

Abstract
The inositol 1,4,5-trisphosphate (InsP3) receptor (InsP3R), a Ca2+-release channel localized to the endoplasmic reticulum, plays a critical role in generating complex cytoplasmic Ca2+ signals in many cell types. Three InsP3R isoforms are expressed in different subcellular locations, at variable relative levels with heteromultimer formation in different cell types. A proposed reason for this diversity of InsP3R expression is that the isoforms are differentially inhibited by high cytoplasmic free Ca2+ concentrations ([Ca2+]i), possibly due to their different interactions with calmodulin. Here, we have investigated the possible roles of calmodulin and bath [Ca2+] in mediating high [Ca2+]i inhibition of InsP3R gating by studying single endogenous type 1 InsP3R channels through patch clamp electrophysiology of the outer membrane of isolated Xenopus oocyte nuclei. Neither high concentrations of a calmodulin antagonist nor overexpression of a dominant-negative Ca2+-insensitive mutant calmodulin affected inhibition of gating by high [Ca2+]i. However, a novel, calmodulin-independent regulation of [Ca2+]i inhibition of gating was revealed: whereas channels recorded from nuclei kept in the regular bathing solution with [Ca2+] ∼400 nM were inhibited by 290 μM [Ca2+]i, exposure of the isolated nuclei to a bath solution with ultra-low [Ca2+] (<5 nM, for ∼300 s) before the patch-clamp experiments reversibly relieved Ca2+ inhibition, with channel activities observed in [Ca2+]i up to 1.5 mM. Although InsP3 activates gating by relieving high [Ca2+]i inhibition, it was nevertheless still required to activate channels that lacked high [Ca2+]i inhibition. Our observations suggest that high [Ca2+]i inhibition of InsP3R channel gating is not regulated by calmodulin, whereas it can be disrupted by environmental conditions experienced by the channel, raising the possibility that presence or absence of high [Ca2+]i inhibition may not be an immutable property of different InsP3R isoforms. Furthermore, these observations support an allosteric model in which Ca2+ inhibition of the InsP3R is mediated by two Ca2+ binding sites, only one of which is sensitive to InsP3.
Keywords: single-channel electrophysiology, patch clamp, calcium, Xenopus oocyte, nucleus
INTRODUCTION
The second messenger, inositol 1,4,5-trisphosphate (InsP3), is generated in many cell types through the hydrolysis of phosphatidylinositol 4,5-bisphosphate by membrane-bound phospholipase C activated by plasma membrane receptors responding to extracellular stimuli. InsP3 then diffuses through the cytoplasm to bind to its receptor (InsP3R) in the ER and activate it as a Ca2+ channel to release Ca2+ stored in the ER lumen. Modulation of the cytoplasmic free Ca2+ concentration ([Ca2+]i) by InsP3R-mediated Ca2+ release is a ubiquitous intracellular signal transduction mechanism that regulates numerous processes (Berridge, 1993).
Three isoforms of the InsP3R, with spliced variants, have been identified (Joseph, 1996). Most mammalian cell types express multiple InsP3R isoforms in distinct and overlapping intracellular locations with their absolute and relative expression levels regulated by gene transcription, alternative splicing and receptor degradation that differ during different stages of cell development and in response to extracellular stimuli (Taylor et al., 1999). Furthermore, formation of hetero-tetrameric channels is possible in cell types expressing more than one InsP3R isoform (Joseph et al., 1995; Monkawa et al., 1995; Wojcikiewicz, 1995; Nucifora et al., 1996). Although this diversity of InsP3R expression is impressive, its functional correlates and physiological implications remain unclear. Studies of the single-channel properties of the various InsP3R isoforms have revealed that whereas their permeation and conductance properties are very similar (Mak et al., 2000; Ramos-Franco et al., 2000), their gating may be differentially inhibited by high [Ca2+]i (Bezprozvanny et al., 1991; Hagar et al., 1998; Mak et al., 1998; Ramos-Franco et al., 1998a,b, 2000; Boehning et al., 2001; Mak et al., 2001a). Because high [Ca2+]i inhibition of InsP3R channel gating may be a pivotal feedback mechanism for the regulation of intracellular Ca2+ signaling (Taylor, 1998), it has been suggested that differential inhibition by high [Ca2+]i of the different InsP3R isoforms may generate distinct Ca2+ signals in different cell types with different patterns of InsP3R isoform expression, and that this may be a reason for the diversity of InsP3R expression (Hagar et al., 1998).
It has been suggested that high [Ca2+]i inhibition of the InsP3R is mediated by calmodulin (CaM), a ubiquitous Ca2+-binding protein that binds to and regulates the functions of many proteins. CaM was found to bind to the InsP3R-1 in the presence of free Ca2+ to a single site in the regulatory domain (Maeda et al., 1991; Yamada et al., 1995; Hirota et al., 1999). Purified InsP3R-1 channels lacking bound CaM were not inhibited by high [Ca2+]i, whereas addition of CaM restored inhibition of channel gating by high [Ca2+]i (Hirota et al., 1999; Michikawa et al., 1999). The notion that high Ca2+ inhibition of channel gating was mediated by CaM was reinforced by observations that the type 3 InsP3R (InsP3R-3) did not bind CaM (Yamada et al., 1995; Cardy and Taylor, 1998; Lin et al., 2000) and was not inhibited by high [Ca2+]i (Hagar et al., 1998). Nevertheless, other data suggest that the role of CaM in high [Ca2+]i inhibition of InsP3R channel gating is far from unequivocal. Despite the absence of detectable interaction between CaM and a mutant InsP3R-1 in which the putative CaM binding site was eliminated (Yamada et al., 1995), more recent studies have demonstrated that this mutant channel is nevertheless still inhibited by high [Ca2+]i (Zhang and Joseph, 2001; Nosyreva et al., 2002). Furthermore, whereas the InsP3R-3 lacks the CaM binding site present in the InsP3R-1 and no interaction between InsP3R-3 and CaM has been detected (Yamada et al., 1995; Cardy and Taylor, 1998; Lin et al., 2000), electrophysiological studies of the recombinant rat InsP3R-3 in its native membrane environment demonstrated that it is nevertheless inhibited by high [Ca2+]i (Mak et al., 2001a) with quantitative features similar to those of inhibition of the InsP3R-1 in the same membrane (Mak et al., 1998).
Here, we investigated the possible effects of CaM on high [Ca2+]i inhibition of the gating of single endogenous InsP3R-1 channels in their native membrane environment using nuclear membrane patch clamp electrophysiology (Mak and Foskett, 1994). Our experiments do not provide evidence supporting any role for CaM in this process. However, we discovered a novel regulation of high [Ca2+]i inhibition of InsP3R-1 channel gating. Inhibition of InsP3R-1 gating by high [Ca2+]i can be reversibly abrogated by exposure of the channel to a bathing solution containing ultra-low [Ca2+] (<5 nM). Our observations indicate that inhibition of InsP3R-1 channel gating by high [Ca2+]i can be disrupted by environmental conditions experienced by the channel, and therefore may not be an invariant property of a specific InsP3R isoform. Furthermore, these observations support an allosteric model in which Ca2+ inhibition of the InsP3R is mediated by two Ca2+ binding sites, only one of which is sensitive to InsP3.
MATERIALS AND METHODS
Heterologous Expression of Calmodulin in Xenopus Oocytes
Maintenance of Xenopus laevis and surgical extraction of ovaries were performed as described previously (Mak and Foskett, 1994, 1997, 1998). Oocytes were defolliculated as described (Jiang et al., 1998). cRNA (1 μg/μl) of rat calmodulin (CaM), either wild-type (w.t.) or a quadruple mutant (q.m.) containing a D→A mutation in each of the four EF hands so that Ca2+ binding in all EF hands was abolished (Xia et al., 1998; Keen et al., 1999), was synthesized in vitro from cDNA provided as a gift by Dr. John P. Adelman (Vollum Institute, Portland, OR). 23 nl of cRNA (either w.t. or q.m.) was injected into the cytoplasm of oocytes 1 d after defolliculation, as described (Mak et al., 2000). cRNA-injected and uninjected control oocytes were maintained under identical conditions in individual wells in 96-well plates containing 200 μl of ASOS (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, pH adjusted to 7.6 with NaOH; with 3 mM Na pyruvate, 100 μg/ml gentamycin, and 100 μM N-acetyl-Leu-Leu-Norleucinal; Sigma-Aldrich). 80 μl of ASOS in each well was changed daily. Nuclear patch clamp experiments and immunoprecipitations were performed 2–4 d after c-RNA injection when the expression level of exogenous CaM was stable as determined by Western analysis.
Western Analysis and Immunoprecipitation
Western analysis was performed on oocyte extracts (cRNA-injected and uninjected), as described in Mak et al. (2000), to ascertain the levels of endogenous and heterologously expressed CaM in the oocytes using a specific antibody (Upstate Biotechnology). Immunoprecipitation of InsP3R (type 1) and CaM was performed using oocyte lysates, as described in (Mak et al., 2000), with a specific type 1 InsP3R antibody (Joseph and Samanta, 1993; Joseph et al., 1995) and protein A agarose (GIBCO BRL), and an antibody to CaM and protein G agarose (GIBCO BRL), respectively.
Solutions for Patch Clamp Experiments
All patch clamp experiments were performed with solutions containing 140 mM KCl and 10 mM HEPES with pH adjusted to 7.1 with KOH. The free Ca2+ concentration ([Ca2+]i) of the pipette solutions (to which the cytoplasmic side of the InsP3R is exposed in patch-clamp experiments) was tightly controlled by buffering various amounts of added CaCl2 (40–400 μM) with 500 μM of the high-affinity Ca2+ chelator, BAPTA (1,2-bis(O-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid; Molecular Probes) and 0.5 mM Na2ATP (100 nM < [Ca2+]i < 2.5 μM); or 500 μM of the low-affinity Ca2+ chelator, 5,5′-dibromo BAPTA (Molecular Probes) and 0.5 mM Na2ATP (5 μM < [Ca2+]i < 15 μM); or 0.5 mM Na2ATP alone (15 μM < [Ca2+]i < 300 μM). Solutions with [Ca2+]i > 300 μM contained no Ca2+ chelator for buffering. The normal Ca2+ bath solution (NCaS) contained 500 μM BAPTA and 250 μM CaCl2 (free [Ca2+] ≈ 400–500 nM), and the physiological Ca2+ bath solution (PCaS) contained 500 μM BAPTA and 70 μM CaCl2 (free [Ca2+] = 48 ± 5 nM). The free [Ca2+] of these solutions was directly measured using Ca2+-selective mini-electrodes (Baudet et al., 1994). The ultra-low Ca2+ bath solution (ULCaS) contained 1 mM BAPTA and no added CaCl2. The contaminating [Ca2+] in the solution was determined by induction-coupled plasma mass spectrometry (Mayo Medical Laboratory) to be ∼6–10 μM. Ca2+-selective minielectrodes were unable to determine accurately the free [Ca2+] in the ULCaS because of the nonlinear response of the electrode in free [Ca2+] < 5 nM. Free [Ca2+] was calculated using the Maxchelator software (C. Patton, Stanford University, Stanford, CA) to be ∼0.9–1.5 nM.
Unless specified otherwise, all pipette solutions contained a saturating concentration (10 μM) of InsP3 (Mak and Foskett, 1994) from Molecular Probes. When specified, the pipette solutions also contained 500 μM W-7 (a CaM binding antagonist; N-(6-aminohexyl)-5-chloro-1-naphthalene-sulfonamide hydrochloride; Calbiochem), or 10 μM purified bovine CaM (Calbiochem). All reagents were used with no further purification.
Oocyte Nucleus Isolation Protocols
A stage V or VI oocyte was gently teased open mechanically in the isolation bathing solution, enabling the translucent nucleus to be isolated from the cytoplasmic material. The isolated nucleus was either directly transferred to the experimental bathing solution (protocol Nd, Ld, and Pd, Fig. 1), or it was transferred through a series of culture dishes containing 4–5 ml of incubation bath solutions (protocol L, LN, and LNL, in Fig. 1) before it was ultimately transferred to the experimental bath. The nucleus remained in each incubation bath for at least 20 min before the next transfer, to ensure that the solution in the perinuclear lumen between the outer and inner nuclear envelope had attained ionic equilibrium with the bath solution (Mak and Foskett, 1994). Approximately 20 μl of the previous bath solution accompanied the nucleus to the new bath in a transfer. The culture dish containing the nucleus in the experimental bath solution was finally moved onto the stage of the inverted microscope where patch clamp experiments were performed.
Figure 1.

Schematic diagram showing the various protocols used to isolate oocyte nuclei for nuclear patch clamp experiments.
Acquisition and Analysis of Single-Channel Patch-clamp Current Records
The isolated nucleus was gently immobilized as described previously (Mak and Foskett, 1994) so that membrane patches could be repeatedly obtained from the same region (±2 μm) of the outer nuclear membrane (Mak and Foskett, 1997). Due to abrupt termination of channel activity (Mak and Foskett, 1994, 1997), patch clamp experiments were performed in “on-nucleus” configuration to maximize the duration of channel activities recorded. To prevent contamination of the pipette solution by the bath solution (especially the Ca2+ chelator in the bath solution) by diffusion through the pipette tip during the time when the pipette was immersed in the bath and before giga-Ohm seal formation, a positive pressure (∼10 mmHg) was maintained inside the pipette until the pipette tip was properly positioned on the nuclear membrane. Then suction was applied in the pipette to obtain the giga-Ohm seal. All experiments were performed at room temperature with the pipette electrode at +20 mV relative to the reference bath electrode unless specifically stated otherwise. Each experiment recorded the InsP3R channel activity at a specific [Ca2+]i and [InsP3], with no change of the pipette or bath solutions during the experiment. Data acquisition was performed as previously described (Mak et al., 1998), with currents recorded with a filtering frequency of 1 kHz and a digitizing frequency of 5 kHz.
The patch clamp current traces were analyzed using MacTac software (Bruxton) to identify channel-opening and -closing events using a 50% threshold. Current traces exhibiting one InsP3R channel, or two InsP3R channels determined to be identical and independently gated (Mak and Foskett, 1997), were used for channel open probability (P o) evaluation. The number of channels in the membrane patch was assumed to be the maximum number of open channel current levels observed throughout the current record. In experimental conditions with P o > 0.1, only current records with longer than 10 s of InsP3R channel activities were used for determination of P o, so there is little uncertainty in the number of channels in the current traces used. In experimental conditions with P o < 0.1, only current records exhibiting one open channel current level with InsP3R channel activities lasting longer than 30 s were used, to ensure that they were truly single-channel records (Mak et al., 2001a). The P o data shown for each set of experimental conditions are the means of results from at least four separate patch-clamp experiments performed under the same conditions. Error bars indicate the SEM.
RESULTS
Lack of Effect of Calmodulin on Ca2+ Inhibition of InsP3R Gating in Endoplasmic Reticulum Membrane
Previous single-channel patch-clamp studies of the endogenous Xenopus type 1 InsP3R (X-InsP3R-1) in its native ER membrane environment revealed a biphasic regulation by [Ca2+]i of the single-channel open probability (P o) (Mak et al., 1998, 2001b). It has been suggested that calmodulin (CaM) bound to the channel mediates inhibition of InsP3R-1 gating by high [Ca2+]i (Michikawa et al., 1999). We therefore investigated the possibility that the high [Ca2+]i inhibition of X-InsP3R-1 channel gating observed in our previous studies was mediated by CaM. Oocyte nuclei were isolated and transferred directly into an experimental bath of NCaS for patch-clamp experiments (protocol Nd in Fig. 1) . By repeated patch clamping over the surface of an isolated nucleus, regions on the outer nuclear envelope were identified in which the probability of detecting InsP3R channel activities in membrane patches (P d) was high (Mak and Foskett, 1997). A series of patch-clamp experiments was performed at these regions with pipette solutions (to which the cytoplasmic side of the InsP3R was exposed) alternately containing either [Ca2+]i = 755 nM, or very high [Ca2+]i (290 μM) with 500 μM of W-7, a CaM binding antagonist. The former solution is one in which the channel gates with a high P o, thereby ascertaining the presence of functional InsP3R channels in the regions selected during the series of experiments. In contrast, the latter solution has [Ca2+]i sufficiently high to inhibit InsP3R channel gating (Mak et al., 1998). Because CaM is endogenously expressed in Xenopus oocytes (Fig. 2 , Lane A and C), we reasoned that if CaM mediated the high [Ca2+]i inhibition of InsP3R channel gating, then inclusion of 500 μM of W-7 in the pipette solution may block high [Ca2+]i inhibition by interfering with CaM binding to the InsP3R channel (Michikawa et al., 1999), making channel gating observable in the 290 μM [Ca2+]i solutions. Nevertheless, no channel activity was detected in any of the five patches with 500 μM W-7 and [Ca2+]i = 290 μM (Fig. 3 B), whereas InsP3R channel activities were readily detected in five out of six patches with [Ca2+]i = 755 nM (Fig. 3 A).
Figure 2.

Western blots of oocyte lysates probed with a CaM antibody (both w.t. and q.m.). Lysates from oocytes injected with CaM cRNA (w.t. or q.m.) or uninjected oocytes were used as labeled. Oocytes used for lanes A and B or C and D were from the same batches, respectively. Top arrow indicates wild-type CaM and the bottom arrow indicates the quadruple mutant CaM. The slightly faster mobility of q.m. CaM is likely a reflection of the known Ca2+-binding dependence of CaM mobility in gels (Xia et al., 1998).
Figure 3.

Typical current traces from nuclei in NCaS bath with pipette solutions containing 10 μM InsP3. Arrows indicate closed channel current levels. (A and B) Uninjected oocytes were used. InsP3R channel activity was observed with [Ca2+]i of 755 nM (A, n = 3), whereas no channel activity was observed in a membrane patch obtained from the same region of the same nucleus with [Ca2+]i of 290 μM and the pipette solution containing 500 μM W-7 (B, n = 5). (C and D) Oocytes injected with CaM q.m. cRNA were used. InsP3R channel activity was observed with [Ca2+]i of 2.1 μM (C, n = 4), whereas no channel activity was observed in a membrane patch obtained from the same region of the same nucleus with [Ca2+]i of 290 μM (D, n = 9).
Whereas this result with W-7 is seemingly inconsistent with the hypothesis that CaM mediates Ca2+ inhibition of InsP3R gating, CaM-dependent regulation of the small-conductance Ca2+-activated K+ (SK) channel gating is insensitive to W-7 and other CaM inhibitors (Xia et al., 1998). However, overexpression of a mutant CaM, in which the Ca2+-binding EF hand motifs were disabled, interfered with the Ca2+ activation of the SK channel gating by competing with the endogenous CaM for the interaction with the channels (Xia et al., 1998; Keen et al., 1999). The effects of mutant CaM expression on SK channel gating provided evidence that endogenous CaM is tightly and constitutively (even in the absence of Ca2+) associated with the SK channel and mediates the effects of Ca2+ on SK channel gating. The ability of high Ca2+ concentrations to inhibit InsP3R channel gating in our in vitro electrophysiological studies can be observed for long times (up to 2 h) after isolation of the nuclei (Mak et al., 1998; Boehning et al., 2001; Mak et al., 2001b). Thus, if CaM mediates the effect of high [Ca2+]i, it must remain associated with the channel in the isolated nuclei, and therefore must be tightly bound to the InsP3R and not free to diffuse away into the large experimental bath. We therefore explored the possibility that Ca2+ inhibition of InsP3R channel gating was mediated by a constitutive tight association of CaM with the channel, by examining the effects of overexpression of the Ca2+-insensitive quadruple mutant (q.m.) CaM on the Ca2+ regulation of the InsP3R.
The q.m. CaM, which has all EF hands mutated and therefore is Ca2+ insensitive, was overexpressed in Xenopus oocytes by cytoplasmic microinjection of cRNA. Western analysis (n = 5) indicated that the exogenous q.m. CaM was expressed to a level that was at least an order of magnitude higher than the endogenous wild-type CaM (Xia et al., 1998; Fig. 2). Patch-clamp experiments using nuclei isolated by protocol Nd (Fig. 1) from q.m. CaM-expressing oocytes revealed that InsP3R channel gating was still inhibited by high [Ca2+]i: InsP3R channel activities were detected in 11 out of 11 patches with pipette solutions containing [Ca2+]i = 2.1 μM (Fig. 3 C), but no channel activity was detected in any of 9 patches with pipette solutions containing 290 μM [Ca2+]i (Fig. 3 D). These results therefore also did not support the hypothesis that Ca2+ inhibition of InsP3R channel gating is mediated by CaM.
The lack of effect of overexpression of the q.m. CaM on Ca2+ inhibition of gating may suggest that endogenous CaM is not normally associated with the InsP3R. We examined the biochemical association between the InsP3R and CaM by coimmunoprecipitation. Using lysates prepared from cRNA-injected oocytes overexpressing either w.t. or q.m. CaM (Fig. 2), immunoprecipitation of the endogenous type 1 InsP3R with a specific antibody did not coimmunoprecipitate either w.t. or q.m. CaM (n = 4; unpublished data). In the converse experiments, immunoprecipitation of CaM with an antibody that binds to both w.t. and q.m. forms did not coimmunoprecipitate the InsP3R (n = 4; unpublished data). These results therefore do not provide evidence of an association between CaM and the InsP3R.
In summary, our single-channel patch clamp experiments revealed that neither high concentrations of a CaM antagonist, nor overexpression of a Ca2+-insensitive q.m. CaM had any effect on [Ca2+]i inhibition of InsP3R channel gating. In addition, coimmunoprecipitation failed to demonstrate an association between CaM and the InsP3R. Thus, our investigations did not provide any evidence supporting the hypothesis that high [Ca2+]i inhibition of InsP3R gating observed in in vitro patch clamp studies is mediated by CaM. These conclusions are therefore in agreement with those reached in some other studies (Zhang and Joseph, 2001; Nosyreva et al., 2002).
Abrogation of Ca2+-dependent Inhibition of InsP3R Channel Gating
Our experimental results suggested that CaM is not involved in the inhibition of InsP3R channel gating by high [Ca2+]i. However, it remained possible that a different molecule may be involved, and that conditions could be identified which would strip such a putative effector from the InsP3R in the isolated nucleus, thereby rendering the InsP3R insensitive to Ca2+ inhibition. We reasoned that the putative effector, as a sensor of [Ca2+]i, might be dependent on normal [Ca2+]i for its association with the InsP3R. We therefore incubated the isolated nuclei in an ultra-low Ca2+ bath solution (ULCaS) before using them for nuclear patch clamp experiments to determine the Ca2+ dependence of the InsP3R gating.
In the first set of experiments, nuclei were isolated by protocol L (Fig. 1) into a bath of ULCaS ([Ca2+] < 5 nM). In the presence of 10 μM cytoplasmic (pipette) [InsP3] and [Ca2+]i < 20 μM, gating of the InsP3R exposed to the ULCaS was very similar to that of InsP3R in nuclei isolated directly into NCaS by protocol Nd (Fig. 4 ; Mak et al., 1998). In both cases, channel P o was low (<0.2) in [Ca2+]i < 150 nM, it increased dramatically to 0.8 as [Ca2+]i was increased from 150 nM to 1μM, and then P o remained at the maximum level of 0.8 when [Ca2+]i was further increased from 1 to 20 μM (Fig. 5) . The InsP3R in nuclei isolated by protocol Nd were inhibited by [Ca2+]i > 20 μM (Mak et al., 1998) but, remarkably, InsP3R in nuclei isolated into ULCaS by protocol L exhibited robust channel activities in [Ca2+]i as high as 1.5 mM (Fig. 4) with no decrease in channel P o (Fig. 5). Thus, a 20-min exposure to the ULCaS containing <5 nM Ca2+ caused the gating of InsP3R channel to be no longer inhibited by high [Ca2+]i. All of the InsP3R channel activities observed in the ultra-low [Ca2+] bath solution also terminated abruptly after ∼30 s, like those previously observed in the regular bath solution (Mak and Foskett, 1994, 1997).
Figure 4.
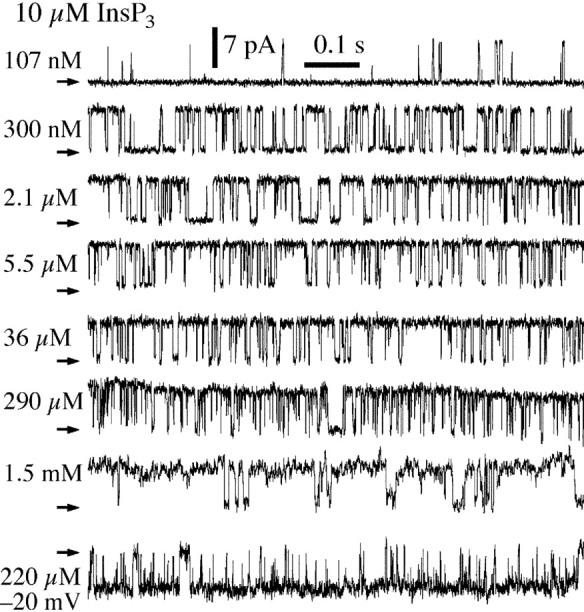
Typical current traces from nuclei in ULCaS bath isolated by protocol L. Arrows indicate closed channel current levels. Pipette solutions contained 10 μM InsP3 and [Ca2+]i as tabulated. The last current trace was obtained with −20 mV applied transmembrane potential. Other current traces were obtained with +20 mV applied potential.
Figure 5.

[Ca2+]i dependencies of the channel P o of the InsP3R in oocyte nuclei isolated using various protocols (Nd, L, and LNL) and applied potentials (±20 mV) as tabulated. All pipette solutions used contained 10 μM InsP3. The dashed curve is a simple activating Hill equation fit for the data from nuclei isolated with protocol L (large open circles). For comparison, the biphasic Hill equation fit (continuous curve) for the data points from nuclei isolated directly into NCaS bath (small filled circles) obtained in a previous study (Mak et al., 1998) are also shown. The InsP3R channel P o was lower in ULCaS than in NCaS at [Ca2+]i ≈ 100 nM. It is possible that this reflects some intrinsic properties of the InsP3R after exposure to the low bath [Ca2+]. Alternately, this may only be an artifact as a result of the movement of free Ca2+ ion across the open channel. With pipette [Ca2+]i ≈ 100 nM, when the oocyte nucleus was in NCaS ([Ca2+] = 400–500 nM), the Nernst reversal potential for Ca2+ ions was ∼35 mV so Ca2+ ions moved across the open InsP3R channel from the lumenal side to the cytoplasmic side despite an applied transmembrane voltage of 20 mV. This could cause the effective [Ca2+]i at the activating Ca2+-binding sites on the cytoplasmic side of the channel to be higher than the free [Ca2+] in the bulk of the pipette solution if the Ca2+-binding sites are close enough to the ion conducting pore. Conversely, when the nucleus was in ULCaS ([Ca2+] < 5 nM), Ca2+ ions moved across the open InsP3R channel in the opposite direction, down the electrical and chemical gradients, possibly lowering the effective [Ca2+]i at the Ca2+-binding sites. In [Ca2+]i < 250 nM, the mean open channel duration (<τo>) of the InsP3R increases with [Ca2+]i (Mak and Foskett, 1998). Therefore, if Ca2+ flux across the open InsP3R channel caused the effective [Ca2+]i at the activating Ca2+-binding sites to deviate from the free [Ca2+] in the bulk pipette solution, then channels in NCaS bath would have longer <τo> and higher channel P o than those in ULCaS bath, as observed. On the other hand, in [Ca2+]i > 300 nM, <τo> does not exhibit any dependence on [Ca2+]i although the mean closed channel duration (<τc>) is still affected by [Ca2+]i (Mak and Foskett, 1998). Deviation of effective [Ca2+] at the Ca2+-binding sites from the bulk free [Ca2+] would dissipate quickly by diffusion once the channel closed and therefore would not affect <τc>. Thus, there would be no difference between the observed P o of InsP3R in ULCaS and NCaS bath in [Ca2+]i > 300 nM, as observed.
Because of the difference between the free Ca2+ concentration in the high [Ca2+]i pipette solutions and ultra-low [Ca2+] bath solutions, it is possible that a potential difference may be established across the membrane and affect the high [Ca2+]i inhibition of the InsP3R and thus its P o. We performed patch clamp experiments with −20 mV applied potential, using high [Ca2+]i pipette solution ([Ca2+]i = 221 μM) with nuclei isolated with protocol L. The InsP3R channel P d (9 out of 20 patches exhibited channel activity), gating kinetics (last current trace in Fig. 4), and P o (Fig. 5) were not detectably different from that recorded at +20 mV (P d = 6 out of 8 patches), indicating that the abrogation of high [Ca2+]i inhibition by exposure to ULCaS is not due to simple electrostatic effects that change the membrane potential.
We previously demonstrated that the Ca2+ dependence of channel P o in nuclei isolated by protocol Nd into NCaS was well fitted by a biphasic Hill equation
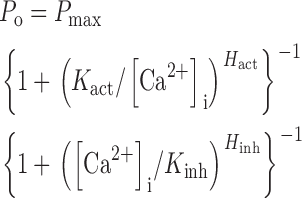 |
(1) |
with maximum channel open probability (P max) = 0.81 ± 0.02, half-maximal activating [Ca2+]i (K act) = 210 ± 20 nM, activation Hill coefficient (H act) = 1.9 ± 0.3, half-maximal inhibitory [Ca2+]i (K inh) = 54 ± 3 μM, and inhibitory Hill coefficient (H inh) = 3.9 ± 0.7 (Mak et al., 1998). Our new data indicated that the InsP3R in nuclei isolated by protocol L into ULCaS exhibited no inhibition by high [Ca2+]i, so that the Ca2+ dependence of channel P o can be fitted by a simple activating Hill equation
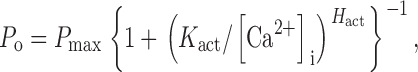 |
(2) |
with maximum open probability P max of 0.84 ± 0.01, half-maximal activating [Ca2+]i (K act) of 280 ± 30 nM, and activation Hill coefficient (H act) of 2.7 ± 0.3 (Fig. 5).
Nuclei isolated directly into a ULCaS bath by protocol Ld were used to determine the minimum duration of exposure to ULCaS bath required to relieve high [Ca2+]i inhibition of InsP3R gating. We found that channel activities could be detected with a pipette solution containing 10 μM InsP3 and 290 μM [Ca2+]i no earlier than 5 min after the nucleus was isolated into the ULCaS bath. Thus, the process involved in the relief of Ca2+ inhibition of InsP3R channel gating by exposure of the isolated nuclei to ULCaS is a slow one, requiring a few minutes.
To determine if normal cytoplasmic [Ca2+] (∼50 nM) is low enough to cause the relief of high [Ca2+]i inhibition of InsP3R gating, we isolated oocyte nuclei directly in PCaS bath (protocol Pd, Fig. 1). In a series of experiments performed in areas of the nuclear membrane identified with very high P d, using pipette solutions with 10 μM InsP3 and 0.5 mM ATP, containing alternately 630 nM or 221 μM [Ca2+]i, InsP3R channels were observed in seven out of seven patches with 630 nM [Ca2+]i, but no InsP3R channel activity was observed in any of 11 patches with 221 μM [Ca2+]i, even when the nucleus was exposed to the PCaS bath for over 160 min. Thus, the normal resting [Ca2+] of the cytoplasm (∼50 nM) is not sufficiently low to induce the relief of Ca2+ inhibition observed in the ultra-low Ca2+ condition.
InsP3 Dependence of the InsP3R in ULCaS Bath
Our previous studies (Mak et al., 1998, 2001a) revealed that InsP3 activates gating by relieving the Ca2+ inhibition of the channel. InsP3 increases K inh, the inhibitory half-maximal [Ca2+]i, with no effect on the values of the channel Ca2+ activation parameters (K act, H act) or P max in Eq. 1. It seemed likely that this mode of InsP3 activation cannot operate if the channel is not inhibited by high [Ca2+]i as observed after the channel had been exposed to the ULCaS bath for a few minutes. We therefore examined whether InsP3 was still required to gate the InsP3R under conditions that abrogated Ca2+ inhibition of the channel.
A series of experiments was performed using nuclei isolated by protocol L into ULCaS bath, patching in regions of the nuclei identified to exhibit high P d, with pipette solutions alternately containing either 10 μM InsP3 and [Ca2+]i = 755 nM, or no InsP3 and [Ca2+]i between 60 nM and 290 μM. Again, the former solution was used to ascertain the presence of functional InsP3R channels in the regions of the isolated nuclei selected for our experiments for the entire duration of the series. InsP3R channel activities were observed in 27 out of 30 membrane patches in the presence of InsP3, but no channel activity was detected in any of the 10 patches without InsP3 (Fig. 6 A). Therefore, even though the InsP3R was no longer inhibited by high [Ca2+]i when the nucleus was isolated into ULCaS, InsP3 was nonetheless still necessary for channel gating.
Figure 6.

(A and B) Typical current traces from nuclei isolated with protocol L. Arrows indicate closed channel current levels. (A) The pipette solutions contained no InsP3 and 290 μM [Ca2+]i as tabulated. (B) The pipette solutions contained 10 nM InsP3 and [Ca2+]i as tabulated. (C) [Ca2+]i dependence of the channel P o of the InsP3R in the presence of various [InsP3] as tabulated. The number of channels used to evaluate each of the data points (n) is tabulated next to the corresponding data point. Oocyte nuclei used were isolated using protocol L. The curves are simple activating Hill equation fits (Eq. 2) with the same K act = 280 nM and H act = 2.7. The dashed, dotted, and continuous curves have P max = 0.18, 0.35, and 0.84 for [InsP3] = 10 nM, 20 nM, and 10 μM, respectively.
Because it seemed paradoxical that InsP3 activates channel gating by modulating the ability of Ca2+ to inhibit the channel, and yet InsP3R channels that exhibit no high [Ca2+]i inhibition still require InsP3 for gating, we examined the effects of subsaturating [InsP3] on channel gating under conditions that abolish high [Ca2+]i inhibition. It was shown previously that in the presence of a subsaturating concentration of InsP3 (10–33 nM), InsP3R channels isolated directly into NCaS (protocol Nd) were much more sensitive to Ca2+ inhibition than those exposed to higher [InsP3] (Mak et al., 1998). In contrast, we observed that channels in nuclei isolated into and incubated in ULCaS (protocol L), and activated by subsaturating concentrations of InsP3 (10–20 nM) exhibited no Ca2+ inhibition. Channel activities were observed in 340 μM [Ca2+]i, a normally inhibiting [Ca2+]i, as well as in 4.2 μM [Ca2+]i (Fig. 6 B) with similar channel P o (Fig. 6 C). Importantly, the maximum P o observed in subsaturating [InsP3] was lower than that observed in saturating [InsP3] (c.f. Fig. 4, [Ca2+]i = 5.5 and 340 μM, and Fig. 6 B). Within the subsaturating range, i.e., [InsP3] <100 nM, increasing [InsP3] affected the channel activity mainly by tuning the value of P max in the simple activating Hill equation (Eq. 2) (Fig. 6 C), instead of affecting the Ca2+ inhibitory parameters (K inh or H inh), but not P max in the biphasic Hill equation (Eq. 1), as normally observed in the InsP3R channel exposed to NCaS (Mak et al., 1998). Thus, the effect of InsP3 on the InsP3R channel in ULCaS was dramatically different from that observed in NCaS.
Reversibility of the Regulation by Bath [Ca2+] of Ca2+ Inhibition of the InsP3R Channel
It is possible, as we stated before, that the inhibition of InsP3R gating by high [Ca2+]i is mediated by some molecule that is tightly bound to the InsP3R in the NCaS bath, and that dissociates from the channel in the presence of extremely low [Ca2+] in the ULCaS bath. Dissociation of this putative effector from the InsP3R channel can then render the channel insensitive to inhibition by high [Ca2+]i. Accordingly, after dissociation, the putative effector molecule could possibly diffuse away into the essentially infinitely large volume of the bath. If this model is correct, the loss of Ca2+ inhibition should be irreversible. To explore the reversibility of the loss of Ca2+ inhibition, we performed patch-clamp experiments on nuclei isolated from the same batch of oocytes using different isolation/incubation protocols. As described above, Ca2+ inhibition was abrogated when the nuclei were isolated into ULCaS bath by protocol L (Fig. 7 A). However, when the nuclei were returned to the NCaS bath for 20 min before patch clamping (protocol LN, Fig. 1), no InsP3R channel activities were detected at [Ca2+]i = 290 μM (Fig. 7 B) in any of the 11 patches obtained, even though channel gating was observed in 4 out of 5 patches using pipette solutions with [Ca2+]i = 5.5 μM (Fig. 7 C). Thus, despite prior exposure to ULCaS, normal Ca2+ inhibition of InsP3R channel gating was restored when the nuclei were transferred back into NCaS. This restoration of normal Ca2+ inhibition was in turn reversible. Reexposure of the nuclei to ULCaS (protocol LNL, Fig. 1) again eliminated normal Ca2+ inhibition of gating (Fig. 7 D). The InsP3R channels in nuclei isolated by protocol LNL exhibited the same P o (Fig. 5, filled square) as those in nuclei isolated into ULCaS by protocol L without ever being exposed to NCaS (Fig. 5, open circles). These experiments indicated, first, that abolition of Ca2+ inhibition of channel gating by exposure of nuclei to ultra-low bath [Ca2+] was fully reversible, and second, that it was affected only by the [Ca2+] of the bathing solution in which the patch-clamp experiments were performed, independent of the history of bath [Ca2+] to which the nuclei were previously exposed. These results suggest either that the sensitivity of Ca2+ inhibition of the InsP3R to the bath [Ca2+] is an intrinsic property of the InsP3R channel, or that it is mediated by some molecule that remains in a stable complex with the channel throughout the multiple transfers of the nucleus into various baths containing different [Ca2+].
Figure 7.

Typical current traces obtained from nuclei isolated using different protocols, all from the same batch of oocytes. Arrows indicate closed channel current levels. All pipette solutions contained 10 μM InsP3. (A) InsP3R channel activity in 290 μM [Ca2+]i in nuclei isolated by protocol L into ULCaS, n = 5. (B) Absence of InsP3R channel activity in 290 μM [Ca2+]i in nuclei isolated by protocol LN, n = 11. (C) InsP3R channel gating in 5.5 μM [Ca2+]i (n = 2) in the same nucleus as used in B. (D) InsP3R channel activity in 290 μM [Ca2+]i in nuclei isolated by protocol LNL, n = 4.
Is CaM Involved in the Regulation by Bath [Ca2+] of Ca2+ Inhibition of InsP3R Channel?
We explored the possible role of CaM in mediating the novel regulation of Ca2+ inhibition of InsP3R gating by the bath [Ca2+]. The working hypothesis was that addition of CaM would restore normal inhibition of channel gating by high [Ca2+]i after it had been relieved by exposure to the low [Ca2+] bath. Patch-clamp experiments were performed on nuclei isolated by protocol L into ULCaS bath, using a pipette solution containing 10 μM purified CaM with 10 μM InsP3 and high [Ca2+]i (290 μM). Nevertheless, InsP3R channel gating was observed in the presence of CaM (Fig. 8 A) that was indistinguishable (P > 0.05, Fig. 8 E) from that observed under the same conditions without CaM (compare Fig. 4, [Ca2+]i = 290 μM). Thus, addition of CaM did not reconstitute normal high [Ca2+]i inhibition of channel gating.
Figure 8.

(A) Typical current traces obtained from nuclei isolated from uninjected oocytes using protocol L with pipette solution containing 10 μM InsP3, 290 μM [Ca2+]i, and 10 μM purified CaM, n = 4. (B–D) Typical current traces obtained from nuclei isolated from oocytes expressing q.m. CaM with pipette solution containing 10 μM InsP3 and 290 μM [Ca2+]i. InsP3R channel activity was observed in 290 μM [Ca2+]i in nuclei isolated by protocol L (B, n = 6) or protocol LNL (D, n = 8), but not in nuclei isolated by protocol LN (C, n = 9). Arrows indicate closed channel current levels. (E) Histogram of InsP3R channel P o at 10 μM InsP3 and 290 μM [Ca2+]i observed in various nuclei under experimental conditions as tabulated.
We also performed a series of patch-clamp experiments on nuclei isolated from oocytes expressing the Ca2+-insensitive q.m. CaM. When the nuclei were isolated by protocol L into ULCaS bath, InsP3R channel activities were observed in high (290 μM) [Ca2+]i (six of eight patches, Fig. 8 B) as frequently as in the normally “permissive” [Ca2+]i between 500 nM and 5.5 μM (seven of eight patches). Furthermore, the channel P o was the same as that observed in the channels in nuclei isolated by protocol L from uninjected oocytes (P > 0.05, Fig. 8 E). In addition, expression of the q.m. CaM had no effect on the reversibility of the low [Ca2+] bath effect. Thus, no InsP3R channels were detected in any of the nine patches from nuclei isolated from mutant CaM-expressing oocytes by protocol LN (Fig. 8 C). Moreover, high [Ca2+]i inhibition of the channel was still completely abrogated in nuclei isolated from mutant CaM-expressing oocytes by protocol LNL. Thus, channel activities were observed in 290 μM [Ca2+]i (Fig. 8 D) with P d (8 of 13 patches) similar (P > 0.05) to that in [Ca2+]i between 500 nM and 5.5 μM (seven of eight patches). InsP3R channel P o in these nuclei from mutant CaM-expressing oocytes was the same as that observed in nuclei isolated from uninjected oocytes by protocol L or LNL (P from t test was >0.05, Fig. 8 E). Therefore, there were no differences between the Ca2+ inhibition (or lack thereof) of InsP3R channel gating observed in nuclei isolated by various protocols from oocytes overexpressing the mutant CaM and that observed in nuclei isolated from uninjected oocytes, under all experimental conditions.
DISCUSSION
Is There a Role of CaM in Inhibition by High [Ca2+]i of InsP3R-1 Gating?
Numerous investigations have explored the interactions between the InsP3R and CaM, but their nature, regulation, and functional effects on intracellular Ca2+ signaling are still far from clear. Although it was reported that CaM binding regulates high [Ca2+]i inhibition of the InsP3R-1 channel (Hirota et al., 1999; Michikawa et al., 1999), subsequent studies using microsomal fluxes or reconstituted channels in lipid bilayers have provided contradictory evidence (Zhang and Joseph, 2001; Nosyreva et al., 2002). In this study, we investigated the possible involvement of CaM in the high [Ca2+]i inhibition of single-channel InsP3R-1 gating using the nuclear patch clamp method (Mak and Foskett, 1994). This approach enables single-channel recording of endogenous and recombinant InsP3R channels in their native membrane environment. Similar biphasic regulation by [Ca2+]i of both the endogenous Xenopus type 1 channel and the recombinant rat type 3 InsP3R channel have been observed in previous nuclear patch clamp studies (Mak et al., 1998, 2001a). In this study, we directly explored the role of CaM in high [Ca2+]i inhibition of InsP3R channel gating. We have found no evidence to support the hypothesis that inhibition of InsP3R-1 channel activities by high [Ca2+]i is mediated by direct interaction between the InsP3R channel and CaM. First, in the presence of 500 μM W-7, a CaM antagonist that was previously reported to alleviate Ca2+ inhibition of InsP3R-1 channels reconstituted into bilayers (Michikawa et al., 1999), the X-InsP3R-1 was still inhibited by high [Ca2+]i in our nuclear patch-clamping experiments (Fig. 3, A and B). Second, overexpression in oocytes of a dominant-negative, Ca2+-insensitive q.m. CaM did not interfere with normal Ca2+ inhibition of the X-InsP3R-1 in the oocyte nuclear envelope (Fig. 3, C and D). Third, addition of CaM (10 μM) to the pipette solution did not reconstitute normal Ca2+ inhibition of InsP3R channel after it was abrogated by exposure of the channel to ULCaS bath (Fig. 8 A). Fourth, overexpression of q.m. CaM in oocytes did not affect the abrogation of Ca2+ inhibition by exposure of the channel to ULCaS bath, nor did it affect the restoration of Ca2+ inhibition when the channel was placed back in NCaS bath (Fig. 8, B–D). Furthermore, coimmunoprecipitation experiments did not detect any association between CaM and InsP3R-1 in the Xenopus oocytes. Therefore, whereas CaM may regulate intracellular Ca2+ signaling through other mechanisms, our experimental results, together with other recent publications (Zhang and Joseph, 2001; Nosyreva et al., 2002), indicate that it does not regulate inhibition of InsP3R-1 channel gating by high [Ca2+]i.
What then could be the mechanism of Ca2+ inhibition? The simplest hypothesis is that the Ca2+ binding sites responsible for Ca2+ inhibition of channel gating are contained within the structure of the InsP3R protein itself. Many regions of the protein have been shown to bind Ca2+ in in vitro studies (Sienaert et al., 1996, 1997). One or more of these or as yet unidentified sites may play a role, although there are no data available that address this issue. Alternately, another molecule could perhaps be involved. The InsP3R interacts with other proteins (Patel et al., 1999; Yang et al., 2002). Of interest, a calmodulin-like protein, CaBP1, interacts with high affinity with the ligand-binding region of the channel (Yang et al., 2002). Whereas it is highly unlikely that CaBP1 and its isoforms mediate Ca2+ inhibition, since they are likely neurally restricted and have been shown to stimulate channel gating (Yang et al., 2002), the identification of noncalmodulin Ca2+-binding protein interactions with the receptor lends credence to the notion that a Ca2+-binding protein could possibly be involved in mediating Ca2+ responses of the channel. Because Ca2+ inhibition of channel activity has been observed in a number of distinct experimental systems from different species, such a putative effector would need to be ubiquitously expressed and tightly associated with the channel.
A Novel Regulation of [Ca2+]i Inhibition of InsP3R-1 Channel Gating
Our investigations have revealed a novel CaM-independent regulation of the InsP3R-1 channel: abrogation of high [Ca2+]i inhibition of InsP3R-1 channel gating by exposure of the channel to ultra-low bath [Ca2+] (<5 nM). The physical location of the low [Ca2+]bath-sensing mechanism on the InsP3R protein is unknown. The abrogation could possibly be caused by low [Ca2+] in the perinuclear space between the inner and outer nuclear envelope, to which the lumenal side of the InsP3R-1 channel is exposed. In this case, exposure to the ultra-low bath [Ca2+] causes the lumenal [Ca2+] to fall to low levels due to uncompensated Ca2+ leak; and the [Ca2+] sensing mechanism responsible for switching high [Ca2+]i inhibition of InsP3R-1 channel on and off is located on the lumenal side of the InsP3R channel. The existence of a Ca2+-binding site on the lumenal side of the InsP3R-1 channel has been reported (Sienaert et al., 1996). Our previous studies indicated that the ionic composition of the solution in the perinuclear space of the isolated oocyte nucleus is likely to be similar to that of the bath solution (Mak and Foskett, 1997). The long lag time (∼300 s) between the isolation of the nucleus into the ultra-low [Ca2+] bath solution and the earliest detection of InsP3R channel activities that could not be inhibited by high [Ca2+]i may reflect the time required for the solution in the perinuclear space to become fully equilibrated with the bath solution, or the time taken for Ca2+ bound to the lumenal Ca2+-binding sites of the InsP3R channel to dissociate from the sites after the drop in lumenal [Ca2+], or a combination of the two.
Alternately, the [Ca2+]-sensing mechanism could possibly be located on the cytoplasmic side of the channel. In this case, the long lag time (∼300 s) between exposure of the channel to ultra-low [Ca2+] and the abrogation of high [Ca2+]i inhibition would imply that dissociation of Ca2+ from the sensing mechanism is slow (rate ∼0.003 s−1). Although such a sensing mechanism would be exposed to high [Ca2+] in the pipette solution as soon as the giga-ohm seal was formed, InsP3R channel activities were nevertheless observed for typically >10 s when the channel was exposed to [Ca2+]i ∼290 μM before the activities abruptly terminated (Mak and Foskett, 1997; Boehning et al., 2001). Thus, binding of Ca2+ to the sensing mechanism to restore normal high [Ca2+]i inhibition must also be a very slow process (rate <0.1 s−1). If the [Ca2+] sensing mechanism is in equilibrium with the cytoplasmic solution, the forward rate constant (k f) for Ca2+ dissociation from the [Ca2+]-sensing mechanism is ≈0.003 s−1 and the reverse rate constant (k r) is such that 0.1 s−1 ≈ k r × 290 μM. If the [Ca2+]-sensing mechanism is a simple Ca2+ binding site, then the equilibrium constant K (k f/k r) for Ca2+ dissociation from the site should then be ≈10 μM. However, abrogation of channel inhibition was not observed in our normal bath solutions that contain 300–500 nM Ca2+ (Mak et al., 1998), or in our physiological Ca2+ bath solution containing 50 nM free Ca2+. It could only be observed when bath [Ca2+] was reduced to very low levels. Thus, if the [Ca2+]bath-sensing mechanism is located on the cytoplasmic side of the channel, it is likely to be a set of cooperative Ca2+-binding sites. Further studies are necessary to distinguish whether cytoplasmic or lumenal [Ca2+] is being sensed in the disruption of high [Ca2+]i inhibition of the InsP3R, and to determine the molecular mechanisms involved in that process.
Mechanism of Regulation of High [Ca2+]i Inhibition of InsP3R-1 Channel Gating by Exposure to Low [Ca2+] Bath
A novel allosteric model, developed in the accompanying manuscript, can account for the effect of ultra-low [Ca2+] bath exposure on the abrogation of high [Ca2+]i inhibition as well as the effect of InsP3 to modulate maximum channel P o, rather than K inh, under these conditions. In brief, this model accounts for our results by postulating the existence of two functional inhibitory Ca2+ binding sites associated with each monomer of the tetrameric channel. One site is only inhibitory when the channel is not liganded with InsP3, because InsP3 binding relieves the Ca2+ inhibition imposed by this site. In contrast, the properties of the other inhibitory site are not affected by InsP3 binding. In normal physiological [Ca2+]i conditions, Ca2+ binding to this InsP3-insensitive site provides the observed high [Ca2+]i inhibition (K inh ∼50 μM) of the fully InsP3-liganded channel. The ability of this InsP3-insensitive site to be inhibitory is reversibly lost after exposure of the channel for >5 min to an ultra-low bath [Ca2+] (<5 nM). Thus, the observed abrogation of high [Ca2+]i inhibition of channel activity in saturating [InsP3] can be accounted for by the fact that there is no longer any functional inhibitory Ca2+-binding site. On the other hand, in the absence of InsP3, the InsP3-sensitive Ca2+ inhibition site is functional and keeps the channel closed. Thus, the channel still requires InsP3 to gate open even when the InsP3-insensitive site has been disrupted by exposure to ultra-low bath [Ca2+]. A detailed description of this model, which can account for these and many other features of ligand regulation of the channel observed in nuclear patch clamp experiments, is developed in the accompanying manuscript (Mak et al., 2003, in this issue).
Are Different Sensitivities to Inhibition by High [Ca2+]i a Fundamental Distinguishing Feature among the InsP3R Isoforms?
The three isoforms of InsP3R have complicated patterns of expression in various tissues with complex regulation by various mechanisms (Taylor et al., 1999). Because the permeation and conductance properties of the InsP3R isoforms are very similar (Mak et al., 2000; Ramos-Franco et al., 2000), differences among the isoforms in localization and channel gating and its regulation are likely to be reasons for the existence of InsP3R diversity. A review of published single-channel studies of various InsP3R isoforms suggests that different sensitivities to inhibition by high [Ca2+]i may be one distinguishing functional feature among the various InsP3R isoforms. Nevertheless, it is not clear whether such differences are intrinsic to the channels, or whether they are perhaps artificially generated by the different experimental protocols used for studying InsP3R channel activity.
In the presence of ∼1 μM InsP3, native and recombinant InsP3R-1 channels (including various splice variants) reconstituted into lipid bilayers exhibited similar strong inhibition by [Ca2+]i with half-maximal inhibitory [Ca2+]i of 0.1–2 μM (Bezprozvanny et al., 1991; Ramos-Franco et al., 1998a,b; Tu et al., 2002), whereas native Xenopus and recombinant rat InsP3R-1 channels studied in their native membrane environment using nuclear patch clamp techniques exhibited inhibition by high [Ca2+]i, but with a significantly higher half-maximal inhibitory [Ca2+]i of ∼50 μM (Mak et al., 1998; Boehning et al., 2001). When reconstituted into planar bilayers, Ca2+ inhibition of InsP3R-1 could be alleviated by very high [InsP3] (180 μM) (Kaftan et al., 1997; Moraru et al., 1999), whereas Ca2+ inhibition of InsP3R-1 studied in the native membrane environment was not further affected by [InsP3] once the channel was saturated with [InsP3] >100 nM (Mak et al., 1998).
InsP3R-2 channels reconstituted in lipid bilayers exhibited variable but low sensitivity to inhibition by high [Ca2+]i, with a half-maximal [Ca2+]i of ∼400 μM for recombinant InsP3R-2 channels (Ramos-Franco et al., 2000) and >1 mM for native channels (Ramos-Franco et al., 1998b, 2000) in 1 μM InsP3.
Native type 3 InsP3R channels reconstituted into lipid bilayers exhibited no detectable inhibition by high [Ca2+]i and its P o remained at its maximum value (∼0.05) in [Ca2+]i between 1 and 100 μM in the presence of 2 μM InsP3 (Hagar et al., 1998). In marked contrast, recombinant r-InsP3R-3 in the nuclear membrane of oocytes is inhibited by high [Ca2+]i in an InsP3-dependent manner very similar to that for X-InsP3R-1 under identical experimental conditions (Mak et al., 2001a).
How can we account for such divergent results? Our studies here demonstrate that Ca2+ inhibition of the X-InsP3R-1 channel in its native membrane environment can be completely, specifically and reversibly abrogated under certain experimental conditions (after exposure to a nominally Ca2+-free bath). Associated with this effect, the InsP3 dependence of the channel P o was also changed—normally, InsP3 affects the apparent affinity of the inhibitory Ca2+-binding sites of the channel (Mak et al., 1998), whereas after ULCaS bath exposure, InsP3 affects the maximum P o observed (Fig. 6 C). Of note, this InsP3 dependence of maximum P o is very similar to the observed effect of InsP3 on the P o of InsP3R-2 channels reconstituted into lipid bilayers (Ramos-Franco et al., 1998b). These observations raise the intriguing possibility that the observed differences in the sensitivities to Ca2+ inhibition of various InsP3R isoforms may be a consequence of the different environment and/or isolation conditions to which the channels were exposed, rather than the result of differences in fundamental intrinsic characteristics of the individual isoforms. For example, the InsP3R-1 and InsP3R-3 channel isoforms exhibited very similar inhibition by high [Ca2+]i when they are studied in a native ER membrane environment (Mak et al., 1998, 2001a), but they behaved differently in reconstitution systems. We suggest that it is worth considering the possibility that procedures employed in the isolation and reconstitution and recording of the InsP3R-3 used in (Hagar et al., 1998) disrupted the normal high [Ca2+]i inhibition of the InsP3R-3, causing the observed lack of Ca2+ inhibition, in very much the same way that exposure to a ULCaS bath abrogated the high [Ca2+]i inhibition of InsP3R-1 observed in this study. Whereas the procedures used in the isolation and reconstitution and recording of InsP3R-1 by themselves did not eliminate high [Ca2+]i inhibition of the channel (Bezprozvanny et al., 1991; Ramos-Franco et al., 1998a,b; Tu et al., 2002), they may account for the ability, observed only in the reconstituted systems, of extremely high [InsP3] to abrogate high [Ca2+]i inhibition (Kaftan et al., 1997; Moraru et al., 1999). By the same token, it is possible that the very low sensitivity to high [Ca2+]i inhibition of the InsP3R-2 channel isoform reconstituted in lipid bilayers (Ramos-Franco et al., 1998b, 2000) was induced by the isolation and reconstitution and recording protocols. Obviously, these issues will need to be resolved in future studies, for example, of the Ca2+ responses of type 2 InsP3R channels in the native ER membrane environment, under the same experimental conditions as those used for the types 1 and 3 InsP3R isoforms; and of the sensitivities of Ca2+ inhibition of the other InsP3R isoforms to exposure to ultra-low bath [Ca2+]. Nevertheless, our identification in this study of conditions that can radically alter the [Ca2+]i inhibition properties of the channel suggests that careful consideration of the isolation protocols and other conditions to which InsP3R channels are exposed before they are examined will be warranted in future studies.
Acknowledgments
This work was supported by grants to J.K. Foskett from the NIH (MH59937, GM56328) and to D.-O.D. Mak from the American Heart Association (9906220U).
Olaf S. Andersen served as editor.
Abbreviations used in this paper: InsP3, inositol 1,4,5-trisphosphate; InsP3R, InsP3 receptor; NCaS, regular [Ca2+] bath solution; PCaS, physiological [Ca2+] bath solution; r-InsP3R-3, rat type 3 InsP3R; CaM, calmodulin; ULCaS, ultra-low [Ca2+] bath solution; X-InsP3R-1, Xenopus type 1 InsP3R.
References
- Baudet, S.B., L. Hove-Madsen, and D.M. Bers. 1994. How to make and use calcium-specific mini- and microelectrodes. A Practical Guide to the Study of Calcium in Living Cells. 1st ed. R. Nuccitelli, editor. Academic Press, San Diego. 94–114.
- Berridge, M.J. 1993. Inositol trisphosphate and calcium signalling. Nature. 361:315–325. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny, I., J. Watras, and B.E. Ehrlich. 1991. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 351:751–754. [DOI] [PubMed] [Google Scholar]
- Boehning, D., S.K. Joseph, D.-O.D. Mak, and J.K. Foskett. 2001. Single-channel recordings of recombinant inositol trisphosphate receptors in mammalian nuclear envelope. Biophys. J. 81:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardy, T.J., and C.W. Taylor. 1998. A novel role for calmodulin: Ca2+-independent inhibition of type-1 inositol trisphosphate receptors. Biochem. J. 334:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar, R.E., A.D. Burgstahler, M.H. Nathanson, and B.E. Ehrlich. 1998. Type III InsP3 receptor channel stays open in the presence of increased calcium. Nature. 396:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota, J., T. Michikawa, T. Natsume, T. Furuichi, and K. Mikoshiba. 1999. Calmodulin inhibits inositol 1,4,5-trisphosphate-induced calcium release through the purified and reconstituted inositol 1,4,5- trisphosphate receptor type 1. FEBS Lett. 456:322–326. [DOI] [PubMed] [Google Scholar]
- Jiang, Q., D.-O.D. Mak, S. Devidas, E.M. Schwiebert, A. Bragin, Y. Zhang, W.R. Skach, W.B. Guggino, J.K. Foskett, and J.F. Engelhardt. 1998. Cystic fibrosis transmembrane conductance regulator-associated ATP release is controlled by a chloride sensor. J. Cell Biol. 143:645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph, S.K., and S. Samanta. 1993. Detergent solubility of the inositol trisphosphate receptor in rat brain membranes. Evidence for association of the receptor with ankyrin. J. Biol. Chem. 268:6477–6486. [PubMed] [Google Scholar]
- Joseph, S.K., C. Lin, S. Pierson, A.P. Thomas, and A.R. Maranto. 1995. Heteroligomers of type-I and type-III inositol trisphosphate receptors in WB rat liver epithelial cells. J. Biol. Chem. 270:23310–23316. [DOI] [PubMed] [Google Scholar]
- Joseph, S.K. 1996. The inositol trisphosphate receptor family. Cell. Signal. 8:1–7. [DOI] [PubMed] [Google Scholar]
- Kaftan, E.J., B.E. Ehrlich, and J. Watras. 1997. Inositol 1,4,5-trisphosphate (InsP3) and calcium interact to increase the dynamic range of InsP3 receptor-dependent calcium signaling. J. Gen. Physiol. 110:529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keen, J.E., R. Khawaled, D.L. Farrens, T. Neelands, A. Rivard, C.T. Bond, A. Janowsky, B. Fakler, J.P. Adelman, and J. Maylie. 1999. Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. J. Neurosci. 19:8830–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C., J. Widjaja, and S.K. Joseph. 2000. The interaction of calmodulin with alternatively spliced isoforms of the type-I inositol trisphosphate receptor. J. Biol. Chem. 275:2305–2311. [DOI] [PubMed] [Google Scholar]
- Maeda, N., T. Kawasaki, S. Nakade, N. Yokota, T. Taguchi, M. Kasai, and K. Mikoshiba. 1991. Structural and functional characterization of inositol 1,4,5- trisphosphate receptor channel from mouse cerebellum. J. Biol. Chem. 266:1109–1116. [PubMed] [Google Scholar]
- Mak, D.-O.D., and J.K. Foskett. 1994. Single-channel inositol 1,4,5-trisphosphate receptor currents revealed by patch clamp of isolated Xenopus oocyte nuclei. J. Biol. Chem. 269:29375–29378. [PubMed] [Google Scholar]
- Mak, D.-O.D., and J.K. Foskett. 1997. Single-channel kinetics, inactivation, and spatial distribution of inositol trisphosphate (IP3) receptors in Xenopus oocyte nucleus. J. Gen. Physiol. 109:571–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., and J.K. Foskett. 1998. Effects of divalent cations on single-channel conduction properties of Xenopus IP3 receptor. Am. J. Physiol. 275:C179–C188. [DOI] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 1998. Inositol 1,4,5-trisphosphate activation of inositol trisphosphate receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc. Natl. Acad. Sci. USA. 95:15821–15825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, V. Raghuram, Y. Yue, S.K. Joseph, and J.K. Foskett. 2000. Single-channel properties in endoplasmic reticulum membrane of recombinant type 3 inositol trisphosphate receptor. J. Gen. Physiol. 115:241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 2001. a. Regulation by Ca2+ and inositol 1,4,5-trisphosphate (InsP3) of single recombinant type 3 InsP3 receptor channels. Ca2+ activation uniquely distinguishes types 1 and 3 InsP3 receptors. J. Gen. Physiol. 117:435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 2001. b. ATP-dependent adenophostin activation of inositol 1,4,5-trisphosphate receptor channel gating: kinetic implications for the durations of calcium puffs in cells. J. Gen. Physiol. 117:299–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak, D.-O.D., S. McBride, and J.K. Foskett. 2003. Spontaneous channel activity of the inositol 1,4,5-triphosphate (InsP3) receptor (InsP3R). Application of allosteric modeling to calcium and InsP3 regulation of InsP3R single-channel gating. J. Gen. Physiol. 122:583–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michikawa, T., J. Hirota, S. Kawano, M. Hiraoka, M. Yamada, T. Furuichi, and K. Mikoshiba. 1999. Calmodulin mediates calcium-dependent inactivation of the cerebellar type 1 inositol 1,4,5-trisphosphate receptor. Neuron. 23:799–808. [DOI] [PubMed] [Google Scholar]
- Monkawa, T., A. Miyawaki, T. Sugiyama, H. Yoneshima, M. Yamamoto-Hino, T. Furuichi, T. Saruta, M. Hasegawa, and K. Mikoshiba. 1995. Heterotetrameric complex formation of inositol 1,4,5-trisphosphate receptor subunits. J. Biol. Chem. 270:14700–14704. [DOI] [PubMed] [Google Scholar]
- Moraru, I.I., E.J. Kaftan, B.E. Ehrlich, and J. Watras. 1999. Regulation of type 1 inositol 1,4,5-trisphosphate-gated calcium channels by InsP3 and calcium: Simulation of single channel kinetics based on ligand binding and electrophysiological analysis. J. Gen. Physiol. 113:837–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva, E., T. Miyakawa, Z. Wang, L. Glouchankova, A. Mizushima, M. Iino, and I. Bezprozvanny. 2002. The high affinity calcium-calmodulin-binding site does not play a role in modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by calcium and calmodulin. Biochem. J. 365:659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucifora, F.C., Jr., A.H. Sharp, S.L. Milgram, and C.A. Ross. 1996. Inositol 1,4,5-trisphosphate receptors in endocrine cells: localization and association in hetero- and homotetramers. Mol. Biol. Cell. 7:949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, S., S.K. Joseph, and A.P. Thomas. 1999. Molecular properties of inositol 1,4,5-trisphosphate receptors. Cell Calcium. 25:247–264. [DOI] [PubMed] [Google Scholar]
- Ramos-Franco, J., S. Caenepeel, M. Fill, and G. Mignery. 1998. a. Single channel function of recombinant type-1 inositol 1,4,5-trisphosphate receptor ligand binding domain splice variants. Biophys. J. 75:2783–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Franco, J., M. Fill, and G.A. Mignery. 1998. b. Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biophys. J. 75:834–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Franco, J., D. Bare, S. Caenepeel, A. Nani, M. Fill, and G. Mignery. 2000. Single-channel function of recombinant type 2 inositol 1,4, 5- trisphosphate receptor. Biophys. J. 79:1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienaert, I., H. De Smedt, J.B. Parys, L. Missiaen, S. Vanlingen, H. Sipma, and R. Casteels. 1996. Characterization of a cytosolic and a luminal Ca2+ binding site in the type I inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 271:27005–27012. [DOI] [PubMed] [Google Scholar]
- Sienaert, I., L. Missiaen, H. De Smedt, J.B. Parys, H. Sipma, and R. Casteels. 1997. Molecular and functional evidence for multiple Ca2+-binding domains in the type 1 inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 272:25899–25906. [DOI] [PubMed] [Google Scholar]
- Taylor, C.W. 1998. Inositol trisphosphate receptors: Ca2+-modulated intracellular Ca2+ channels. Biochim. Biophys. Acta. 1436:19–33. [DOI] [PubMed] [Google Scholar]
- Taylor, C.W., A.A. Genazzani, and S.A. Morris. 1999. Expression of inositol trisphosphate receptors. Cell Calcium. 26:237–251. [DOI] [PubMed] [Google Scholar]
- Tu, H., T. Miyakawa, Z. Wang, L. Glouchankova, M. Iino, and I. Bezprozvanny. 2002. Functional characterization of the Type 1 inositol 1,4,5-trisphosphate receptor coupling domain SII(+/−) splice variants and the Opisthotonos mutant form. Biophys. J. 82:1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcikiewicz, R.J. 1995. Type I, II, and III inositol 1,4,5-trisphosphate receptors are unequally susceptible to down-regulation and are expressed in markedly different proportions in different cell types. J. Biol. Chem. 270:11678–11683. [DOI] [PubMed] [Google Scholar]
- Xia, X.M., B. Fakler, A. Rivard, G. Wayman, T. Johnson-Pais, J.E. Keen, T. Ishii, B. Hirschberg, C.T. Bond, S. Lutsenko, et al. 1998. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 395:503–507. [DOI] [PubMed] [Google Scholar]
- Yamada, M., A. Miyawaki, K. Saito, T. Nakajima, M. Yamamoto-Hino, Y. Ryo, T. Furuichi, and K. Mikoshiba. 1995. The calmodulin-binding domain in the mouse type 1 inositol 1,4,5-trisphosphate receptor. Biochem. J. 308:83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J., S. McBride, D.O. Mak, N. Vardi, K. Palczewski, F. Haeseleer, and J.K. Foskett. 2002. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc. Natl. Acad. Sci. USA. 99:7711–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., and S.K. Joseph. 2001. Effect of mutation of a calmodulin binding site on Ca2+ regulation of inositol trisphosphate receptors. Biochem. J. 360:395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]