 mM MgCl2, and 25 mM HEPES (pH 7.3, 345 mOsm) (Edgar, 1995). Adult carcasses were separated from washed eggs by density centrifugation in 30% sucrose. The egg layer was removed by pipette and washed one time with egg buffer and then pelleted. Eggshells were removed by resuspending pelleted eggs in egg buffer containing 1–2.5 U/ml of chitinase for 45–90 min at room temperature. After digestion of the eggshell, the suspension was gently pipetted up and down several times to dissociate the cells. Cells were washed two times with L-15 cell culture medium (Life Technologies) containing 10% FBS (Hyclone), 50 U/ml penicillin, and 50 μg/ml streptomycin and adjusted to 345 mOsm with sucrose.
Dissociated embryo cells were filtered through a sterile 5-μm Durapore syringe filter (Millipore) to remove undissociated embryos and newly hatched larvae. Filtered cells were plated on 12-mm-diameter glass coverslips coated with 0.5 mg/ml peanut lectin agglutinin. Cultures were maintained at 24°C in a humidified incubator in L-15 cell culture medium.
Patch Clamp Recordings
Coverslips with cultured embryo cells were placed in the bottom of a bath chamber (model R-26G; Warner Instrument Corp.) that was mounted onto the stage of a Nikon TE300 inverted microscope. Cells were visualized by fluorescence and video-enhanced differential interference contrast (DIC) microscopy.
Patch electrodes were pulled from soft glass capillary tubes (PG10165-4; World Precision Instruments) that had been silanized with dimethyl-dichloro silane. The standard pipette solution for whole-cell recording from intestinal cells contained (mM) 147 sodium gluconate (NaGluconate), 0.6 CaCl2, 1 MgCl2, 10 EGTA or 10 BAPTA, 10 HEPES, 2 Na2ATP, 0.5 Na2GTP, pH 7.2 (adjusted with CsOH), 325 mOsm. The standard bath solution contained (mM) 145 NaCl, 1 CaCl2, 5 MgCl2, 10 HEPES, 20 Glucose, pH 7.2 (adjusted with NaOH). Osmolality was adjusted to 340–345 mOsm with sucrose. Free Ca2+ and free Mg2+ levels in the various solutions used were calculated using MaxChelator software WINMAXC v.2.1 (www.stanford.edu/~cpatton/maxc.html).
Whole-cell currents were recorded using an Axopatch 200B (Axon Instruments, Inc.) patch clamp amplifier. Command voltage generation, data digitization, and data analysis were performed on a 1.6-GHz Pentium computer (Dimension 4400; Dell Computer Corp.) using a Digidata 1322A AD/DA interface with pClamp 8.2 and Clampfit 8.2 software (Axon Instruments, Inc.). Currents were filtered at 5 kHz and digitized at 20–40 kHz. Electrical connections to the amplifier were made using Ag/AgCl wires and 3-M KCl/agar bridges.
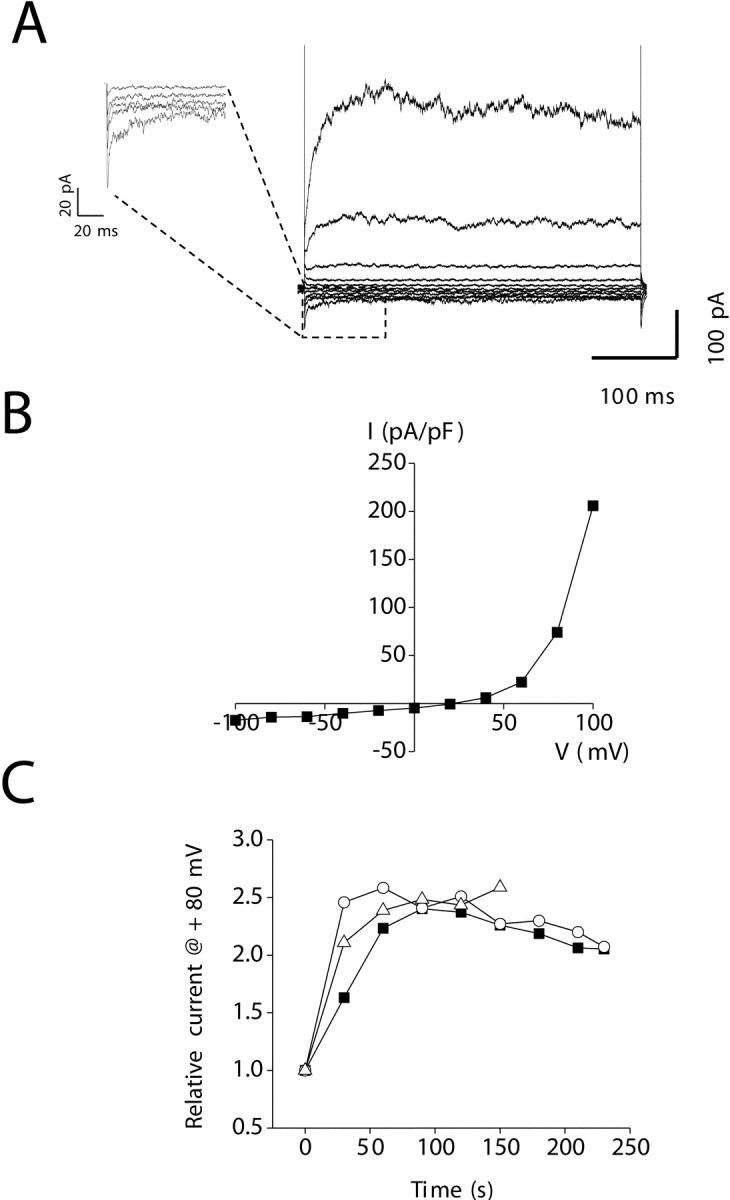
Ion substitution studies were performed by replacement of bath Na+ with various test cations. For all ion substitution experiments, changes in liquid junction potentials were measured directly using a free-flowing 3-M KCl electrode. Reversal potentials were corrected for these changes. Relative permeabilities were calculated using the following equations derived from the Goldman-Hodgkin-Katz equation (Lewis, 1979; Hille, 2001):
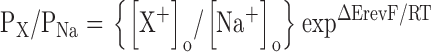 |
(1) |
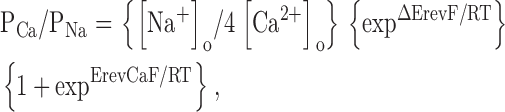 |
(2) |
where [X+]o is the extracellular concentration of the monovalent substitute cation; [Na+]o is the extracellular Na+ concentration; [Ca2+]o is the extracellular Ca2+ concentration; ΔErev is the change in reversal potential; and F, R, and T have their usual meanings. In studies of the intestinal cell store-operated current, we defined leak current as the current observed immediately after obtaining whole-cell access. This leak current was subtracted from all subsequent current records obtained in the cell.
Chemicals
Thapsigargin, ionomycin, and BAPTA were purchased from Molecular Probes. IP3 and 2-APB were purchased from Calbiochem. All other chemicals were obtained from Sigma-Aldrich.
Statistical Analyses
Data are presented as means ± SEM. Statistical significance was determined using a two-tailed t test or ANOVA followed by a Bonferroni multiple comparisons test. P values of <0.05 were taken to indicate statistical significance.
RESULTS
Identification of C. elegans Intestinal Epithelial Cells in Primary Culture
As described previously, isolated C. elegans embryo cells undergo terminal differentiation when cultured in vitro (Christensen et al., 2002). Culturing embryo cells from worm strains expressing cell-specific GFP reporters allows identification of differentiated cell types. ELT-2 is a GATA transcription factor expressed exclusively in intestinal cells and is required for intestinal morphogenesis beginning at the 44–46-cell stage of embryonic development (Fukushige et al., 1999). Primary cell cultures were prepared from worm strains expressing an elt-2–GFP transcriptional fusion construct (worm strains provided by J. Rothman [University of California, Santa Barbara, CA] and J. McGhee [University of Calgary, Calgary, Alberta, Canada]). Fig. 1 shows combined DIC and fluorescence micrographs of a transgenic worm expressing elt-2–GFP and an intestinal epithelial cell cultured from elt-2–GFP-expressing worms.
Figure 1.

Expression of elt-2–GFP reporter in C. elegans intestinal cells. Images are overlays of DIC and fluorescence micrographs of a transgenic worm (left) and cultured intestinal cell (right) expressing elt-2–GFP in the cell nucleus. GFP fluorescence is shown in green. Bars: (left) 10 μm; (right) 2.5 μm. Em, developing embryo in uterus; Oo, oocyte in proximal gonad; IC, intestinal cell; N, intestinal cell nucleus. Arrowheads denote refractile granules that are most likely intracellular storage granules.
Our previous studies have shown that various muscle and neuronal cell types in primary culture are present at a frequency remarkably close to that observed in a newly hatched L1 larva (Christensen et al., 2002). An L1 larva is comprised of 558 cells of which 20, or 3.6%, are intestinal cells. Intestinal cells represented 4.1 ± 0.1% (n = 3) of the total cells in culture. elt-2–GFP expression was concentrated in the nuclei of cultured intestinal cells (Fig. 1, right), similar to that observed in vivo (Fig. 1, left). In addition, the cytoplasm of the cultured cells contained numerous refractile granules (Fig. 1, right, arrowheads) that were also highly autofluorescent (unpublished data). These most likely represent storage granules, which are a prominent characteristic of intestinal cells in the intact worm (Kostich et al., 2000; Fig. 1, left).
Intestinal Cells Express an Outwardly Rectifying Cation Current
Cultured intestinal cells were patch clamped readily in the whole-cell mode. An outwardly rectifying whole-cell current (Fig. 2, A and B) was observed when cells were bathed and dialyzed with control bath (145 mM NaCl) and pipette (147 mM NaGluconate) solutions. Whole-cell current amplitude typically increased approximately two- to threefold and then stabilized within 1–2 min after obtaining the whole-cell configuration (Fig. 2 C).
Figure 2.
Characteristics of outwardly rectifying intestinal cell whole-cell current. (A) Whole-cell currents recorded from a cultured intestinal cell using standard bath (145 mM NaCl) and pipette (147 mM NaGluconate) solutions. Currents were elicited by stepping membrane voltage from −100 to +100 mV in 20-mV steps from a holding potential of 0 mV. Voltage steps were 400 ms long. Inset shows inactivation behavior observed at strongly hyperpolarized voltages. (B) Steady-state I–V relationship for the whole-cell currents shown in A. (C) Whole-cell current activation observed after membrane rupture. Current recordings from three intestinal cells are shown. Values shown are relative to those observed at time 0, which is defined as the time at which whole-cell access was obtained. Whole-cell current typically increased two- to threefold within 1–3 min after membrane rupture and then stabilized.
The whole-cell current was voltage and time dependent (Fig. 2, A and B). Strong depolarization and hyperpolarization activated and inactivated the current, respectively. At +100 mV, current activation was well fit by a double exponential describing mean ± SEM fast (τf) and slow (τs) time constants of 19 ± 2 and 154 ± 29 ms (n = 43), respectively. Current inactivation was also well fit by a double exponential. Mean ± SEM τf and τs at −100 mV were 14 ± 2 and 195 ± 36 ms (n = 43), respectively.
The ionic nature of the outwardly rectifying whole-cell current was determined by ion substitution studies. Replacement of 147 mM Na+ in the pipette solution with NMDG+ dramatically inhibited outward current (Fig. 3 A) and increased Erev significantly (P < 0.005) from a mean ± SEM value of 21 ± 3 (n = 5) to 37 ± 3 mV (n = 5).1 The shift in Erev and reduction in outward current are consistent with a cation current.
Figure 3.

Ionic dependence of outwardly rectifying whole-cell current. (A) Replacement of 147 mM Na+ (Nai, □) in the pipette solution with NMDG+ (▪) increased reversal potential (Erev) by +16 mV (P < 0.005) and dramatically reduced outward current.1 Values are means ± SEM (n = 5). (B) Reduction of bath Cl− concentration (Clo) from 157 (□) to 10 mM (▪) (gluconate substitution) caused a small increase in outward current and decrease in Erev of −3 ± 2 mV. The changes in current amplitude and Erev were not statistically significant (P > 0.1). Values are means ± SEM (n = 3). Changes in Erev and whole-cell current amplitude indicate that the current is carried primarily by cations. Voltage clamp protocol was the same as described in Fig. 2.
Reduction of bath Cl− from 157 to 10 mM (gluconate replacement2) induced a small increase in outward current and shift in mean ± SEM Erev from 29 ± 3 to 24 ± 1 mV (n = 3; Fig. 3 B). The small changes in current amplitude and Erev induced by reduction of bath Cl− were not statistically significant (P > 0.1) and are opposite to those expected for anion-selective channels. Taken together, the data in Fig. 3 demonstrate that the whole-cell current is carried predominantly by cations.
The Outwardly Rectifying Cation Conductance Has a High Selectivity for Ca2+ over Monovalent Cations
The relative permeability to various cations of the channel responsible for the outwardly rectifying whole-cell current was determined using the Goldman-Hodgkin-Katz equation after complete substitution of bath Na+ with various test cations. Removal of bath Ca2+ and Mg2+ and addition of 1 mM EGTA (nominally divalent-free medium) caused an immediate and substantial increase in whole-cell current (Fig. 4) . The mean ± SEM increase observed at +80 mV was 286 ± 58 pA (n = 27; P < 0.0001). Removal of Ca2+ and Mg2+ also caused a significant (P < 0.0001) decrease in Erev, from 24 ± 1 (n = 48) to 11 ± 0.5 mV (n = 27), and altered current voltage and time dependence (Fig. 4 A). At potentials of +80 mV and above, currents observed in nominally divalent-free medium typically showed partial inactivation (Fig. 4 A). Current inactivation was fit by a single exponential describing a mean ± SEM time constant (τ) at +100 mV of 194 ± 42 ms (n = 17).
Figure 4.

Effect of extracellular Ca2+ and Mg2+ removal on amplitude of the outwardly rectifying cation current. (A) Whole-cell currents observed under control (1 mM Ca2+ and 5 mM Mg2+) and nominally divalent-free (0 mM Ca2+, 0 mM Mg2+, and 1 mM EGTA) conditions. Voltage clamp protocol was the same as described in Fig. 2. (B) Steady-state I–V relationships of outwardly rectifying currents observed under control conditions (•), in nominally divalent-free medium (buffered with 1 mM EGTA; ○) and in divalent-free medium (buffered with 1 mM EDTA; □). Values are means ± SEM (n = 6–33).
Micromolar concentrations of extracellular Mg2+ block channels such as the NMDA receptor (Mayer et al., 1984; Nowak et al., 1984) and the recently described Mg2+-inhibited cation (MIC) channel (Hermosura et al., 2002; Kozak et al., 2002). To determine whether a similar block occurs in the channel responsible for the outwardly rectifying cation current, we exposed cells to divalent-free medium containing 1 mM EDTA in order to fully chelate extracellular Mg2+. As shown in Fig. 4 B, the current-to-voltage relationship of the outwardly rectifying current was largely unaffected by this maneuver.
Replacement of bath Na+ with K+, Cs+, or NMDG+ shifted Erev to more negative values (Table I) . The calculated relative permeabilities (i.e., Pcation/PNa; Table I) determined from the changes in Erev yielded a monovalent cation selectivity sequence of Na+ > K+ > Cs+ ≫ NMDG+.
TABLE I.
Relative Cation Permeability of the Outwardly Rectifying Conductance
| Cation | ΔErev | Pcation/PNa |
|---|---|---|
| mV | ||
| K+ | −10 ± 1 | 0.67 ± 0.02 (7) |
| Cs+ | −34 ± 2 | 0.27 ± 0.02 (7) |
| NMDG+ | −90 ± 5 | 0.03 ± 0.01 (7) |
| Ca2+ | 29 ± 1 | 64 ± 2 (6)a |
Whole-cell currents were elicited by stepping membrane voltage from −100 to +100 mV in 20-mV steps from a holding potential of 0 mV. Voltage steps were 400 ms long. Steady-state current-to-voltage relationships were plotted for determination of Erev in the presence of Na+ and various test cations. Relative permeabilities were calculated using equations derived from the Goldman-Hodgkin-Katz equation (see materials and methods). Values are means ± SEM (number of cells). All changes in Erev are statistically significant (P < 0.0001).
See footnote 3.
The increase in whole-cell current upon removal of extracellular divalent cations (Fig. 4) suggested strongly that the channel is permeable to Ca2+ and/or Mg2+ and that permeation by divalents blocks monovalent cation flux. To examine this possibility directly, we measured whole-cell currents in the presence of extracellular solutions containing varying proportions of Ca2+ and Na+. Addition of 1 mM Ca2+ to a solution containing 74 mM Na+ caused a dramatic reduction in whole-cell current (Fig. 5 A). As the proportion of Ca2+ was elevated, the current passed through a minimum and then increased (Fig. 5 A). This anomalous mole-fraction behavior is characteristic of multi-ion channels where two or more ions simultaneously occupy and move through the channel pore (Hille, 2001). Both the number and type of ions in the pore determine the overall permeability properties of multi-ion channels (Hille, 2001). Many highly Ca2+-selective cation channels exhibit anomalous mole-fraction behavior (Hoth, 1995; Vennekens et al., 2001). High affinity Ca2+ binding in the channel pore appears to be responsible for their high selectivity for Ca2+ versus monovalent cations (Almers and McCleskey, 1984; Lepple-Wienhues and Cahalan, 1996).
Figure 5.
Calcium selectivity of the outwardly rectifying cation conductance. (A) Relationship between relative whole-cell current amplitude and fractional concentration of extracellular Ca2+ ([Ca2+]o/([Na+]o + [Ca2+]o). As the proportion of Ca2+ is elevated, the current passes through a minimum and then increases. This anomalous mole-fraction behavior is characteristic of highly Ca2+-selective cation channels (Hoth, 1995; Vennekens et al., 2001). All solutions were nominally Mg2+ free. The Ca2+-free solution was buffered with 1 mM EGTA. Values are means ± SEM (n = 6–11). (B) I–V relationships of whole-cell current in the presence of 150 mM Na+ in the bath (□) and when Na+ was replaced with 130 mM NMDG+ and 10 mM Ca2+ (▪). Elevation of extracellular Ca2+ increased Erev by 29 ± 1 mV (P < 0.0001). Calculated relative Ca2+ permeability (PCa/PNa) is 64:1 (Table I). Voltage clamp protocol was the same as described in Fig. 2.
Given the results in Fig. 4 and Fig. 5 A, we measured relative Ca2+ permeability by replacing bath Na+ with 130 mM NMDG+ and 10 mM Ca2+. Elevation of bath Ca2+ increased Erev significantly (P < 0.0001), by 29 ± 1 mV (n = 6) (Fig. 5 B; Table I). The calculated relative permeability of Ca2+ to Na+ is 64:1 (Table I).3 Together, the data in Figs. 4 and 5 demonstrate that a highly Ca2+-selective cation channel carries the outwardly rectifying whole-cell current. We hereafter refer to the outwardly rectifying Ca2+ current as IORCa.
Pharmacological Characteristics of IORCa
Lanthanum is a trivalent cation that inhibits voltage-gated and store-operated Ca2+ channels as well as some members of the transient receptor potential (TRP) cation channel superfamily (Aussel et al., 1996; Halaszovich et al., 2000; Beedle et al., 2002). Exposure of cultured intestinal cells to 100 μM La3+ in the bath solution virtually abolished the outwardly rectifying current (P < 0.05; Fig. 6 A). Current inhibition by 100 μM La3+ was fully reversible (Fig. 6 A). The concentration of La3+ required for 50% inhibition of IORCa is 3.4 μM (Fig. 6 B).
Figure 6.
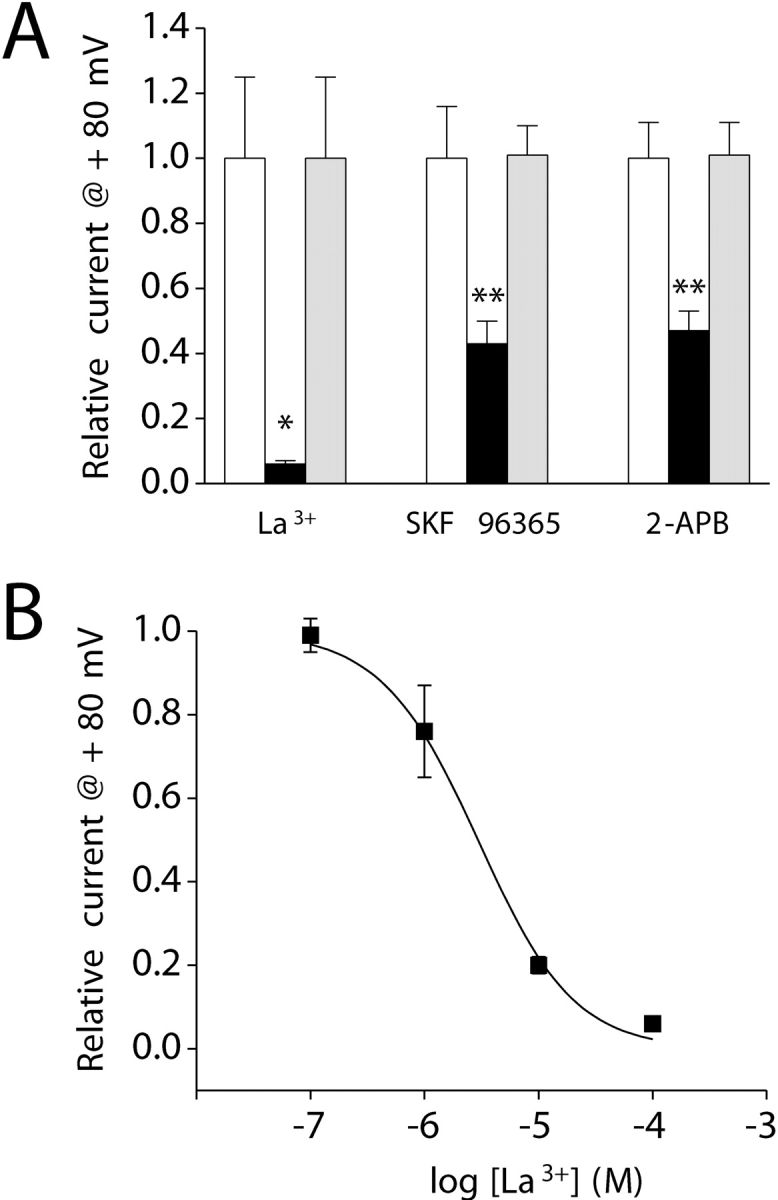
Pharmacology of the outwardly rectifying cation conductance. (A) Addition of 100 μM La3+, 100 μM SKF 96365, or 100 μM 2-APB (black bar) to the bath inhibited whole-cell current by 50–90%. *, P < 0.05; **, P < 0.01 (compared with control, white bar). Inhibitory effects of La3+, SKF 96365, and 2-APB were fully reversible. Current levels observed after drug washout (gray bar) were not significantly (P > 0.05) different from those observed before drug addition (white bar). Values are means ± SEM (n = 4–5). (B) Dose–response relationship for the inhibitory effect of La3+. Data were fit using the equation I = 1/1 + ([Mg2+]/K1/2)n. K1/2 and n are 3.4 μM and 1.1, respectively. Values are means ± SEM (n = 5–9). Voltage clamp protocol was the same as described in Fig. 2.
SKF 96365 inhibits receptor-activated and voltage-gated calcium influx (Merritt et al., 1990) as well as the Ca2+ release–activated channel (CRAC) (Kozak et al., 2002; Prakriya and Lewis, 2002) and TRP channel activity (Halaszovich et al., 2000). 100 μM SKF 96365 inhibited IORCa ∼60% (P < 0.01) in a reversible manner (Fig. 6 A). 2-APB inhibits IP3 receptor Ca2+ channel activity (Bilmen and Michelangeli, 2002) and plasma membrane Ca2+ entry channels, including CRAC and TRP channels (Prakriya and Lewis, 2001; Bootman et al., 2002; Hermosura et al., 2002; Prakriya and Lewis, 2002). 100 μM 2-APB reversibly inhibited IORCa by ∼50% (P < 0.01; Fig. 6 A). Lanthanum, SKF 96365, and 2-APB all inhibited IORCa rapidly; current inhibition was maximal within 30–60 s after adding the drugs to the bath (unpublished data). Washout of the drugs and recovery of IORCa occurred with a similar time course (unpublished data).
Depletion of Intracellular Ca2+ Stores Does Not Regulate IORCa
As shown in Fig. 2 C, IORCa activated two- to threefold during the first 1–2 min after obtaining whole-cell access. Two observations suggested that current activation might be due to depletion of intracellular Ca2+ stores. First, the biophysical characteristics of the current resemble those of members of the TRP cation channel superfamily. Evidence suggests that some TRPs may be regulated by store depletion (Clapham et al., 2001; Elliott, 2001; Montell, 2001; Zitt et al., 2002). Second, the pipette solutions used in the studies shown in Fig. 2 C contained 10 mM EGTA and low Ca2+, which could lead to passive store depletion (Hoth and Penner, 1993).
We performed three experiments to directly examine the role of store depletion in regulation of IORCa. First, cells were patch clamped with a Ca2+-free pipette solution to passively deplete stores or a solution containing 175 nM free Ca2+. Calcium levels >90 nM are expected to maintain store Ca2+ levels (Hermosura et al., 2002; Kozak et al., 2002). As shown in Fig. 7 A, there was no significant (P > 0.2) difference in either the rate of current activation or the peak current amplitude observed in these two experiments.
Figure 7.
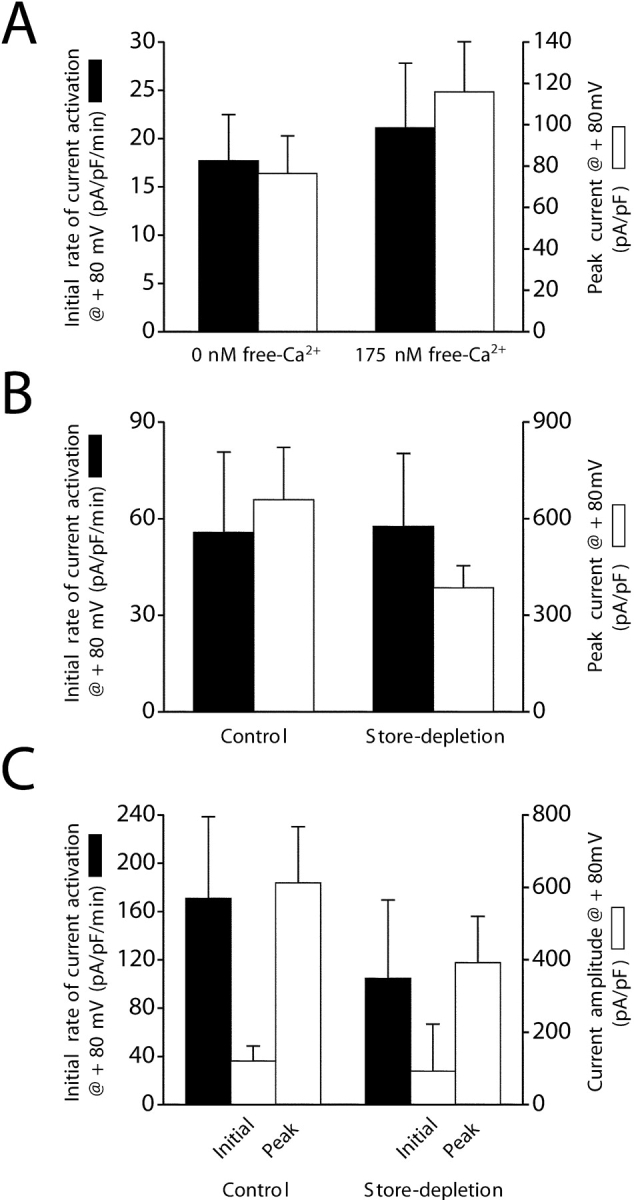
The outwardly rectifying cation current is not regulated by depletion of Ca2+ stores. (A) Rates of whole-cell current activation (black bar) and peak current amplitudes (white bar) in cells dialyzed with a Ca2+-free pipette solution to passively deplete intracellular Ca2+ stores and a pipette solution containing 175 nM free Ca2+. Removal of intracellular Ca2+ had no significant (P > 0.2) effect on the rate of current activation or the peak current amplitude. Intracellular Ca2+ was buffered with 1 mM EGTA. Currents were measured using the standard bath medium containing both Ca2+ and Mg2+. Whole-cell currents were elicited by ramping membrane potential from −80 to +80 mV at 160 mV/s every 5 s. (B) Rates of whole-cell current activation (black bar) and peak current amplitudes (white bar) in control cells and cells treated with 10 μM IP3 and 1 μM thapsigargin. Depletion of intracellular stores with IP3 and thapsigargin had no significant (P > 0.1) effect on the rate of current activation or the peak current amplitude. Intracellular Ca2+ was buffered to 11 nM with 10 mM EGTA. Cells were exposed to nominally divalent-free bathing medium immediately after obtaining whole-cell access. Voltage clamp protocol was the same as described in Fig. 2. (C) Effect of depletion of Ca2+ stores in intact cells on rate of current activation (black bar) and initial and peak current amplitude (white bar). Experimental cells were preincubated for 9–17 min in nominally divalent-free bathing medium containing 1 μM thapsigargin and then patch clamped with a pipette solution containing 10 μM IP3. Control cells were exposed to nominally divalent-free bathing medium immediately after obtaining whole-cell access and were not treated with IP3 or thapsigargin. Store depletion had no significant (P > 0.1) effect on initial whole-cell current or subsequent current activation. Intracellular Ca2+ was buffered to 11 nM with 10 mM EGTA. Values are means ± SEM (n = 3–6). Voltage clamp protocol was the same as described in A. Initial rates of current activation were quantified by performing linear regression analysis on whole-cell currents measured during the first 60–180 s after obtaining whole-cell access.
Second, we patch clamped cells with a pipette solution containing 10 μM IP3. Immediately after obtaining whole-cell access, IP3-treated cells were exposed to a nominally divalent-free bath solution containing 1 μM thapsigargin to inhibit store Ca2+ uptake. Neither the rate of current activation nor the peak current amplitude were significantly (P > 0.1) altered by IP3 and thapsigargin.
Finally, we attempted to activate IORCa in intact cells by depleting intracellular Ca2+ stores before patch clamping. Cells were incubated in nominally divalent-free bath solution containing 1 μM thapsigargin for 9–17 min and then were patch clamped with a pipette solution containing 10 μM IP3. Control cells were patch clamped with a standard pipette solution without IP3 and were exposed to nominally divalent-free bath immediately after obtaining whole-cell access. If store depletion activates IORCa, initial current levels should be higher in cells pretreated with Ca2+- and Mg2+-free bath containing thapsigargin. However, as shown in Fig. 7 C, initial whole-cell current amplitudes were not significantly (P > 0.1) different in control and experimental cells. The outward currents in both groups of cells showed gradual activation, but the rates of current activation and peak current amplitudes were not significantly (P > 0.5) different (Fig. 7 C). We conclude from the data shown in Fig. 7 that IORCa is not activated by depletion of intracellular Ca2+ stores.
IORCa Is Inhibited by Intracellular Mg2+
We examined the effect of intracellular Mg2+ concentration on the activity of IORCa. Cells were patch clamped with an ATP- and GTP-free pipette solution in which EGTA was replaced by 10 mM BAPTA. The concentration of MgCl2 added to the pipette solution varied between 0 and 6 mM. As shown in Fig. 8 (A and B), increasing free Mg2+ concentrations inhibited IORCa in a saturable manner. The estimated K1/2 value for free Mg2+ is 692 μM (Fig. 8 B).
Figure 8.
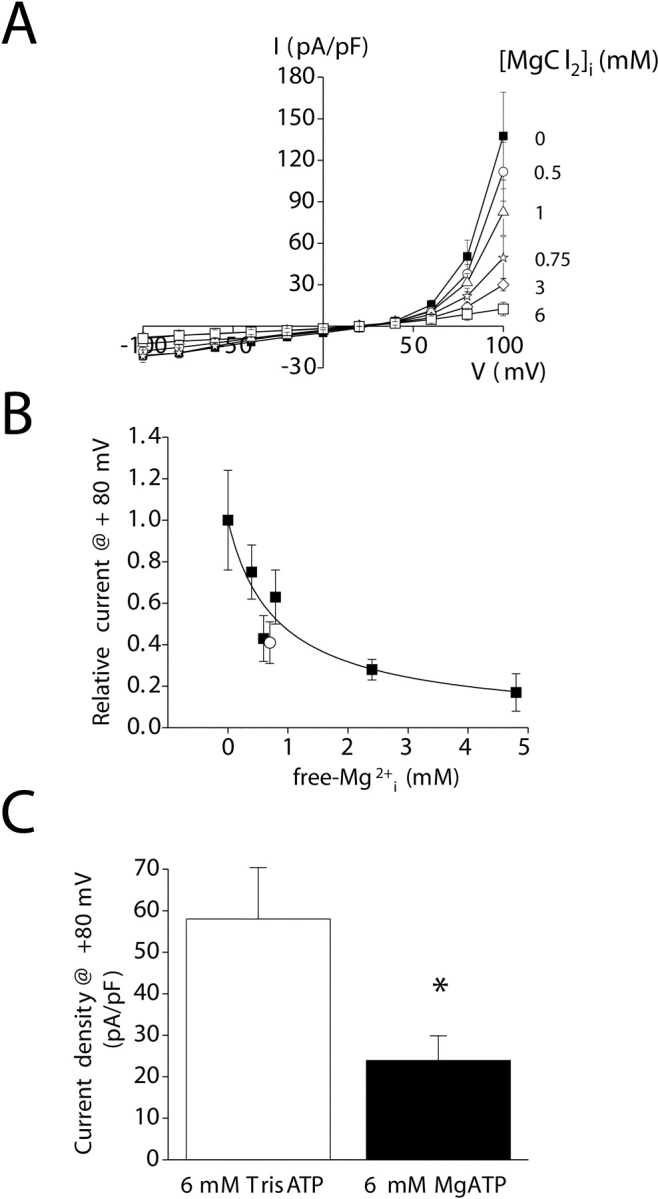
Inhibition of the outwardly rectifying cation current by intracellular Mg2+. (A) I–V relationships for cells patch clamped with ATP- and GTP-free pipette solutions containing 0–6 mM MgCl2. Chloride concentrations in the solutions were maintained constant by addition of 0–12 mM NMDGCl. EGTA was replaced with 10 mM BAPTA to buffer intracellular Ca2+ at 14 nM. Note that the Mg2+ concentrations indicated on the figure are those added to the pipette solution. Therefore, 0 Mg2+ should be considered nominally Mg2+ free. (B) Dose–response relationship for inhibition of the outwardly rectifying current by intracellular free Mg2+. Data were fit using the equation I = 1/1 + ([Mg2+]/K1/2)n. K1/2 and n are 692 μM and 0.8, respectively. Open circle is inhibition observed when free Mg2+ concentration is elevated by addition of 6 mM MgATP. (C) Effect of Mg2+ nucleotides on the outwardly rectifying cation current. Whole-cell current is inhibited ∼60% by 6 mM MgATP. Calculated concentration of free Mg2+ in the pipette solution containing 6 mM MgATP is 700 μM. The degree of inhibition is similar to that observed when free Mg2+ is elevated by addition of MgCl2 (see B). *, P < 0.05 (compared with 6 mM TrisATP). Values are means ± SEM (n = 5–9). Currents were measured in standard bath medium containing both Ca2+ and Mg2+ 3–4 min after obtaining whole-cell access when activation was complete. Voltage clamp protocol was the same as described in Fig. 2.
To determine if IORCa is also inhibited by Mg-nucleotides, 6 mM MgATP or 6 mM TrisATP was added to the pipette solution. As shown in Fig. 8 C, 6 mM MgATP inhibited whole-cell current ∼60% at +80 mV (P < 0.05). The calculated free Mg2+ concentration in a solution containing 6 mM MgATP is 700 μM. When plotted as a function of calculated free Mg2+ concentration (Fig. 8 B, open circle), the degree of inhibition observed with 6 mM MgATP is very similar to that observed with Mg2+ alone. These results indicate that IORCa is inhibited by free Mg2+ but is insensitive to MgATP.
Intracellular Ca2+ Store Depletion Activates an Inwardly Rectifying Current
SOCCs play important roles in IP3-dependent intracellular Ca2+ signaling pathways (Putney and McKay, 1999; Lewis, 2001; Taylor and Thorn, 2001). Given the dependence of the C. elegans defecation cycle on IP3 and oscillatory Ca2+ signaling in the intestine (Dal Santo et al., 1999), we performed patch clamp studies to determine if intestinal cells expressed store-operated channels.
IORCa, which dominates whole-cell recordings, was inhibited by inclusion of 5 mM free Mg2+ in the pipette solution (Kozak et al., 2002; Prakriya and Lewis, 2002). To prevent Ca2+ store depletion, we patch clamped cells with a pipette solution containing ATP, GTP, and 200 nM free Ca2+ buffered with 10 mM BAPTA. The bath solution contained 145 mM Na+ and 20 mM Ca2+. Using these solutions, we observed that whole-cell current remained stable for at least 5–7 min after obtaining whole-cell access in three out of three cells (Fig. 9 C). The mean ± SEM current observed just before loss of the whole-cell seal was −5.5 ± 2.7 pA/pF at −120 mV (n = 3).
Figure 9.
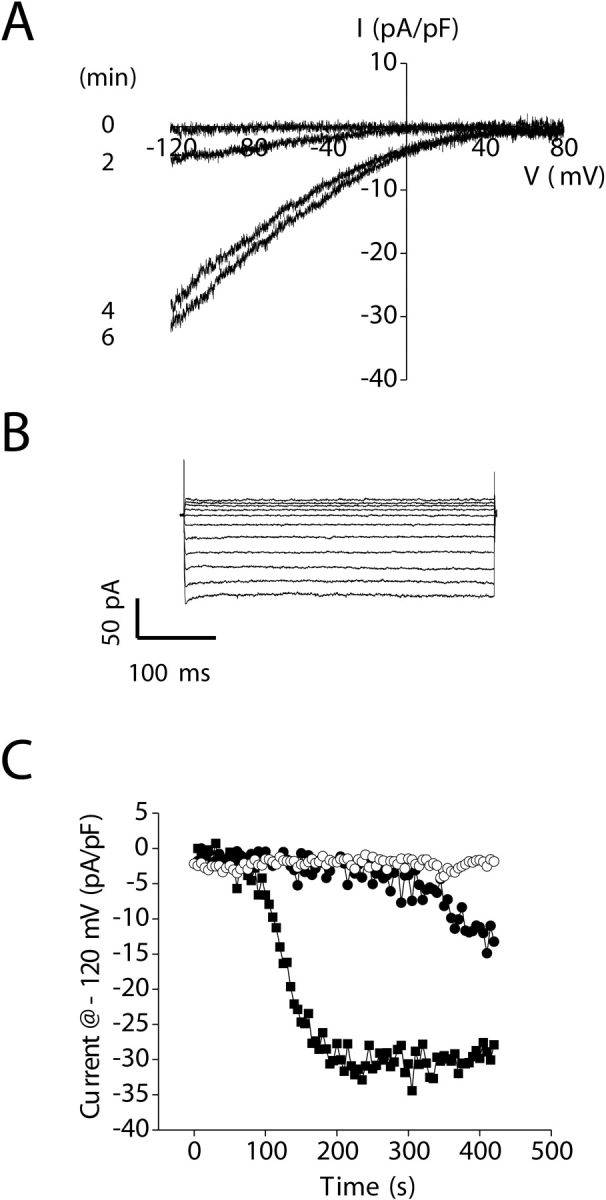
Activation of an inwardly rectifying store-operated current by depletion of intracellular Ca2+ stores. (A) An inwardly rectifying current activates when store depletion is induced by addition of 10 μM IP3 to a pipette solution containing 18 nM free Ca2+. Currents were elicited by ramping membrane voltage from −120 to +80 mV at 200 mV/s every 5 s. Leak current was subtracted from the traces shown. (B) Inwardly rectifying whole-cell currents elicited by stepping membrane voltage from −120 to +80 mV from a holding potential of 0 mV. Steps were 400 ms in duration. Currents were measured after activation induced by IP3 was complete. (C) Changes in whole-cell current observed in the absence of store depletion (○), during passive store depletion (•), and during active store depletion induced by IP3 (▪). Leak current was subtracted from the currents induced by active and passive store depletion. All experiments shown were performed in the presence of 5 mM free Mg2+ in the pipette solution to inhibit IORCa and 145 mM Na+/20 mM Ca2+ in the bath.
Active depletion of Ca2+ stores was induced by dialyzing cells with a nucleotide-free pipette solution containing 18 nM free Ca2+ and 10 μM IP3. A strongly inwardly rectifying, voltage-independent cation current was activated under these conditions (Fig. 9, A–C). The mean time to the start of current activation after membrane rupture was 84 s, and the current activated to a peak value of −31 pA/pF at −120 mV at an initial rate of −26 pA/pF/min (Table II) .
TABLE II.
Store Depletion–induced Activation Characteristics of the Inwardly Rectifying Current
| Store depletion protocol | Time to start of current activationa | Rate of current activationb | Time to peak current | Peak current |
|---|---|---|---|---|
| s | pA/pF/min | s | pA/pF | |
| Passive | 163 ± 26 (8) | −3.3 ± 0.5 (8) | 462 ± 52 (6) | −16 ± 3 (6) |
| Active | ||||
| 10 μm IP3 | 84 ± 12c (81) | −26 ± 3e (81) | 230 ± 17d (77) | −31 ± 2d (77) |
| Active | ||||
| 2 μm ionomycin | −72 ± 17e (8) | −38 ± 14 (8) |
Currents were elicited by ramping membrane voltage from −120 to +80 mV at 200 mV/s every 5 s. Peak current and rate of current activation were measured at −120 mV.
Time to start of current activation is the time after obtaining whole-cell access that current activation began.
Rate of current activation was quantified by performing linear regression analysis on whole-cell currents measured during the first 60–120 s after initiation of current activation. The time course of current activation in many cells was sigmoidal in nature (e.g., Fig. 9 C), displaying an initial very slowly rising phase followed by a much steeper and rapid linear increase in current amplitude. To avoid ambiguity in characterizing current activation, we measured the rate of activation during the steepest initial part of this sigmoidal curve. The first point to fall within this region was defined as the start of current activation. Values are means ± SEM (number of cells).
P < 0.05, compared with passive store depletion.
P < 0.01, compared with passive store depletion.
P < 0.001, compared with passive store depletion.
It is conceivable that the inwardly rectifying current is activated by IP3 rather than store depletion per se. To test for this possibility, we passively depleted intracellular Ca2+ stores by dialyzing cells with a pipette solution containing 18 nM free Ca2+ alone. Passive store depletion also activated an inwardly rectifying current (Fig. 9 C), albeit at a rate ∼13% of that observed when stores were depleted actively. The mean time to the start of current activation after membrane rupture was 163 s (Table II). This delay in current activation was significantly (P < 0.05) longer than that observed in the presence of IP3 (Table II). Mean initial rate of current activation and peak current at −120 mV were −3.3 pA/pF/min and −16 pA/pF, respectively (Table II). The rate of current activation and peak current were significantly (P < 0.01) decreased compared with that observed with active store depletion induced by IP3 (Table II). These results are consistent with store depletion being the mechanism responsible for activation of the inwardly rectifying current.
As a final test for the involvement of store depletion in current activation, we dialyzed cells with a pipette solution containing 18 nM free Ca2+ and then exposed them 30–150 s after obtaining whole-cell access to 2 μM ionomycin for 30–60 s. Ionomycin is a Ca2+ ionophore that is expected to induce efflux of Ca2+ from intracellular stores and activation of store-dependent cation channels (Hoth and Penner, 1993; Parekh, 1998; Voets et al., 2001). In the presence of ionomycin, the inwardly rectifying current activated rapidly. The initial rate of current activation and peak current in the presence of ionomycin were −72 pA/pF/min and −38 pA/pF, respectively, at −120 mV. The rate of current activation was ∼22-fold faster (P < 0.001) than that observed with passive store depletion (Table II). Taken together, the results shown in Fig. 9 and Table II demonstrate clearly that depletion of IP3-dependent intracellular Ca2+ stores activates an inwardly rectifying store-operated current. Currents activated by the three store depletion protocols had the same I–V relationships (unpublished data), indicating that they were likely carried by the same channel.
Selectivity of the Store-operated Channel
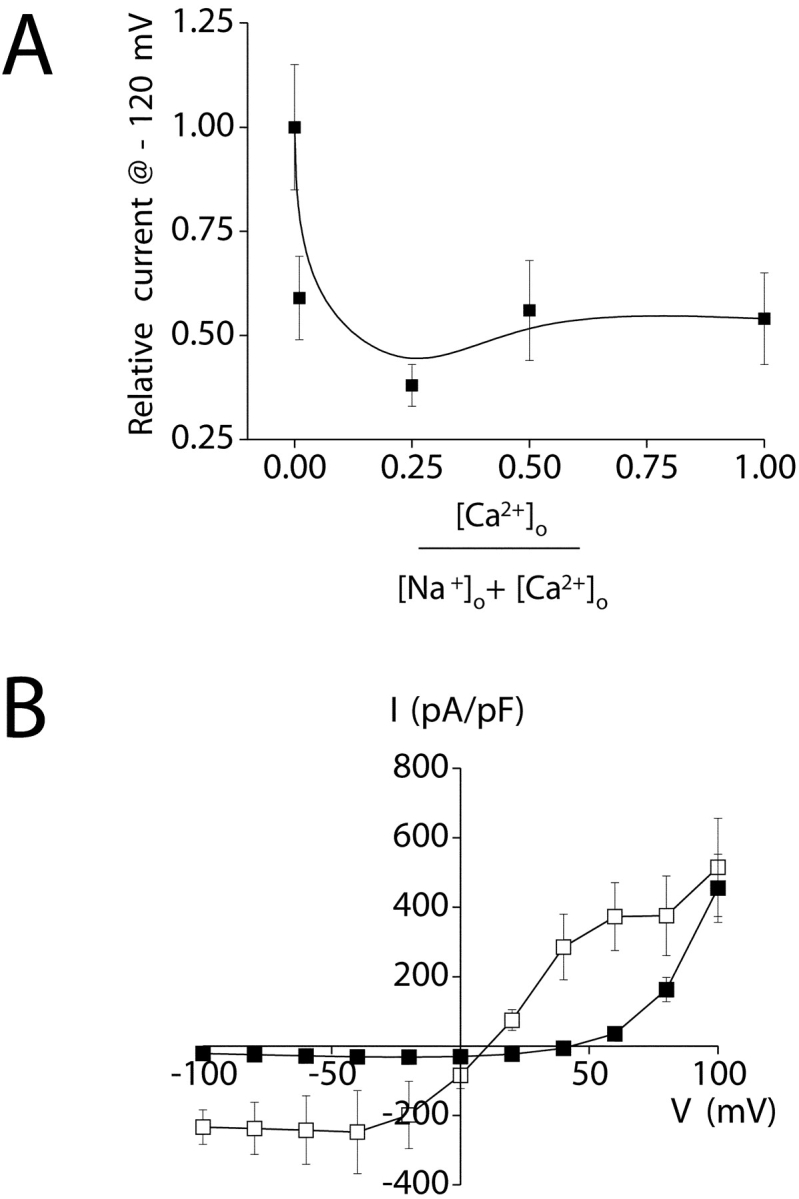
In the presence of 145 mM Na+ and 20 mM Ca2+ in the bath, the store-operated current showed very strong inward rectification (Fig. 9, A and B) and voltage-independent gating (Fig. 9 B). For the majority of cells examined, the slope of the I–V plot at positive voltages was extremely shallow, and a clear reversal of current direction was not detectable (Fig. 9 A).
Replacement of bath Na+ and Ca2+ with NMDG+ completely blocked inward current (Fig. 10 A), demonstrating that the channel is highly cation selective. In the presence of 130 mM NMDG+ and 10 mM Ca2+, a strongly inwardly rectifying current was observed (Fig. 10 A). Mean ± SEM current density at −120 mV was −31 ± 5 pA/pF (n = 4), which is similar to that observed in the presence of Na+ and Ca2+ (Table II). These results demonstrate that the store-operated channel has a high selectivity for Ca2+ or Na+. The inability to accurately measure Erev at positive voltages precludes accurate calculation of relative Ca2+ to Na+ permeability. However, if Erev is >+80 mV, the estimated PCa/PNa is >1,000:1. Based on these results, we hereafter refer to the inwardly rectifying store-operated Ca2+ channel as SOCC and the channel current as ISOC.
Figure 10.
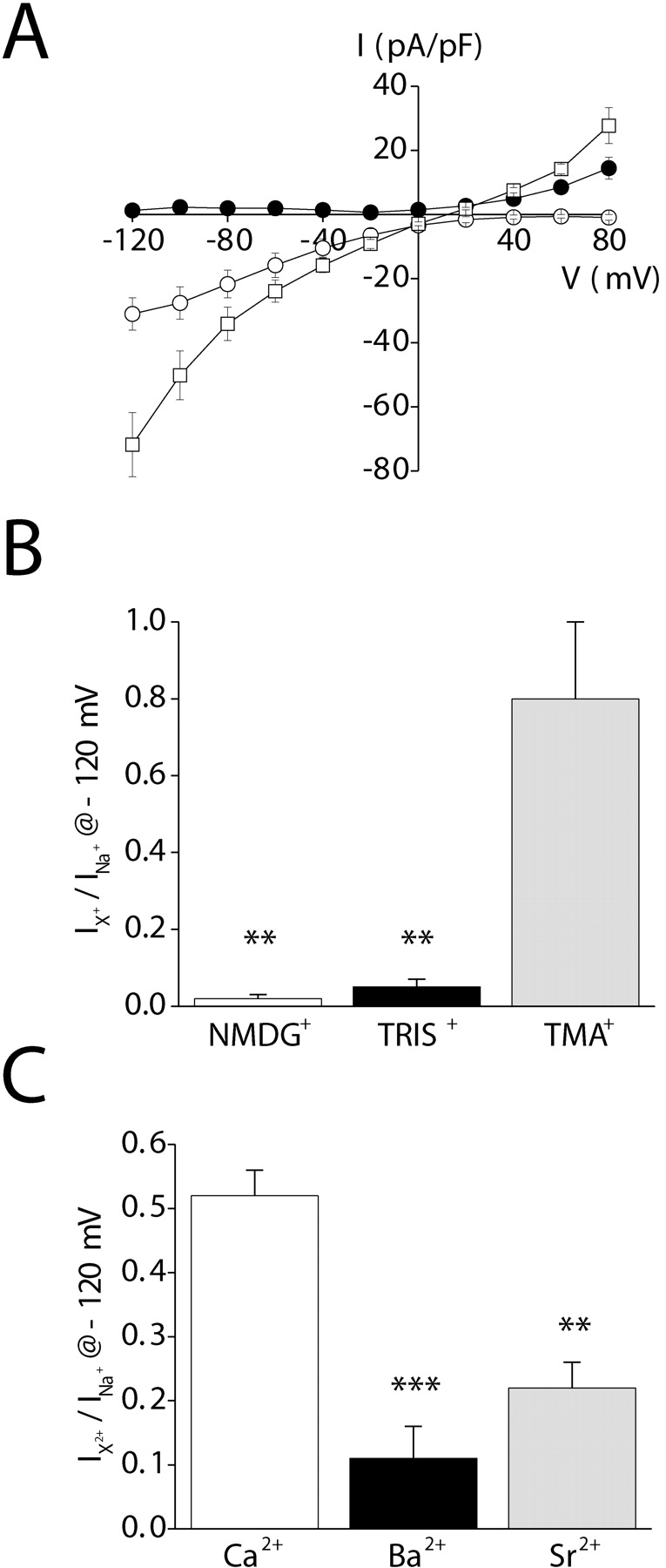
Cation selectivity of the inwardly rectifying current. (A) Whole-cell currents observed in the presence of various extracellular cations. Exposure of cells to divalent-free (buffered with 1 mM EDTA) 150 mM Na+ bath increases inward and outward monovalent current (□; compare with Fig. 9 A). Replacement of bath Na+ with 150 mM NMDG+ (•) eliminates inward current. In the presence of 10 mM Ca2+ and 130 mM NMDG+ (○), inward current is restored. (B) Relative organic cation currents (i.e., Ication/INa). Whole-cell currents were measured at −120 mV in divalent-free (buffered with 1 mM EDTA) 150 mM Na+-containing bath and after complete replacement of Na+ with various organic cations. **, P < 0.01 (compared with current observed with Na+). (C) Relative divalent cation currents (i.e., Idivalent/INa). Whole-cell currents were measured at −120 mV in a divalent-free (buffered with 1 mM EDTA) 150 mM Na+-containing bath and in a bath containing 130 mM NMDG+ and 10 mM Ca2+, Ba2+, or Sr2+. **, P < 0.01; ***, P < 0.001 (compared with current observed with Ca2+). Values are means ± SEM (n = 4–13). Whole-cell currents were elicited by stepping membrane voltage from −120 to +80 mV from a holding potential of 0 mV. Steps were 400 ms in duration. Whole-cell current was activated by inclusion of 10 μM IP3 in the patch pipette solution. Leak currents were subtracted from all current records. Experiments were performed in the presence of 5 mM free Mg2+ in the pipette solution to inhibit IORCa.
In the presence of divalent-free (buffered with 1 mM EDTA) 150 mM Na+-containing bath, significant inward and outward current was detected (Fig. 10 A). Mean ± SEM Erev of the Na+ current was 13 ± 1 mV (n = 28). Replacement of bath Na+ with Cs+ or Rb+ shifted Erev to more negative values (Table III) . Substitution of Na+ with Li+ had no significant (P > 0.2) effect on Erev (Table III). The calculated relative cation permeabilities (i.e., Pcation/PNa; Table III) determined from the changes in Erev yielded a monovalent inorganic cation selectivity sequence of Na+ ≈ Li+ > Rb+ ≈ Cs+.
TABLE III.
Relative Inorganic Monovalent Cation Permeability of the Inwardly Rectifying Conductance
| Cation | ΔErev | Pcation/PNa |
|---|---|---|
| mV | ||
| Li+ | 1.4 ± 0.9 | 1.06 ± 0.04 (5) |
| Cs+ | −16 ± 5a | 0.60 ± 0.10 (7) |
| Rb+ | −16 ± 4b | 0.57 ± 0.08 (6) |
Whole-cell currents were elicited by stepping membrane voltage from −120 to +80 mV in 20-mV steps from a holding potential of 0 mV. Voltage steps were 400 ms long. Steady-state current-to-voltage relationships were plotted for determination of Erev in the presence of Na+ and various test cations. Relative permeabilities were calculated using equations derived from the Goldman-Hodgkin-Katz equation (see materials and methods). Values are means ± SEM (number of cells).
P < 0.03.
P < 0.02.
We attempted to measure relative organic cation permeability of SOCC by replacing bath Na+ with TMA+, TRIS+, or NMDG+. Reversal potentials could not be accurately estimated for NMDG+ and TRIS+. We therefore quantified organic cation current relative to Na+ current (i.e., Ication/INa) at −120 mV. TMA+ and TRIS+ currents were ∼80 and 5%, respectively, of those observed with Na+; NMDG+ was effectively impermeant (Fig. 10, A and B). The diameters of TMA+, TRIS+, and NMDG+ are 0.55 nm, 0.64 nm, and 0.68 nm, respectively (Hille, 2001). This suggests that the pore diameter of SOCC is ∼0.6–0.7 nm.
To determine whether the observed organic solute permeabilities are reflective of SOCC rather than nonspecific leak, we measured TMA+ and TRIS+ currents relative to Na+ in the absence of store depletion and ISOC activation. Mean ± SEM Ication/INa for TMA+ and TRIS+ were 1.0 ± 0.2 (n = 5) and 1.6 ± 0.9 (n = 6), respectively. These values were not significantly different (P > 0.6), indicating that the leak does not discriminate between the two cations. In contrast, SOCC discriminates strongly between TMA+ and TRIS+ (Fig. 10 B).
We also attempted to measure relative divalent cation permeability of the channel. Cells were bathed initially with a divalent-free solution containing 150 mM Na+. Sodium was then replaced by a solution containing 130 mM NMDG+ and 10 mM Ca2+, Ba2+, or Sr2+. Relative divalent cation currents (i.e., Idivalent/INa) at −120 mV are shown in Fig. 10 C. Both Ba2+ and Sr2+ were significantly (P < 0.01) less permeable than Ca2+. The divalent cation selectivity sequence of SOCC is Ca2+ > Ba2+ ≈ Sr2+.
Pharmacological Characteristics of ISOC
We compared the pharmacological properties of ISOC to IORCa. Exposure of intestinal cells to 100 μM La3+ inhibited ISOC ∼90% (P < 0.01; Fig. 11, A and B) . This inhibitory effect was partially reversible (Fig. 11 A). The concentration of La3+ required to inhibit ISOC 50% was 9 μM (Fig. 11 B), a value twofold higher than that observed for IORCa (Fig. 6 A).
Figure 11.
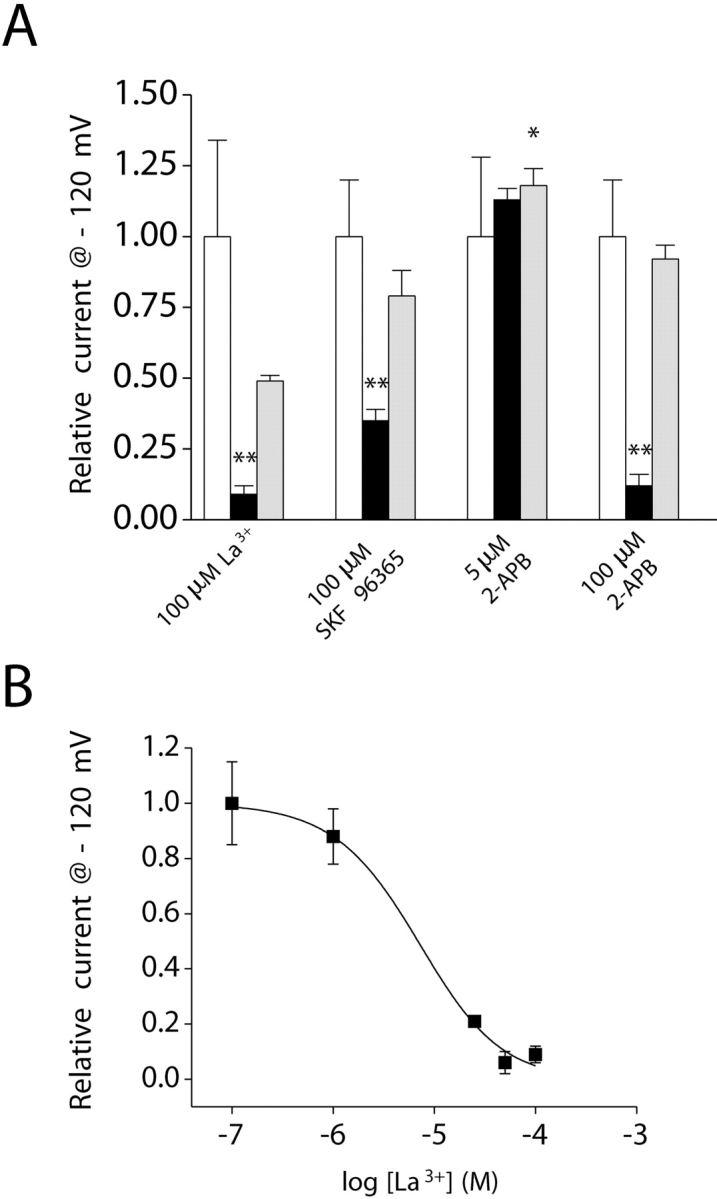
Pharmacology of the inwardly rectifying current. (A) Addition of 100 μM La3+, 100 μM SKF 96365, or 100 μM 2-APB (black bar) to the bath inhibited whole-cell current by 65–90%. *, P < 0.05; **, P < 0.01 (compared with control, white bar). 5 μM 2-APB had no effect on whole-cell current (black bar). Inhibitory effects of SKF 96365 and 2-APB were fully reversible (gray bar). Current levels observed after washout of SKF 96365 and 100 μM 2-APB (gray bar) were not significantly (P > 0.05) different from those observed before drug addition (white bar). Washout of 5 μM 2-APB caused a small but significant (*, P < 0.05) stimulation of whole-cell current. Values are means ± SEM (n = 4–6). (B) Dose–response relationship for the inhibitory effect of La3+. Data were fit using the equation I = 1/1 + ([Mg2+]/K1/2)n. K1/2 and n are 9 μM and 1.2, respectively. Values are means ± SEM (n = 4–8). Voltage clamp protocol was the same as described in Fig. 9 B. Whole-cell current was activated by inclusion of 10 μM IP3 in the patch pipette solution. Leak currents were subtracted from all current records. Experiments were performed in the presence of 5 mM free Mg2+ in the pipette solution to inhibit IORCa and 145 mM Na+/20 mM Ca2+ in the bath.
100 μM SKF 96365 inhibited ISOC ∼65% (P < 0.01) in a reversible manner (Fig. 11 A). Concentrations of 2-APB >10 μM irreversibly inhibit CRAC (Prakriya and Lewis, 2001, 2002; Voets et al., 2001; Hermosura et al., 2002). However, at lower concentrations (5 μM), the drug stimulates CRAC activity (Prakriya and Lewis, 2001, 2002). The intestinal cell SOCC has some characteristics that resemble those of CRAC (see discussion). We therefore tested the effects of low and high concentrations of 2-APB on the current. Exposure to 5 μM 2-APB had no significant (P > 0.05) effect on ISOC (Fig. 11 A). Washout of the drug induced a small, but statistically significant (P < 0.05), increase in current. Exposure to 100 μM 2-APB inhibited ISOC by ∼90% (Fig. 11 A). Inhibition was completely reversed by drug washout (Fig. 11 A).
Inactivation of ISOC
ISOC activity was relatively stable in the presence of a bath solution containing 145 mM Na+ and 20 mM Ca2+ (Fig. 12 A). Mean ± SEM relative current observed 3 min after store depletion–induced activation was complete was 0.93 ± 0.02 (n = 18). In contrast, the SOCC-mediated Na+ current observed in divalent-free bath inactivated immediately after Ca2+ removal (Fig. 12 B). The mean ± SEM rate of Na+ current inactivation was −12 ± 2 pA/pF/min or −13 ± 2%/min (n = 9).
Figure 12.

Inactivation of the inwardly rectifying current. (A) Example of active store depletion–induced activation of whole cell in a bath solution containing 145 mM Na+ and 20 mM Ca2+. Pipette solution contained 10 μM IP3 and 18 nM free Ca2+. Current remains stable after store depletion–induced activation is complete. (B) Effect of divalent-free (buffered with 1 mM EDTA) bath solution on whole-cell current. Current was activated by active store depletion with 10 μM IP3. Sodium current begins to inactivate immediately after removal of divalent cations from the bath. Voltage clamp protocol was the same as described in Fig. 9 A. Leak currents were subtracted from all current records. Experiments were performed in the presence of 5 mM free Mg2+ in the pipette solution to inhibit IORCa.
DISCUSSION
Functional Properties of C. elegans Intestinal Epithelial Cell Ca2+ Conductances
The C. elegans intestine provides a unique model system in which to characterize the molecular details of IP3-dependent oscillatory Ca2+ signaling. To begin defining the functional roles and regulation of cation channels involved in Ca2+ signaling events, we performed patch clamp analysis of intestinal cells cultured in vitro (Christensen et al., 2002). Intestinal epithelial cells develop and survive in culture (Fig. 1) and are present at a frequency similar to that observed in newly hatched L1 larvae (see results).
Our initial patch clamp studies on intestinal cells were performed using “physiological” bath (5 mM K+ and 145 mM Na+) and pipette (143 mM K+ and 4 mM Na+) solutions described originally by Lockery and coworkers (Goodman et al., 1998). Under these conditions, whole-cell current showed strong outward rectification (unpublished data). Ion substitution studies demonstrated that an outwardly rectifying cation channel carries this current (Figs. 2–4; Table I). The channel conducts both monovalent and divalent cations and has high selectivity for Ca2+ over Na+ (PCa/PNa = 64:1; Table I). Lanthanum, 2-APB, and SKF 96365 reversibly inhibited the current (Fig. 6), and intracellular Mg2+ inhibited channel activity with a K1/2 of 692 μM (Fig. 8).
The outwardly rectifying Ca2+ current (IORCa) was constitutively active in all cells examined. Currents typically increased two to threefold after whole-cell access was obtained. Activation is not mediated by depletion of intracellular Ca2+ stores (Fig. 7). Channel activation may be mediated by washout of intracellular Mg2+, changes in protein phosphorylation, and/or other as yet undefined mechanisms.
IORCa shares some characteristics with MIC, Mg2+-nucleotide–regulated metal ion (MagNuM), and TRPM7 currents. These shared characteristics include cation selectivity, permeability to Ca2+, strong outward rectification, gradual activation after obtaining whole-cell access, and insensitivity to store depletion (Nadler et al., 2001; Hermosura et al., 2002; Kozak et al., 2002; Prakriya and Lewis, 2002). Importantly, the degree of inhibition of ORCa by Mg2+ is similar to that of the recently described MIC/MagNuM currents in RBL and Jurkat T cells (Hermosura et al., 2002; Kozak et al., 2002; Prakriya and Lewis, 2002).
TRPM7 and/or other TRP genes have been proposed to encode MIC/MagNuM (Nadler et al., 2001; Clapham, 2002; Prakriya and Lewis, 2002). The biophysical similarities between MIC/MagNuM and ORCa suggest that the channels may have a common molecular origin. However, there are also a number of significant differences between the channel types. For example, removal of extracellular Mg2+ and Ca2+ causes the I–V relationship to become linear for MIC, MagNuM, and TRPM7 (Nadler et al., 2001; Hermosura et al., 2002; Kozak et al., 2002; Prakriya and Lewis, 2002), but does not alter IORCa rectification (Fig. 4). IORCa shows voltage- and time-dependent gating (Fig. 4), whereas gating of TRPM7 is largely voltage insensitive (Runnels et al., 2001). MIC, MagNuM, and TRPM7 discriminate poorly between Ca2+ and Na+ (Nadler et al., 2001; Runnels et al., 2001) and Cs+ and Na+ (PCs/PNa ≈ 1) (Runnels et al., 2001; Kozak et al., 2002; Prakriya and Lewis, 2002). In contrast, IORCa is highly selective for Ca2+ over Na+ and PCs/PNa is 0.27 (Table I). Finally, Na+ current through ORCa is half blocked by ∼1 mM Ca2+ (Fig. 5 A), whereas MIC is half blocked by <5 μM Ca2+ (Kerschbaum and Cahalan, 1998). Studies focused on identifying the gene or genes that encode ORCa are currently underway.
Given the role of IP3 and Ca2+ signaling in regulating defecation rhythm in C. elegans (Dal Santo et al., 1999), we performed a series of studies to determine whether intestinal cells also express SOCCs. To observe SOCC activity, we inhibited IORCa by addition of millimolar concentrations of free Mg2+ (Hermosura et al., 2002; Kozak et al., 2002) to the patch pipette solution.
In the absence of store depletion, whole-cell current was stable (Fig. 9 C). Active or passive depletion of Ca2+ stores activated a strongly inwardly rectifying current (Fig. 9, A and B). When stores were depleted actively by inclusion of 10 μM IP3 in the patch pipette solution or by addition of 2 μM ionomycin to the bath, the rates of current activation were increased ∼8- and ∼22-fold compared with passive store depletion (Fig. 9 C; Table II). These results demonstrate clearly that Ca2+ store depletion activates an inwardly rectifying store-operated channel.
The inwardly rectifying channel is highly cation selective. Inward current was undetectable when bath Na+ and Ca2+ were replaced by NMDG+ (Fig. 10 A). Addition of Ca2+ to an NMDGCl bath solution induced inward current with an amplitude similar to that observed with Na+ and Ca2+ (Fig. 9 A; Fig. 10 A; Table II). In a divalent-free Na+-containing bath, Erev was 13 mV (Fig. 10 A). Addition of 20 mM Ca2+ shifted Erev to more positive potentials. However, it was not possible to measure Erev accurately under these conditions because the slope of the I–V plot from 0 to +80 mV was extremely shallow (Fig. 9 A). Nevertheless, these results demonstrate that the store-operated channel is highly selective for Ca2+ over monovalent cations. If Erev > +80 mV, the channel would have a Ca2+ to Na+ selectivity of at least 1,000:1.
CRAC is the most extensively characterized SOCC and is probably expressed ubiquitously in vertebrate cells (Parekh and Penner, 1997). The intestinal cell SOCC shares a number of characteristics with CRAC, including activation by passive and active store depletion, very strong inward rectification, and an apparent high selectivity for Ca2+ over monovalent cations (Hoth and Penner, 1993). Cation selectivity sequences of CRAC vary somewhat between cell types and are possibly altered by intracellular Ca2+ buffering (Zhang and McCloskey, 1995; Fierro and Parekh, 2000). Hoth and Penner (1992) and Zweifach and Lewis (1993) reported a CRAC divalent cation selectivity sequence of Ca2+ > Ba2+ ≈ Sr2+ in mast and Jurkat T cells. In RBL cells, Fierro and Parekh (1999) reported a slightly different divalent cation selectivity sequence of Ca2+ > Sr2+ > Ba2+. The selectivity sequence observed in mast and T cells is similar to that of the intestinal cell SOCC (Fig. 10 C).
CRAC monovalent cation selectivity sequences of Na+ ≈ Li+ > K+ > Cs+ (Voets et al., 2001) and Na+ ≈ Li+ > Rb+ > Cs+ (Bakowski and Parekh, 2002) have been observed in RBL cells. In T cells, Lepple-Wienhues and Cahalan (1996) reported a monovalent cation selectivity for CRAC of Na+ > Li+ = K+ > Rb+ ≫ Cs+. The monovalent selectivity sequence for the intestinal cell SOCC is Na+ ≈ Li+ > Rb+ ≈ Cs+ (Table III) and resembles that reported for RBL cells (Voets et al., 2001; Bakowski and Parekh, 2002). The most significant difference in CRAC and SOCC cation selectivity is their relative Cs+ permeabilities. CRAC has a PCs/PNa of ∼0.1, whereas PCs/PNa for SOCC is 0.6 (Table III).
TMA+ permeated SOCC nearly as well as Na+, whereas TRIS+ and NMDG+ had very low or negligible permeability (Fig. 10, A and B), suggesting that the channel has a minimum pore diameter of ∼0.6–0.7 nm. TMA+ permeation through CRAC in RBL cells is undetectable, and a pore diameter of 0.32–0.55 nm has been estimated (Bakowski and Parekh, 2002). In Jurkat T cells, TMA+ permeates CRAC, albeit poorly (Kerschbaum and Cahalan, 1998). Kerschbaum and Cahalan (1998) have estimated a minimum pore diameter for the T cell CRAC of at least 0.58 nm.
When exposed to divalent-free Na+ medium, CRAC undergoes a rapid inactivation (Christian et al., 1996; Lepple-Wienhues and Cahalan, 1996; Voets et al., 2001; Kozak et al., 2002; Prakriya and Lewis, 2002). In Jurkat T cells for example, CRAC activity declines up to ∼80% within ∼20 s after removal of bath Ca2+ (Prakriya and Lewis, 2002). Inactivation of CRAC may reflect extracellular Ca2+-dependent changes in channel gating (Zweifach and Lewis, 1995; Christian et al., 1996).
The C. elegans SOCC also inactivates in divalent-free medium (Fig. 12). However, this inactivation is considerably slower than that observed with CRAC. The SOCC-mediated Na+ current inactivates at a rate of −13%/min.
At present, it is not possible to conclude that homologous genes encode CRAC and the intestinal SOCC because the molecular identities of both channels are unknown. However, C. elegans clearly provides unique experimental advantages and opportunities for identifying SOCC-encoding genes. Identification of these genes may ultimately provide clues into the molecular identity of CRAC.
Role of Store-independent and Store-operated Ca2+ Channels in Oscillatory Ca2+ Signaling
Extracellular agonist–induced Ca2+ signaling in nonexcitable cells requires the release of Ca2+ from IP3-regulated intracellular stores and the influx of Ca2+ across the plasma membrane via SMOCCs and SOCCs (Elliott, 2001; Zitt et al., 2002). High concentrations of agonists typically trigger sustained elevation of cytoplasmic Ca2+ levels. It is generally accepted that SOCCs play important roles in maintaining globally elevated Ca2+ concentrations and in refilling depleted Ca2+ stores (Parekh and Penner, 1997; Elliott, 2001). However, in the presence of lower, physiologically relevant agonist concentrations, Ca2+ changes are more complex, occurring in oscillations and waves and in localized areas of the cell (Shuttleworth, 1999; Berridge et al., 2000). The mechanisms responsible for generating Ca2+ oscillations are varied and depend on passive Ca2+ buffering, the spatial distribution of Ca2+ stores, rates of Ca2+ transport across the plasma membrane, and mitochondrial and ER Ca2+ uptake (Shuttleworth, 1999; Berridge et al., 2000; Bootman et al., 2001; Petersen, 2002). The specific roles played by SMOCCs and SOCCs in oscillatory Ca2+ signaling are unclear. Calcium oscillations in some cell types continue for long periods in the absence of extracellular Ca2+ (Lechleiter and Clapham, 1992), whereas oscillatory Ca2+ signals in other cell types are strictly dependent on Ca2+ influx (Torihashi et al., 2002; Wu et al., 2002).
Dolmetsch and Lewis (1994) have proposed that Ca2+ oscillations in T lymphocytes are driven primarily by pulsatile Ca2+ influx via CRAC. These investigators suggest that the main function of the intracellular Ca2+ stores is to control the extent and timing of CRAC activity (Dolmetsch and Lewis, 1994). Pharmacological studies in rat hepatocytes (Gregory and Barritt, 2003) and astrocytes (Pizzo et al., 2001) also suggest that Ca2+ oscillations are dependent on CRAC-mediated Ca2+ influx.
Shuttleworth and coworkers (Shuttleworth, 1999; Mignen et al., 2003) have noted that evidence for the involvement of CRAC specifically and SOCCs in general in generating Ca2+ oscillations is limited. Shuttleworth has also argued that CRAC possesses neither the sensitivity to store depletion nor the activation kinetics required for oscillatory Ca2+ signaling (Shuttleworth, 1999). Instead, he has suggested that the function of CRAC may be primarily to mediate plasma membrane Ca2+ influx required for refilling ER stores under conditions of sustained store depletion (Shuttleworth, 1999).
Recently, an arachidonic acid–regulated Ca2+ channel (ARC) has been described in many cell types (Mignen and Shuttleworth, 2000; Moneer and Taylor, 2002). It has been proposed that ARC is a major Ca2+ entry pathway required for oscillatory Ca2+ signaling (Shuttleworth, 1999; Mignen et al., 2001, 2003; see also Lankisch et al., 1999). In contrast, Luo et al. (2001) have suggested that an influx pathway distinct from both ARCs and SOCCs mediates Ca2+ entry that drives Ca2+ oscillations in HEK cells. The disparate conclusions of these studies underscore the need for extensive additional work to define the mechanisms of Ca2+ entry in nonexcitable cells, to define the role of SMOCCs and SOCCs in oscillatory Ca2+ signaling, and to identify molecular mechanisms of SMOCC and SOCC regulation.
As noted earlier, intracellular Ca2+ levels in the nematode intestine oscillate with a periodicity of 45–50 s (Dal Santo et al., 1999). These oscillations drive rhythmic contraction of body wall muscles and are dependent on IP3 receptor function (Dal Santo et al., 1999). Both the ORCa and SOC channels could play central roles in generating and maintaining oscillatory Ca2+ signaling in the intestine. However, determination of their physiological functions requires in vitro and in vivo characterization of intestinal cell Ca2+ signaling events and identification of the genes that encode both channels. These studies are currently underway and will likely be facilitated by the genetic and molecular tractability of C. elegans as well as by the physiological accessibility of cultured intestinal cells.
Genetic and Molecular Analysis of the C. elegans Defecation Cycle
Mutagenesis and forward genetic analysis in C. elegans has to date identified ∼12 genes that disrupt normal defecation rhythm when they are mutated (Iwasaki et al., 1995). itr-1 and flr-1, which encodes a putative DEG/ENaC cation channel (Take-Uchi et al., 1998), are the only genes that have been mapped and characterized in detail. In addition, mutations in calcium/calmodulin-dependent serine/threonine kinase type II (CaMKII) have been shown to disrupt the defecation cycle (Reiner et al., 1999). CaMKII plays essential roles in oscillatory Ca2+ signaling (De Koninck and Schulman, 1998; Dupont and Goldbeter, 1998) and is expressed in multiple C. elegans cell types, including intestinal epithelial cells (unpublished observation cited in De Koninck and Schulman, 1998).
Mapping and characterization of mutant genes that disrupt defecation rhythm as well as isolation of other defecation mutants will likely provide unique insights into intestinal cell oscillatory Ca2+ signaling. It will also be important to utilize reverse genetic approaches to identify genes that encode the ORCa and SOC channels. At present, it is reasonable to postulate that the channels are encoded by one or more TRP genes (Montell, 2001; Clapham, 2002). The C. elegans genome contains 13 predicted TRP-encoding genes (3 TRPC, 5 TRPV, 4 TRPM, and 1 TRPN). Gene function in C. elegans can be rapidly and economically disrupted either by the use of chemical deletion mutagenesis or RNA interference (Barr, 2003; Strange, 2003). Deletion mutagenesis should allow definitive testing of the hypothesis that TRP genes encode the ORCa and SOC channels.
In conclusion, we have identified two highly Ca2+-selective cation conductances in C. elegans intestinal epithelial cells. One conductance is store independent, and the other is activated by store depletion. Our studies provide the first detailed electrophysiological characterization of voltage-independent and store-operated Ca2+ conductances in C. elegans. The ability to combine patch clamp electrophysiological measurements on intestinal epithelial cells with forward and reverse genetic analyses provides a powerful new approach for defining the cellular and molecular mechanisms of IP3-dependent oscillatory Ca2+ signaling and its role in controlling rhythmic biological processes.
Acknowledgments
We thank Drs. Joel Rothman and James McGhee for providing the elt-2–GFP-expressing worm strains JR1838 and JM63, respectively. We also thank Andrew M. Beld for technical assistance and Dr. Louis J. DeFelice for helpful discussions.
This work was supported by National Institutes of Health (NIH) grant DK51610. A.Y. Estevez was supported by a National Science Foundation postdoctoral fellowship. R.K. Roberts was supported by NIH Vanderbilt University Bridges Program grant GM60190-01S1.
Olaf S. Andersen served as editor.
Footnotes
Abbreviations used in this paper: ARC, arachidonic acid–regulated Ca2+ channel; CRAC, Ca2+ release–activated channel; DIC, differential interference contrast; IP3, inositol-1,4,5-trisphosphate; MagNuM, Mg2+-nucleotide–regulated metal ion; MIC, Mg2+-inhibited cation; SMOCC, second messenger–operated Ca2+ channel; SOCC, store-operated Ca2+ channel; TRP, transient receptor potential.
The NMDG+ pipette solution contained 3.5 mM CsOH and 5 mM Na+ derived from 0.5 mM Na2GTP and 2 mM Na2ATP.
Because gluconate is a strong calcium buffer, Ca2+ activity was measured with a calcium-sensitive electrode and adjusted to the same level present in NaCl bath.
Removal of Ca2+ from the bath is likely to alter seal resistance. Changes in seal resistance would alter the magnitude of leak current and may affect estimates of PCa/PNa. To assess the effect of leak on ORCa Ca2+ selectivity measurements, we patch clamped cells with a pipette solution containing 5 mM free Mg2+ to inhibit ORCa and 200 nM Ca2+ to prevent store depletion and ISOC activation. Mean (n = 4) leak currents were estimated in bath solutions containing either 150 mM Na+ or 130 mM NMDG+ and 10 mM Ca2+. Leak currents were subtracted from ORCa currents measured under the same conditions. The mean ± SEM shift in Erev observed after replacement of bath Na+ with NMDG+ and Ca2+ was 29 ± 1 mV (n = 6) in the absence of leak subtraction versus 33 ± 1 mV (n = 6) after correction for leak. This small change was significant (P < 0.02) and increased the estimated PCa/PNa from 64 ± 2 to 71 ± 2.
References
- Almers, W., and E.W. McCleskey. 1984. Non-selective conductance in calcium channels of frog muscle: calcium selectivity in a single-file pore. J. Physiol. 353:585–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aussel, C., R. Marhaba, C. Pelassy, and J.P. Breittmayer. 1996. Submicromolar La3+ concentrations block the calcium release-activated channel, and impair CD69 and CD25 expression in CD3- or thapsigargin-activated Jurkat cells. Biochem. J. 313:909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski, D., and A.B. Parekh. 2002. Monovalent cation permeability and Ca2+ block of the store-operated Ca2+ current I CRAC in rat basophilic leukemia cells. Pflügers Arch. 443:892–902. [DOI] [PubMed] [Google Scholar]
- Barr, M.M. 2003. Super models. Physiol. Genomics. 13:15–24. [DOI] [PubMed] [Google Scholar]
- Beedle, A.M., J. Hamid, and G.W. Zamponi. 2002. Inhibition of transiently expressed low- and high-voltage-activated calcium channels by trivalent metal cations. J. Membr. Biol. 187:225–238. [DOI] [PubMed] [Google Scholar]
- Berridge, M.J., P. Lipp, and M.D. Bootman. 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1:11–21. [DOI] [PubMed] [Google Scholar]
- Bilmen, J., and F. Michelangeli. 2002. Inhibition of the type 1 inositol 1,4,5-trisphosphate receptor by 2-aminoethoxydiphenylborate. Cell. Signal. 14:955–960. [DOI] [PubMed] [Google Scholar]
- Bootman, M.D., P. Lipp, and M.J. Berridge. 2001. The organisation and functions of local Ca2+ signals. J. Cell Sci. 114:2213–2222. [DOI] [PubMed] [Google Scholar]
- Bootman, M.D., T.J. Collins, L. Mackenzie, H.L. Roderick, M.J. Berridge, and C.M. Peppiatt. 2002. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 16:1145–1150. [DOI] [PubMed] [Google Scholar]
- Brenner, S. 1974. The genetics of Caenorhabditis elegans. Genetics. 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, M., A. Estevez, X. Yin, R. Fox, R. Morrison, M. McDonnell, C. Gleason, D.M. Miller, and K. Strange. 2002. A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron. 33:503–514. [DOI] [PubMed] [Google Scholar]
- Christian, E.P., K.T. Spence, J.A. Togo, P.G. Dargis, and J. Patel. 1996. Calcium-dependent enhancement of depletion-activated calcium current in Jurkat T lymphocytes. J. Membr. Biol. 150:63–71. [DOI] [PubMed] [Google Scholar]
- Clapham, D.E. 2002. Sorting out MIC, TRP, and CRAC ion channels. J. Gen. Physiol. 120:217–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham, D.E., L.W. Runnels, and C. Strubing. 2001. The TRP ion channel family. Nat. Rev. Neurosci. 2:387–396. [DOI] [PubMed] [Google Scholar]
- Dal Santo, P., M.A. Logan, A.D. Chisholm, and E.M. Jorgensen. 1999. The inositol trisphosphate receptor regulates a 50-second behavioral rhythm in C. elegans. Cell. 98:757–767. [DOI] [PubMed] [Google Scholar]
- De Koninck, P., and H. Schulman. 1998. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 279:227–230. [DOI] [PubMed] [Google Scholar]
- Dolmetsch, R.E., and R.S. Lewis. 1994. Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. J. Gen. Physiol. 103:365–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont, G., and A. Goldbeter. 1998. CaM kinase II as frequency decoder of Ca2+ oscillations. Bioessays. 20:607–610. [DOI] [PubMed] [Google Scholar]
- Edgar, L.G. 1995. Blastomere culture and analysis. Methods Cell Biol. 48:303–321. [DOI] [PubMed] [Google Scholar]
- Elliott, A.C. 2001. Recent developments in non-excitable cell calcium entry. Cell Calcium. 30:73–93. [DOI] [PubMed] [Google Scholar]
- Fierro, L., and A.B. Parekh. 1999. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. J. Membr. Biol. 168:9–17. [DOI] [PubMed] [Google Scholar]
- Fierro, L., and A.B. Parekh. 2000. Substantial depletion of the intracellular Ca2+ stores is required for macroscopic activation of the Ca2+ release-activated Ca2+ current in rat basophilic leukaemia cells. J. Physiol. 522:247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige, T., M.J. Hendzel, D.P. Bazett-Jones, and J.D. McGhee. 1999. Direct visualization of the elt-2 gut-specific GATA factor binding to a target promoter inside the living Caenorhabditis elegans embryo. Proc. Natl. Acad. Sci. USA. 96:11883–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, M.B., D.H. Hall, L. Avery, and S.R. Lockery. 1998. Active currents regulate sensitivity and dynamic range in C. elegans neurons. Neuron. 20:763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, R.B., and G.J. Barritt. 2003. Evidence that Ca2+-release-activated Ca2+ channels in rat hepatocytes are required for the maintenance of hormone-induced Ca2+ oscillations. Biochem. J. 370:695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaszovich, C.R., C. Zitt, E. Jungling, and A. Luckhoff. 2000. Inhibition of TRP3 channels by lanthanides. Block from the cytosolic side of the plasma membrane. J. Biol. Chem. 275:37423–37428. [DOI] [PubMed] [Google Scholar]
- Hermosura, M.C., M.K. Monteilh-Zoller, A.M. Scharenberg, R. Penner, and A. Fleig. 2002. Dissociation of the store-operated calcium current I CRAC and the Mg-nucleotide-regulated metal ion current MagNuM. J. Physiol. 539:445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. 3rd ed. Sinauer Associates, Inc., Sunderland, MA. 814 pp.
- Hoth, M. 1995. Calcium and barium permeation through calcium release-activated calcium (CRAC) channels. Pflügers Arch. 430:315–322. [DOI] [PubMed] [Google Scholar]
- Hoth, M., and R. Penner. 1992. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 355:353–356. [DOI] [PubMed] [Google Scholar]
- Hoth, M., and R. Penner. 1993. Calcium release-activated calcium current in rat mast cells. J. Physiol. 465:359–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki, K., and J.H. Thomas. 1997. Genetics in rhythm. Trends Genet. 13:111–115. [DOI] [PubMed] [Google Scholar]
- Iwasaki, K., D.W.C. Liu, and J.H. Thomas. 1995. Genes that control a temperature-compensated ultradian clock in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 92:10317–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschbaum, H.H., and M.D. Cahalan. 1998. Monovalent permeability, rectification, and ionic block of store-operated calcium channels in Jurkat T lymphocytes. J. Gen. Physiol. 111:521–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostich, M., A. Fire, and D.M. Fambrough. 2000. Identification and molecular-genetic characterization of a LAMP/CD68-like protein from Caenorhabditis elegans. J. Cell Sci. 113:2595–2606. [DOI] [PubMed] [Google Scholar]
- Kozak, J.A., H.H. Kerschbaum, and M.D. Cahalan. 2002. Distinct properties of CRAC and MIC channels in RBL cells. J. Gen. Physiol. 120:221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankisch, T.O., F. Nozu, C. Owyang, and Y. Tsunoda. 1999. High-affinity cholecystokinin type A receptor/cytosolic phospholipase A2 pathways mediate Ca2+ oscillations via a positive feedback regulation by calmodulin kinase in pancreatic acini. Eur. J. Cell Biol. 78:632–641. [DOI] [PubMed] [Google Scholar]
- Lechleiter, J.D., and D.E. Clapham. 1992. Molecular mechanisms of intracellular calcium excitability in X. laevis oocytes. Cell. 69:283–294. [DOI] [PubMed] [Google Scholar]
- Lepple-Wienhues, A., and M.D. Cahalan. 1996. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (I CRAC) in Jurkat T cells. Biophys. J. 71:787–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, C.A. 1979. Ion-concentration dependence of the reversal potential and the single channel conductance of ion channels at the frog neuromuscular junction. J. Physiol. 286:417–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, J.A., and J.T. Fleming. 1995. Basic culture methods. Methods Cell Biol. 48:3–29. [PubMed] [Google Scholar]
- Lewis, R.S. 2001. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 19:497–521. [DOI] [PubMed] [Google Scholar]
- Luo, D., L.M. Broad, J. Bird, and J.W. Putney, Jr. 2001. Signaling pathways underlying muscarinic receptor-induced [Ca2+]i oscillations in HEK293 cells. J. Biol. Chem. 276:5613–5621. [DOI] [PubMed] [Google Scholar]
- Mayer, M.L., G.L. Westbrook, and P.B. Guthrie. 1984. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 309:261–263. [DOI] [PubMed] [Google Scholar]
- Merritt, J.E., W.P. Armstrong, C.D. Benham, T.J. Hallam, R. Jacob, A. Jaxa-Chamiec, B.K. Leigh, S.A. McCarthy, K.E. Moores, and T.J. Rink. 1990. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 271:515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen, O., and T.J. Shuttleworth. 2000. I ARC, a novel arachidonate-regulated, noncapacitative Ca2+ entry channel. J. Biol. Chem. 275:9114–9119. [DOI] [PubMed] [Google Scholar]
- Mignen, O., J.L. Thompson, and T.J. Shuttleworth. 2001. Reciprocal regulation of capacitative and arachidonate-regulated noncapacitative Ca2+ entry pathways. J. Biol. Chem. 276:35676–35683. [DOI] [PubMed] [Google Scholar]
- Mignen, O., J.L. Thompson, and T.J. Shuttleworth. 2003. Ca2+ selectivity and fatty acid specificity of the noncapacitative, arachidonate-regulated Ca2+ (ARC) channels. J. Biol. Chem. 278:10174–10181. [DOI] [PubMed] [Google Scholar]
- Moneer, Z., and C.W. Taylor. 2002. Reciprocal regulation of capacitative and non-capacitative Ca2+ entry in A7r5 vascular smooth muscle cells: only the latter operates during receptor activation. Biochem. J. 362:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montell, C. 2001. Physiology, phylogeny, and functions of the TRP superfamily of cation channels. Sci. STKE. 90:RE1–RE17. [DOI] [PubMed] [Google Scholar]
- Nadler, M.J., M.C. Hermosura, K. Inabe, A.L. Perraud, Q. Zhu, A.J. Stokes, T. Kurosaki, J.P. Kinet, R. Penner, A.M. Scharenberg, and A. Fleig. 2001. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 411:590–595. [DOI] [PubMed] [Google Scholar]
- Nowak, L., P. Bregestovski, P. Ascher, A. Herbet, and A. Prochiantz. 1984. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 307:462–465. [DOI] [PubMed] [Google Scholar]
- Parekh, A.B. 1998. Slow feedback inhibition of calcium release-activated calcium current by calcium entry. J. Biol. Chem. 273:14925–14932. [DOI] [PubMed] [Google Scholar]
- Parekh, A.B., and R. Penner. 1997. Store depletion and calcium influx. Physiol. Rev. 77:901–930. [DOI] [PubMed] [Google Scholar]
- Petersen, O.H. 2002. Calcium signal compartmentalization. Biol. Res. 35:177–182. [DOI] [PubMed] [Google Scholar]
- Pizzo, P., A. Burgo, T. Pozzan, and C. Fasolato. 2001. Role of capacitative calcium entry on glutamate-induced calcium influx in type-I rat cortical astrocytes. J. Neurochem. 79:98–109. [DOI] [PubMed] [Google Scholar]
- Prakriya, M., and R.S. Lewis. 2001. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J. Physiol. 536:3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya, M., and R.S. Lewis. 2002. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J. Gen. Physiol. 119:487–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney, J.W., Jr., and R.R. McKay. 1999. Capacitative calcium entry channels. Bioessays. 21:38–46. [DOI] [PubMed] [Google Scholar]
- Reiner, D.J., E.M. Newton, H. Tian, and J.H. Thomas. 1999. Diverse behavioural defects caused by mutations in Caenorhabditis elegans unc-43 CaM kinase II. Nature. 402:199–203. [DOI] [PubMed] [Google Scholar]
- Runnels, L.W., L. Yue, and D.E. Clapham. 2001. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 291:1043–1047. [DOI] [PubMed] [Google Scholar]
- Shuttleworth, T.J. 1999. What drives calcium entry during [Ca2+]i oscillations? - challenging the capacitative model. Cell Calcium. 25:237–246. [DOI] [PubMed] [Google Scholar]
- Strange, K. 2003. From genes to integrative physiology: Ion channel and transporter biology in Caenorhabditis elegans. Physiol. Rev. 83:377–415. [DOI] [PubMed] [Google Scholar]
- Take-Uchi, M., M. Kawakami, T. Ishihara, T. Amano, K. Kondo, and I. Katsura. 1998. An ion channel of the degenerin/epithelial sodium channel superfamily controls the defecation rhythm in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 95:11775–11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, C.W., and P. Thorn. 2001. Calcium signalling: IP3 rises again, and again. Curr. Biol. 11:R352–R355. [DOI] [PubMed] [Google Scholar]
- Torihashi, S., T. Fujimoto, C. Trost, and S. Nakayama. 2002. Calcium oscillation linked to pacemaking of interstitial cells of Cajal. Requirement of calcium influx and localization of TRP4 in caveolae. J. Biol. Chem. 277:19191–19197. [DOI] [PubMed] [Google Scholar]
- Vennekens, R., J. Prenen, J.G. Hoenderop, R.J. Bindels, G. Droogmans, and B. Nilius. 2001. Pore properties and ionic block of the rabbit epithelial calcium channel expressed in HEK 293 cells. J. Physiol. 530:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets, T., J. Prenen, A. Fleig, R. Vennekens, H. Watanabe, J.G.J. Hoenderop, R.J.M. Bindels, G. Droogmans, R. Penner, and B. Nilius. 2001. CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. J. Biol. Chem. 276:47767–47770. [DOI] [PubMed] [Google Scholar]
- Wu, X., G. Babnigg, T. Zagranichnaya, and M.L. Villereal. 2002. The role of endogenous human TRP4 in regulating carbachol-induced calcium oscillations in HEK-293 cells. J. Biol. Chem. 277:13597–13608. [DOI] [PubMed] [Google Scholar]
- Zhang, L., and M.A. McCloskey. 1995. Immunoglobulin E receptor-activated calcium conductance in rat mast cells. J. Physiol. 483:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitt, C., C.R. Halaszovich, and A. Luckhoff. 2002. The TRP family of cation channels: probing and advancing the concepts on receptor-activated calcium entry. Prog. Neurobiol. 66:243–264. [DOI] [PubMed] [Google Scholar]
- Zweifach, A., and R.S. Lewis. 1993. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA. 90:6295–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach, A., and R.S. Lewis. 1995. Rapid inactivation of depletion-activated calcium current (I CRAC) due to local calcium feedback. J. Gen. Physiol. 105:209–226. [DOI] [PMC free article] [PubMed] [Google Scholar]