

Abstract
The potentiation by Ca2+ of inositol 1,4,5-trisphosphate (IP3)-induced Ca2+ release was studied by measuring luminal Ca2+ concentrations of the Ca2+ stores using a fluorescent Ca2+ indicator, furaptra, in permeabilized smooth muscle cells.
Ca2+ release at 10 μM IP3 was potentiated by an increase in the cytoplasmic Ca2+ concentration in the presence of 10 mM EGTA. This effect was not due to the pharmacological effect of EGTA, because changes in the EGTA concentration at a constant Ca2+ concentration had no effect on the Ca2+ release rate.
With an increase in the cytoplasmic Ca2+ concentration from 30 to 630 nM, the Ca2+ release rate at a saturating IP3 concentration increased 110-fold and the EC50 for IP3 increased from 0.07 to 1.0 μM. It was also indicated that the relationship between Ca2+ concentration and Ca2+ release rate was shifted towards higher Ca2+ concentrations at higher IP3 concentrations.
These results suggest that IP3 and submicromolar concentrations of Ca2+ allosterically lower the affinity of the IP3 receptor for each other and are both required for IP3 receptor activation. These properties enable the IP3 receptors to detect simultaneous increases in IP3 and Ca2+ concentrations.
Agonist-activated cells often show intermittent increases in intracellular Ca2+ concentration, or Ca2+ oscillations, which are usually accompanied by intracellular and/or intercellular Ca2+ waves. These properties of the Ca2+ signalling in stimulated cells resemble the membrane action potentials that exhibit temporal oscillations and propagate as spatial waves. In analogy to the generation of action potentials in excitable cells, it has been thought that the underlying mechanism of the Ca2+ waves and oscillations is the presence of a Ca2+-based excitable medium, in which an increase in Ca2+ concentration activates release of Ca2+ from its stores in a regenerative manner. Although the mechanism of these complex Ca2+ mobilization patterns is not fully understood, Ca2+-mediated feedback regulation of ryanodine receptor (RyR) or IP3 receptor (IP3R) activity is likely to be a key component of the excitable medium in many types of cells (Berridge, 1993). Indeed, RyR has been shown to function by Ca2+-induced Ca2+ release (CICR) and this seems to be the mechanism of Ca2+ oscillations and waves in striated muscle cells (Endo, Tanaka & Ogawa, 1970; Takamatsu & Wier, 1990). It has been shown that IP3R activity is also controlled by changes in the cytoplasmic Ca2+ concentration, in a biphasic manner: Ca2+ at submicromolar concentrations potentiates IP3-induced Ca2+ release, while Ca2+ at higher concentrations inhibits it (Iino, 1990; Bezprozvanny, Watras & Ehrlich, 1991; Finch, Turner & Goldin, 1991; Bootman, Missiaen, Parys, De Smedt & Casteels, 1995). Furthermore, it has been demonstrated that IP3 induces Ca2+ release from the Ca2+ stores in permeabilized smooth muscle cells in a regenerative manner, via the Ca2+-mediated feedback regulation of IP3R activity (Iino & Endo, 1992). However, the quantitative aspects of the IP3R activation are not fully understood and it is not yet known how Ca2+ potentiates IP3R activity.
To elucidate the mechanism of the Ca2+-mediated potentiation of IP3R activity, we analysed the Ca2+ dependence of IP3-induced Ca2+ release using a recently developed method for real-time measurement of the luminal Ca2+ concentrations of the Ca2+ stores (Hirose & Iino, 1994). Our results show that Ca2+ potentiates IP3R activity not by increasing IP3R affinity for IP3, but because it is absolutely required for IP3R activity. This property enables the IP3R to detect a simultaneous increase in the concentrations of two different intracellular signals: Ca2+ and IP3.
METHODS
Preparation
Guinea-pigs (weight, ∼300 g each) were stunned and exsanguinated. Segments of the portal vein were dissected from the liver and transferred to a dissecting chamber containing physiological salt solution (PSS). The vein was longitudinally cut open, and the endothelium was removed by rubbing with a small piece of paper. Thin smooth muscle bundles (3 mm in length, 150–250 μm in width and 50–60 μm in thickness) were obtained after removal of the adventitial tissue, and were tied to a thin stainless steel wire. We employed furaptra as an fluorescent indicator of Ca2+ concentration within the Ca2+ stores, because furaptra is a low-affinity Ca2+ indicator (∼48 μM at ionic strength of 0.2 M, pH 7; K. Hirose & M. Iino, unpublished observation) and is thus suitable for measurement of the high Ca2+ concentrations expected inside the Ca2+ stores. The specimens were incubated in PSS containing 20–40 μM furaptra-AM (the acetoxymethyl ester form of furaptra) for 3–5 h at 35°C. The furaptra-loaded fibre bundles were then permeabilized by incubation with 40 μM β-escin in relaxing solution to wash out the furaptra in the cytoplasm. This procedure enables measurement of the concentrations of Ca2+ within the intracellular organelles.
Experimental apparatus
The apparatus used for the experiments has been described previously (Hirose & Iino, 1994). Briefly, the specimen tied to the stainless steel wire was inserted into a small glass capillary (i.d. 400 μm) which was attached to the cuvette holder of a fluorescence spectrophotometer (CAF110, JASCO, Tokyo, Japan). The specimen was then illuminated with 340 nm and 375 nm wavelength light alternately and the emitted light was directed to a photomultiplier tube. The ratio of the fluorescence intensity at 340 nm excitation to that at 380 nm excitation was used as an indicator of the luminal Ca2+ concentration. One end of the glass capillary was connected to the common output of an electrically driven valve which had sixteen channel inputs.
The other end of the capillary was connected to two peristaltic pumps. One of the pumps was used to change the solution around the specimen rapidly (within 1 s). Continuous slow flow of the solution over the specimen was driven by another peristaltic pump.
Protocol
The solution within the capillary was sequentially exchanged to load and release Ca2+ from the Ca2+ stores of the specimen. After the fibre bundle was bathed in relaxing solution for 60 s, Ca2+ was loaded into the Ca2+ stores by application of loading solution, containing 100 nM Ca2+ and 0.5 mM MgATP, for 25 s. The Ca2+ loading was terminated by the exchange of loading solution with G10RMg0 solution, which contained 10 mM EGTA with neither Ca2+ nor MgATP. (All the solutions used in the subsequent steps also contained no MgATP.) The G10RMg0 solution was exchanged with G1RMg0 solution in which the concentration of EGTA was reduced to 1 mM. After 30 or 60 s, the concentration of Ca2+ was increased by application of assay solution containing 10 mM EGTA and various amounts of Ca2+. IP3 (30 nM-10 μM) was subsequently applied for 120 s in the same solution. The Ca2+ stores were then completely depleted by 120 s application of 10 μM IP3 in the presence of 300 nM Ca2+. After IP3 and Ca2+ were washed out for 60 s, the specimen was again bathed in the relaxing solution to start a new run. The Ca2+ loading and Ca2+ release sequences were repeated several times for each specimen.
Measurement of IP3R activity
The IP3R activity was evaluated in terms of the initial Ca2+ release rate as described previously (Hirose & Iino, 1994). The observed changes in ratio of fluorescence intensities were normalized so that 1 and 0 corresponded, respectively, to the values just before the application of IP3 and after complete depletion by 10 μM IP3 for 120 s.
The initial part of the normalized time course, where the normalized fluorescence ratio signal remained between 1.0 and 0.8, was fitted by a single exponential function, e-rt. The rate constant, r, thus estimated was used as an index of the extent of IP3R activation.
Solutions
The solutions used for the present experiments were essentially the same as those previously described (Iino, 1990; Hirose & Iino, 1994). PSS contained (mM): 150 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, 5.6 glucose and 5 Hepes (pH 7.4). Relaxing solution contained (mM): 116 potassium methanesulphonate, 3.31 ATP, 0.554 magnesium methanesulphonate and 1 EGTA. Loading solution contained (mM): 112 potassium methanesulphonate, 3.32 ATP, 0.564 magnesium methanesulphonate, 1.96 CaEGTA and 8.04 EGTA. G10RMg0 solution contained 112 mM potassium methanesulphonate and 10 mM EGTA. CaG10RMg0 solution contained 112 mM potassium methanesulphonate and 10 mM CaEGTA. G1RMg0 contained 139 mM potassium methanesulphonate and 1 mM EGTA. All solutions except for PSS contained (mM): 20 NaN3, 1 EGTA and 20 Pipes (pH 7.0). Assay solutions containing Ca2+ at various concentrations were prepared by mixing CaG10RMg0 solution and G10RMg0 solution at appropriate ratios. The ratios of the volume of CaG10RMg0 solution to the total volume of the assay solution were: 0.072, 0.196, 0.329, 0.434 and 0.608 for 30, 100, 200, 300 and 630 nM Ca2+, respectively.
Chemicals
Furaptra-AM (mag-fura-2 AM) was purchased from Molecular Probes, and IP3 from Dojin-do (Kumamoto, Japan). All other drugs were purchased either from Sigma or from Wako Chemicals (Tokyo, Japan).
RESULTS
Ca2+-mediated potentiation of IP3-induced Ca2+ release
Figure 1A shows the time courses of Ca2+ release from the Ca2+ stores of permeabilized smooth muscle cells, following application of 10 μM IP3 at various cytoplasmic Ca2+ concentrations in the absence of MgATP. The Ca2+ release rate increased as the concentration of Ca2+ was increased, within the range used in the experiments (30–630 nM). Figure 1B shows the results compiled from five preparations. It can be seen that the Ca2+ release rate at 10 μM IP3 increased markedly with increasing cytoplasmic Ca2+ concentration. This observation is consistent with previous reports based on different methods (Iino, 1990; Bezprozvanny et al. 1991; Finch et al. 1991). In the absence of IP3, changes in cytoplasmic Ca2+ concentration had no effect on the time course of Ca2+ release (not shown). Thus, the observed potentiation of Ca2+ release by Ca2+ did not involve Ca2+-induced Ca2+ release (CICR) via ryanodine receptors (RyRs). This result is consistent with the results of a previous study showing CICR occurs only when the cytoplasmic Ca2+ concentration exceeds 1 μM in smooth muscle cells (Iino, 1989).
Figure 1. IP3-induced Ca2+ release at various cytoplasmic Ca2+ concentrations.
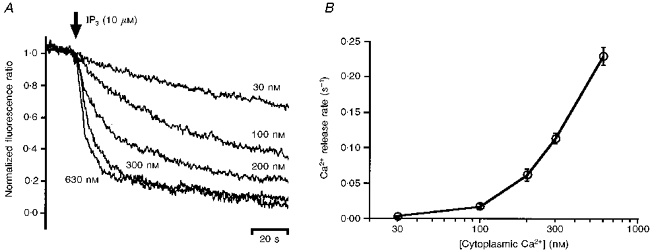
A, time courses of Ca2+ release at the indicated cytoplasmic Ca2+ concentrations. Application of 10 μM IP3 was started at the time point indicated by the arrow. B, the initial rate of Ca2+ release was plotted against the cytoplasmic Ca2+ concentration (mean ±s.e.m., n = 5).
We did not measure the Ca2+ release rate at higher cytoplasmic Ca2+ concentrations (> 1 μM), at which Ca2+ has been reported to inhibit IP3-induced Ca2+ release (Iino, 1990). This is because application of high concentrations of Ca2+ (> 1 μM) alone increased the fluorescence intensity ratio of furaptra even in the absence of MgATP. The change in the fluorescence intensity ratio was not due to passive influx of Ca2+ into the Ca2+ stores, because the increase in the fluorescence intensity ratio was reversed upon removal of Ca2+ in the absence of IP3. Therefore, the change in the furaptra fluorescence intensity ratio at high concentrations of Ca2+ (> 1 μM) is likely to be due to the binding of Ca2+ to compartmentalized furaptra which is accessed only at high cytoplasmic Ca2+ concentrations. Although this compartment might be mitochondria, which are known to respond to high concentrations of Ca2+ and have been shown to accumulate Ca2+-sensitive fluorescent dyes including furaptra (Hoffer, Schlue, Curci & Machen, 1995; Golovina & Blaustein, 1997), we did not study the possibility further. We thus analysed the Ca2+ release only at < 1 μM Ca2+, ensuring the observed time courses were free from such effects.
Effect of EGTA on Ca2+ release rate
Ca2+ chelating agents such as BAPTA have been reported to inhibit the binding of IP3 to its receptor, resulting in attenuation of Ca2+ release even at a constant Ca2+ concentration (Richardson & Taylor, 1993). This pharmacological effect of Ca2+ chelating agents might interfere with the observed potentiation by changing the Ca2+ concentration, because the concentration of Ca2+-unbound EGTA, which has been reported to inhibit IP3 binding more strongly than Ca2+-bound EGTA, decreased when the Ca2+ concentration was raised in the experiments presented in Fig. 1. Although the inhibitory effect of EGTA on IP3 binding has been reported to be weaker than those of other chelators, we tested the possibility that the potentiation of Ca2+ release was due to the pharmacological effects of EGTA.
We found that the potentiating effect of Ca2+ (300 nM) did not change when the total EGTA concentration was increased from 10 mM to 14.3 mM or decreased to 4.3 mM so that the concentration of Ca2+-unbound or Ca2+-bound EGTA, respectively, was the same as that at 100 nM Ca2+ with a 10 mM total EGTA concentration (Fig. 2). The results show that the extent of potentiation depends only on the free Ca2+ concentration and is independent of the concentration of Ca2+-bound and Ca2+-unbound EGTA, and indicate that the observed potentiation by Ca2+ of IP3-induced Ca2+ release is not the result of the inhibitory effect of EGTA on IP3 binding.
Figure 2. Extent of Ca2+-mediated potentiation of IP3-induced Ca2+ release is independent of EGTA concentration.
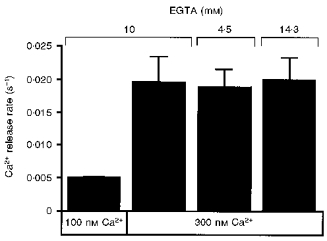
The total EGTA concentration at 300 nM Ca2+ was changed from 10 mM to either 4.5 mM or 14.3 mM for adjustment of the concentration of Ca2+-bound or Ca2+-unbound EGTA, respectively, to that at 100 nM Ca2+. The IP3 concentration was 100 nM. Means ±s.e.m., n = 5.
Effect of Ca2+ on IP3 sensitivity of Ca2+ release via IP3R
We then attempted to elucidate the mechanism by which Ca2+ potentiates IP3R activity. We first considered two extreme cases. One possible mechanism is that Ca2+ increases the affinity of IP3R for IP3. The other is that Ca2+ increases the maximal level of IP3R channel activity without changing the affinity of IP3R for IP3.
In the former mechanism, the maximal Ca2+ release rate at saturating IP3 concentrations should be independent of the Ca2+ concentration, although the EC50 for IP3 should decrease with increasing Ca2+ concentration. However, in the latter case, the maximum level of IP3R activation cannot be attained at low Ca2+ concentrations even with saturating concentrations of IP3, and the EC50 for IP3 should be constant regardless of the Ca2+ concentration.
To test the above hypotheses, we compared the IP3 dependence of Ca2+ release at high and low concentrations of Ca2+. Ca2+ was released at various concentrations of IP3 at both 300 and 100 nM Ca2+ (Fig. 3A). At 300 nM Ca2+, the rate of Ca2+ release increased markedly with increasing concentration of IP3, whereas at 100 nM Ca2+, the effect of an increase in the concentration of IP3 was far weaker. Figure 3B shows the relationship between IP3 concentration and initial rate of Ca2+ release at 100 and 300 nM Ca2+. We fitted Hill's equation to the data points,
where rmax, K and n represent maximal Ca2+ release rate, Ca2+ concentration required for half-maximal activation of Ca2+ release (EC50), and Hill coefficient, respectively. rmax was ∼6.8 times higher at 300 nM Ca2+ than at 100 nM Ca2+. Vertical scaling of the IP3 dependence curve at 100 nM Ca2+ (dotted line in Fig. 3B) by 6.8 times shows that the EC50 for IP3 was higher at 300 nM Ca2+ than at 100 nM Ca2+.
Figure 3. Ca2+ concentration dependence of IP3-induced Ca2+ release.
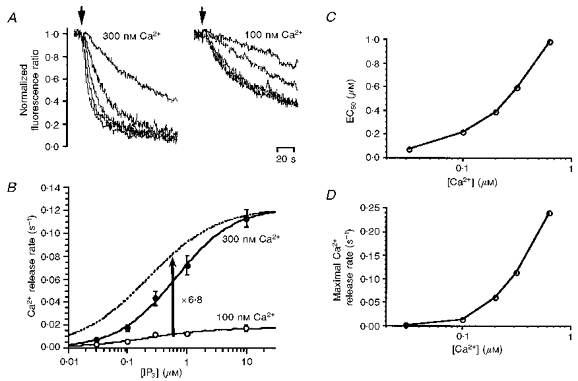
A, time course of Ca2+ release induced by IP3 at cytoplasmic Ca2+ concentrations of 300 nM (left) and 100 nM (right). B, the Ca2+ release rate was calculated from experimental results similar to those in A and plotted against the IP3 concentration. Open and filled circles represent Ca2+ release rates at 100 and 300 nM Ca2+, respectively. The data were fitted by Hill's equation to give continuous curves. The fitted curve for 100 nM Ca2+ was scaled up 6.8-fold (dotted curve). C, the IP3 concentration required for half-maximal activation of Ca2+ release (EC50) was plotted against the cytoplasmic Ca2+ concentration. D, the maximal Ca2+ release rate was plotted against the Ca2+ concentration.
We conducted similar experiments at various cytoplasmic Ca2+ concentrations and derived the relationship between the EC50 for IP3 and the Ca2+ concentration shown in Fig. 3C. The EC50 for IP3 increased from 0.07 to 1.0 μM with increasing cytoplasmic Ca2+ concentration from 30 to 630 nM. The maximal Ca2+ release rate also increased 110-fold with increasing concentration of Ca2+ (Fig. 3D), indicating that the activity of Ca2+ release channels is enhanced by Ca2+ even at a saturating concentration of IP3. In the presence of a low cytoplasmic Ca2+ concentration (below 100 nM), IP3 very weakly induced Ca2+ release. These results are inconsistent with the two extreme cases that we initially considered.
Both Ca2+ and IP3 are required for maximal IP3R activation
To obtain a further understanding of how IP3R is activated, we replotted the Ca2+ release rate against the Ca2+ and IP3 concentrations in a three-dimensional graph (Fig. 4A, spheres). It is obvious that the Ca2+ release rate increased markedly only when both the Ca2+ and the IP3 concentration were increased. Neither Ca2+ nor IP3 induced Ca2+ release in the absence of the other. These results indicate that Ca2+ and IP3 are simultaneously required for IP3R activation in smooth muscle cells.
Figure 4. Allosteric activation of IP3 receptors by Ca2+ and IP3.
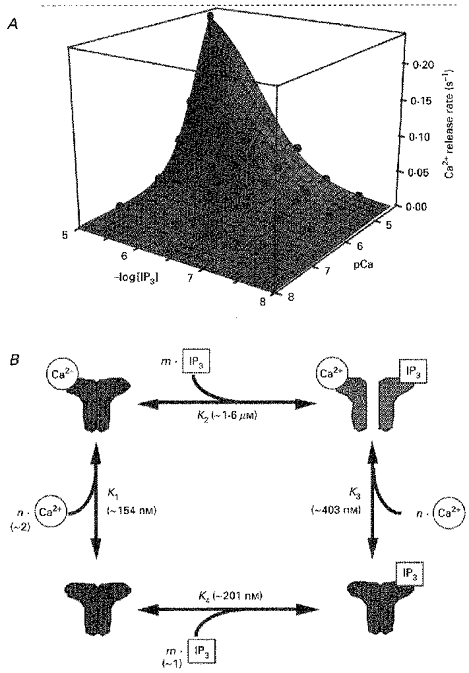
A, the Ca2+ release rate was plotted against the Ca2+ and IP3 concentrations. Experimental data (spheres) are fitted by the equation described in B, as shown by the curved surface. B, a possible mechanism of IP3 receptor activation. The channel has n Ca2+ binding sites and m IP3 binding sites. K1 and K3 represent Kd for Ca2+ binding to IP3-unbound and IP3-bound forms of the receptor, respectively. K2 and K4 represent Kd for IP3 binding to Ca2+-unbound and Ca2+-bound forms of the receptor, respectively.
DISCUSSION
In this study, we found that the submicromolar Ca2+-mediated potentiation of Ca2+ release via IP3R is not caused by an increase in the affinity of IP3R for IP3, and that cytoplasmic Ca2+ and IP3 are simultaneously required for the gating of the IP3R Ca2+ release channel. We showed that the cytoplasmic Ca2+ concentration in a submicromolar range determines the level of IP3R activity at near-saturating IP3 concentrations and that at the same time the increasing Ca2+ concentration lowers the affinity of IP3R for IP3 (Fig. 3).
Four-state model of IP3R activation
We constructed a minimum steady-state model to account for the observed features of IP3-induced Ca2+ release. The essential property of the model is that the binding of either Ca2+ or IP3 alone to the IP3R does not induce IP3R activation, but simultaneous binding of both Ca2+ and IP3 to IP3R leads to IP3R activation. We speculated that this simultaneous binding is essential, based on our finding that the rate of Ca2+ release is extremely low, even at an IP3 concentration as high as 10 μM, when the Ca2+ concentration is low (Figs 3D and 4A). Another important feature of our model is that the binding of Ca2+ and IP3 to IP3R is allosterically controlled by the binding of IP3 and Ca2+, respectively. We included this in our model, based on our finding that the EC50 for IP3 increased with increasing Ca2+ concentration (Fig. 3C). We noted a similar change in the Ca2+ dependence of Ca2+ release with an increase in the IP3 concentration (data not shown). A four-state model that accommodates the above considerations is shown in Fig. 4B. The stoichiometry of IP3 and Ca2+ binding was assumed to be m and n, respectively. The channel is open only in the Ca2+- and IP3-bound state. The Ca2+ release rate is thus described as:
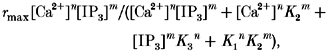 |
where rmax is the maximal Ca2+ release rate, K1 and K3 are the Kd for Ca2+ binding to IP3-unbound and -bound IP3R, respectively, and K2 is the Kd for IP3 binding to Ca2+-unbound IP3R. We found that the model describes the important features of the experimental data (Fig. 4A, curved surface). Using a least-squares fitting method of a mathematics software package (Mathematica, Worfram Research, Inc., IL, USA) running on a personal computer, we obtained the following parameter values for each reaction: K1, ∼154 nM; K2, ∼1.6 μM; K3, ∼403 nM; K4 (the Kd for IP3 binding to Ca2+-bound IP3R), ∼201 nM; m, ∼1; and n, ∼2. According to this model, a Ca2+-bound IP3R has about 8-fold lower affinity for IP3 than does a Ca2+-unbound IP3R. The affinity of the IP3R for Ca2+ also decreased about 2.5-fold upon binding of IP3.
Although our model successfully describes the present experimental data, it does not describe all the features of the Ca2+ dependence of IP3R within the entire physiological range of Ca2+ concentrations. At micromolar concentrations, Ca2+ has an inhibitory effect on IP3R activity (Iino, 1990; Bezprozvanny et al. 1991; Finch et al. 1991; Bootman et al. 1995). Unfortunately we could not study the Ca2+-mediated inhibition due to current experimental limitations. There are several possible ways in which the present model could be modified to include the inhibitory effect of Ca2+. One model is that Ca2+ binding to an inhibitory site alters the equilibrium of the four states in our model, so lowering the affinity of Ca2+ and/or IP3 for their respective activation sites, resulting in a decreased population of active IP3Rs (allosteric inhibition). In another model, the binding of Ca2+ to the inhibitory site occurs only in the IP3R that binds Ca2+ at activation sites and IP3 (sequential inhibition). Clearly we need more information to discriminate between these and other possibilities.
Comparison of the present results with those of the IP3 binding studies
Several biochemical reports regarding Ca2+-mediated regulation of IP3 binding to the IP3R have been published. Benevolensky et al. demonstrated the presence of high- and low-affinity IP3 binding sites in sarcoplasmic reticulum vesicles from aortic smooth muscle (Benevolensky, Moraru & Watras, 1994). The Kd of the low-affinity binding sites, accounting for > 90% of the total IP3 binding sites, depended on the cytoplasmic Ca2+ concentration. The Kd was 49.3 nM in the absence of Ca2+ and 155 nM in the presence of 2 μM Ca2+. Similar Ca2+-mediated inhibition of IP3 binding has been detected in cerebellar IP3R (Worley, Baraban, Supattapone, Wilson & Snyder, 1987; Hannaert-Merah, Coquil, Combettes, Claret, Mauger & Champeil, 1994). Only a single class of IP3 binding sites was detected in IP3R purified from human IP3R type 1 synthesized in a baculovirus expression vector system, and a Ca2+-dependent increase in Kd for IP3, from 79 nM at 3 nM Ca2+ to 312 nM at 1.4 μM Ca2+, has been reported (Yoneshima, Miyawaki, Michikawa, Furuichi & Mikoshiba, 1997). There are, however, contradictory results for several cell types, in which an increase in the Ca2+ concentration causes a decrease in Kd for IP3 (Marshall & Taylor, 1994). The effect of Ca2+ is complicated in rat basophilic leukaemia (RBL) cells, which have low- and high-affinity IP3 binding sites. An increase in the Ca2+ concentration from 100 to 500 nM results both in the conversion of a portion of the low-affinity sites to high-affinity sites and an increase in the Kd of the low-affinity sites (Watras, Moraru, Costa & Kindman, 1994). The Kd for IP3 binding to IP3R type 3 decreased with an increase in Ca2+ concentration (Yoneshima et al. 1997). Thus, the mode of Ca2+-dependent IP3 binding may depend on the subtype of the IP3R and/or cell type-specific post-translational modification of the IP3R. IP3R type 1 seems to be dominantly expressed in smooth muscle cells (Chadwick, Saito & Fleischer, 1990; De Smedt et al. 1994; Islam, Yoshida, Koga, Kojima, Kangawa & Imai, 1996). If this is also the case in guinea-pig portal vein, the present results seem to be in general agreement with the results of binding studies, in that the lowering of the sensitivity for IP3 is accompanied by an increase in the Ca2+ concentration. It would be interesting to examine the Ca2+ dependence of IP3-induced Ca2+ release in cells expressing only IP3R type 3.
Physiological significance of the requirement for both Ca2+ and IP3 for IP3R activation
The properties of the IP3R revealed in the present study are consistent in many ways with the role of IP3R as one of the key components for inducing Ca2+ oscillations. Firstly, in the presence of low concentrations of Ca2+, high concentrations of IP3 activate IP3R only weakly. This is consistent with the baseline Ca2+ oscillations which are a characteristic pattern of Ca2+ oscillations seen in many cell types (Berridge, 1993). In these baseline Ca2+ oscillations, after each Ca2+ spike the Ca2+ concentration returns to the baseline level. During the period between two successive spikes, the Ca2+ release rate should remain low in the presence of IP3. This may be enabled by the low level of IP3R activity in the presence of a low concentration of Ca2+. Secondly, the Ca2+ concentration dependence of the Ca2+ release rate had a Hill coefficient of ∼2 (also, n≈ 2 in our model fitting), suggesting that IP3R activation requires simultaneous binding of two Ca2+ ions. The strong Ca2+ concentration dependence is advantageous for generation of Ca2+ spikes. Thirdly, the extent of Ca2+-mediated control of IP3R activity is IP3 concentration-dependent. In the presence of a low concentration of IP3, Ca2+ alone activates the IP3R so weakly that Ca2+ spikes are not generated. With an increase in the IP3 concentration, Ca2+ becomes effective in inducing Ca2+ release. This IP3 requirement may explain the requirement for receptor activation to start the cytoplasmic Ca2+ oscillation.
It seems pertinent here to mention mechanisms other than Ca2+-mediated potentiation of IP3R activity that participate in the generation of Ca2+ oscillations. Firstly, the falling phase of the Ca2+ oscillation clearly requires inhibitory mechanisms of Ca2+ release that are still elusive. One possible mechanism for the termination of each Ca2+ oscillation may be the depletion of the Ca2+ stores, which results in a decreased Ca2+ flux rate and a decrease in the extent of Ca2+-mediated positive feedback. Another candidate mechanism may be Ca2+-mediated inhibition of IP3R (Iino, 1990; Bezprozvanny et al. 1991; Finch et al. 1991; Bootman et al. 1995). After Ca2+ spikes are generated, the Ca2+ concentration around the Ca2+ stores may become high enough to terminate Ca2+ release via the Ca2+-mediated inhibitory mechanism. Secondly, CICR mechanism via RyRs may be involved in the Ca2+ oscillations and waves, as is the case in striated muscle cells (Endo et al. 1970; Takamatsu & Wier, 1990). Because activation of CICR has been shown to require a higher Ca2+ concentration than activation of IP3R (Iino, 1989), CICR has the potential to play a role in the amplification of Ca2+ release after the IP3R is activated. However, RyRs are not always expressed in cells in which the production of IP3 is required for the generation of Ca2+ oscillations and waves. Furthermore, CICR in smooth muscle cells that express RyRs contributes little in the generation of Ca2+ waves (Iino, Yamazawa, Miyashita, Endo & Kasai, 1993).
In conclusion, the requirement for both Ca2+ and IP3 for IP3R activation is important for the generation of Ca2+ spikes and Ca2+ oscillations, in which a multitude of molecular mechanisms are involved.
Acknowledgments
The authors thank Y. Hirose for technical assistance. This work was supported by grants from the Ministry of Education, Science and Culture of Japan and the Uehara Memorial Foundation.
References
- Benevolensky D, Moraru II, Watras J. Micromolar calcium decreases affinity of inositol trisphosphate receptor in vascular smooth muscle. Biochemical Journal. 1994;299:631–636. doi: 10.1042/bj2990631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curve of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Missiaen L, Parys JB, De Smedt H, Casteels R. Control of inositol 1,4,5-trisphosphate-induced Ca2+ release by cytosolic Ca2+ Biochemical Journal. 1995;306:445–451. doi: 10.1042/bj3060445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick CC, Saito A, Fleischer S. Isolation and characterization of the inositol trisphosphate receptor from smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1990;87:2132–2136. doi: 10.1073/pnas.87.6.2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smedt H, Missiaen L, Parys JB, Bootman MD, Mertens L, Van Den Bosch L, Casteels R. Determination of relative amounts of inositol trisphosphate receptor mRNA by ratio polymerase chain reaction. Journal of Biological Chemistry. 1994;269:21691–21698. [PubMed] [Google Scholar]
- Endo M, Tanaka M, Ogawa Y. Calcium induced release of calcium from the sarcoplasmic reticulum of skinned skeletal muscle fibres. Nature. 1970;228:34–36. doi: 10.1038/228034a0. [DOI] [PubMed] [Google Scholar]
- Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–1648. doi: 10.1126/science.275.5306.1643. 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- Hannaert-Merah Z, Coquil JF, Combettes L, Claret M, Mauger JP, Champeil P. Rapid kinetics of myo-inositol trisphosphate binding and dissociation in cerebellar microsomes. Journal of Biological Chemistry. 1994;269:29642–29649. [PubMed] [Google Scholar]
- Hirose K, Iino M. Heterogeneity of channel density in inositol-1,4,5-trisphosphate-sensitive Ca2+ stores. Nature. 1994;372:791–794. doi: 10.1038/372791a0. [DOI] [PubMed] [Google Scholar]
- Hoffer AM, Schlue WR, Curci S, Machen TE. Spatial distribution and quantitation of free luminal [Ca] within the InsP3-sensitive internal store of individual BHK-21 cells: ion dependence of InsP3-induced Ca release and reloading. FASEB Journal. 1995;9:788–798. doi: 10.1096/fasebj.9.9.7601343. [DOI] [PubMed] [Google Scholar]
- Iino M. Calcium-induced calcium release mechanism in guinea pig taenia caeci. Journal of General Physiology. 1989;94:363–383. doi: 10.1085/jgp.94.2.363. 10.1085/jgp.94.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. Journal of General Physiology. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M, Endo M. Calcium-dependent immediate feedback control of inositol 1,4,5-triphosphate-induced Ca2+ release. Nature. 1992;360:76–78. doi: 10.1038/360076a0. 10.1038/360076a0. [DOI] [PubMed] [Google Scholar]
- Iino M, Yamazawa T, Miyashita Y, Endo M, Kasai H. Critical intracellular Ca2+ concentration for all-or-none Ca2+ spiking in single smooth muscle cells. EMBO Journal. 1993;12:5287–5291. doi: 10.1002/j.1460-2075.1993.tb06224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MO, Yoshida Y, Koga T, Kojima M, Kangawa K, Imai S. Isolation and characterization of vascular smooth muscle inositol 1,4,5-trisphosphate receptor. Biochemical Journal. 1996;316:295–302. doi: 10.1042/bj3160295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall IC, Taylor CW. Two calcium-binding sites mediate the interconversion of liver inositol 1,4,5-trisphosphate receptors between three conformational states. Biochemical Journal. 1994;301:591–598. doi: 10.1042/bj3010591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson A, Taylor CW. Effects of Ca2+ chelators on purified inositol 1,4,5-trisphosphate (InsP3) receptors and InsP3-stimulated Ca2+ mobilization. Journal of Biological Chemistry. 1993;268:11528–11533. [PubMed] [Google Scholar]
- Takamatsu T, Wier WG. Calcium waves in mammalian heart: quantification of origin, magnitude, waveform, and velocity. FASEB Journal. 1990;4:1519–1525. doi: 10.1096/fasebj.4.5.2307330. [DOI] [PubMed] [Google Scholar]
- Watras J, Moraru I, Costa DJ, Kindman LA. Two inositol 1,4,5-trisphosphate binding sites in rat basophilic leukemia cells: relationship between receptor occupancy and calcium release. Biochemistry. 1994;33:14359–14367. doi: 10.1021/bi00251a050. [DOI] [PubMed] [Google Scholar]
- Worley PF, Baraban JM, Supattapone S, Wilson VS, Snyder SH. Characterization of inositol trisphosphate receptor binding in brain. Regulation by pH and calcium. Journal of Biological Chemistry. 1987;262:12132–12136. [PubMed] [Google Scholar]
- Yoneshima H, Miyawaki A, Michikawa T, Furuichi T, Mikoshiba K. Ca2+ differentially regulates the ligand-affinity states of type 1 and type 3 inositol 1,4,5-trisphosphate receptors. Biochemical Journal. 1997;322:591–596. doi: 10.1042/bj3220591. [DOI] [PMC free article] [PubMed] [Google Scholar]