

Abstract
The InsP3R Ca2+ release channel has a biphasic dependence on cytoplasmic free Ca2+ concentration ([Ca2+]i). InsP3 activates gating primarily by reducing the sensitivity of the channel to inhibition by high [Ca2+]i. To determine if relieving Ca2+ inhibition is sufficient for channel activation, we examined single-channel activities in low [Ca2+]i in the absence of InsP3, by patch clamping isolated Xenopus oocyte nuclei. For both endogenous Xenopus type 1 and recombinant rat type 3 InsP3R channels, spontaneous InsP3-independent channel activities with low open probability P o (∼0.03) were observed in [Ca2+]i < 5 nM with the same frequency as in the presence of InsP3, whereas no activities were observed in 25 nM Ca2+. These results establish the half-maximal inhibitory [Ca2+]i of the channel to be 1.2–4.0 nM in the absence of InsP3, and demonstrate that the channel can be active when all of its ligand-binding sites (including InsP3) are unoccupied. In the simplest allosteric model that fits all observations in nuclear patch-clamp studies of [Ca2+]i and InsP3 regulation of steady-state channel gating behavior of types 1 and 3 InsP3R isoforms, including spontaneous InsP3-independent channel activities, the tetrameric channel can adopt six different conformations, the equilibria among which are controlled by two inhibitory and one activating Ca2+-binding and one InsP3-binding sites in a manner outlined in the Monod-Wyman-Changeux model. InsP3 binding activates gating by affecting the Ca2+ affinities of the high-affinity inhibitory sites in different conformations, transforming it into an activating site. Ca2+ inhibition of InsP3-liganded channels is mediated by an InsP3-independent low-affinity inhibitory site. The model also suggests that besides the ligand-regulated gating mechanism, the channel has a ligand-independent gating mechanism responsible for maximum channel P o being less than unity. The validity of this model was established by its successful quantitative prediction of channel behavior after it had been exposed to ultra-low bath [Ca2+].
Keywords: single-channel electrophysiology, patch clamp, calcium, Xenopus oocyte, nucleus
INTRODUCTION
In many cell types, the second messenger inositol 1,4,5-trisphosphate (InsP3) is generated in the cytoplasm in response to the binding of extracellular ligands to plasma membrane receptors. InsP3 binds to its receptor, the InsP3R, in the ER and activates it as a Ca2+ channel to liberate stored Ca2+ from the ER lumen into the cytoplasm. This rapid release of Ca2+ modulates the cytoplasmic free Ca2+ concentration ([Ca2+]i), which serves as a ubiquitous cellular signal that can be manifested temporally as repetitive spikes or oscillations, and spatially as propagating waves or highly localized events (Meyer and Stryer, 1991; Berridge, 1993; Toescu, 1995). The temporal and spatial complexity of this signaling system involves sophisticated regulation of the activity of the InsP3R by various mechanisms, including cooperative activation by InsP3 (Meyer et al., 1988; Mak et al., 1998) and biphasic feedback from the permeant Ca2+ ion (Iino, 1990; Bezprozvanny et al., 1991; Mak et al., 1998).
A family of three InsP3 receptor isoforms has been identified—types 1, 2, and 3, with different primary sequences derived from different genes (Patel et al., 1999). Recent studies have demonstrated that channel P o of both the types 1 and 3 InsP3R isoforms is modulated with biphasic dependencies on cytoplasmic free Ca2+ concentration ([Ca2+]i), suggesting that the channels have two distinct types of functional Ca2+-binding sites: activating and inhibitory (Mak et al., 1998, 2001b). InsP3 activates the InsP3R by tuning the sensitivity of the channel to Ca2+ inhibition, with increases in the cytoplasmic concentration of InsP3 ([InsP3]) causing a decrease in the apparent Ca2+ affinity of the inhibitory binding sites of the channel. Nevertheless, the fully InsP3-liganded channel can still be inhibited by Ca2+, albeit at sufficiently high concentrations (Mak et al., 1998, 2001b). Importantly, InsP3 has little apparent effect on activation parameters (half-maximal activation [Ca2+]i, K act; and activation Hill coefficient, H act) of the biphasic Hill equation that describes the Ca2+ response of the channel, nor does it affect the robust maximum open probability exhibited by either InsP3R isoform under optimal activating conditions.
Whereas previous studies provided estimates of the affinity of the inhibitory Ca2+-binding sites in subsaturating and saturating concentrations of InsP3 (Mak et al., 1998, 2001b), the apparent affinity of the inhibitory Ca2+-binding sites of an InsP3R channel in the absence of InsP3 has not been determined. The effects of InsP3 have been modeled empirically assuming infinitely high affinity of the Ca2+ inhibition sites in a channel not bound to InsP3 (Mak et al., 1998, 2001b), but it is more reasonable to expect that the inhibitory Ca2+-binding sites adopt a finite maximal Ca2+ affinity in the absence of InsP3.
Here, we examined activities of the types 1 and 3 InsP3R channels in the absence of InsP3 to characterize the apparent affinity of the inhibitory Ca2+-binding site of the InsP3R not bound to InsP3. We reasoned that since InsP3 activates the channel by preventing Ca2+ from inhibiting it, it might be possible to activate the channel in the absence of InsP3 by removing Ca2+ from the inhibitory site by simply reducing [Ca2+]i to very low levels. We demonstrate that the InsP3R channel opens spontaneously in the absence of InsP3 when the channel is exposed to [Ca2+]i < 5 nM, but not when [Ca2+]i is elevated to 25 nM. These observations establish the apparent affinity of the Ca2+ inhibition sites of an InsP3R channel not bound to InsP3, and they support an allosteric model of InsP3R activation by Ca2+.
Many models have been developed to account for InsP3R-mediated [Ca2+]i signals, but all previously proposed models of InsP3R single-channel gating (De Young and Keizer, 1992; Swillens et al., 1994; Kaftan et al., 1997; Marchant and Taylor, 1997; Swillens et al., 1998; Adkins and Taylor, 1999; Moraru et al., 1999) assumed that only the InsP3-bound state(s) of the receptor is active. Thus, they fail to account for the spontaneous, InsP3-independent activities of the InsP3R observed in our study. To provide insights into the mechanisms underlying ligand regulation of InsP3R channel activity, we have developed an allosteric molecular model that can quantitatively account for not only the spontaneous, InsP3-independent channel activities in low [Ca2+]i, but all other characteristics of InsP3 and [Ca2+]i regulation of both types 1 and 3 InsP3R isoforms observed in nuclear patch clamp experiments (Mak et al., 1998, 2001b, 2003).
MATERIALS AND METHODS
Selection and Microinjection of Xenopus Oocytes
Maintenance of Xenopus laevis and surgical extraction of ovaries were performed as previously described (Jiang et al., 1998). The level of endogenous InsP3R channel activity was determined for each new batch of oocytes by patch clamping at least 3 isolated nuclei, obtaining 4–6 patches from each (Mak et al., 2000, 2001b). Rat type 3 InsP3R (r-InsP3R-3) channels were expressed by cRNA injection into oocytes ascertained to have extremely low level of endogenous InsP3R activities. In these studies, one endogenous Xenopus oocyte type 1 InsP3R (X-InsP3R-1) channel was observed in 100 patches from 5 batches of oocytes used for r-InsP3R-3 cRNA injection. In contrast, 544 channels were detected in 330 membrane patches, with 108 patches containing multiple InsP3R channels, from nuclei of r-InsP3R-3-expressing oocytes 4–5 d after cRNA injection. Assuming that the types 1 and 3 InsP3R associate randomly to form tetrameric channels, 97.6% of InsP3R channels detected in these experiments were contributed by type 3 homotetramers (Mak et al., 2000).
The endogenous X-InsP3R-1 was studied using batches of oocytes with high level of endogenous InsP3R activities, up to four days after ovary extraction (Mak and Foskett, 1994, 1997, 1998).
Patch Clamp Data Acquisition and Analysis
Patch clamp electrophysiology of isolated nuclei was performed as described (Mak and Foskett, 1994, 1997, 1998; Mak et al., 2000) in “on-nucleus” configuration at room temperature with the pipette electrode at +20 mV (unless stated otherwise) relative to the reference bath electrode. Transmembrane currents were amplified, filtered at 1 kHz, digitized at 5 kHz and recorded directly onto hard disk.
Channel opening and closing events were identified with a 50% threshold, and channel open probabilities and mean open and closed durations, were evaluated using MacTac software (Bruxton). The number of channels in the membrane patch was assumed to be the maximum number of open channel current levels observed throughout the current record (Mak et al., 2001b). When low channel open probability (P o < 0.1) was observed, generally only current records lasting >30 s and exhibiting only one open channel current level were used in our analyses to avoid under-estimating the total number of active InsP3R channels present in the membrane patch, which would lead to over-estimation of channel P o.
The data points shown for each set of experimental conditions are the means of results from at least four separate patch-clamp experiments performed under the same conditions. Error bars indicate the SEM.
Iterative fitting of the experimentally obtained channel P o in various [InsP3] and [Ca2+]i by the different molecular models were performed using Igor Pro software (WaveMetrics) with a nonlinear least-square fit (Levenberg-Marquardt) algorithm.
Solutions for Patch Clamp Experiments
All pipette solutions used in patch clamp experiments contained 140 mM KCl and 10 mM HEPES, except the low KCl solutions, which contained 14 mM KCl and 1 mM HEPES. The pipette solutions were pH adjusted to 7.3 with KOH.
By using K1 as the current carrier and appropriate quantities of the high-affinity Ca2+ chelator, BAPTA (1,2-bis(O-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid; Molecular Probes) (500–1,000 μM), Ca2+ concentrations in our experimental solutions were tightly controlled (Mak et al., 2003). For solutions with free [Ca2+] > 10 nM, free [Ca2+] was directly measured using Ca2+-selective minielectrodes (Baudet et al., 1994). For experimental solutions with [Ca2+] < 10 nM, the total [Ca2+] was determined by induction-coupled plasma mass spectrometry (Mayo Medical Laboratory) to be 6–10 μM. In the presence of 1 mM BAPTA in 140 mM KCl, 10 mM HEPES and 0.5 mM ATP at pH 7.3, the [Ca2+]i was calculated to be 0.9–1.5 nM using the Maxchelator software (C. Patton, Stanford University, Stanford, CA). Direct measurement by Ca2+-selective electrode confirmed the free [Ca2+] to be <5 nM, but the accuracy of this measurement was limited by the nonlinearity of the calibration curve of the electrode in such low free [Ca2+].
Pipette solutions contained various concentrations of Na2ATP, either 0 or 10 μM of InsP3 (Molecular Probes) and either 0 or 100 μg/ml heparin (Sigma-Aldrich) as stated.
The bath solutions used in all experiment had 140 mM KCl, 10 mM HEPES, 300 μM CaCl2, 500 μM BAPTA (measured [Ca2+] ≈ 400–500 nM), and pH 7.3.
Online Supplemental Material
The online supplemental material provides details, descriptions, and derivations of the allosteric models (both Monod-Wyman-Changeux [MWC] based and non-MWC–based models) that were considered to describe the ligand regulation of the InsP3R channel gating. The mathematical derivations from first principles of the equations used to calculate the theoretical InsP3R channel P o according to each of those models are presented, and comparisons between the calculated channel P o and experimental data under selected conditions are discussed. Online supplemental material is available at http://www.jgp.org/cgi/content/full/jgp.200308809/DC1.
RESULTS
Regulation of Types 1 and 3 InsP3R Channel Po by Cytoplasmic Ca2+, InsP3 and ATP
Single X-InsP3R-1 and r-InsP3R-3 channels observed in the same nuclear membrane system exhibit biphasic regulation by [Ca2+]i, with open probabilities (P o) well described by the empirical biphasic Hill equation (Mak et al., 1998, 2001b):
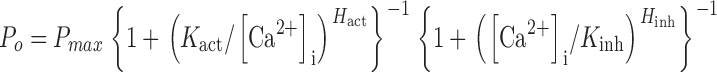 |
(1) |
where P max is the maximum channel open probability that can be achieved by the InsP3R channel under optimal [Ca2+]i and saturating [InsP3], K act is the half-maximal activating [Ca2+]i, H act is the activation Hill coefficient, K inh is the half-maximal inhibitory [Ca2+]i, and H inh is the inhibition Hill coefficient.
In nuclear patch clamp experiments, both InsP3R isoforms achieve a robust P max of 0.8 under optimal conditions. X-InsP3R-1 and r-InsP3R-3 channels both exhibit similar inhibition by Ca2+: K inh in the presence of saturating [InsP3] is ∼40–50 μM, and H inh is ∼3–4, indicating that the Ca2+ inhibition process is highly cooperative. InsP3 activates both channel isoforms by increasing K inh, i.e., decreasing the sensitivity of the channel to Ca2+ inhibition, with no effect on the other Hill equation parameters (P max, H inh, K act or H act) (Mak et al., 1998, 2001b). In the presence of 0.5 mM free ATP, the InsP3-concentration dependence of K inh of each InsP3R isoform can be described empirically by a simple Hill equation (Mak et al., 1998, 2001b):
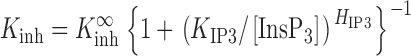 |
(2) |
with similar parameters: the half-maximal activating [InsP3] (K IP3) ∼50 nM, the Hill coefficient (H IP3) ∼4, and the maximum half-maximal inhibitory [Ca2+]i at saturating [InsP3] (K inh ∞) ∼45 μM.
Since the affinity of the inhibitory Ca2+-binding site must be finite even in the absence of InsP3, we hypothesized that the InsP3 requirement for channel activities could be waived if Ca2+ could be dissociated from the inhibitory Ca2+-binding site by an InsP3-independent method. Although one straight-forward method to accomplish this would be by lowering [Ca2+]i, channel opening requires Ca2+ binding to Ca2+ activation sites. We speculated that InsP3-independent channel activities should occur in low [Ca2+]i conditions in which Ca2+ would dissociate only from the inhibitory sites but not from the activating sites. Thus, we attempted to determine if simply dissociating Ca2+ from the inhibitory sites would be sufficient to activate channel opening, by using experimental conditions in which the affinity of the activating Ca2+-binding site was as high as possible. It was previously demonstrated that cytoplasmic free ATP acid (ATP3− and ATP4−) markedly enhances the Ca2+ affinity of the activation sites in both isoforms (Mak et al., 1998, 2001b). In saturating (10 μM) InsP3, the r-InsP3R-3 in 0.5 mM ATP and the X-InsP3R-1 in 9.5 mM ATP both exhibit a moderate P o of 0.2–0.4 in the presence of very low (25 nM) [Ca2+]i (Mak et al., 1999, 2001a). Thus, the requirement for Ca2+ binding to the Ca2+ activation site is satisfied at this Ca2+ concentration under these conditions. We reasoned that if the minimum value of K inh of the channel is not too low (for example, >20 nM), then InsP3R channel activity should be observable at 25 nM Ca2+ in appropriate [ATP] even in the absence of InsP3.
Lack of Channel Activity at 25 nM Ca2+ for InsP3R Not Bound to InsP3
A series of experiments was performed with membrane patches obtained from the same areas (±2 μm) of isolated nuclei from uninjected oocytes, where clustering of endogenous X-InsP3R-1 channels gave an exceptionally high probability of observing channel activity in membrane patches (Mak and Foskett, 1997). The pipette solutions alternately contained either 25 nM Ca2+, no InsP3, and 9.5 mM free ATP; or 1,150 nM Ca2+, 10 μM InsP3, and 0.5 mM ATP. The latter solution is one that maximizes the P o of the channel (Mak et al., 1998), and was therefore used to ensure that lack of channel activities in the former solution was not due to absence of InsP3R in the patched membranes. Whereas X-InsP3R-1 channel activities were detected in all eight patches with pipette solutions containing 10 μM InsP3, no channel activity was observed in any of the 26 patches with pipette solutions lacking InsP3. Thus, the type 1 channel cannot open in the absence of InsP3 in 25 nM Ca2+. In a parallel series of experiments using nuclei from r-InsP3R-3–expressing oocytes, in which the expressed recombinant channels exhibit similar clustering (Mak et al., 2000), no r-InsP3R-3 channel activity was detected in any of the eight patches with pipette solutions lacking InsP3, even though r-InsP3R-3 channel activities were observed in all seven patches with pipette solutions that contained 10 μM InsP3. These results therefore suggested that the apparent K inh in the absence of InsP3 (K inh 0) for both the X-InsP3R-1 and r-InsP3R-3 channel isoforms is lower than 25 nM.
InsP3-independent Activity of X-InsP3R-1 at Ultra-low [Ca2+]i
When [Ca2+]i was further decreased to <5 nM (calculated to be 0.9–1.5 nM, see materials and methods), with no InsP3 and 0.5 mM ATP in the pipette solution, channel activities with low open probability of ∼0.03, and with conduction and gating properties very similar to those of the InsP3R were observed in nuclei from uninjected oocytes (Fig. 1 A). Even though these channel activities were observed in the absence of InsP3, several characteristics identified them as being contributed by the endogenous X-InsP3R-1. First, the most frequently observed (>90%) channel conductance was 330 ± 15 pS (Fig. 2 A), indistinguishable from that of the InsP3-activated X-InsP3R-1 channels observed in the same system (Mak and Foskett, 1998). Importantly, no channel activities with conductances between 100 and 450 pS have been observed previously in the absence of InsP3 in thousands of nuclear patch clamp recordings on isolated oocyte nuclei (Mak and Foskett, 1998; Mak et al., 1998). Second, although the P o of the X-InsP3R-1 channel has a strong [Ca2+]i-dependence, the mean channel open duration <τo> lies within a narrow range (3–12 ms) in all the various experimental conditions previously investigated (Mak et al., 1998, 1999). <τo> of the channel activities observed in < 5 nM [Ca2+]i was 3.4 ± 0.4 ms (Fig. 1 A), which is within the normal narrow range of <τo> exhibited by X-InsPR3-1 channels (Fig. 1 F). Third, the probability of observing this channel activity in a membrane patch (P d) in these experimental conditions was similar to that in the same nucleus with pipettes containing an optimal activating solution (1,150 nM Ca2+, 10 μM InsP3, and 0.5 mM ATP; Fig. 3 A). Fourth, both the slope conductance (365 ± 20 pS) and reversal potential (56 ± 2 mV) of the channel observed in <5 nM [Ca2+]i and 0 InsP3 (Fig. 2 B) in the presence of asymmetric KCl conditions (14 mM KCl in the pipette and 140 mM KCl in bath) were very similar to those values (312 ± 20 pS and 54 ± 2 mV, respectively) observed for InsP3R channel under similar ionic conditions in the presence of 1 μM [Ca2+]i and 10 μM [InsP3] (Fig. 2 C). This indicates that the channel responsible for the activities observed in the absence of InsP3 has the same channel conductance and cation selectivity as the InsP3R channel under the same ionic conditions. Together, these observations render it highly unlikely that the channel activities observed in <5 nM [Ca2+]i were due to channels other than the X-InsP3R-1.
Figure 1.

Typical single-channel current traces of X-InsP3R-1 channels in various [Ca2+]i, [ATP] and [InsP3], as labeled. Arrows indicate closed channel current levels. 100 μg/ml heparin was used in +heparin experiments.
Figure 2.
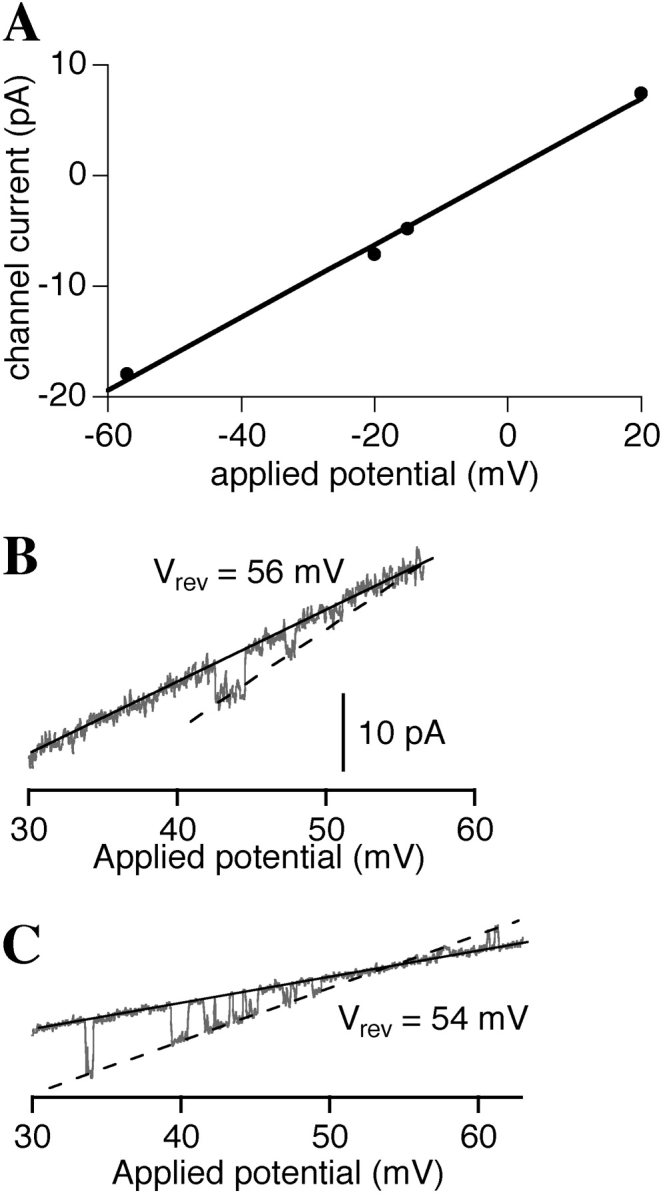
(A) Channel current versus applied transmembrane potential curve for InsP3R channels (n = 4) observed in symmetric 140 mM KCl in the presence of [Ca2+]i < 5 nM, 0.5 mM ATP and no InsP3. (B and C) Current traces with InsP3R channel observed under an applied potential ramp in 140 mM KCl bath and 14 mM KCl pipette solutions. For B, pipette solution contained [Ca2+]i < 5 nM, 0.5 mM ATP and no InsP3, whereas for C, pipette solution contained 1 μM [Ca2+]i, 0.5 mM ATP and 10 μM InsP3. The slope conductances of the channels were evaluated as the difference between the slopes of the open (dashed line) and closed (solid line) channel current levels. The positive reversal potentials (as tabulated in the graphs) indicate that the InsP3R channels observed are cation selective.
Figure 3.

(A) P d and (B) P o histograms of the X-InsP3R-1 channel in various experimental conditions. In the P d graph, numbers above each bar represent the fraction of nuclear membrane patches obtained that exhibited X-InsP3R-1 channel activities. In the P o graph, the number above each bar is the number of single-channel current records used to evaluate P o. Given the variance of the channel P o in experiments performed under the same experimental conditions, the channel P o observed under the various set of experimental conditions are not statistically different (P > 0.05 from t test) from P o observed under control conditions (0 InsP3, 0.5 mM ATP, [Ca2+]i < 5 nM).
The observations of InsP3-independent channel activities support our working hypothesis that ligand-independent channel activity can be achieved under conditions that dissociate Ca2+ from the inhibitory Ca2+-binding site. A further prediction of this hypothesis is that not only is InsP3 not necessary for channel activities under ultra-low [Ca2+]i conditions, but that channel activities will in fact be insensitive to InsP3. To investigate the dependence on InsP3 of InsP3R-1 channel activity in <5 nM [Ca2+]i, we used pipette solutions containing either 10 μM InsP3, or no InsP3. To rule out effects of possible contaminating InsP3 present in our system, 100 μg/ml heparin, a competitive inhibitor of InsP3 binding to the InsP3R (Worley et al., 1987; Cullen et al., 1988), was used in the pipette solution with no InsP3. Similar channel activities were observed (Fig. 1, B and C) with comparable P d as in the absence of InsP3 (Fig. 3 A). In addition, there was no systematic or statistically significant difference in the single-channel P o (∼0.03) in the presence or absence of InsP3 and heparin (Fig. 3 B). Thus, the X-InsP3R-1 has a low but nonzero P o at <5 nM [Ca2+]i regardless of whether the InsP3-binding site is occupied or not. This result suggests that the inhibitory Ca2+-binding sites of the channel was mostly unoccupied at [Ca2+]i < 5 nM regardless of the [InsP3].
Of note, because K act = 190 nM in 0.5 mM ATP (Mak et al., 1998), the activating Ca2+-binding site of the X-InsP3R-1 channel was also effectively unoccupied when [Ca2+]i < 5 nM. This result suggests that the spontaneous channel activity can occur when both the activating as well as the inhibitory Ca2+ sites are un-liganded. Because ATP stimulates channel activities by enhancing the functional affinity of the activating Ca2+-binding sites (Mak et al., 1999), the fact that the activating Ca2+-binding sites remain effectively unoccupied in <5 nM Ca2+ predicts that the InsP3-independent channel activities should be unaffected by ATP. In agreement, channel activities with similar conductances were observed regardless of [ATP] (0–9.5 mM; Fig. 1, A, D, and E). Neither P d nor P o of the X-InsP3R-1 channel in the absence of InsP3 were significantly affected by [ATP] (Fig. 3, P > 0.05). Together, these results demonstrate that the X-InsP3R-1 channel has an intrinsic, low P o even when its InsP3-binding sites and activating Ca2+-binding sites are not occupied, as long as its inhibitory Ca2+-binding sites are unoccupied.
InsP3-independent Activity of r-InsP3R-3 at Ultra-low [Ca2+]i
Similar results were obtained for the recombinant r-InsP3R-3 channels. In <5 nM [Ca2+]i, channel activities with conductances very similar to those of the X-InsP3R-1 were also observed in nuclei from r-InsP3R-3 cRNA-injected oocytes, independent of [InsP3] (0 or 10 μM), [ATP] (0 or 0.5 mM), or the presence of heparin (100 μg/ml) (Fig. 4 , A–D). Also similar to the X-InsP3R-1, the P o of the r-InsP3R-3 channel at <5 nM [Ca2+]i exhibited no systematic or statistically significant dependence (P > 0.05) on [InsP3] or [ATP] (Fig. 5 B). <τo> of the observed type 3 channels were 3.5–9 ms, very similar to the <τo> of 3–20 ms exhibited by the channel in the presence of InsP3 (comparing Fig. 4, A–D, with Fig. 4 E). P d of the channel activities at [Ca2+]i < 5 nM under various [InsP3] and [ATP] were similar to that for experiments using the same cRNA-injected oocyte nuclei with a pipette solution containing 1,150 nM Ca2+, 10 μM InsP3, and 0.5 mM ATP (Fig. 5 A). These results indicate that, similar to the X-InsP3R-1, the inhibitory Ca2+-binding sites of r-InsP3R-3 are mostly unoccupied in [Ca2+]i < 5 nM, and the r-InsP3R-3 channel has a low but nonzero P o even when its InsP3-binding sites and activating Ca2+-binding sites are not occupied, as long as its inhibitory Ca2+-binding sites are also unoccupied.
Figure 4.

Typical single-channel current traces of r-InsP3R-3 channels in various [Ca2+]i, [ATP] and [InsP3], as labeled. Arrows indicate closed channel current levels. 100 μg/ml heparin was used in +heparin experiments.
Figure 5.

(A) P d and (B) P o histograms of the r-InsP3R-3 channel in various experimental conditions. Numbers tabulated in the graphs have the same meanings as in Fig. 3. Given the variance of the channel P o in experiments performed under the same experimental conditions, the channel P o observed under the various set of experimental conditions are not statistically different (P > 0.05 from t test) from P o observed under control conditions (0 InsP3, 0.5 mM ATP, [Ca2+]i < 5 nM).
DISCUSSION
Finite Affinity of the Inhibitory Ca2+-binding Sites in InsP3R Channels
This study has revealed that InsP3R channels can be active spontaneously in the absence of InsP3 when [Ca2+]i is lowered to <5 nM, but not when it is lowered only to 25 nM. These observations suggest that the inhibitory Ca2+ sites were mostly unoccupied at <5 nM [Ca2+]i, whereas they were occupied when [Ca2+]i was 25 nM. The lack of occupancy of the inhibitory Ca2+ sites at <5 nM [Ca2+]i obviated the requirement for InsP3 binding, enabling the channel to open in the absence of the physiological ligand. These results support a model in which InsP3 binding activates InsP3R channel by reducing the apparent affinity of inhibitory Ca2+-binding sites (Mak et al., 1998), and they have implications for our understanding of the molecular mechanisms that regulate channel activity.
To provide a better empirical description of the tuning by [InsP3] of the channel sensitivity to Ca2+ inhibition that incorporates our present observations, the simple Hill equation describing the effects of InsP3 (Eq. 2) has to be modified to:
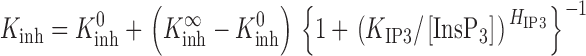 |
(3) |
where K inh 0 is the nonzero minimum K inh in the absence of InsP3. The empirical biphasic Hill equation describing the Ca2+ dependence of the P o of the InsP3R (Eq. 1) also has to be modified to:
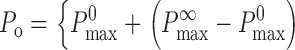 |
(4) |
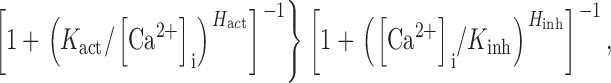 |
with P max 0 and P max ∞ being the maximum P o when the activating Ca2+ sites are unoccupied or fully occupied, respectively. Because the values of P max ∞, H act, H inh, and K act in the presence of various [ATP] have already been obtained in our previous studies for X-InsP3R-1 (Mak et al., 1998, 1999) and r-InsP3R-3 (Mak et al., 2001a,b), the channel P o for various [Ca2+]i, [InsP3] and [ATP] can be evaluated using Eqs. 3 and 4. Therefore, even though the values of K inh 0 and P max 0 are not precisely defined by our observations of spontaneous ligand-independent InsP3R channel activities, they can nevertheless be estimated using the constraints derived from our observations. First, even in the presence of optimal ATP concentrations, there were no detectable InsP3R channel activities in 25 nM [Ca2+]i in the absence of InsP3. Because of the technical limitations of the experimental system, channel activity with P o < 0.001 is not detectable in our experiments. Thus, the P o must be lower than 0.001 (marked by the horizontal dashed lines in Fig. 6) in 25 nM [Ca2+]i (marked by the vertical dashed lines in Fig. 6) in the absence of InsP3. Second, both InsP3R isoforms exhibited channel activities in 0.9–1.5 nM [Ca2+]i in various InsP3 and ATP concentrations with P o as shown in Figs. 3 B and 5 B. The observed X-InsP3R-1 channel P o are consistent with those calculated from Eqs. 3 and 4 using P max 0 = 0.02–0.07, and K inh 0 = 1.2–5.5 nM (Fig. 6 A). Similarly, experimental r-InsP3R-3 channel P o agree with those calculated from Eqs. 3 and 4 using P max 0 = 0.005–0.018, and K inh 0 = 1.2–3.8 nM (Fig. 6 B).
Figure 6.
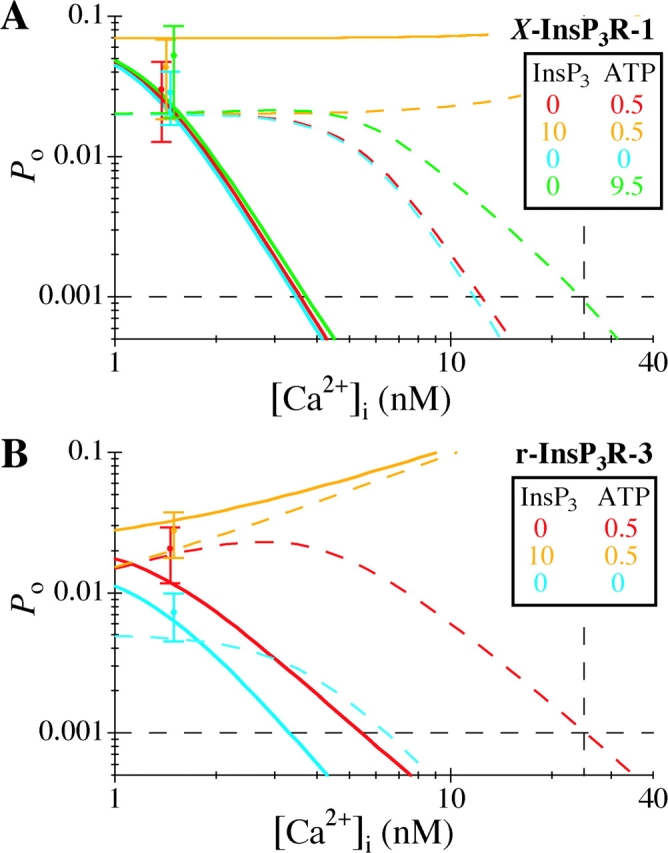
Estimating K inh 0 and P max 0 from channel activities at low [Ca2+]i for (A) X-InsP3R-1 and (B) r-InsP3R-3. Different colors correspond to different InsP3 and ATP concentrations as tabulated in the graphs. InsP3R channel P o observed in calculated [Ca2+]i of 0.9–1.5 nM are plotted as data points at [Ca2+]i = 1.5 nM (c.f. Figs. 3 B and 5 B). InsP3R channel P o at various [Ca2+]i can be calculated with Eq. 4 using the values of P max ∞, K act, H act, and H inh obtained in our previous studies for X-InsP3R-1 (Mak et al., 1998, 1999) and r-InsP3R-3 (Mak et al., 2001a,b). The values of parameters K inh 0 and P max 0 in Eq. 4, which were not determined in previous experiments, must be constrained so that: (a) the calculated channel P o at various InsP3 and ATP concentrations agree with experimental observations (i.e., lie within the error limits of the data points at 1.5 nM [Ca2+]i), and (b) the calculated channel P o in the absence of InsP3 is <0.001 at 25 nM [Ca2+]i, so that no channel activity was detected at 25 nM [Ca2+]i. The continuous and dashed curves represent channel P o calculated using two extreme sets of values for K inh 0 and P max 0 that satisfy those requirements. For X-InsP3R-1, the continuous curves are calculated with P max 0 = 0.07 and K inh 0 = 1.2 nM; and the dashed curves are calculated with P max 0 = 0.02 and K inh 0 = 5.5 nM. For r-InsP3R-3, the continuous curves are calculated with P max 0 = 0.018 and K inh 0 = 1.2 nM; and the dashed curves are calculated with P max 0 = 0.005 and K inh 0 = 3.8 nM. The observed channel P o data points in both graphs and the continuous curves in A are slightly offset along the [Ca2+]i axis for easier visualization.
If the InsP3R channel can be active in the absence of InsP3 binding, how can high fidelity Ca2+ release responses be achieved during cellular signaling? Although InsP3R channels can exhibit InsP3-independent activities, such spontaneous activities only occur in the presence of ultra-low [Ca2+]i (<25 nM), levels unlikely to be achieved under physiological conditions. Therefore, although the detection of the InsP3-independent spontaneous channel activities provides insights into the molecular bases for the complex regulation of the channel by Ca2+ and InsP3 (discussed below), such spontaneous activities by themselves probably have limited physiological implications in intracellular Ca2+ signaling. However, the regulation of the channel can now be viewed as a complex strategy designed to prevent spontaneous Ca2+ release while satisfying competing requirements of the channel. First, the channel requires Ca2+-induced Ca2+ release (CICR) properties to enable it to amplify and propagate [Ca2+]i signals. Conversely, the activity of the channel must be highly controlled to enable it to provide signals with high temporal and spatial specificity and fidelity. By using Ca2+ as a high-affinity inhibitor of channel activity, the channel is provided with a mechanism to prevent spontaneous channel activity from triggering inappropriate CICR. By using InsP3 as a negative regulator of Ca2+ inhibition, the channel is provided with a mechanism to ensure graded Ca2+ release activity with high temporal specificity in response to cellular signals.
Toward an Allosteric Model for the Regulation of InsP3R Channel Activities by [Ca2+]i and InsP3
Although Eqs. 3 and 4 can describe the regulation of InsP3R channel P o by its ligands Ca2+ and InsP3, enabling the channel P o at any [Ca2+]i and [InsP3] to be evaluated in terms of a set of parameters (P max 0, P max ∞, K act, H act, H inh, K inh 0, K inh ∞, K IP3, and H IP3) that are deduced from experimental data, the equations are empirical and they do not provide insights into the specific molecular mechanisms underlying ligand regulation of InsP3R activity. Therefore, it is desirable to develop a molecular model for ligand regulation of InsP3R activity that can, in terms of simple molecular mechanisms, account for all the features of the regulation of InsP3R channels (both types 1 and 3 isoforms) by [Ca2+]i and [InsP3] observed in extensive nuclear patch-clamp studies (Mak et al., 1998, 2001b, 2003; Boehning et al., 2001; and this study), as well as satisfy constraints imposed by the known structure of the InsP3R molecule and channel.
The observations, for both types 1 and 3 isoforms, that must be accounted for in such a molecular model are as follows:
(i) The InsP3R channel can be active when none of its ligand-binding sites are occupied ([InsP3]= 0 and [Ca2+]i = 1.5–2 nM << K act and K inh). Spontaneous activities of the InsP3R channel in the absence of all ligands observed in the present study are reminiscent of the spontaneous activities observed in the acetylcholine receptor channel (Jackson, 1984) and cyclic nucleotide–gated channels (Picones and Korenbrot, 1995). In those channels, ligand-independent gating suggested that allosteric models, in which the channel has a nonzero probability of being open even when its ligand-binding sites are unoccupied, were more appropriate than schemes that assume ligand binding to be necessary for channel opening. The ligand-independent opening of the InsP3R channels observed here cannot be accounted for by previously proposed models of InsP3R single-channel gating (De Young and Keizer, 1992; Swillens et al., 1994; Kaftan et al., 1997; Marchant and Taylor, 1997; Swillens et al., 1998; Adkins and Taylor, 1999; Moraru et al., 1999), in which only the InsP3-bound state(s) of the receptor is assumed to be active. Instead, our new observations suggest that an allosteric model in which the InsP3R channel has a finite probability of being open even when its activating Ca2+ and InsP3 binding sites are unoccupied (Monod et al., 1965) probably offers a better molecular picture for the ligand activation of the InsP3R. Furthermore, the model must also account for the absence of any spontaneous InsP3-independent channel activities in [Ca2+]i = 25 nM.
(ii) When the channel is studied in regular bath [Ca2+] (400–500 nM), InsP3 has no effect on Ca2+ activation parameters (specifically K act and H act) in the empirical Hill equation (Eq. 2 or 4) of the channel (both isoforms). At a low [Ca2+]i (for example, 100 nM), the channel P o remains unchanged at either sub-saturating (33 nM) or saturating (10 μM) concentrations of InsP3. InsP3 activates the InsP3R by reducing the sensitivity of the channel to high [Ca2+]i inhibition (i.e., increasing K inh in Eq. 2 or 4) (Mak et al., 1998, 2001b). This lack of effect of InsP3 on K act and H act cannot be accounted for by any previously proposed model for the InsP3R channel (De Young and Keizer, 1992; Swillens et al., 1994; Kaftan et al., 1997; Marchant and Taylor, 1997; Swillens et al., 1998; Adkins and Taylor, 1999; Moraru et al., 1999), in which InsP3 binding to the channel affects Ca2+ binding to the activating site, and vice versa.
(iii) When studied in the presence of regular bath [Ca2+] (400–500 nM), InsP3R channel P o exhibits biphasic regulation by [Ca2+]i in the presence of both saturating (10 μM) as well as subsaturating (≤100 nM) [InsP3] (Mak et al., 1998, 2001b).
(iv) Ca2+ inhibition of InsP3R channel activity is extremely sensitive to small changes in [InsP3] when 10 nM < [InsP3] < 100 nM. When the [InsP3] is raised from 10 to 100 nM, the K inh value for InsP3R-1 increases by over two orders of magnitude (Mak et al., 1998). Fitting the experimentally derived K inh for types 1 and 3 isoforms by Eq. 3 indicates that the empirical Hill coefficient for the InsP3 dependence of K inh is ∼4.
(v) The response of InsP3R channel activity to InsP3 saturates very abruptly. InsP3R-1 channel activity is already maximal when InsP3 = 100 nM, so that the sensitivity of the channel to Ca2+ inhibition exhibits no discernible change when [InsP3] is further increased by over three orders of magnitude from 100 nM to 180 μM. Despite the effect of InsP3 on the apparent affinity of the inhibitory Ca2+ sites of the InsP3R, once the InsP3R is fully activated by InsP3 (i.e., [InsP3] > 100 nM), the presence of a higher [InsP3] does not necessitate a higher [Ca2+]i to inhibit the channel. Indeed, the P o of InsP3R-1 is equally low at 60 μM [Ca2+]i in the presence of 180 μM or 10 μM InsP3 (Mak et al., 1998).
(vi) The maximum channel P o (P max) attained when the InsP3R is optimally activated is ∼0.8, less than 1 (Mak et al., 1998, 2001b).
(vii) The regulation of the InsP3R channel P o by Ca2+ and InsP3 mainly affects the mean closed channel duration <τc>, which correlates inversely with the channel P o, decreasing when the channel is activated and increasing when the channel is inhibited (Mak et al., 1998, 2001b,c). On the other hand, <τo> remains within a narrow range (5–15 ms) over all [Ca2+]i and [InsP3] until the channel P o drops to <0.1 (Mak et al., 1998, 2001b,c).
(viii) In addition to the observed properties of the ligand regulation of single-channel InsP3R activity, a molecular model of the regulation of the InsP3R channel must also take into consideration the molecular structure of the channel. It is well established that a functional InsP3R channel is a tetrameric unit (Mikoshiba et al., 1993). Although different isoforms of InsP3R can assemble to form heterotetramers (Joseph et al., 1995), the InsP3R channels (both types 1 and 3 isoforms) studied in our nuclear patch clamp experiments were overwhelmingly homotetrameric (Mak and Foskett, 1994; Mak et al., 2000), made up of four identical InsP3R molecules. Thus, the molecular model for InsP3R channel should exhibit either a fourfold symmetry or a twofold symmetry (dimer of dimers: Liu et al., 1998; Richards and Gordon, 2000).
(ix) Biochemically, multiple (>3) Ca2+-binding regions in the InsP3R sequences have been identified experimentally (Sienaert et al., 1996, 1997). These sequences, which are located mostly on the cytoplasmic side of the InsP3R molecule with one exposed to the lumen of the ER, may regulate InsP3R channel activities. In contrast, only one InsP3-binding region has been identified in the InsP3R sequence (Yoshikawa et al., 1996).
Allosteric Models Considered for Describing the Ligand Regulation of the InsP3R Channel
Because previously proposed models of InsP3R gating, in which only the InsP3-bound state(s) of the receptor can be active, fail to account for the spontaneous, InsP3-independent channel activities of the InsP3R, we systematically examined a series of allosteric models in increasing levels of complexity to find the simplest molecular model that can account for all the characteristics of the regulation by [InsP3] and [Ca2+]i of the InsP3R channel tabulated in the previous section. We started with allosteric schemes based on the Monod-Wyman-Changeux (MWC) model. As outlined in (Monod et al., 1965), the four identical InsP3R molecules in the homotetrameric channel occupy equivalent positions (condition viii) with an axis of rotational symmetry along the axis of the pore of the channel (as depicted in Mikoshiba et al., 1993), and the four monomers in the channel always adopt the same conformation, changing from one conformation to another concertedly. The InsP3R channel can change from one conformation with any number of ligands bound to its ligand-binding sites to another conformation with the same number of ligands bound. The equivalent ligand-binding sites of all the identical monomers in an InsP3R channel have the same affinity. Furthermore, whereas the affinities of the ligand-binding sites can differ in different conformations of the channel, they are not affected by the state of occupation of any other ligand-binding site (Monod et al., 1965; Changeux and Edelstein, 1998). The following MWC-based models were examined:
(a) MWC models in which the InsP3R tetramer can assume two conformations (one open and one closed), and each InsP3R monomer has two or more Ca2+-binding sties (at least one activating and one inhibitory);
(b) MWC-based models in which the InsP3R tetramer has three conformations (one open and two closed conformations, or two open and one closed conformations), and each InsP3R monomer has two Ca2+-binding sites;
(c) an MWC-based model in which the InsP3R tetramer has four conformations (two open and two closed conformations), and each InsP3R monomer has two Ca2+-binding sites;
(d) an MWC-based model in which the InsP3R tetramer has four conformations (two open and two closed conformations), and each InsP3R monomer has three Ca2+-binding sites;
(e) a variation of model (d) in which the InsP3R tetramer has two extra closed conformations.
Besides MWC-based models, we also examined allosteric models in which the constraints assumed in the MWC-based models were relaxed to various extents to allow more degrees of freedom to describe the gating behaviors of the InsP3R channel. In those non-MWC models we considered, the constraint that all the InsP3R monomers in the tetrameric channel change conformation concertedly is retained. However, the constraints that the equivalent ligand-binding sites of all the monomers in an InsP3R channel have the same affinity, and that the affinities of the ligand-binding sites are not affected by the state of occupation of any other ligand-binding site, are selectively relaxed. We examined the following non-MWC models:
(f) a “type I” non-MWC model—an allosteric model in which the affinity of the inhibitory Ca2+-binding site is affected by the binding status of the InsP3-binding site on the same InsP3R monomer—with the InsP3R tetramer having two conformations, and each InsP3R monomer having one activating Ca2+-binding site and one inhibitory Ca2+-binding site;
(g) a type I non-MWC model with the InsP3R tetramer having two conformations, and each InsP3R monomer having one activating and two inhibitory Ca2+-binding sites, with only one of the inhibitory Ca2+-binding sites affected by InsP3 binding;
(h) a “type II” non-MWC model—an allosteric model in which InsP3 binding to the InsP3-binding sites in the tetramer affects the affinities of all the inhibitory Ca2+-binding sites and InsP3-binding sites in the tetramer—with the InsP3R tetramer having two conformations, and each InsP3R monomer having one activating and one inhibitory Ca2+-binding sites;
(i) a type II non-MWC model with the InsP3R tetramer having two conformations, and each InsP3R monomer having one activating and two inhibitory Ca2+-binding sites;
(j) a variation of model (i) in which the InsP3R tetramer has three conformations.
In all the models considered, each InsP3R monomer has only one InsP3-binding site because of condition (ix). Detailed descriptions of all the models considered, mathematical derivation of analytical formulas to calculate the InsP3R channel P o at various [InsP3] and [Ca2+]i, the rationales for selecting those models to be studied and not considering other possible allosteric models, and comparisons of experimental InsP3R channel P o with those calculated according to the various models, are provided in either the (model (e)), or the online supplemental material section (all other models) available at http://www.jgp.org/cgi/content/full/jgp.200308809/DC1.
Basic Features of the Simplest Allosteric Model That Can Describe the Ligand Regulation of InsP3R Channel Activity
Among all the models considered, the simplest model, defined as the one involving the fewest number of free parameters (Jones, 1999), that can account for all our observations of the regulation by [Ca2+]i and [InsP3] of InsP3R channel activity, and can satisfy the constraints imposed by the structure of the InsP3R channel, is the MWC-based, four-plus-two-conformation model (model e above). This model postulates that the InsP3R monomers, and therefore the InsP3R tetrameric channel as a whole, can adopt six different conformations (Fig. 7) . The channel is open when it is in the A* and C* conformations. The B, D, A′, and C′ conformations are closed. The equilibria between A*, B, C*, and D conformations are dependent on InsP3 and Ca2+ binding to the channel, which confers regulation of channel activity by [InsP3] and [Ca2+]i. In contrast, the equilibrium A*↔A′ is not affected by [InsP3] or [Ca2+]i, i.e., the affinities of the InsP3 and Ca2+ sites of the InsP3R channel are the same in A* and A′. The ratio of the total durations an InsP3R channel spends in the A* conformation and in the A′ conformation is the same regardless of [InsP3] and [Ca2+]i. Thus, the A* and A′ conformations can be grouped together as the “active” A conformation (a conformation in which the channel can open, denoted by a green box in Fig. 7) when we consider the effects of InsP3 and Ca2+ on channel conformations. Similarly, C′ and C* are grouped together as the active C conformation (denoted by a green box with dashed border in Fig. 7) because the equilibrium C*↔C′ is likewise not affected by [InsP3] or [Ca2+]i. Thus, even in the presence of optimal [InsP3] and [Ca2+]i, when the InsP3R channel hardly exists in the closed B and D conformations, the maximum observed channel P o is <1 because the channel exists a fraction of the time in the closed A′ and C′ conformations. This accounts for the observation that the maximum InsP3R channel P o in saturating [InsP3] (10 μM) and optimal [Ca2+]i is only ∼0.8 (<1) (Mak et al., 1998). The model also postulates that each of the four InsP3R monomers has one InsP3-binding site (Q) and three different functional Ca2+-binding sites (F, G, and H) on the cytoplasmic side of the channel. Because of its tetrameric structure, an InsP3R channel can bind a maximum of four InsP3 molecules in its Q sites and four Ca2+ in each of the three types (F, G, and H) of Ca2+ sites. The affinities of these ligand-binding sites are different in the different channel conformations (A, B, C, and D). InsP3 and Ca2+ regulate channel activity because binding of InsP3 or Ca2+ to these sites will stabilize those conformations in which the sites have higher affinities, thereby affecting the equilibria among the A, B, C, and D conformations, as outlined in Monod et al. (1965).
Figure 7.
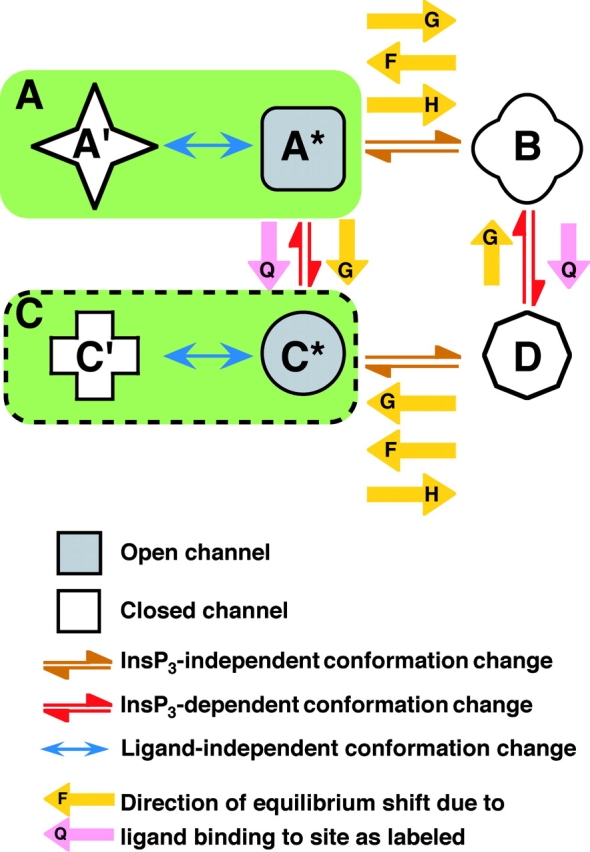
The MWC-based four-plus-two-conformation model for InsP3R channel gating. Only conformation transitions are represented in the schemes. Reactions involving binding of InsP3 and Ca2+ to the InsP3R channel and the state of occupation of the various ligand-binding sites of the channel are omitted from the schemes for clarity. The green boxes represent the grouping of the open A* and closed A′ conformations into the active A conformation, and the grouping of the C* and C′ conformations into the active C conformation.
An important feature of a MWC-based model of ligand regulation is that the effect of ligand binding on the equilibrium between two channel conformations is determined by the affinities of the site in the two conformations. At ligand concentrations << the lower dissociation constant of the site, there is not enough ligand binding to the site to shift the equilibrium position. At ligand concentrations >> the higher dissociation constant, the ligand binding site is saturated and ligand concentration is no longer relevant to the equilibrium position since the ligand will bind to the site regardless of what conformation the channel is in. Thus, the difference between the higher and lower dissociation constants of the site corresponds approximately to the range of ligand concentrations over which the effects of the site can be felt. Importantly, the magnitude of the difference between the two affinities of the site determines the full extent of the effect of the site, i.e., how much activation or inhibition the site produces between zero and saturating ligand concentrations. (Of course, this cannot be the case if the difference between the dissociation constants is so large that the equilibrium position is already totally shifted to the favorable conformation before the ligand concentration becomes >> higher dissociation constant.)
The mechanisms for Ca2+ and InsP3 regulation are mostly segregated in this model (see the for more detailed reasoning behind this assertion), allowing further reduction in the number of free parameters involved. This means that in our model, InsP3 binding to the Q sites only affects the equilibria A↔C, and B↔D (red double arrows in Fig. 7). In the absence of InsP3, the equilibria overwhelmingly favor the A and B conformations. InsP3 regulates the InsP3R channel solely by stabilizing the C conformation relative to the A conformation; and stabilizing the D conformation relative to the B conformation (indicated by the pink arrows in Fig. 7). Thus, in saturating [InsP3], the channel exists mostly in the C and D conformations. The equilibria A↔B and C↔D (brown double arrows in Fig. 7) are InsP3-independent, i.e., the affinities of the Q sites in A and B conformations are the same, and so are those of the Q sites in C and D conformations.
The F sites are responsible for the InsP3-independent Ca2+ activation of the channel. Ca2+ binding to the F sites only affects the InsP3-independent A↔B and C↔D equilibria (brown double arrows in Fig. 7), stabilizing the active A and C conformations (indicated by the yellow arrows in Fig. 7). The affinities of the F sites are the same in A and C conformations, and so are the affinities of those in B and D conformations. Thus, Ca2+ binding to the F sites does not affect the InsP3-dependent A↔C, or B↔D equilibria (red double arrows in Fig. 7).
The H sites are responsible for inhibition of the channel by high [Ca2+]i. The affinities of the H sites are the same in the A and C conformations and are the same in the B and D conformations. Thus, InsP3-induced shifts (pink arrows in Fig. 7) in the A↔C and B↔D equilibria (red double arrows in Fig. 7) do not affect Ca2+ binding to the H sites. Ca2+ binding to the H sites only affects the InsP3-independent equilibria A↔B and C↔D (brown double arrows in Fig. 7), stabilizing the closed B and D conformations relative to the active A and C conformations (indicated by the yellow arrows).
Regulation of the InsP3R by the G sites is more complex because the G sites have different affinities (Table I) in the four conformations (A, B, C, and D). The G sites in the closed B conformation have higher Ca2+ affinity than those in the active A conformation, so that the G sites are inhibitory Ca2+-binding sites (as indicated by the top yellow arrow in Fig. 7) in the A↔B equilibrium, which is the dominating equilibrium in the absence of InsP3. Most interestingly, however, the G sites in the active C conformation have higher Ca2+ affinity than those in the closed D conformation, so in the C↔D equilibrium, which is the dominating equilibrium under saturating [InsP3], the G sites are activating Ca2+-binding sites (as indicated by the yellow arrow in the lower half of Fig. 7). Between zero and saturating [InsP3], InsP3 binding to the channel shifts it from the A and B conformations toward the C and D channel. Thus, in subsaturating [InsP3], the “effective” dissociation constant of the G sites in the closed channel lies between those in the B and D conformations, according to the equilibrium position of the channel among the conformations as dictated by [InsP3]. Similarly, the “effective” dissociation constant of the G sites in the active conformations lies between those in the A and C conformations. As [InsP3] increases, not only do the G sites change from being inhibitory to activating, the difference between the effective affinities of the G sites in the closed and active channel also changes. As discussed earlier, the InsP3-induced change in the magnitude of the affinity difference of the G sites alters the full extent of the effect of the G sites on the channel, i.e., how much activation (or inhibition) the G sites produce between zero and saturating [Ca2+]i. It should be noted that since the F and H sites are both InsP3 independent, the G site is the only one modulated by InsP3 binding to the channel. Thus, all InsP3 regulation of the InsP3R stems from the effect of InsP3 binding on the properties of the G site.
TABLE I.
Parameters Used to Calculate the Po for InsP3R-1 and InsP3R-3 in Fig. 8 According to the MWC-based Four-Plus-Two-Conformation Model with Three Ca2+-binding Sites per InsP3R Monomer
| Parameters | InsP3R-1a | InsP3R-3 |
|---|---|---|
| L BA | 29.2 | 38.7 |
| L DB | 2.50 × 10−5 | 0.412 |
| L CA | 1.86 × 10−5 | 9.12 × 10−3 |
| K FA = K FC = K F1 | 223 nM | 1.01 nM |
| K FB = K FD = K F2 | 271 nM | 5.47 nM |
| K GA | >200 nMb | >100 nMb |
| K GB | 58.63 nM | 2.69 nM |
| K GC | 153 nM | 261 nM |
| K GD | 1.37 μM | 661 nM |
| K HA = K HC = K H1 | >1 mMb | >1 mMb |
| K HB = K HD = K H2 | 19 μM | 32 μM |
| K QA = K QB = K Q1 | >3 μMb | >3 μMb |
| K QC = K QD = K Q2 | 0.28 nM | 0.30 nM |
| R = [A*]/[A′] = [C*]/[C′] | 5.74 | 8.31 |
It should be noted that although the calculated channel P o derived from these sets of parameters agree reasonably well with experimental data, these sets of parameters may not be unique. Other sets of parameters that give good fits may exist in the huge parameter space.
The same set of parameters can be used to fit X-InsP3R-1 channel P o in ultra-low bath [Ca2+] except K H1 = K H2. As long as K H1 = K H2, Ca2+ binding to H sites will not stabilize one conformation relative to another and the H sites will no longer be functional.
These parameters can only be determined to be greater than the tabulated values because the calculated P o are not very sensitive to these parameters.
Since InsP3 binding to the channel affects Ca2+ binding to G sites, microreversibility dictates that Ca2+ binding to G sites should affect the InsP3-dependent equilibria A↔C and B↔D (as indicated by the vertical yellow arrows in Fig. 7). However, this effect is much weaker than the effect of InsP3 binding to the Q sites and so is not noticeable in our experiments.
Considering the InsP3-independent equilibria A↔B and C↔D, the affinities of the Ca2+-binding sites are in the order G ∼ F > H. For the C↔D equilibrium, Ca2+ will tend to bind first to the G sites and the F sites, both stabilizing the open C conformation, and then to the H sites, stabilizing the closed D conformation. For the A↔B equilibrium, as [Ca2+]i increases, Ca2+ will tend to first bind to the G sites, stabilizing the closed B conformation, and to the F sites, stabilizing the open A conformation. However, Ca2+ binding to the F sites cannot overcome the inhibitory effects of the G sites, so the channel remains mostly in the closed conformation. This is because the magnitude of the difference between the affinities of the G site in the closed B and active A conformations is greater than that of the F sites (Table I). Thus, the F site is less effective at activating the channel than the G site is at inhibiting it.
It should be noted that this molecular model does not take into consideration the kinetically abrupt termination of the InsP3R channel activities that causes the channel activities observed in our patch clamp experiments to disappear over time under constant [InsP3] and [Ca2+]i (Mak and Foskett, 1997). Therefore, it also does not account for any possible Ca2+ dependence of the termination of the channel activities (Boehning et al., 2001). Furthermore, this model does not consider other ligands that bind at or near the InsP3 binding site and activate channel gating, including the fungal metabolite adenophostin (Takahashi et al., 1994; Marchant et al., 1997; Mak et al., 2001c) and the neuronal CaBP1 protein (Yang et al., 2002). We have restricted our analyses to InsP3 because the dataset is much more extensive. To a first approximation, however, we believe that our conclusions regarding the effects of InsP3 can likely be generalized to these other ligands as well.
Agreement between the Selected Simplest Allosteric Model and Features of InsP3 and Ca2+ Regulation of InsP3R Channel Observed in Regular Bath Solution
We extended the mathematical treatment for the MWC allosteric model outlined in (Monod et al., 1965) to derive analytical equations to evaluate the channel P o in the presence of various [InsP3] and [Ca2+]i according to the MWC-based four-plus-two-conformation model (see for derivation of the equations). The theoretical channel P o values calculated from these equations (Fig. 8) with the optimized set of physical parameters (dissociation constants of the various ligand-binding sites in different channel conformations, and the equilibrium constants of the transitions between different channel conformations in the absence of any ligands, as listed in Table I) fit reasonably well the experimental channel P o observed in extensive nuclear patch-clamp studies for both types 1 and 3 isoforms (Mak et al., 1998, 2001b, 2003; and this study).
Figure 8.
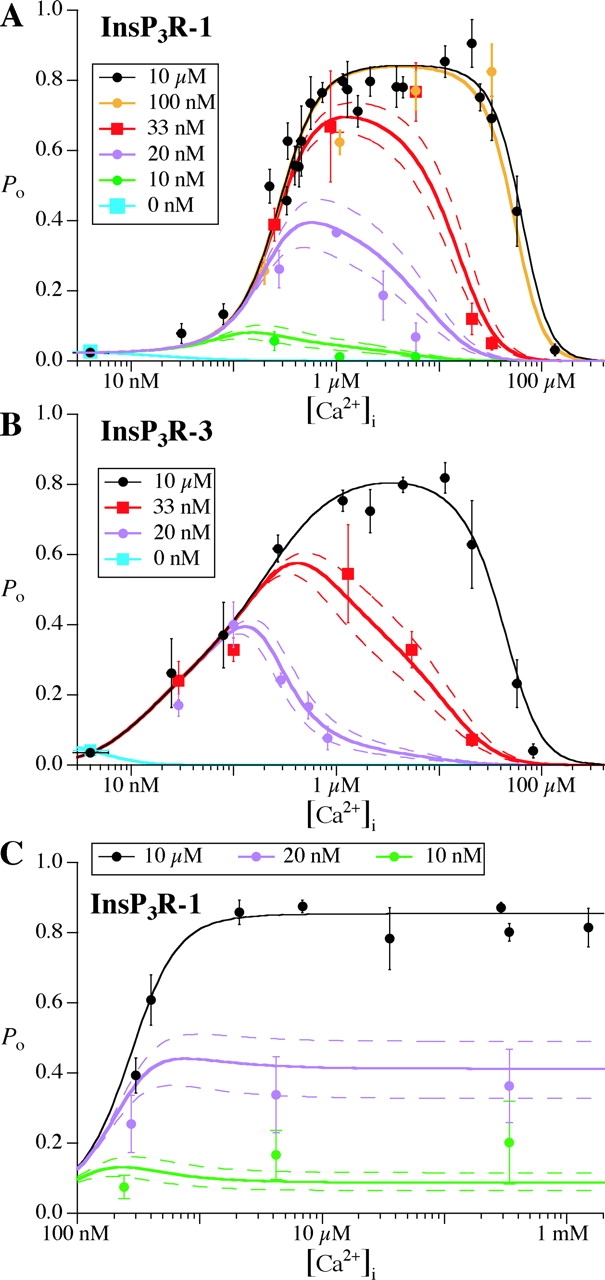
Fitting of the InsP3R channel P o in various [Ca2+]i and [InsP3] by the MWC-based four-plus-two-conformation model. (A) InsP3R-1 in regular bath (300 nM [Ca2+]), (B) InsP3R-3 in regular bath (300 nM [Ca2+]), and (C) InsP3R-1 in bath containing <5 nM Ca2+. The symbols represent the experimental P o in the tabulated [InsP3]. The continuous curves are the theoretical P o calculated from the model. The dashed curves indicate the range of calculated P o for ± 10% of the tabulated [InsP3]. Parameters used for the P o calculations are tabulated in Table I.
It should be noted that the agreement between theoretical and experimental channel P o is remarkable considering the multitude of distinctive features of ligand regulation of InsP3R channel activities the model had to account for, and the wide range of [InsP3] and [Ca2+]i examined for two distinct channel isoforms from two species.
Specifically, the model accounts for the following experimentally observed features.
The InsP3R Channel Can Be Active When None of its Ligand Binding Sites Is Occupied (Condition i)
The spontaneous InsP3R channel activities observed in this study are accounted for in the model as they are in the standard MWC model. In the absence of any ligand binding ([Ca2+]i < 5 nM and 0 InsP3), the channel is mostly in the closed B conformation. However, there is a nonzero probability for the channel to adopt the open A* conformation, giving rise to the spontaneous channel activities observed.
Why does raising the [Ca2+]i inhibit spontaneous opening? In the absence of InsP3, the channel exists overwhelmingly in the A and B conformations. In these conformations the Ca2+-binding G sites are inhibitory and they are more effective than the activating Ca2+-binding F sites, as discussed above. Therefore, no channel activity is observed at [Ca2+]i = 25 nM because, at that concentration, cytoplasmic Ca2+ will bind to the G sites and stabilize the closed B conformation strongly, thus inhibiting channel activity. Ca2+ binding to the activating F sites also occurs, but F site occupancy is insufficient to counter the inhibitory effect of the G sites.
InsP3 has No Effect on Ca2+ Activation Parameters (Kact and Hact) of the InsP3R Channel (Condition ii)
In our model, Ca2+ binding to the F sites activates the InsP3R channel by stabilizing the active A and C conformations relative to the closed B and D conformations. [InsP3] has no effect on this Ca2+ activation of channel activity because:
(1) the A↔B and C↔D (active↔closed) equilibria (brown double arrows in Fig. 7) that are driven by Ca2+ binding are InsP3 independent; and
(2) in the InsP3-dependent A↔C and B↔D equilibria (red double arrows in Fig. 7), Ca2+ affinities of the F sites are not affected by [InsP3] because the affinities of F sites are the same in the A and C conformations, and also the same in the B and D conformations (Table I).
Nevertheless, a novel insight emerges from our model: there is another, distinct contribution to Ca2+ activation that is indeed provided by InsP3. This InsP3-dependent contribution to the Ca2+ activation of the channel arises because InsP3 changes the G sites from inhibitory to activating. However, empirically, this effect is not clearly distinguishable from the activation of the channel by the F sites because of the similar affinities of the activating F and G sites in the C conformation of the InsP3R channel (i.e., K FC ≈ K GC in Table I). This can account for why just one set of Ca2+ activation parameters (K act and H act) in the empirical Hill equation was required to fit the experimental observations, and why those parameters exhibited no InsP3 dependence (Mak et al., 1998, 2001b).
Biphasic [Ca2+]i Regulation of InsP3R Channel Activity Is Observed at all [InsP3] (Condition iii)
A distinguishing feature of our allosteric model is that a third type of Ca2+-binding site, the H site, is postulated to exist. Besides the InsP3-independent activating F sites, and the G sites that are entirely responsible for the InsP3 dependence of the channel, inclusion of this novel InsP3-insensitive inhibitory site in the model was necessary to account for Ca2+ inhibition of the channel. The H sites are independent of [InsP3] because their affinities are the same in the B and D channel conformations, and in the A and C conformations (Table I).
In low [InsP3] at which the G sites are inhibitory, the activating F sites and the inhibitory G sites together produce the biphasic Ca2+ regulation observed. The inhibitory effect of the H sites is not observable. As [InsP3] increases, the G sites become activating. Then the InsP3-independent H sites are the only inhibitory Ca2+-binding sites. The activating F and G sites, together with the inhibitory H sites, produce the biphasic Ca2+dependence of the channel P o. Thus, the biphasic Ca2+ regulation is observed at all [InsP3].
Ca2+ Inhibition of InsP3R Channel Activity Is Sensitive to Small Changes in [InsP3] (Condition iv)
Several factors contribute to the exquisite sensitivity of the channel P o to small changes in [InsP3] at low [InsP3]. The affinity of the Q sites for InsP3 in the C and D channel conformations is extremely high (Table I) so that even at very low concentrations, InsP3 starts to bind to the channel. Furthermore, as [InsP3] increases, the strong binding of InsP3 to the Q sites rapidly shifts the equilibrium toward the C and D conformations. As discussed above, this shift changes the effective affinities of the G sites in the closed and active channel, thereby changing the nature of the G sites from inhibitory to activating. Consequently, the mechanism of Ca2+ inhibition of the channel changes from being mediated by Ca2+ binding to the high-affinity G sites to being mediated by Ca2+ binding to the low-affinity H sites. This switch results in a substantial change in the ability of Ca2+ to inhibit the channel. Consequently, the apparent half-maximal inhibitory [Ca2+]i (K inh) of the type 1 InsP3R changes >300-fold when [InsP3] increases just 10-fold (Mak et al., 1998), even though each InsP3R monomer has only one InsP3-binding site.
Response of InsP3R to InsP3 Saturates Rapidly and Abruptly by [InsP3] = 100 nM so That Higher [InsP3] Does Not Require Higher [Ca2+]i for Inhibition (Condition v)
The abrupt saturation of the response of the InsP3R to InsP3 cannot be due to saturation of the InsP3 binding site because the apparent affinity of Ca2+ to inhibit the type 1 InsP3R channel (K inh) is still highly sensitive to changes in [InsP3] near 100 nM where the response saturates. The G sites are activating at 100 nM InsP3, so the only mechanism available for Ca2+ inhibition of the channel is that mediated by Ca2+ binding to the H sites. The abrupt saturation of the response to InsP3 is due to the fact that properties of the H sites are InsP3 independent. Even as [InsP3] is further increased over three orders of magnitude, the same [Ca2+]i is required to inhibit the InsP3R channel (Mak et al., 1998).
The Maximum Channel Po Is Always ∼0.8 (Condition vi)
Even when the experimental conditions are optimized to bias the equilibria among the A, B, C, and D channel conformations in favor of the active conformations, the observed channel P o is still limited by the fact that the InsP3R channel in the active A (or C) conformation spends only a fraction of its time being open (in the A* or C* conformation), resulting in channel P max < 1. Furthermore, because the ligand-independent equilibria A*↔A′ and C*↔C′ have the same equilibrium constant in our model, the theoretical channel P max is not affected by any of the experimental conditions that may shift the equilibria among the A, B, C, and D conformations. This feature accounts for the observation that the channel P max remains the same in all experiments. It is possible that the ligand-independent conformation transitions A*↔A′ and C*↔C′ are controlled by a gating mechanism different from that controlling the ligand-dependent conformation transitions among A*, C*, B, and D.
The Mean Channel Open Duration <τo> Is Ligand Independent over a Wide Range of [Ca2+]i and [InsP3], Whereas the Mean Channel Closed Duration Is Ligand Dependent (Condition vii)
According to the model, an open channel in the A* or C* conformations can close either through a ligand-dependent transition into the B or D conformations, or through a ligand-independent transition into the A′ or C′ conformations (Fig. 7). The observed mean channel open duration <τo> is determined by the fastest one of the transitions out of the open A* and C* conformations. Our model postulates that the rates of the ligand-independent conformation transitions, A*↔A′ and C*↔C′, are substantially higher than the rates of the ligand-dependent transitions among the A*, C*, B, and D conformations under most [InsP3] and [Ca2+]i examined. Thus, once the channel opens into the A* (or C*) conformation from the B (or D) conformation, it undergoes many ligand-independent A*↔A′ (or C*↔C′) transitions before it closes via a ligand-dependent transition back to the B or D conformations. This limits the open channel duration. The rates of channel closing via the ligand-dependent transitions (A*→B, A*→D, C*→B and C*→D) become comparable to the ligand-independent transitions (A*→A′ and C*→C′) only in conditions ([Ca2+]i << K act, or [Ca2+]i >> K inh) when channel activity is significantly inhibited (P o < 0.1). Therefore, the observed <τo> of InsP3R channel remain within a narrow range even under various conditions of [Ca2+]i and [InsP3] in which the channel P o changes dramatically (Mak et al., 1998, 2001b,c). <τo> only decreases when channel activity is substantially inhibited (P o < 0.1), when one of the ligand-dependent channel-closing transitions becomes more frequent than the ligand-independent transitions.
We rejected the possibility that the channel conformations are connected such that A*↔A′↔B because in this case, the channel can only exit the open A* and C* conformations by entering the closed A′ and C′ conformations, respectively, through ligand-independent conformation transitions. In that case, <τo> would not be affected by [Ca2+]i or [InsP3] at all, contrary to observations.
On the other hand, a closed channel in the B and D conformations opens only through ligand-dependent transitions, whereas a closed channel in the A′ and C′ conformations opens only through ligand-independent transitions. The mean channel closed duration <τc>, the mean of the durations of the channel being in the B, D, A′, and C′ conformations, is dominated by the slowest of the channel opening transition rates, which is ligand dependent in our model. Hence, <τc> exhibits ligand dependence with a trend opposite to that of the channel P o, i.e., <τc> decreases as channel P o increases and vice versa, as observed in our experiments (Mak et al., 1998, 2001b).
Ligand Regulation of the InsP3R Channel after Exposure to Ultra-low Bath [Ca2+] Can be Accounted for by the MWC-based Four-Plus-Two-Conformation Allosteric Model
The MWC-based four-plus-two-conformation allosteric model postulates that the InsP3R channel has three types of regulatory Ca2+-binding sites that are mutually independent. The model predicts, therefore, that it could be theoretically possible, by mutagenesis or other experimental or physiological means, to specifically modify any one of the Ca2+-binding sites without affecting the other ligand binding sites. Furthermore, the model enables quantitative predictions to be made about the behavior of a channel with any specific Ca2+ site so modified. We therefore considered whether the novel InsP3R-1 channel behaviors observed following exposure of nuclei to an ultra-low bath [Ca2+] (Mak et al., 2003) could be predicted from our model by assuming that the experimental treatment specifically rendered the H site nonfunctional, because this site is responsible for high Ca2+ inhibition. In other words, we simply assumed that the only effect of exposure to ultra-low bath [Ca2+] is to make the affinities of the H sites the same in the A, B, C, and D conformations. Remarkably, the observed channel behaviors are well-predicted by this assumption.
First, with the inhibitory H sites rendered nonfunctional by exposure to ultra-low bath [Ca2+], the model predicts that the channel will exhibit no Ca2+ inhibition in [InsP3] that is high enough (≥10 nM), such that the combined effect of Ca2+ binding to the F and G sites is activating. Indeed, in all [InsP3] used (10 nM, 20 nM, and 10 μM), the channel P o observed in our experiments after the nuclei were exposed to ultra-low bath [Ca2+] increased as [Ca2+]i was raised from 100 nM to 2 μM due to the combined activating effect of the F and G sites. Then the channel P o remained at the same plateau value for all [Ca2+]i > 2 μM (up to 1.5 mM) with no detectable inhibition by [Ca2+]i (Mak et al., 2003). The model predicts this because with the F and G sites being activating and no functional H sites, there is no more Ca2+-binding sites in the InsP3R channel to generate any inhibitory effect.
Second, the model predicts that rendering the H sites nonfunctional by exposure to ultra-low bath [Ca2+] should not affect the function of the F and G sites because the Ca2+-binding sites are independent in our model. Thus, the model predicts that exposure to ultra-low bath [Ca2+] should have no effect on the Ca2+ activation properties of the channel in saturating [InsP3]. Indeed, in 10 μM [InsP3], Ca2+ activation (100 nM < [Ca2+]i < 1 μM) of the channel exposed to ultra-low bath [Ca2+] was very similar to that of channels exposed to regular bath [Ca2+] (400–500 nM) (Mak et al., 2003).
Third, the model predicts that even with the H sites nonfunctional, the InsP3R channel activity should nevertheless remain InsP3 dependent because the G sites remain inhibitory in the absence of InsP3. This is indeed what was observed. Even though the major apparent effect of InsP3 is to relieve high [Ca2+]i inhibition of the channel exposed to regular bath [Ca2+] (Mak et al., 1998), and exposure of the channel to the ultra-low bath [Ca2+] eliminates high [Ca2+]i inhibition in the presence of saturating InsP3, InsP3 nevertheless is still required to activate channel activity (Mak et al., 2003).
Fourth, the model predicts that even with the H site nonfunctional, and with the experimental conditions overwhelmingly favoring the channel being in the active C conformation, the channel will still exist for a ligand-independent fraction of time in the closed C′ conformation. Indeed, even after exposure to ultra-low bath [Ca2+], the channel still exhibited a P max of ∼0.8, (<1) in saturating [InsP3] and high [Ca2+]i (Mak et al., 2003).
More importantly, the model predicts that distinct and novel channel behavior should be observed in subsaturating [InsP3] after exposure to an ultra-low bath [Ca2+] renders the H sites nonfunctional. Specifically, in the absence of functional H sites, the model indicates that the effect of InsP3 on the channel should be manifested as a change in the maximum channel P o, a behavior distinguished from the behavior of the channel with the H site functional, where the effect of InsP3 is manifested as a change in the apparent K inh, with no effect on the parameter P max used in the biphasic Hill equation (Eq. 1). The observed behavior of the channel in various [InsP3] after exposure to an ultra-low bath [Ca2+] (Mak et al., 2003) is in very good agreement with this prediction. Understanding this novel behavior requires consideration of the effects of InsP3 on the properties of the G site. In subsaturating [InsP3], increases in [InsP3] shifts the channel toward the C and D conformations. This not only changes the nature of the G site from being inhibitory to activating, but also changes the difference between the effective affinities of the G site in the closed and active channel, thereby alters how much activation or inhibition the G site produces. At ∼10 nM, the equilibria of the X-InsP3R-1 is shifted sufficiently toward the C and D conformation that the G sites become activating (Fig. 8). Thus, as [Ca2+]i increases from 0.1 to 2 μM, Ca2+ binding to the F and G sites activates the channel and raises the channel P o (Fig. 8 C). However, the extent of this activation is limited because at 10 nM [InsP3], the difference between the effective affinities of the G sites in the closed and active channel is small. Thus, the channel P o is only increased to a maximum of 0.2 (Mak et al., 2003), substantially lower than P max ≈ 0.8. With no functional H sites, there is no Ca2+ inhibition so the channel P o remains at that maximum level even as [Ca2+]i increases. Further increases in [InsP3] further favor the C and D conformations, increasing the difference between the effective affinities of the G sites, thereby enhancing the extent of activation of the channel. This enhancement is manifested as an increase in the maximum P o the channel exhibits. Increases in [InsP3] continue to raise the maximum channel P o until it reaches 0.8—P max, which is dictated by the C′↔C* equilibrium.
Thus, with a single simple assumption that the exposure of the InsP3R channel to ultra-low bath [Ca2+] renders the H sites in the channel nonfunctional, the MWC-based, four-plus-two-conformation allosteric model can quantitatively account for the ligand regulation of the channel exposed to ultra-low bath [Ca2+] observed in (Mak et al., 2003), without involving any additional free parameters. This is significant, because the model we have developed here was devised to account for the regulation by [Ca2+]i and [InsP3] of the InsP3R channel in regular bath [Ca2+] (400–500 nM). The fact that it successfully quantitatively predicts independent and distinct experimental data (regulation by [Ca2+]i and [InsP3] of the channel after exposure to ultra-low bath [Ca2+]) provides strong support for its validity.
We would like to point out that in the extension of our model described above, we use our model to separately account for the behaviors of the InsP3R channel when it is exposed to regular Ca2+ bath, and when it has been exposed to low Ca2+ bath. Thus, we limit the description of the sensing mechanism that detects the exposure of the channel to ultra-low bath [Ca2+] to a qualitative one, as a switch that turns on and off the inhibition of channel gating mediated by the H sites, depending on the bath [Ca2+] the channel has been exposed to. We did not attempt to quantitatively incorporate the sensing mechanism into our model for the following reasons. First, our allosteric model is derived based on the behavior of the InsP3R channel in steady-state conditions. Thus, it cannot, in its present form, provide a quantitative description for the kinetic behavior of InsP3R channels in response to changes in [InsP3] and [Ca2+]i, including the time course of the disruption of high-[Ca2+]i inhibition of the channel after it was exposed to ultra-low bath [Ca2+], or the reversal of the disruption when the nucleus was returned to regular bath [Ca2+]. Second, because we do not know the physical location (cytoplasmic or lumenal) of the sensing mechanism in the InsP3R channel, we cannot be sure of the exact experimental conditions (lumenal or cytoplasmic free [Ca2+]) that trigger the disruption of the high [Ca2+]i inhibition of the channel. Trying to describe this Ca2+ sensing mechanism quantitatively will entail developing two alternative models, one for each possible scenario, which is premature at this point. Third, the application of our model to the understanding of the physiological regulation of InsP3R by [Ca2+]i and [InsP3], the main reason for developing the model, is not significantly limited by our qualitative description of the sensing mechanism. This is because at present, disruption of the high [Ca2+]i inhibition of InsP3R channels was only observed when the channels were exposed to a very low [Ca2+] (nM), in either cytoplasmic or lumenal sides. Neither of these cases is likely to occur under physiologically relevant situations. The model can be modified later to better incorporate the Ca2+ sensing mechanism when further information about the mechanism becomes available, and if physiological conditions are found to disrupt the high [Ca2+]i inhibition of InsP3R channel activity.
Conclusions
Examination of InsP3R channel activity (both Xenopus type 1 and rat type 3) in extremely low [Ca2+]i revealed that InsP3 is not necessary for InsP3R channel opening. Spontaneous InsP3R channel activity was observed because the inhibitory Ca2+-binding sites of the channel have a finite affinity even in the absence of InsP3 so that in [Ca2+]i < 5 nM, the inhibitory Ca2+-binding sites are not occupied and there is no Ca2+ inhibition of the channel. The observation of spontaneous, ligand-independent activity suggested that the Ca2+ and InsP3 regulation of the InsP3R channel could be described by an allosteric model for channel gating in which a channel that is not bound to Ca2+ or InsP3 nevertheless has a finite, nonzero, probability of adopting an open conformation. In contrast, all previous models have assumed that channel opening has a strict requirement for InsP3 binding. Thus, our modeling effort is the first one to incorporate this spontaneous activity into an allosteric model to describe the InsP3R channel. Furthermore, it is the first quantitative model that takes into consideration the tetrameric structure of the InsP3R channel, and thus addresses fully and quantitatively the cooperative nature of the activation and inhibition of InsP3R channel gating by [Ca2+]i, and the cooperative nature of InsP3R channel regulation by InsP3.
We examined various allosteric models to find one that could describe channel-gating characteristics observed in extensive electrophysiological studies of the InsP3R in native endoplasmic reticulum membrane. The MWC-based four-plus-two-conformation model with one InsP3- and three different Ca2+-binding sites in each InsP3R monomer in a tetrameric channel can account for the nine distinct observations that we explicitly defined, including the spontaneous activities observed here, for both the types 1 and 3 InsP3R, over a wide observed range of [Ca2+]i (∼3 nM to 200 μM) and [InsP3] (0 to 180 μM). This model can account for the experimental observations with the minimum number of free parameters (14), and is therefore considered most likely. Importantly, the model derived from these data can also account for independent observations regarding the lack of Ca2+ inhibition (up to 1.5 mM) of channel activity and the InsP3 regulation of the maximum channel P o exhibited by X-InsP3R-1 exposed to ultra-low bath [Ca2+] (< 5 nM) described in the preceding paper (Mak et al., 2003). Of note, it quantitatively did so, and without involving more parameters, by simply assuming that the exposure to ultra-low bath [Ca2+] specifically renders one of the Ca2+-binding sites nonfunctional. The ability of the model to predict this complex behavior strongly validates it, and suggests that it will be useful for interpreting the molecular basis for other channel behaviors observed in future studies.
The model provides insights into the possible molecular mechanisms that enable the InsP3R channel to be so precisely regulated by InsP3 and Ca2+. It has remained difficult to understand how the InsP3R channel can be regulated so exquisitely by InsP3. Small changes in [InsP3] over a narrow range (10–100 nM) cause the apparent K inh to change by over 2 orders of magnitude (from 160 nM to 60 μM), even though it is well established that there are only four InsP3-binding sites in each InsP3R tetrameric channel. Furthermore, the mechanisms that can account for the total saturation of the channel response to [InsP3] once [InsP3] goes beyond 100 nM have also been unclear. Insights into these properties of InsP3R regulation are highly relevant for understanding the mechanisms that generate rapid and well-controlled Ca2+ signals in cells. These properties can now be accounted for in our model, by positing three different functional Ca2+-binding sites in each InsP3R monomer that directly affect the equilibria among active and closed conformations of the channel. One of these sites is activating, whereas another is inhibitory, but both are independent of InsP3. In contrast, a third Ca2+-binding site is affected by InsP3, being inhibitory in the absence of InsP3 but becoming activating as [InsP3] increases. All previous models of Ca2+ regulation of InsP3R function have assumed that each channel monomer possessed a single inhibitory Ca2+-binding site, including our previous empirical description of the effects of InsP3 on channel gating (Mak et al., 1998, 2001b). Our previous description assumed a single inhibitory Ca2+binding site whose apparent affinity was allosterically reduced by InsP3 binding. The model derived here now suggests that two Ca2+-binding sites present in each monomer contribute to Ca2+ inhibition. Ca2+ binding to an InsP3-dependent G site inhibits InsP3R activity in the absence of InsP3, which is responsible for the lack of channel activity in the absence of InsP3 in normal [Ca2+]i. InsP3 binding to the channel changes the effective affinities of this site, and in so doing transforms it into an activating site. This InsP3-mediated transformation of the nature of this Ca2+-binding site is responsible for all the InsP3-dependence of the channel, accounting for the extremely high sensitivity of Ca2+ inhibition of InsP3R channel gating to small changes in [InsP3]. The second Ca2+-binding site (H site) is strictly inhibitory with a lower Ca2+ affinity (10–30 μM) that is not modulated by InsP3 binding. The InsP3 independence of this site is responsible for the lack of further effect of InsP3 on the channel once [InsP3] > 100 nM, accounting for the observation that the effects of InsP3 abruptly saturate around this concentration. Furthermore, Ca2+ binding to the H site is responsible for the observed inhibition of the channel even at lower [Ca2+]i (<10–30 μM), when [InsP3] is <100 nM. Whereas the properties of the H sites are insensitive to InsP3 binding, they are rendered nonfunctional by a nonphysiological protocol: exposure to an ultra-low bath [Ca2+]. Nevertheless, the channel exposed to ultra-low bath [Ca2+] remains dependent on InsP3 because the other Ca2+-binding sites, specifically the InsP3-dependent G site, are not affected by the low bath [Ca2+]. Ca2+ binding to the G site inhibits InsP3R activity in the absence of InsP3.
Our molecular model suggests that not all conformation transitions of the InsP3R that affect the channel opening are regulated by InsP3 and [Ca2+]i. In our model, P max of the InsP3R channel is limited to ∼0.8 (<1) by conformation transitions that affect channel opening but are independent of [Ca2+]i and [InsP3]. This can account for the observed constancy of the mean InsP3R channel open durations over a wide range of [Ca2+]i and [InsP3]. Such conformation transitions probably arise from a channel gating mechanism different from the one regulated by ligands (InsP3 and Ca2+).
A critical insight that has emerged from analysis of the behavior of the model is that the major effect of InsP3 in regulating the activity of the InsP3R channel is to tune the nature of the G sites. In contrast, we previously interpreted the effect of InsP3 as tuning the sensitivity of the channel to Ca2+ inhibition (Mak et al., 1998, 2001b). How can we reconcile the empirical observation that K inh is tuned by [InsP3] with this insight from the model? Normally, the functional H site has a dissociation constant in the closed B and D conformations of 20–30 μM (Table I). However, the inhibitory effect of the H sites is not only manifested at such high [Ca2+]i. The [Ca2+]i at which H site-mediated Ca2+ inhibition is manifested depends on the properties of the G sites. In low [InsP3] at which the G sites have just become activating, the difference between the effective affinities of the G sites in the closed and active channel is small so that the extent of G site-mediated activation is limited. On the other hand, there is finite Ca2+ binding to the H site even at [Ca2+]i << K HD (200–300 nM), which strongly stabilizes the closed conformations. This inhibitory effect can be sufficient to counter the activating effect of the F and G sites. Therefore, in the presence of low [InsP3] with weak G site activation, the H site inhibition is manifested even at low [Ca2+]i. This results in a narrow bell-shape dependence of channel P o on [Ca2+]i with the channel achieving a low maximum P o, as observed (Mak et al., 1998). As [InsP3] increases, G site activation is enhanced, so Ca2+ binding to the G sites stabilizes the active conformations more strongly. The inhibitory effect of H-site binding is then only manifested at higher [Ca2+]i. This generates a wider bell-shape dependence of channel P o on [Ca2+]i with the channel exhibiting a higher maximum P o centered at higher [Ca2+]i.
The maximum channel P o reaches 0.8 (P max) at [InsP3] ∼30 nM. Further increases in [InsP3] beyond this cause no further increase in the maximum channel P o. However, higher [InsP3] continues to shift the channel equilibria toward the C and D conformations, resulting in stronger G site activation. This delays the onset of observable H site–mediated inhibition to even higher [Ca2+]i, broadening the biphasic dependence of channel P o on [Ca2+]i into a plateau shape (Mak et al., 1998). In this manner, InsP3 tuning of the extent of G site activation is empirically manifested as an apparent InsP3-dependent shift in the ability of Ca2+ to inhibit the channel.
This model developed here will be useful in guiding future experimental investigations as well as providing insights for understanding existing InsP3R channel data. First, it will be important for providing a quantitative framework for understanding the roles of other channel regulators. For example, insights into the mechanisms of Mg2+ effects on BK channels were greatly facilitated by having available the previously developed complex allosteric schemes that account for Ca2+ and voltage regulation of the channel (see Magleby, 2001). In the case of InsP3R, the allosteric model may provide a framework for modeling the effect of ATP, phosphorylation, and other modulators on channel gating. Second, use of the model will be important as mutagenesis is applied to this channel in attempts to discover the molecular bases for ligand regulation. Because effects of mutagenesis may be allosterically coupled to the ligand-binding sites through long-range effects, the model will be important for analyzing mutant channel behavior to discriminate mutations that are truly at the binding sites from those that are allosterically coupled to the binding sites. For example, if a mutation is observed to change the properties of InsP3 activation of channel gating, the target of the mutation could be the InsP3-binding Q site itself, but our model suggests that the mutation could possibly modify the InsP3-dependent, Ca2+-binding G sites instead. With our model, changes in channel behavior resulting from any kind of modulation of the properties of the ligand-binding sites can now be interpreted within the context of the model to make inferences regarding the molecular mechanisms involved, as we have done in our analysis of the effects of ultra-low bath [Ca2+] exposure. For instance, experimental or physiological modulation of the affinity of the G sites in just the C conformation of the channel can affect all the parameters (P max, K act, H act, K inh, H inh) in the empirical Hill equation (Eq. 1). Without a molecular model, it would be extremely difficult to understand the underlying mechanisms just from the effects of the modulation. Indeed, a study of the effects of a point mutation on the gating of the type 1 channel (Tu et al., 2003) was limited to phenomenological description because of a lack of a model by which to quantitatively account for the results. Third, although our model is a general one based on observations made under steady-state conditions, with all the ligand-binding reactions assumed to be possible in any sequential order, it can nevertheless incorporate sequential binding models in which certain ligand binding sequences are “forbidden”, like that proposed in (Marchant and Taylor, 1997; Adkins and Taylor, 1999) in which Ca2+ cannot bind to the activating sites before InsP3 binds to the InsP3-binding sites. Our model can incorporate sequential schemes because it only explicitly involves the equilibrium constants of ligand binding and conformation transitions. Thus, “forbidden” ligand binding sequences can be incorporated simply by assuming that certain ligand binding and conformation transitions have much slower reaction rates than other reactions in the ligand binding scheme. The model may also be useful in predicting which transitions are “forbidden”. Fourth, when transient kinetic responses of channels are measured in response to ligand concentration changes, our model with its specified equilibrium constants may help to constrain the set of possible schemes and values of reaction rate constants that need to be considered. Fifth, application of the model to datasets obtained from various InsP3R isoforms may prove useful in identifying the properties that distinguish them and account for any observed distinct behaviors. For example, the type 2 InsP3R channel was reported to be distinct from the type 1 channel in its relative lack of high Ca2+ inhibition (Ramos-Franco et al., 2000). Our model suggests that this difference could be accounted for by a less effective inhibitory H site in the type 2 channel.
Finally, besides its application to enhance our understanding of the regulation of InsP3R channel gating, the modeling effort described here has extended substantially the basic MWC model upon which it is based. Our systematic mathematical treatment of not only the MWC-based four-plus-two-conformation model, but also of the other MWC-based and non-MWC models (presented in the online supplemental material section) may be useful in future modeling of other allosteric processes involving multiple ligands.
Supplemental Material
Acknowledgments
We would like to thank Dr. F.T. Horrigan (University of Pennsylvania) and Dr. S. Ponce-Dawson (University of Buenos Aires, Argentina) for reviewing our manuscript before submission and giving us many helpful comments.
This work was supported by grants to J.K. Foskett from the NIH (MH59937, GM56328) and to D.-O.D. Mak from the American Heart Association (9906220U).
Olaf S. Andersen served as editor.
APPENDIX
MWC-based Four-Plus-Two-Conformation Model—Model e
In this allosteric model, the homotetrameric InsP3R channel can assume six conformations: four (A*, B, C*, and D) that are connected by ligand dependent transitions, plus two (A′ and C′) that are connected to the others by ligand independent transitions (hence the name). The channel is open in two conformations (A* and C*), and closed in the others (B, D, A′ and C′). However, since the transitions A*↔A′ and C*↔C′ are ligand independent, we consider the conformations A* and A′ as one active conformation A; and C* and C′ as one active conformation C when we examine the regulation of InsP3R channel activity by ligands InsP3 and Ca2+ (Fig. 7).
Each InsP3R monomer has three Ca2+-binding sites (F, G, and H) and one InsP3-binding site (Q). The ligand-binding status of an InsP3R tetrameric channel can be represented using the convention in which q hAf g represents the channel in the A conformation with f Ca2+ bound to the F sites, g Ca2+ bound to the G sites, h Ca2+ bound to the H sites, and q InsP3 molecules bound to the Q sites (0 ≤ f, g, h, q ≤ 4).
Based on the simplifications assumed in the MWC model (Monod et al., 1965), all the F sites in an InsP3R channel in conformation A have the same dissociation constant for Ca2+ binding (represented as K FA in this discussion), regardless of the ligand-binding status of the channel. Other dissociation constants are represented similarly, like K GA and K QC. After the derivation in Monod et al. (1965), these dissociation constants (a total of 16 for 4 sites in 4 conformations) together with the three independent equilibrium constants (L BA, L CA, and L DB) for conformation transitions between unliganded channels (0 0A0 0↔0 0B0 0, 0 0A0 0↔0 0C0 0, and 0 0B0 0↔0 0D0 0respectively) constitute the full set of parameters that completely describes the regulation by InsP3 and Ca2+ of the conformation changes of the InsP3R channel. Using the symbol conventions described above, the concentration of the InsP3R channel in any specific ligand-binding state, q hAf g, can be expressed in terms of [InsP3], [Ca2+]i, 0 0A0 0, and the set of parameters:
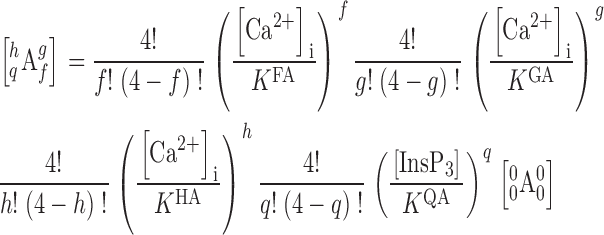 |
(5) |
Similar equations can be derived to express ligand-binding states of other conformations.
For our model of ligand regulation of InsP3R gating, we postulate that [Ca2+]i mainly regulates the equilibrium A↔B and C↔D. Furthermore, we postulate that F is an activating Ca2+-binding site, whereas H is an inhibitory Ca2+-binding site. This means that K FA and K FC < K FB and K FD, so that Ca2+ binding to the activating F sites stabilizes the active conformations. In contrast, K HA and K HC > K HB and K HD, so that Ca2+ binding to the inhibitory H sites stabilizes the closed conformations.
On the other hand, [InsP3] regulates the A↔C and B↔D equilibria, modulating channel P o by stabilizing the C and D channel conformations relative to the A and B conformations (K QA and K QB >> K QC and K QD).
The observed independence of the Ca2+ activation of the channel on [InsP3] (condition ii) constrains the number of free parameters involved in this model. Because K FA ≠ K FB and K FC ≠ K FD, the InsP3 affinity of the Q sites must be the same for the A and B conformations (K QA = K QB), and for the C and D conformations (K QC = K QD) so that InsP3 binding to the channel does not affect the equilibria A↔B and C↔D. This prevents InsP3 binding to the channel from shifting the equilibria A↔B, or C↔D, thus leaving Ca2+ activation of the channel unaffected by [InsP3]. To emphasize this constraint, we define K Q1 = K QA = K QB, and K Q2 = K QC = K QD. Furthermore, because InsP3 binding to the Q sites does affect the equilibria A↔C and B↔D (that is how InsP3 regulates the channel), the affinity of the F sites must be the same in the A and C conformations (K FA = K FC) and the same in the B and D conformations (K FB = K FD), so that shifts in the equilibria A↔C and B↔D due to InsP3 binding will not affect Ca2+ activation either. Again, to emphasize the constraint on these dissociation constants, we define K F1 = K FA = K FC and K F2 = K FB = K FD.
Even though Ca2+ inhibition of InsP3R is very sensitive to change in [InsP3] from 10 to 100 nM, Ca2+ inhibition does not change any more once [InsP3] reaches 100 nM despite a further three orders of magnitude increase in [InsP3] (from 100 nM to 180 μM). Thus, higher [InsP3] beyond 100 nM does not require higher [Ca2+]i for inhibition (condition v). This indicates that Ca2+ binding to the H sites is not affected by [InsP3]. Therefore, much like the relation between the dissociation constants of the F sites (K FA = K FC = K F1 and K FB = K FD = K F2), the dissociation constants of the H sites in the A and C channel conformations are the same (K HA = K HC = K H1), and those in the B and D conformations are the same (K HB = K HD = K H2).
The concentrations of the InsP3R channel in one conformation regardless of its ligand-binding status is shown in Eq. 6.
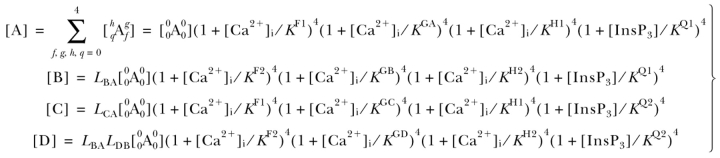 |
(6) |
The open probability P o of a single InsP3R channel is the fraction of time the channel spends in the open A* and C* conformations. In a stationary system, this is the same as the relative abundance of channels that adopt the open conformations in an ensemble of many (ideally infinite) channels, i.e.,
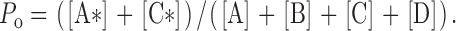 |
(7) |
Whereas the equilibria between the B, D, A*, and C* conformations are ligand dependent, the equilibrium constants R A and R C for the transitions A*↔A′ and C*↔C′, respectively, are independent of [InsP3] and [Ca2+]i. Because the experimental parameter P max is not affected by [InsP3] (Mak et al., 1998, 2001b), R A = R C = R. Thus,
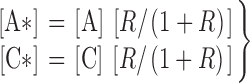 |
(8) |
This means that the channel is open for only a fraction [R/(1 + R)] of the time it is in the A or C conformation. This allows the model an extra degree of freedom to fit the experimentally observed channel P max, found to be <1 (condition ix). Together, Eqs. 6–8 describe the channel P o under all [Ca2+]i and [InsP3] with 14 free parameters (Table I).
The online version of this paper contains supplemental material.
Abbreviations used in this paper: InsP3, inositol 1,4,5-trisphosphate; InsP3R, InsP3 receptor; X-InsP3R-1, Xenopus type 1 InsP3R; r-InsP3R-3, rat type 3 InsP3R; P o, open probability.
References
- Adkins, C.E., and C.W. Taylor. 1999. Lateral inhibition of inositol 1,4,5-trisphosphate receptors by cytosolic Ca2+. Curr. Biol. 9:1115–1118. [DOI] [PubMed] [Google Scholar]
- Baudet, S.B., L. Hove-Madsen, and D.M. Bers. 1994. How to make and use calcium-specific mini- and microelectrodes. A Practical Guide to the Study of Calcium in Living Cells. 1st ed. R. Nuccitelli, editor. Academic Press, San Diego. 94–114.
- Berridge, M.J. 1993. Inositol trisphosphate and calcium signalling. Nature. 361:315–325. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny, I., J. Watras, and B.E. Ehrlich. 1991. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 351:751–754. [DOI] [PubMed] [Google Scholar]
- Boehning, D., S.K. Joseph, D.-O.D. Mak, and J.K. Foskett. 2001. Single-channel recordings of recombinant inositol trisphosphate receptors in mammalian nuclear envelope. Biophys. J. 81:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux, J.P., and S.J. Edelstein. 1998. Allosteric receptors after 30 years. Neuron. 21:959–980. [DOI] [PubMed] [Google Scholar]
- Cullen, P.J., J.G. Comerford, and A.P. Dawson. 1988. Heparin inhibits the inositol 1,4,5-trisphosphate-induced Ca2+ release from rat liver microsomes. FEBS Lett. 228:57–59. [DOI] [PubMed] [Google Scholar]
- De Young, G.W., and J. Keizer. 1992. A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. USA. 89:9895–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino, M. 1990. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca2+ release in smooth muscle cells of the guinea pig taenia caeci. J. Gen. Physiol. 95:1103–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, M.B. 1984. Spontaneous openings of the acetylcholine receptor channel. Proc. Natl. Acad. Sci. USA. 81:3901–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Q., D.-O.D. Mak, S. Devidas, E.M. Schwiebert, A. Bragin, Y. Zhang, W.R. Skach, W.B. Guggino, J.K. Foskett, and J.F. Engelhardt. 1998. Cystic fibrosis transmembrane conductance regulator-associated ATP release is controlled by a chloride sensor. J. Cell Biol. 143:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S.W. 1999. Commentary: a plausible model. J. Gen. Physiol. 114:271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph, S.K., C. Lin, S. Pierson, A.P. Thomas, and A.R. Maranto. 1995. Heteroligomers of type-I and type-III inositol trisphosphate receptors in WB rat liver epithelial cells. J. Biol. Chem. 270:23310–23316. [DOI] [PubMed] [Google Scholar]
- Kaftan, E.J., B.E. Ehrlich, and J. Watras. 1997. Inositol 1,4,5-trisphosphate (InsP3) and calcium interact to increase the dynamic range of InsP3 receptor-dependent calcium signaling. J. Gen. Physiol. 110:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, D.T., G.R. Tibbs, P. Paoletti, and S.A. Siegelbaum. 1998. Constraining ligand-binding site stoichiometry suggests that a cyclic nucleotide-gated channel is composed of two functional dimers. Neuron. 21:235–248. [DOI] [PubMed] [Google Scholar]
- Magleby, K.L. 2001. Kinetic gating mechanisms for BK channels: when complexity leads to simplicity. J. Gen. Physiol. 118:583–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., and J.K. Foskett. 1994. Single-channel inositol 1,4,5-trisphosphate receptor currents revealed by patch clamp of isolated Xenopus oocyte nuclei. J. Biol. Chem. 269:29375–29378. [PubMed] [Google Scholar]
- Mak, D.-O.D., and J.K. Foskett. 1997. Single-channel kinetics, inactivation, and spatial distribution of inositol trisphosphate (IP3) receptors in Xenopus oocyte nucleus. J. Gen. Physiol. 109:571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., and J.K. Foskett. 1998. Effects of divalent cations on single-channel conduction properties of Xenopus IP3 receptor. Am. J. Physiol. 275:C179–C188. [DOI] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 1998. Inositol 1,4,5-trisphosphate activation of inositol trisphosphate receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc. Natl. Acad. Sci. USA. 95:15821–15825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 1999. ATP regulation of type 1 inositol 1,4,5-trisphosphate receptor channel gating by allosteric tuning of Ca2+ activation. J. Biol. Chem. 274:22231–22237. [DOI] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, V. Raghuram, Y. Yue, S.K. Joseph, and J.K. Foskett. 2000. Single-channel properties in endoplasmic reticulum membrane of recombinant type 3 inositol trisphosphate receptor. J. Gen. Physiol. 115:241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 2001. a. ATP regulation of recombinant type 3 inositol 1,4,5-trisphosphate receptor gating. J. Gen. Physiol. 117:447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 2001. b. Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 InsP3 receptors. J. Gen. Physiol. 117:435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 2001. c. ATP-dependent adenophostin activation of inositol 1,4,5-trisphosphate receptor channel gating: kinetic implications for the durations of calcium puffs in cells. J. Gen. Physiol. 117:299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, N.B. Petrenko, and J.K. Foskett. 2003. Novel regulation of calcium inhibition of the inositol 1,4,5-trisphosphate receptor calcium-release channel. J. Gen. Physiol. 122:583–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant, J.S., M.D. Beecroft, A.M. Riley, D.J. Jenkins, R.D. Marwood, C.W. Taylor, and B.V. Potter. 1997. Disaccharide polyphosphates based upon adenophostin A activate hepatic D-myo-inositol 1,4,5-trisphosphate receptors. Biochemistry. 36:12780–12790. [DOI] [PubMed] [Google Scholar]
- Marchant, J.S., and C.W. Taylor. 1997. Cooperative activation of IP3 receptors by sequential binding of IP3 and Ca2+ safeguards against spontaneous activity. Curr. Biol. 7:510–518. [DOI] [PubMed] [Google Scholar]
- Meyer, T., D. Holowka, and L. Stryer. 1988. Highly cooperative opening of calcium channels by inositol 1,4,5-trisphosphate. Science. 240:653–655. [DOI] [PubMed] [Google Scholar]
- Meyer, T., and L. Stryer. 1991. Calcium spiking. Annu. Rev. Biophys. Biophys. Chem. 20:153–174. [DOI] [PubMed] [Google Scholar]
- Mikoshiba, K., T. Furuichi, A. Miyawaki, S. Yoshikawa, T. Nakagawa, N. Yamada, Y. Hamanaka, I. Fujino, T. Michikawa, Y. Ryo, et al. 1993. Inositol trisphosphate receptor and Ca2+ signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 340:345–349. [DOI] [PubMed] [Google Scholar]
- Monod, J., J. Wyman, and J.-P. Changeux. 1965. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12:88–118. [DOI] [PubMed] [Google Scholar]
- Moraru, I.I., E.J. Kaftan, B.E. Ehrlich, and J. Watras. 1999. Regulation of type 1 inositol 1,4,5-trisphosphate-gated calcium channels by InsP3 and calcium - Simulation of single channel kinetics based on ligand binding and electrophysiological analysis. J. Gen. Physiol. 113:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, S., S.K. Joseph, and A.P. Thomas. 1999. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 25:247–264. [DOI] [PubMed] [Google Scholar]
- Picones, A., and J.I. Korenbrot. 1995. Spontaneous, ligand-independent activity of the cGMP-gated ion channels in cone photoreceptors of fish. J. Physiol. 485:699–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Franco, J., D. Bare, S. Caenepeel, A. Nani, M. Fill, and G. Mignery. 2000. Single-channel function of recombinant type 2 inositol 1,4, 5- trisphosphate receptor. Biophys. J. 79:1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, M.J., and S.E. Gordon. 2000. Cooperativity and cooperation in cyclic nucleotide-gated ion channels. Biochemistry. 39:14003–14011. [DOI] [PubMed] [Google Scholar]
- Sienaert, I., H. De Smedt, J.B. Parys, L. Missiaen, S. Vanlingen, H. Sipma, and R. Casteels. 1996. Characterization of a cytosolic and a luminal Ca2+ binding site in the type I inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 271:27005–27012. [DOI] [PubMed] [Google Scholar]
- Sienaert, I., L. Missiaen, H. De Smedt, J.B. Parys, H. Sipma, and R. Casteels. 1997. Molecular and functional evidence for multiple Ca2+-binding domains in the type 1 inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 272:25899–25906. [DOI] [PubMed] [Google Scholar]
- Swillens, S., L. Combettes, and P. Champeil. 1994. Transient inositol 1,4,5-trisphosphate-induced Ca2+ release: A model based on regulatory Ca2+-binding sites along the permeation pathway. Proc. Natl. Acad. Sci. USA. 91:10074–10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillens, S., P. Champeil, L. Combettes, and G. Dupont. 1998. Stochastic simulation of a single inositol 1,4,5-trisphosphate-sensitive Ca2+ channel reveals repetitive openings during ‘blip-like’ Ca2+ transients. Cell Calcium. 23:291–302. [DOI] [PubMed] [Google Scholar]
- Takahashi, M., K. Tanzawa, and S. Takahashi. 1994. Adenophostins, newly discovered metabolites of Penicillium brevicompactum, act as potent agonists of the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 269:369–372. [PubMed] [Google Scholar]
- Toescu, E.C. 1995. Temporal and spatial heterogeneities of Ca2+ signaling: Mechanisms and physiological roles. Am. J. Physiol. 269:G173–G185. [DOI] [PubMed] [Google Scholar]
- Tu, H., E. Nosyreva, T. Miyakawa, Z. Wang, A. Mizushima, M. Iino, and I. Bezprozvanny. 2003. Functional and biochemical analysis of the type 1 inositol (1,4,5)-trisphosphate receptor calcium sensor. Biophys. J. 85:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley, P.F., J.M. Baraban, S. Supattapone, V.S. Wilson, and S.H. Snyder. 1987. Characterization of inositol trisphosphate receptor binding in brain. Regulation by pH and calcium. J. Biol. Chem. 262:12132–12136. [PubMed] [Google Scholar]
- Yang, J., S. McBride, D.O. Mak, N. Vardi, K. Palczewski, F. Haeseleer, and J.K. Foskett. 2002. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc. Natl. Acad. Sci. USA. 99:7711–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa, F., M. Morita, T. Monkawa, T. Michikawa, T. Furuichi, and K. Mikoshiba. 1996. Mutational analysis of the ligand binding site of the inositol 1,4,5- trisphosphate receptor. J. Biol. Chem. 271:18277–18284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.