

Abstract
Inositol 1,4,5-tris-phosphate (IP3) binding to its receptors (IP3R) in the endoplasmic reticulum (ER) activates Ca2+ release from the ER lumen to the cytoplasm, generating complex cytoplasmic Ca2+ concentration signals including temporal oscillations and propagating waves. IP3-mediated Ca2+ release is also controlled by cytoplasmic Ca2+ concentration with both positive and negative feedback. Single-channel properties of the IP3R in its native ER membrane were investigated by patch clamp electrophysiology of isolated Xenopus oocyte nuclei to determine the dependencies of IP3R on cytoplasmic Ca2+ and IP3 concentrations under rigorously defined conditions. Instead of the expected narrow bell-shaped cytoplasmic free Ca2+ concentration ([Ca2+]i) response centered at ≈300 nM–1 μM, the open probability remained elevated (≈0.8) in the presence of saturating levels (10 μM) of IP3, even as [Ca2+]i was raised to high concentrations, displaying two distinct types of functional Ca2+ binding sites: activating sites with half-maximal activating [Ca2+]i (Kact) of 210 nM and Hill coefficient (Hact) ≈2; and inhibitory sites with half-maximal inhibitory [Ca2+]i (Kinh) of 54 μM and Hill coefficient (Hinh) ≈4. Lowering IP3 concentration was without effect on Ca2+ activation parameters or Hinh, but decreased Kinh with a functional half-maximal activating IP3 concentration (KIP3) of 50 nM and Hill coefficient (HIP3) of 4 for IP3. These results demonstrate that Ca2+ is a true receptor agonist, whereas the sole function of IP3 is to relieve Ca2+ inhibition of IP3R. Allosteric tuning of Ca2+ inhibition by IP3 enables the individual IP3R Ca2+ channel to respond in a graded fashion, which has implications for localized and global cytoplasmic Ca2+ concentration signaling and quantal Ca2+ release.
Modulation of cytoplasmic free Ca2+ concentration ([Ca2+]i) is involved in the regulation of numerous cell physiological processes in which the second messenger inositol 1,4,5-tris-phosphate (IP3) plays a central role in most cell types (1). Binding of extracellular ligands to G protein- or tyrosine kinase-linked receptors in the plasma membrane activates phospholipase C to generate IP3, which binds to its receptors (IP3R) in the endoplasmic reticulum (ER), activating them as Ca2+ channels to release stored Ca2+ from the ER lumen into the cytoplasm. The complex control of [Ca2+]i is manifested temporally as repetitive spikes or oscillations (with frequencies often tuned to the level of stimulation) and spatially as propagating waves (1–3) and displays “adaptation” and “quantal release”, which are poorly understood (4, 5). IP3-mediated Ca2+ release is regulated by [Ca2+]i, with positive and negative feedback acting in a bell-shaped manner with peak Ca2+ release activity at ≈300 nM Ca2+ (6, 7); however, the mechanisms underlying this biphasic response—a fundamental component of [Ca2+]i oscillations and wave propagation models (1, 3, 8)—are still poorly defined. The intracellular location of the IP3R channel has necessitated the use of indirect measurements to infer its activity and has restricted studies of its single-channel properties (9–11). For this report, we systematically investigated the dependencies of single IP3R channels on [Ca2+]i and IP3 concentration under rigorously defined conditions by using nuclear patch clamp of the IP3R in its native ER membrane environment (12–15). Our results provide insights into the mechanism of channel activation by IP3: whereas Ca2+ is a true receptor agonist, the sole function of IP3 is to relieve Ca2+ inhibition of the channel. Ligand tuning of feedback inhibition by the permeant ion endows individual IP3R Ca2+ channels with the ability to respond in a graded fashion to stimulus intensity.
METHODS
Patch clamp of the outer membrane of individual nuclei mechanically isolated from Xenopus laevis oocytes was performed as described (12–14). The oocyte expresses only a single IP3R isoform (type 1) and lacks other (e.g., ryanodine receptor) Ca2+ release channels (16). The cytoplasmic aspect of the IP3R channel faced into the patch pipette. All experimental solutions contained 140 mM KCl, 10 mM Hepes (pH adjusted to 7.1 with KOH), and 0 or 0.5 mM Na2ATP as indicated. By using K+ as the current carrier and appropriate quantities of the high-affinity Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA), the low-affinity Ca2+ chelator, 5,5′-dibromoBAPTA, or ATP alone to buffer Ca2+ in the experimental solutions, concentrations were tightly controlled in our experiments. Total Ca2+ content in the solutions was determined by induction-coupled plasma mass spectrometry (Mayo Medical Laboratory, Rochester, MN). Free Ca2+ concentrations were calculated by using the maxchelator software (C. Patton, Stanford University, Palo Alto, CA). Pipette solutions contained various concentrations of IP3 as stated. All experiments were performed at room temperature with the pipette electrode at +20 mV relative to the reference-bath electrode. Each data point shown is the mean of results from at least four separate patch-clamp experiments performed under the same conditions. Error bars indicate the SEM. Single-channel currents were amplified by an Axopatch-1D amplifier (Axon Instruments, Foster City, CA), filtered at 1 kHz, digitized at 5 kHz, and recorded directly on hard disk by using Pulse+PulseFit 8.02 (HEKA Electronics, Lambrecht/Pfalz, Germany) on a PowerMac 8100 with an ITC-16 interface (Instrutech, Great Neck, NY). Channel dwell times and Pos were obtained by using tac 3.03 (Bruxton, Seattle, WA). Data were fitted and modeled by using igor pro 3.12 (WaveMetrics, Lake Oswego, OR).
RESULTS AND DISCUSSION
[Ca2+]i Dependence of the Gating of the IP3R Channel.
To examine the effects of [Ca2+]i on IP3R channel activity without possible Ca2+ effects on IP3 binding, a functionally saturating IP3 concentration of 10 μM (17) was applied to the cytoplasmic (pipette) side of the channel to fully stimulate it at various [Ca2+]i. With [Ca2+]i corresponding to resting levels in cells (10–100 nM), the Po of the IP3R was low (<0.2, Fig. 1 a and c), with short open intervals (τo < 3 ms) separated by long closed intervals (τc ≈ 100 ms; Fig. 1b). When [Ca2+]i was increased from 100 nM to 1 μM, Po increased dramatically to 0.8 as the result of a marked decrease of τc to ≈2 ms, whereas τo increased moderately to ≈10 ms. Similar [Ca2+]i in the absence of IP3 did not activate the channel (data not shown). Because saturating IP3 concentrations were used, the Ca2+ requirement is not caused by Ca2+ enhancement of IP3 affinity (18). Surprisingly, instead of the expected narrow bell-shaped [Ca2+]i response centered at ≈300 nM–1 μM (9, 10, 15, 19), Po remained very high (≈0.8) even as [Ca2+]i was raised to quite high levels (1–20 μM); with long open bursts lasting >1 sec, during which the channel only closed very briefly. As [Ca2+]i was increased beyond 20 μM, Po decreased sharply, mainly the result of τc increasing to ≈100 ms, whereas τo decreased to ≈1 ms. In the presence of saturating concentrations of IP3, there were no systematic effects on the Po-vs.-[Ca2+]i response of the species or concentration of Ca2+ chelator used (Fig. 1c), or the luminal Ca2+ (between 0.2 and 1.5 μM) or ATP (0 or 0.5 mM; Fig. 1d) concentrations.
Figure 1.

[Ca2+]i dependence of the IP3R in the presence of 10 μM IP3. (a) Typical single-channel current traces of the IP3R at various [Ca2+]i. Arrows indicate closed-channel current level in all traces. (b) Dependence on [Ca2+]i of mean closed-channel duration τc and open-channel duration τo of the IP3R. (c) Dependence of IP3R Po on [Ca2+]i. Experiments were performed with different Ca2+ chelators (□, BAPTA; ▵, 5,5′-dibromo BAPTA, ○, ATP) in the pipette solutions. Bath solution contained 1.5 μM Ca2+ and 0.5 mM ATP. Numerical labels indicate calculated concentrations (in μM) of free Ca2+ chelators present in the pipette solutions for the corresponding data points. The solid curve is the Hill equation fit for the biphasic [Ca2+]i effect on IP3R Po. (d) IP3R Po vs. [Ca2+]i in various bath solutions. Small open symbols: 0.8 μM luminal [Ca2+], 0.5 mM ATP; large open symbols: 0.2 μM luminal [Ca2+], 0 ATP; solid symbols: 0.2 μM luminal [Ca2+], 0.5 mM ATP. Convention for symbols and the solid curve are the same as in c.
The IP3R Po-vs.-[Ca2+]i response in 10 μM IP3 could be fitted to a biphasic Hill equation (Fig. 1c) so that:
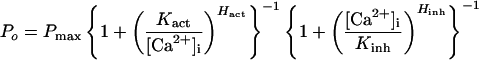 |
1 |
This suggests that a tetrameric IP3R channel can achieve a maximum open probability Pmax of 0.81, with two distinct types of functional Ca2+ binding sites, activating sites with half-maximal activating [Ca2+]i, Kact, of 210 ± 20 nM and Hill coefficient Hact of 1.9 ± 0.3, and inhibitory sites with half-maximal inhibitory [Ca2+]i, Kinh, of 54 ± 3 μM and Hill coefficient Hinh of 3.9 ± 0.7. The large Hill coefficients, Hact and Hinh, indicate that activation and inhibition of the IP3R by Ca2+ are both highly cooperative processes and suggest a requirement for Ca2+ binding to two of four monomers to open the channel in the presence of IP3 and for Ca2+ binding to all four monomers to inhibit channel opening.
IP3 Dependence of the [Ca2+]i Sensitivity.
The striking insensitivity of Po to increases of [Ca2+]i until quite high levels results in a Po-vs.-[Ca2+]i curve with a broad plateau. Previous models of complex temporal and spatial [Ca2+]i signaling have assumed a much higher affinity of the inhibitory Ca2+ binding sites than is suggested by our data. We examined whether this discrepancy arose from our use of saturating IP3 concentrations. However, there was no significant change in the [Ca2+]i dependence when the IP3 concentration was varied between 0.1 and 180 μM (Fig. 2). Thus, there is no evidence that the IP3R channel possesses a low-affinity IP3 binding site with binding coefficient >100 nM, consistent with biochemical determinations (17, 18, 20, 21). Contrary to earlier observations (22–24), no inhibitory effects of the Ca2+ chelator BAPTA on Ca2+ activation of the IP3R were detected, even at low (20–33 nM) IP3 concentrations (Fig. 2). At IP3 concentrations <100 nM, however, the IP3R became more sensitive to Ca2+ inhibition: at 33 nM IP3, Kinh decreased to 9.5 μM, although Hinh and Ca2+ activation were not affected. Further decreases in IP3 to 10 and 20 nM caused pronounced reductions of both the maximum Po and the range of [Ca2+]i over which the IP3R was active. The enhanced Ca2+ inhibition at low IP3 concentrations cannot be explained by inhibition of IP3 binding, because a ≈300-fold increase in [Ca2+]i (from 1 to 300 μM) was required to counter the effect of only a 5-fold increase in IP3 from 20 to 100 nM, indicating that the IP3 relief of [Ca2+]i inhibition is a highly cooperative process.
Figure 2.

IP3-concentration dependence of the [Ca2+]i sensitivity of the IP3R. Different symbols denote data for various IP3 concentrations as tabulated. Data points for 10 μM IP3 were combined from experiments using various bath solutions with the same pipette [Ca2+]. Data points for other concentrations of IP3 were obtained in bath solutions containing 220 nM Ca2+ and no ATP. The curves are Hill equation fits using Eq. 1, with Kinh varying with IP3 concentration with values as listed in the graph, whereas Pmax, Kact, Hact, and Hinh remained independent of IP3 concentration, with the values tabulated in the graph. Numerical labels indicate calculated concentrations (in μM) of free Ca2+ chelator BAPTA present in pipette solutions used for the corresponding data points.
The measured Po at all concentrations of IP3 were fitted well by using the biphasic Hill equation (1), with Kinh being the only IP3-concentration-sensitive parameter (Fig. 2). Even the observed reductions of maximum Po and the range of [Ca2+]i over which the channel is active at very low IP3 concentrations can both be accounted for in this model by a continuous decrease in Kinh with decreasing IP3, whereas the other parameters: Pmax, Kact, Hact, and Hinh all remain unchanged by IP3 concentration. These data lead to an unexpected conclusion: the effect of IP3 binding is not to enable activation of the IP3R by Ca2+, as expected for co-agonist ligands and as generally assumed (17, 18, 20, 21), but rather it is to ameliorate inhibition of the channel by Ca2+. The derived values of Kinh ranged from 160 nM at 10 nM IP3 to 59 μM at 100 nM IP3. This IP3 dependence of Kinh was well fitted with a simple Hill equation so that
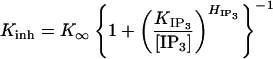 |
2 |
This equation, represented in Fig. 3a, implies that the IP3R has a single class of functional IP3 binding sites with a half-maximal activating IP3 concentration, KIP3, of 50 ± 4 nM, a Hill coefficient HIP3 of 4 ± 0.5, and a maximum inhibitory Ca2+ binding coefficient K∞ of 52 ± 4 μM at a saturating IP3 concentration. The KIP3 derived from this model is similar to both the dissociation constant KD (10–100 nM) in IP3 binding assays and IP3 concentration required (≈10–100 nM) for stimulation of Ca2+ release (17, 20, 21, 25). The large Hill coefficient HIP3 of 4 indicates that IP3 activation of the IP3R is highly cooperative (25–28) even in the absence of positive feedback from cytoplasmic Ca2+, requiring IP3 binding to perhaps all four monomers of the channel to relieve the Ca2+ inhibition and gate the channel open. Because IP3 binding to the IP3R is not cooperative (17, 18, 20, 21), the similar Hill coefficients HIP3 and Hinh (both ≈4) may suggest that IP3 binding to each IP3R monomer influences the inhibitory Ca2+ site in that same monomer.
Figure 3.

(a) Dependence on IP3 concentration of Kinh as derived from Fig. 2. The curve is a simple Hill equation fit. (b) Bell-shaped normalized Po-vs.-[Ca2+]i curves calculated by using Eqs. 1 and 2 for various low concentrations of IP3 (dotted curve, 10 nM IP3 with maximum Po of 0.09; dashed curve, 15 nM IP3 with maximum Po of 0.49; and solid curve, 20 nM IP3 with maximum Po of 0.71). (c) Theoretical Po (calculated by using the model) vs. IP3 concentration at various [Ca2+]i, as labeled. Dashed curves, [Ca2+]i ≤ 1 μM; thin solid curves, [Ca2+]i ≥ 10 μM.
According to these results, cytoplasmic Ca2+ at low concentrations and IP3 both activate the IP3R channel (Fig. 4), but they affect the channel in fundamentally different ways. Ca2+ binding to the IP3R at low [Ca2+]i directly activates the channel—like a conventional agonist. In contrast, IP3 binding to the channel activates it indirectly, solely by decreasing the affinity of the Ca2+ inhibitory site, with no direct stimulatory effect itself. Under conditions of low IP3 concentration, Ca2+ preferentially binds to the inhibitory site because of its higher affinity (Kinh < Kact), causing the channel to be inactive. Under conditions of stimulation associated with higher IP3 concentration, Kinh becomes >Kact, so Ca2+ binds preferentially to the Ca2+ activation site, activating the channel. A recent study of reconstituted IP3R also observed an IP3 dependence of Ca2+ inhibition (11). However, the results and interpretations are fundamentally different from ours. First, the IP3 required to observe relief from Ca2+ inhibition was ≈180 μM, three orders of magnitude higher than that measured here. Furthermore, Pmax was 0.03 vs. 0.81 in our studies. Second, inhibitory effects of Ca2+ on the channel were not observed at 180 μM IP3, in contrast to our results. Third, an additional low-affinity (10 μM) IP3 binding site was invoked to account for the IP3 relief of Ca2+ inhibition, whereas our data indicate that only one functional IP3 binding site is involved, with the affinity expected (≈50 nM) from biochemical studies. Importantly, our data indicate that IP3 mediates its effects by modulating the affinity of Ca2+ inhibitory sites, which is conceptually distinct from agonist activity caused by IP3 binding to a second low affinity site.
Figure 4.

Calculated Po vs. [Ca2+]i and IP3 concentration according to our model. (a) Projection showing Po-vs.-IP3 concentration curves at various activating [Ca2+]i. (b) Projection showing Po-vs.-[Ca2+]i curves at various IP3 concentrations. (c) Projection showing Po vs. IP3 concentration curves at various inhibitory [Ca2+]i.
Implications for IP3-Mediated [Ca2+]i Signaling.
Our results provide another interpretation of the role of IP3, thus warranting reconsideration of previous observations of IP3-mediated [Ca2+]i signals in cells. Our model predicts a bell-shaped relationship between Po and [Ca2+]i, with a sharp peak at [Ca2+]i <1 μM (9, 15, 19), although only under conditions of low (<20 nM) IP3 (Fig. 4b). The low Po and the bell shape, as well as the range of [Ca2+]i over which the channel is active observed for reconstituted IP3R channels at high (2 μM) IP3 (9–11), are remarkably similar to the predicted channel behaviors according to our model, albeit at much lower (IP3 ≈10 nM) concentration, suggesting an IP3 insensitivity of the reconstituted channels in those studies. Although neither the IP3 binding coefficient KIP3 nor the Hill coefficient HIP3 is [Ca2+]i-dependent in our model, the interplay between the effects of [Ca2+]i and IP3 concentration on Po can be manifested as an apparent increase in IP3 sensitivity as [Ca2+]i decreases at low [Ca2+]i (Fig. 3c) and an apparent decrease in cooperativity of IP3 activation as [Ca2+]i increases (Fig. 3c). Thus, our results may qualitatively explain previous observations that submicromolar [Ca2+]i lowered the functional sensitivity of the IP3R for IP3 (29, 30) and that the Hill coefficient for IP3 activation of Ca2+ release decreased as [Ca2+]i increased from the submicromolar to tens of μM range (27, 28, 31).
A major paradox has been [Ca2+]i responses that are graded in proportion to the intensity of the stimulus, because the process of Ca2+-induced Ca2+ release should lead to an all-or-none signal (32, 33). One proposed mechanism suggests that increased IP3 concentration recruits more individual elementary release units that each contribute a quantized amount of Ca2+ (32–35). Our model suggests another mechanism for achieving graded responses involving the IP3R itself. Because IP3 binding to the IP3R relieves Ca2+ inhibition of the channel, higher IP3 concentration shifts the peak of the Po-vs.-[Ca2+]i curve to higher [Ca2+]i (Fig. 3b). A [Ca2+]i that can inhibit channel activity at low IP3 will be insufficient to inhibit it when IP3 is increased (Fig. 4b). Higher [Ca2+]i at the mouth of the channel pore can therefore be achieved before reaching levels that terminate release. This process therefore generates graded Ca2+ release from IP3-sensitive stores through a mechanism intrinsic to the individual IP3R. By enabling higher [Ca2+]i to be achieved at the mouth of individual channels, higher IP3 concentration associated with more intense stimuli would promote greater diffusive spread of the local [Ca2+]i signal to other sites, thereby transforming highly localized signals at low levels of stimulation to more global coordinated Ca2+ release signals as the intensity of the stimulus is increased, without the need to invoke channels with different IP3 sensitivities or clusters of higher channel densities (13, 32–34), although our results in no way exclude these or other mechanisms. By similar reasoning, our model can also account for the transformation of oscillating [Ca2+]i at relatively low levels of agonist stimulation to sustained [Ca2+]i increases during intense stimulation. Quantal Ca2+ release elicited by IP3 (4, 5) has been described as a unique signaling mechanism that enables cells to retain complete responsiveness to changes in stimulus intensity (36). Our model suggests that this property can be intrinsic to the IP3R. Because our experiments investigated the steady-state properties of the IP3R, future kinetic studies will be required to determine the relevance of this model for quantal release in cells, as well as for other aspects of [Ca2+]i signaling, including IP3R desensitization (37) and inactivation (12, 13, 38).
Acknowledgments
We thank Dr. S. K. Joseph for reading the manuscript. This work was supported by grants from the Cystic Fibrosis Foundation and the National Institutes of Health.
ABBREVIATIONS
- IP3
inositol 1,4,5-tris-phosphate
- IP3R
IP3 receptor
- [Ca2+]i
cytoplasmic free Ca2+ concentration
- Po
open probability
- ER
endoplasmic reticulum
- BAPTA
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Berridge M J. Nature (London) 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 2.Meyer T, Stryer L. Annu Rev Biophys Biophys Chem. 1991;20:153–174. doi: 10.1146/annurev.bb.20.060191.001101. [DOI] [PubMed] [Google Scholar]
- 3.Toescu E C. Am J Physiol. 1995;269:G173–G185. doi: 10.1152/ajpgi.1995.269.2.G173. [DOI] [PubMed] [Google Scholar]
- 4.Bootman M D. Mol Cell Endocrinol. 1994;98:157–166. doi: 10.1016/0303-7207(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 5.Parys J B, Missiaen L, De Smedt H, Sienaert I, Casteels R. Pflügers Arch. 1996;432:359–367. doi: 10.1007/s004240050145. [DOI] [PubMed] [Google Scholar]
- 6.Iino M, Tsukioka M. Mol Cell Endocrinol. 1994;98:141–146. doi: 10.1016/0303-7207(94)90132-5. [DOI] [PubMed] [Google Scholar]
- 7.Taylor C W, Marshall I C B. Trends Biochem Sci. 1992;17:403–407. doi: 10.1016/0968-0004(92)90009-x. [DOI] [PubMed] [Google Scholar]
- 8.Putney J W, Jr, Bird G S. Endocr Rev. 1993;14:610–631. doi: 10.1210/edrv-14-5-610. [DOI] [PubMed] [Google Scholar]
- 9.Bezprozvanny I, Watras J, Ehrlich B E. Nature (London) 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 10.Bezprozvanny I, Ehrlich B E. J Membr Biol. 1995;145:205–216. doi: 10.1007/BF00232713. [DOI] [PubMed] [Google Scholar]
- 11.Kaftan E J, Ehrlich B E, Watras J. J Gen Physiol. 1997;110:529–538. doi: 10.1085/jgp.110.5.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mak D-O D, Foskett J K. J Biol Chem. 1994;269:29375–29378. [PubMed] [Google Scholar]
- 13.Mak D-O D, Foskett J K. J Gen Physiol. 1997;109:571–587. doi: 10.1085/jgp.109.5.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mak D-O D, Foskett J K. Am J Physiol. 1998;275:C179–C188. doi: 10.1152/ajpcell.1998.275.1.C179. [DOI] [PubMed] [Google Scholar]
- 15.Stehno-Bittel L, Lückhoff A, Clapham D E. Neuron. 1995;14:163–167. doi: 10.1016/0896-6273(95)90250-3. [DOI] [PubMed] [Google Scholar]
- 16.Kume S, Muto A, Aruga J, Nakagawa T, Michikawa T, Furuichi T, Nakade S, Okano H, Mikoshiba K. Cell. 1993;73:555–570. doi: 10.1016/0092-8674(93)90142-d. [DOI] [PubMed] [Google Scholar]
- 17.Mauger J-P, Lièvremont J-P, Piétri-Rouxel F, Hilly M, Coquil J-F. Mol Cell Endocrinol. 1994;98:133–139. doi: 10.1016/0303-7207(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 18.Taylor C W, Traynor D. J Membr Biol. 1995;145:109–118. doi: 10.1007/BF00237369. [DOI] [PubMed] [Google Scholar]
- 19.Iino M. J Gen Physiol. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor C W, Richardson A. Pharmacol Ther. 1991;51:97–137. doi: 10.1016/0163-7258(91)90043-l. [DOI] [PubMed] [Google Scholar]
- 21.Joseph S K. Cell Signalling. 1995;8:1–7. doi: 10.1016/0898-6568(95)02012-8. [DOI] [PubMed] [Google Scholar]
- 22.Richardson A, Taylor C W. J Biol Chem. 1993;268:11528–11533. [PubMed] [Google Scholar]
- 23.Combettes L, Champeil P. Science. 1994;265:813. doi: 10.1126/science.8047889. [DOI] [PubMed] [Google Scholar]
- 24.Combettes L, Hannaert-Merah Z, Coquil J-F, Rousseau C, Claret M, Swillens S, Champeil P. J Biol Chem. 1994;269:17561–17571. [PubMed] [Google Scholar]
- 25.Meyer T, Holowka D, Stryer L. Science. 1988;240:653–655. doi: 10.1126/science.2452482. [DOI] [PubMed] [Google Scholar]
- 26.Carter T D, Ogden D. J Physiol (Lond) 1997;504:17–33. doi: 10.1111/j.1469-7793.1997.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finch E A, Turner T J, Goldin S M. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- 28.Dufour J F, Arias I M, Turner T J. J Biol Chem. 1997;272:2675–2681. doi: 10.1074/jbc.272.5.2675. [DOI] [PubMed] [Google Scholar]
- 29.Hirose K, Kadowaki S, Iino M. J Physiol (Lond) 1998;506:407–414. doi: 10.1111/j.1469-7793.1998.407bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Missiaen L, De Smedt H, Droogmans G, Casteels R. J Biol Chem. 1992;267:22961–22966. [PubMed] [Google Scholar]
- 31.Combettes L, Claret M, Champeil P. FEBS Lett. 1992;301:287–290. doi: 10.1016/0014-5793(92)80258-i. [DOI] [PubMed] [Google Scholar]
- 32.Bootman M D, Berridge M J. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 33.Berridge M J. J Physiol (Lond) 1997;499:291–306. doi: 10.1113/jphysiol.1997.sp021927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horne J H, Meyer T. Science. 1997;276:1690–1693. doi: 10.1126/science.276.5319.1690. [DOI] [PubMed] [Google Scholar]
- 35.Callamaras N, Marchant J S, Sun X P, Parker I. J Physiol (Lond) 1998;509:81–91. doi: 10.1111/j.1469-7793.1998.081bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kindman L A, Meyer T. Biochemistry. 1993;32:1270–1277. doi: 10.1021/bi00056a011. [DOI] [PubMed] [Google Scholar]
- 37.Oancea E, Meyer T. J Biol Chem. 1996;271:17253–17260. doi: 10.1074/jbc.271.29.17253. [DOI] [PubMed] [Google Scholar]
- 38.Hajnóczky G, Thomas A P. Nature (London) 1994;370:474–477. doi: 10.1038/370474a0. [DOI] [PubMed] [Google Scholar]