

Abstract
The experiments were performed on sensorimotor cortex using current-clamp intracellular recordings in layer V pyramidal neurones and whole-cell voltage-clamp recordings in dissociated pyramidal neurones. The intracellularly recorded neurones were classified on the basis of their firing characteristics as intrinsically bursting (IB) and regular spiking (RS). The RS neurones were further subdivided into adapting (RSAD) or non-adapting (RSNA), depending on the presence or absence of spike frequency adaptation. Since burst firing in neocortical pyramidal neurones has previously been suggested to depend on the persistent fraction of Na+ current (INa,p), pharmacological manipulations with drugs affecting INa inactivation have been employed.
ATX II, a toxin derived from Anemonia sulcata, selectively inhibited INa fast inactivation in dissociated neurones. In current-clamp experiments on neocortical slices, ATX II enhanced the naturally occurring burst firing in IB neurones and revealed the ability of RSNA neurones to discharge in bursts, whereas in RSAD neurones it increased firing frequency, without inducing burst discharges. During the ATX II effect, in all the three neuronal subclasses, episodes of a metastable condition occurred, characterized by long-lasting depolarizing shifts, triggered by action potentials, which were attributed to a peak in the toxin-induced inhibition of INa inactivation. The ATX II effect on IB and RSNA neurones was compared with that induced by veratridine and iodoacetamide. Veratridine induced a small increase in the INa and a large shift to the left in the voltage dependence of INa activation. Accordingly, its major effect on firing characteristics was the induction of prolonged tonic discharges, associated with burst facilitation less pronounced than that induced by ATX II. The alkylating agent iodoacetamide was able to induce a selective small increase in the INa,p, with a similar but less pronounced effect than ATX II on firing behaviour.
The present results show that pharmacological manipulations capable of slowing down INa inactivation significantly enhance burst behaviour in IB neurones and promote burst firing in otherwise non-bursting RSNA neurones. We suggest that IB and, to a lesser extent, RSNA neurones are endowed with a relatively large fraction of INa,p which, in physiological conditions, is sufficient to sustain bursting in IB but not in RSNA neurones.
Pyramidal neurones of layer V in rat sensorimotor neocortex are functionally heterogeneous in terms of their firing properties, and can be divided into different subtypes on the basis of their physiological and morphological features (Connors, Gutnick & Prince, 1982; Connors & Gutnick, 1990; Tseng & Prince, 1993). These different intrinsic properties play a crucial role in determining both the subthreshold behaviour and the firing characteristics of the various neuronal subclasses. As in other mammalian and non-mammalian nervous structures, a remarkable subpopulation of layer V pyramidal neurones, currently referred to as intrinsically bursting (IB) cells, is set to discharge with ‘burst’ firing characterized by a short series of action potentials (APs) superimposed on a slow depolarizing potential. It is assumed that the burst discharge generated by neocortical IB neurones significantly contributes to information processing and neocortical rhythm generation (Silva, Amitai & Connors, 1991; Lisman, 1997); moreover in disinhibited neocortical slices, intrinsically bursting neurones have been found to play an important role in epileptic synchronization (Connors, 1984; Chagnac-Amitai & Connors, 1989).
Previous data, obtained from both neocortical slices (Franceschetti, Guatteo, Panzica, Sancini, Wanke & Avanzini, 1995) and acutely dissociated neocortical pyramidal neurones (Guatteo, Franceschetti, Bacci, Avanzini & Wanke, 1996) have demonstrated that neocortical burst generation substantially depends on a tetrodotoxin-sensitive current, and is unaffected by Ca2+ channel blockers. Similar results have been reported for other cortical structures, such as the subiculum (Mattia, Hwa & Avoli, 1993) and CA1 hippocampal region (Azouz, Jensen & Yaari, 1996), in which both the fast spikes and the slow underlying depolarizing potential have been shown to be blocked only by the Na+ channel blocker tetrodotoxin (TTX). On the basis of the results of these experiments, it has been suggested that the persistent fraction of the sodium current (INa,p) (Taylor, 1993; Crill, 1996), which has been shown to be able to sustain plateau potentials (Stafstrom, Schwindt, Chubb & Crill, 1985; Hoehn, Watson & MacVicar, 1993; Fleidervish & Gutnick, 1996), is the best candidate for burst generation in neocortical pyramidal neurones, but no clear proof that it is sufficient for burst generation has been provided. In order to verify this hypothesis we used pharmacological manipulations capable of modifying the kinetics of the Na+ current (INa) to test the consequence of a selective enhancement of INa,p on firing behaviour. The following drugs were employed: anemone toxin II (ATX II), veratridine and iodoacetamide (IAA).
Anemone toxin II (ATX II) was chosen because of its selective inhibition of INa fast inactivation, which has previously been demonstrated in other cell types (Romey, Abita, Schweitz, Wunderer & Lazdunski, 1976; Bergman, Dubois, Rojas & Rathmayer, 1976; Hartung & Rathmayer, 1985; Neumcke, Schwarz & Stämpfli, 1987). ATX II is a small polypeptide produced by Anemonia sulcata that binds to an extracellular site on domain IV of the voltage-dependent Na+ channel α-subunit (Rogers, Qu, Tanada, Scheuer & Catterall, 1996). Since the effects of ATX II on central mammalian neurones have never been studied, a subset of experiments was performed on dissociated pyramidal neurones, designed to confirm the efficacy and selectivity of ATX II on the inactivation properties of the INa in neocortical neurones. In both neocortical slices and dissociated pyramidal neurones, the effects of ATX II were compared with those induced by veratridine, which is known to affect both activation and inactivation of INa (Hille, 1992) and IAA, an alkylating agent potentially active in slowing down INa inactivation (Ulbricht, 1990). The preliminary results have been previously published in abstract form (Mantegazza, Avanzini & Franceschetti, 1996).
METHODS
Wistar rats aged from 9 to 35 days were deeply anaesthetized with ether, killed by decapitation and their brains rapidly removed and placed in cold (4°C) artificial cerebrospinal fluid (ACSF), bubbled with 95 % O2 and 5 % CO2. Coronal slices, with a thickness of 250-350 μm, were prepared from the sensorimotor cortex, using a vibratome (FTB Vibracut 3). For most of the current-clamp experiments, the slices were immediately transferred to an interface recording chamber (Haas type, modified; Haas, Schaerer & Vosmansky, 1979) kept at 35°C and perfused by ACSF. The slices were allowed to equilibrate for 1-1.5 h before recordings were made. The composition of the ACSF was (mM): NaCl, 124; NaHCO3, 26.5; CaCl2, 2; NaH2PO4, 1.25; MgSO4, 2; KCl, 5; glucose, 10; bubbled with 95 % O2 and 5 % CO2. The composition of the low-Ca2+ solution was (mM): NaCl, 120; NaHCO3, 24; CaCl2, 1; NaH2PO4, 1; MgCl2, 4; KCl, 2.5; glucose, 20; bubbled with 95 % O2 and 5 % CO2. Veratridine was added to the superfusion medium. ATX II was dissolved in ACSF and locally applied, using a pipette with a tip diameter of 10-15 μm. Local application of ATX II was used in order to minimize the quantity of perfused drug; local administration of ACSF alone was tested in preliminary control experiments.
In some of the experiments veratridine and IAA were tested using a submersion recording chamber. In that case, slices were maintained in oxygenated low-Ca2+ ACSF until recording, when slices were individually transferred to a submersion recording chamber and perfused with ACSF at a temperature of 30°C.
Kynurenic acid at 2 mM was added to the perfusing solution in most ATX II experiments. Firing behaviour was characterized before and after addition of the excitatory amino acid blocker to the ACSF and postsynaptic potentials were monitored to confirm the block of the excitatory synaptic transmission. Intracellular recordings were made using an IR-283 (Neurodata Inst. Corp.) or Axoclamp-2A (Axon Instruments, Inc.) amplifier. We used voltage follower microelectrode amplifiers, since they have been found to provide more accurate membrane voltage measurements than patch-clamp amplifiers (Magistretti, Mantegazza, Guatteo & Wanke, 1996).
Intracellular recordings were mainly performed using borosilicate glass electrodes pulled with an horizontal puller, and filled with 3 M potassium acetate (resistance, 80-90 MΩ). In some experiments, the electrodes were filled with 1 M IAA and 1 M KCl, in order to inject IAA into the cytoplasm iontophoretically.
Only healthy neurones with a stable resting membrane potential (Vrest) more negative than -60 mV, a stable firing level and overshooting action potentials were selected for the analysis. Voltage and current signals were continuously displayed on a Tektronix storage oscilloscope.
The duration of APs was measured at a level 40 mV depolarized from the Vrest. Membrane input resistance (Rin) was measured at the peak of the voltage deflection of the membrane potential (Vm) induced by the injection of hyperpolarizing 0.4 nA current pulses.
Voltage-clamp experiments were performed by patch clamping acutely dissociated pyramidal neurones prepared from single slices kept for 10-15 min in the following solution (mM): NaCl, 123; NaHCO3, 15; CaCl2, 0.1; EGTA, 0.1; MgCl2, 2; KCl, 3; glucose, 20; kynurenic acid, 1; Hepes-NaOH, 10; pH 7.4. The solution contained 1 mg ml−1 of protease type XIV (Sigma) to digest the extracellular matrix, and was bubbled with 95 % O2 and 5 % CO2 at 35°C. After enzyme treatment, the slices were washed in an enzyme-free solution and kept in a holding chamber as above. One single slice at a time was mechanically dissociated using fire-polished glass pipettes with progressively smaller tips.
The neurones were plated onto Petri dishes (Costar, Cambridge, MA, USA), left 5-10 min to allow attachment and then bath perfused with the following solution (mM): NaCl, 114; NaHCO3, 25; CaCl2, 2; NaH2PO4, 1.25; MgSO4, 2; KCl, 5; Hepes-NaOH, 10; glucose, 10; bubbled with 95 % O2 and 5 % CO2, pH 7.4. Only large neurones showing a pyramidal shape were selected for patch-clamp recordings.
In order to isolate the Na+ currents, the following solution (physiological Na+ solution), was locally perfused (mM): NaCl, 120; CaCl2, 1.3; MgCl2, 2; CdCl2, 0.4; NiCl2, 0.3; TEACl, 20; Hepes-NaOH, 10; glucose, 10; pH 7.4. In the experiments aimed at evaluating the properties of the fast Na+ current (see Results), NaCl was partially replaced with choline chloride (low-Na+ solution: NaCl, 10 mM; choline chloride, 110 mM) in order to reduce voltage-clamp errors. The drugs (ATX II, TTX, veratridine) were dissolved in physiological Na+ or low-Na+ solutions and locally applied. The borosilicate glass capillary recording pipettes were pulled using a horizontal puller and filled with the following solution (mM): CsF, 120; MgCl2, 1; EGTA-CsOH, 10; Na2ATP, 2; Hepes-CsOH 10; phosphocreatine-diTris, 10; with 20 units ml−1 creatine phosphokinase; pH 7.2. With this solution the resistance of the electrodes was 2-3 MΩ. In some experiments 1 mM IAA was added to the internal solution.
In order to avoid contaminating currents, the traces recorded in the presence of both 1 μM TTX and the test drug were subtracted from those recorded in the presence of the drug alone. When INa was measured using a step voltage stimulus, the INa,p amplitude was determined as the mean current between 100 and 200 ms after the beginning of the stimulus.
In order to record both voltage-activated and Ca2+-activated K+ currents the following external solution was locally perfused (mM): NaCl, 140; CaCl2, 2; MgCl2, 2; KCl, 3; Hepes-NaOH, 10; glucose, 10; pH 7.4. TTX was added at 1 μM to block INa, and ATX II was dissolved in that solution. Recording pipettes were filled with (mM): potassium gluconate, 119; KCl, 13; MgCl2, 2; EGTA-KOH, 5; Na2ATP, 2; Hepes-KOH, 10; NaGTP, 0.2; phosphocreatine-diTris, 10; with 20 units ml−1 creatine phosphokinase; pH 7.2.
The following external solution was used to isolate Ca2+ currents (mM): choline chloride, 80; CaCl2, 2; MgCl2, 2; KCl, 3; Hepes-NaOH, 10; TEA-Cl, 40; 4AP, 5; glucose, 10; pH 7.4. ATX II was dissolved in this solution. The recording pipettes were filled with (mM): CsCl, 78; TEACl, 40; EGTA-CsOH, 10; MgATP, 2; Hepes-CsOH, 10; GTP-Na, 0.2; cAMP, 1; phosphocreatine-diTris, 10; with 20 units ml−1 creatine phosphokinase; pH 7.2.
The voltage-clamp recordings were made at room temperature (21-24°C), using a Biologic RK-400 patch-clamp amplifier; junction potential was not corrected. Capacitative currents were minimized by means of the amplifier circuitry, and 70-80 % series resistance compensation was routinely used. The remaining transients and leakage currents were eliminated using P/4 subtraction.
The pCLAMP 6.0.3 software and a Digidata 1200 interface (Axon Instruments) were used to generate the stimulus protocols and to acquire the signals; the voltage signals were filtered at 3 kHz, and the current signals at 5 kHz (voltage steps) or 0.5 kHz (voltage ramps). The data were analysed using pCLAMP and Origin 4.0 (Microcal Inc.) software on a Pentium 166 PC. Voltage dependency was fitted to Boltzmann relationships in the form:
using the Levenberg-Marquardt algorithm, where V½ is membrane potential at half-maximal inactivation. The statistical results are given as means ± standard error of the mean (s.e.m.); statistical significance was evaluated using the two-tailed Student's test for paired data (only P values < 0.01 were accepted).
ATX II was purchased from Calbiochem-Novabiochem Corp. (La Jolla, CA, USA); veratridine, IAA, TTX, kynurenic acid, phosphocreatine, creatine phosphokinase, GTP and cAMP were purchased from Sigma.
RESULTS
Voltage-clamp recordings, using the whole-cell configuration of patch-clamp technique were obtained from forty-five pyramidal neurones (36 tested with ATX II and 9 with veratridine or IAA) acutely dissociated from rat sensorimotor cortex slices. The isolated neurones had a main membrane capacitance of 12 ± 2 pF and a main membrane resistance of 8 ± 3 GΩ. Based on neuronal shape and size we assume that a large percentage of the isolated neurones belonged to layer V, although the inclusion of some pyramidal neurones from middle layers cannot be ruled out.
Intracellular recordings in current-clamp configuration were carried out in slices of sensorimotor cortex, in thirty-nine pyramidal neurones of layer V (30 tested with ATX II and 9 tested with veratridine or IAA). The mean Vrest was -64 ± 3 mV, and the mean Rin was 48 ± 10 MΩ, with no significant differences among neurones showing, under control conditions, different firing characteristics.
Effects of ATX II on the ionic currents of neocortical neurones
Since neocortical pyramidal neurones have a large membrane area and, in physiological saline, a large fast Na+ current (INa,f), giving rise to voltage- and space-clamp errors, we recorded the INa,f in neurones perfused with a low-Na+ saline (15 mM Na+; see Methods) to minimize voltage errors. Under these conditions, the peak amplitude never exceeded 1 nA; with a maximal series resistance of 15 MΩ and standard 70 % compensation, the voltage error was always < 5 mV.
The action of ATX II on INa,f, evoked with a step potential in a representative cell perfused with low-Na+ external solution, is shown in Fig. 1A. The persistent fraction of the current was greatly enhanced in comparison with the control trace, without any effects on the initial rising rate of the current (inset), and returned to control values after toxin wash-out (not shown). In the presence of ATX II, INa,f first showed a slight increase in amplitude (Fig. 1A, inset) but it subsequently decreased. This phenomenon added up to the slight run-down of the peak current which we always observed under control conditions. This run-down was evaluated by fitting the experimental data, recorded before the ATX II application, to an exponential decaying function that yielded a time constant of 50 ± 4 min. The ATX II-induced decrease in the peak current was greater than that expected from the run-down exponential function; the recovery of the INa,f after the wash-out of the toxin was corrected for the run-down, but it was never complete (not shown). The current-voltage (I-V) relationship of INa,f is shown in Fig. 1B. The control INa,f started to activate near -60 mV and reached maximum amplitude at about -15 mV; in the presence of ATX II, the curve shifted in a hyperpolarizing direction. The voltage dependence of the activation and steady-state inactivation of INa,f fitted a single Boltzmann relationship under control conditions and during ATX II perfusion, as shown in Fig. 1C (see Fig. 1 legend for the description of estimation of the curves). In the presence of ATX II, INa,f activated at more hyperpolarized potentials than under control conditions, and the mid-point (V½) was significantly shifted by 6 mV in the hyperpolarizing direction (paired t test, P < 0.01). The steady-state inactivation curve was slightly shifted in the hyperpolarizing direction and less steep, but these changes did not reach statistical significance. In low-Na+ saline, INa,p was difficult to evaluate because its amplitude was a minimal fraction of the INa,f (about 0.1 %), consequently it was evaluated in a physiological solution (125 mM Na+) using slow depolarizing ramps (40 mV s−1) as voltage stimuli. The ramp stimulus provides the I-V relationship of INa,p while avoiding the concomitant activation of the INa,f. Figure 1D shows the current traces recorded in a representative cell. The control trace had an inward component that activated near -60 mV and peaked near -30 mV, whose amplitude was greatly enhanced by ATX II and completely recovered after toxin wash-out (not shown). The inward deflection due to INa,p was completely abolished in the presence of TTX plus ATX II; under these conditions, a low amplitude outward current remained, whose nature was not investigated, but which was assumed to correspond to the cationic current (Icat) described by Alzheimer (1994). After addition of ATX II, the INa,p amplitude was 6 ± 2 times (n= 10) greater than the control amplitude, with a maximum increase in amplitude of 13-fold (see Fig. 1D) and a minimum increase of 3-fold. The voltage dependence of INa,p activation under control conditions and in the presence of ATX II (calculated on ten cells) is shown in Fig. 1E: the values have not been normalized in order to show the toxin-induced increase. The activation curves did not reach a maximal plateau but declined from a peak to a lower value. Up to peak conductance, they were fitted to a single Boltzmann relationship in order to compare the initial phase of the two curves. The mid-point of activation (V½) was not significantly different between the two conditions, although the slope was steeper for the ATX II curve.
Figure 1. Effects of ATX II on Na+ currents in dissociated neocortical neurones.
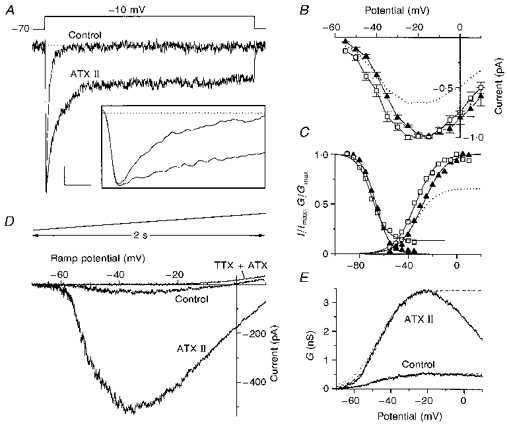
A, whole-cell Na+ currents recorded in low-Na+ external solution (see Methods), under control conditions and with 1 μM ATX II; the stimulus was the step potential shown above; the TTX-insensitive currents were subtracted. Inset: the same traces on an expanded time scale. Calibration bars: 50 pA and 25 ms for the main panel; 100 pA and 1.6 ms for the inset. B, normalized I-V curves from six cells, constructed by measuring INa,f elicited with depolarizing steps at the indicated potentials, starting from a holding potential of -70 mV, under control conditions (▴) and with ATX II (□). The dotted line is the curve in the presence of ATX II, resized to show the average reduction in INa,f (see text). C, voltage dependence of activation (right) and steady-state inactivation (left) under control conditions (▴) and with ATX II (□). The voltage dependence of activation is the normalized conductance (G/Gmax) curve for INa,f calculated as G=I/(V - ENa), where I is the INa of the I-V plots in Fig. 1B, ENa is the calculated Nernst equilibrium potential and V the Vm. The data were fitted to Boltzmann relationships (continuous lines) and yielded the following parameters: V½= -27.5 mV, k= 9.3, A1= 0, A2= 1 (control); V½= -34 mV, k= 8.6, A1= 0, A2= 1 (ATX II). The dotted line is the ATX II Boltzmann relationship resized to show the induced decrease in the current (A2= 0.66). The voltage dependence of steady-state inactivation is the normalized current (I/Imax) curve, where I is the INa,f measured using a test pulse to -10 mV; the cells were held at prepulse potentials over the range of -90 mV to -35 mV for 325 ms before the test pulse. The normalized current is plotted against the voltage values of the prestimuli. The continuous lines are the Boltzmann fits that yielded the following parameters: V½= -66.3; k= 5.7; A1= 1; A2= 0 (control); V½= -69.2; k= 6; A1= 1; A2= 0.14 (ATX II). D, whole-cell INa recorded in physiological Na+ solution (see Methods) under control conditions and with 1 μM ATX II; the displayed curves did not have the TTX-insensitive currents subtracted and the curve recorded in the presence of TTX plus ATX II is also shown; the remaining outward current was assumed to be the Icat (see text). The stimulus was the slow voltage ramp displayed above. E, voltage dependence of activation of INa,p. The curves represent the non-normalized conductances (G) calculated as above, where I is the average current evoked by a voltage ramp (10 cells) under control conditions and with 1 μM ATX II. The curves were fitted up to the peak value for the Boltzmann relationships (dashed lines) using the following parameters: V½= -46.9; k= 7.2; A1= 0; A2= 0.54 (control); V½= -45.8; k= 6.3; A1= 0; A2= 3.45 (ATX II). The data were not normalized in order to show the ATX II-induced increase in INa,p, but the control Boltzmann fit is also shown at higher magnification (dotted line), in order to compare the two curves.
To rule out a possible aspecific ATX II effect due to unexpected activity on other membrane conductances, control experiments were carried out in order to evaluate total K+ and Ca2+ currents in acutely dissociated neurones that were extracellularly perfused with the toxin. ATX II was ineffective on K+ currents: Fig. 2A shows that the total K+ currents recorded in a representative neurone under control conditions and in the presence of ATX II, as well as the I-V relationships, almost completely overlapped. The current traces shown in the figure include Ca2+-activated K+ currents, as the Ca2+ currents were not blocked but hidden by the outward current. In a like manner, ATX II was found to have no effect on Ca2+ currents. The current traces and I-V relationship of the total Ca2+ currents under control conditions and during ATX II perfusion are shown in Fig. 2B, which also includes a T-type current, as the recording steps were preceded by a 100 ms hyperpolarizing prepulse at -100 mV.
Figure 2. Effects of ATX II on K+ and Ca2+ currents in dissociated neocortical neurones.
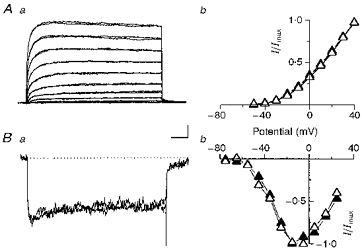
Aa, whole-cell total K+ currents evoked using 10 mV stepped stimuli from -50 to +40 mV, in 10 mV increments, starting from a holding potential of -70 mV. The currents recorded under control conditions and with 1 μM ATX II are superimposed. Calibration bars: 500 pA and 25 ms. Ab, normalized I-V plots from five cells obtained using the voltage stimuli described above, under control conditions (▴) and with 1 μM ATX II (▵). Error bars are not shown. Ba, whole-cell total Ca2+ currents evoked using test voltage steps up to -15 mV, starting from a holding potential of -70 mV and preceded by a 100 ms prestimulus shifting the Vm to -100 mV. The currents recorded under control conditions and with ATX II are superimposed. Calibration bars: 75 pA and 50 ms. Bb,I-V plot of the peak current from five cells, obtained using the stimulus protocol described above and the test potentials indicated in the graph, under control conditions (▴) and with 1 μM ATX II (▵).
Effects of ATX II on the firing of neocortical neurones
The neurones recorded in slices were classified as intrinsically bursting (IB), or regular spiking (RS) (Connors & Gutnick, 1990) on the basis of their firing characteristics in response to the injection of suprathreshold depolarizing current pulses. The RS neurones were further subdivided into adapting (RSAD) or non-adapting (RSNA) depending on the presence or absence of spike frequency adaptation (Silva et al. 1991; Franceschetti et al. 1995).
The firing behaviour of three representative neurones and the changes induced by locally applied ATX II (1-10 μM) are shown in Figs 3 and 4. In almost two-thirds of the IB neurones (Fig. 3A), the typical response to just-supra-threshold long-lasting depolarizing current pulses consisted of a single initial burst of two to five closely spaced APs, followed by a regular train of non-accommodating individual APs, each followed by a prominent depolarizing after-potential (DAP); in the remaining IB neurones the bursts rhythmically recurred throughout the duration of the depolarizing current pulse. ATX II induced recurrent burst firing in the neurones that discharged with only an initial burst under control conditions, and increased the number of APs included in the individual bursts. Occasionally, burst discharges were found to be irregularly and reversibly substituted by individual APs, each followed by a shoulder arising from the repolarizing phase at a potential ranging from -10 to -20 mV (Figs 3A and 4). An ATX II-induced slight increase in firing frequency was observed only in response to the injection of rather high amplitude current pulses (0.4-0.6 nA), depolarizing the Vm by about 15 mV with respect to Vrest. In this case individual action potentials appeared to be intermingled with recurring bursts, and displayed a high recurrence rate (around 15 Hz) (Fig. 4). The RSNA (Fig. 3B) neurones discharged in response to a just-suprathreshold depolarizing current pulse with a regular train of individual APs, each followed by a consistent DAP (Fig. 3B, arrow), these neurones failed to show any adaptation after the first interspike interval, which was commonly slightly shorter than the subsequent ones. An early and transient spike frequency adaptation was detectable only by injecting larger current pulses that led to a considerable depolarization of Vm (by more than 12 mV in comparison with Vrest), but burst firing could never be evoked under control conditions. After local ATX II administration, the RSNA neurones changed their firing properties and developed repetitive burst discharges. As in the IB neurones, the recurrent bursts induced by ATX II perfusion were often substituted by short-lasting shoulders arising from the repolarizing phase of individual APs.
Figure 3. Effects of ATX II on the firing of neocortical layer V neurones in brain slices.
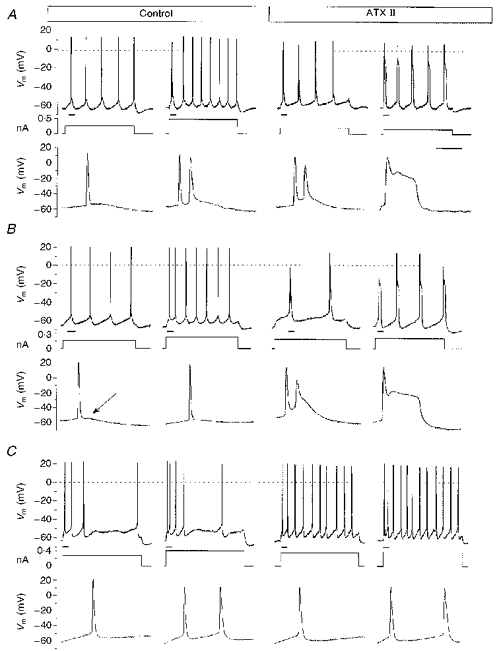
In each example the upper trace is the Vm recorded in a representative neurone and the lower trace is the display of the underlined segment of the upper trace on an expanded time scale; the middle trace is the injected current pulse. A, firing characteristics in a representative IB neurone (from a total of five), under control conditions (left) and after extracellular perfusion with 1 μM ATX II (right). B, firing characteristics of a representative RSNA neurone (from a total of five) recorded under control conditions (left) and with 5 μM ATX II (right), which was capable of inducing repetitive burst discharges. The lower traces better display the DAP (arrow) following each AP in this neuronal type and the subsequent ATX-induced qualitative change in firing behaviour. C, firing characteristics of a representative RSAD neurone (from a total of four), under control conditions (left) and with 5 μM ATX II (right). Calibration bar: 13 ms for the expanded voltage traces; 200 ms for the other voltage traces and current pulses.
Figure 4. Changes in the firing of an IB neurone in the presence of ATX II.
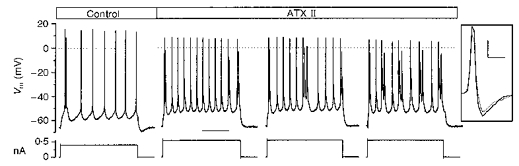
A, a representative IB neurone which, in the presence of 5 μM ATX II, developed a recurrent burst discharge, increased its firing frequency and the number of APs making up the individual bursts. The represented sweeps refer to different consecutive time points during the local perfusion (3, 3.5 and 4 min). Calibration bar: 200 ms. The inset shows the decrease in the AP slopes with ATX II (dotted line) in comparison with the control condition (continuous line). Calibration bars: 70 V s−1 and 2 ms.
The RSAD neurones discharged in response to suprathreshold depolarizing current pulses with a train of individual APs showing prominent spike frequency adaptation (Fig. 3C); the firing frequency markedly increased during ATX II perfusion and the effect of spike frequency adaptation appeared to be simultaneously reduced; the individual APs slightly increased in duration (from 1.7 ± 0.3 ms under control conditions to 1.9 ± 0.3 ms; n= 5), but depolarizing shoulders following repetitive APs were never found.
During ATX II perfusion, in all the recorded neurones, regardless of their basal firing characteristics, the Vm occasionally displayed a metastable condition that was characterized by the intermittent appearance of long-lasting depolarizing events quickly reaching a new depolarized equilibrium potential ranging from -20 to -35 mV. These large depolarizing events were invariably triggered by one AP evoked by the injection of a low amplitude depolarizing current; their duration outlasted that of the injected pulse and prevented regenerative firing. A rather fast repolarization returned the Vm to its previous resting values (Fig. 5A) after hundreds of milliseconds. Even longer depolarizing Vm shifts (Fig. 5B) could occur in unstimulated neurones when minimal depolarizing deflections of the Vrest led to the generation of one or a few spontaneous APs (Fig. 5B). As with the evoked ones, these depolarizing events spontaneously recovered after tens of seconds.
Figure 5. Metastable depolarized condition of Vm in the presence of ATX II.
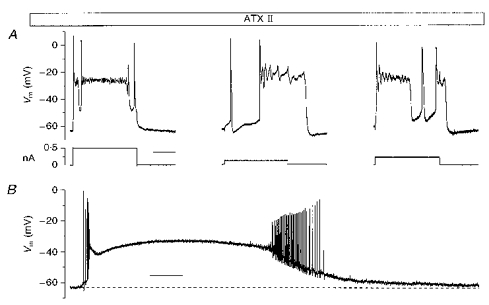
A, the ‘metastable’ condition of Vm induced by ATX II in an IB neurone (left, 5 μM ATX II), a RSNA neurone (middle, 1 μM ATX II) and a RSAD neurone (right, 5 μM ATX II). Calibration bar: 200 ms. B, a spontaneous depolarizing shift in a representative RSNA neurone. Calibration bar: 5 s.
Although the different ATX II-induced changes in firing behaviour were not found to be clearly related to toxin concentration, we considered the appearance of the metastable condition as the extreme toxin-induced effect, due to a peak in its blocking action on INa inactivation. The lack of a clear dose dependency for the different degrees of the ATX II effect could be due to the fact that we perfused the drug locally. This mode of administration, which allows us to minimize the required dose, limits the value of quantitative pharmacological evaluation in slice experiments.
At the same time as the appearance of the metastable condition, the AP amplitude and its positive and negative slopes (Fig. 5) slightly decreased. A similar but less evident effect on APs was seldom found during the ATX II-induced enhancement of burst activity.
To exclude a circuit contribution to ATX II-induced membrane excitability, the glutamate receptor antagonist kynurenic acid was simultaneously added to ACSF in order to block the glutamatergic inputs. The addition of 2 mM of kynurenic acid invariably blocked the excitatory synaptic responses to white matter stimulation without inducing any change in control firing behaviour (not shown) and was unable to change the above-mentioned ATX II effects.
Effects of other agents acting on Na+ current inactivation
To explore the effects of other agents known to act on Na+ current inactivation, we used the alkaloid veratridine, which has been extensively studied in other preparations (Hille, 1992). This agent has several actions on Na+ channels, including (1) the inhibition of inactivation; (2) a leftward shift (up to 90 mV) in the voltage dependence of activation; and (3) a reduction in single-channel conductance. The conspicuous shift of activation gives rise to the opening of Na+ channels at Vrest, leading to large and long-lasting ‘tail’ currents after the return to the holding potential in voltage-clamp experiments. The modifications in the INa induced by 1 μM veratridine in a representative dissociated neocortical neurone are shown in Fig. 6Aa. Since the action of veratridine is use dependent, its effect was tested (n= 4) by injecting voltage pulses at a frequency of 1 Hz: after 15 min INa,p had increased to 2.5 ± 0.5 times the control value. The veratridine-induced plateau tail conductance (GT, measured 25 ms after the end of the stimulus) was compared with the increased persistent Na+ conductance (GNa,p), in order to give an index of their size that was independent of the driving force; the conductance was calculated as G=I/(V - ENa), where G is conductance, I is current (INa,p or plateau tail current), V is membrane voltage, and ENa is the calculated Nernst Na+ equilibrium potential. After 15 min of stimulation in the presence of 1 μM veratridine, GT was 1.8 ± 0.6 larger than the GNa,p. In current-clamp experiments in neocortical slices (n= 5: 3 RSNA, 2 IB), we were able to follow the progressive development of the veratridine effect during the injection of 700 ms depolarizing current pulses at a frequency of 0.4 Hz; the fully expressed effect was always reached after 20-30 min. The foremost effect of the toxin on a RSNA neurone is shown in Fig. 6Ab, and was invariably found in all the five recorded neurones. It consisted of a marked increase in firing frequency during the depolarizing current pulse injection, and of a concomitant appearance of a tonic discharge of individual APs persisting beyond the end of the pulse injection. Subsequently, the RSNA neurones developed few burst discharges at the beginning of the stimulus followed by individual APs showing a further increase in firing frequency. The duration of the post-stimulus discharges ranged between seconds and tens of seconds. The AP shoulders and long-lasting Vm depolarizing shift that characterized the ATX II effect were never observed during veratridine perfusion.
Figure 6. Effects of veratridine and IAA on neocortical firing.
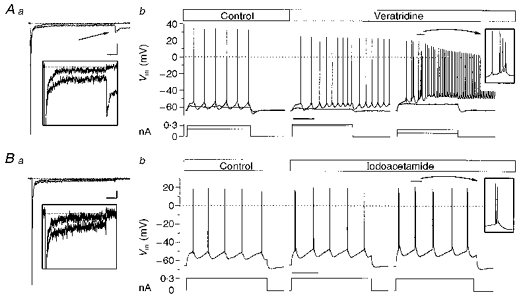
Aa, Na+ currents in a representative dissociated neurone, under control conditions and with 1 μM veratridine. The arrow shows the veratridine-induced tail current. Inset: the same current traces displayed on an expanded scale. Calibration bar: 1 nA, 50 ms for the main panel; 200 pA, 35 ms for the inset. Ab, the firing of a representative RSNA neurone in a neocortical slice under control conditions (left) and in the presence of 0.6 μM veratridine (right). Calibration bar: 200 ms. Ba, Na+ currents in a representative dissociated neurone, under control conditions and with 1 mM IAA in the recording patch pipette. The inset shows the same traces on an expanded current scale. Calibration bar: 500 pA, 50 ms for the full trace; 50 pA, 35 ms for the inset. Bb, the firing of a representative RSNA neurone in a neocortical slice, under control conditions (left) and after the iontophoretic injection of IAA (right). Calibration bar: 200 ms.
We also tested the alkylating agent IAA, which in our preparation was capable of inhibiting the inactivation of the INa by acting from the intracellular side, as is shown in Fig. 6Ba for a representative neurone The amplitude of INa,p after 10 min of intracellular administration with 1 mM IAA (n= 5) was 2.0 ± 0.1 times the amplitude of the current at the beginning of the experiment. Like ATX II, IAA did not cause any major leftward shift in the activation of the current, and did not induce any tail current. Figure 6Bb shows the effect of iontophoretically injected IAA in a representative RSNA neurone (n= 4: 3 RSNA, 1 IB): the cell first developed an initial single burst discharge, and then recurrent burst firing without any post-stimulus discharge, thus mimicking the effect of extracellular ATX II perfusion; however, IAA was never found to induce long-lasting depolarizing events.
DISCUSSION
Our experiments show that ATX II selectively affects INa in isolated neocortical pyramidal neurones, with neither the K+ nor the Ca2+ currents being modified, and directly demonstrate that an increase in INa,p greatly enhances bursting in IB neurones and reveals burst discharges in a subpopulation of RS neurones.
Effects of ATX II on INa
The main action of ATX II was a consistent inhibition of INa fast inactivation, so that a larger persistent component could be evoked by depolarizing stimuli. The voltage dependence of the activation of the enhanced INa,p fraction was substantially unaffected. Also unaffected was the conductance decay after a peak that was observed under control conditions and which has been previously described by other authors (Brown, Schwindt & Crill, 1994). ATX II can therefore be considered to selectively increase INa,p amplitude, without inducing any other major modifications in its properties.
To a lesser extent, ATX II was also found to affect the characteristics of INa,f, including (1) the voltage dependence of the activation, (2) the steady-state inactivation and (3) the peak current amplitude.
The observed leftward shift in the mid-point of the voltage dependence of activation can be predicted on the basis of the model of Aldrich, Corey & Stevens (1983) and is similar to that induced in dissociated neocortical neurones after the removal of Na+ channel inactivation by including the proteolytic enzyme papaine in the recording pipette (Brown et al. 1994). Concurrently with the increase in amplitude of the persistent INa fraction, the activation curve of the INa,f tends to be shifted leftward towards that of the INa,p and to decrease after a peak (compare Fig. 1C with Fig. 1E), in a similar way to the INa,p activation curve. This finding is consistent with the hypothesis that a modal gating of the Na+ channel may be the mechanism generating the INa,p (Brown et al. 1994; Crill, 1996). The decrease in the slope of the voltage dependence of steady-state INa,f inactivation appears to be negligible, since the observed shift in the mid-point of the curve was very small and did not reach statistical significance; nevertheless the observed change is consistent with that previously reported in amphibious myelinated nerve (Bergman et al. 1976).
The observed early and slight ATX II-induced increase in INa,f amplitude was found to be followed by a consistent decrease. This later effect apparently conflicts with the large increase predicted by the model of Aldrich et al. (1983) and previously observed in neocortical neurones exposed to intracellular papaine in order to remove the inactivation (Brown et al. 1994). A decrease in INa,f has been previously found in rat peripheral nerve and in skeletal muscle exposed to ATX II (Neumcke et al. 1987; Cannon & Corey, 1993). A possible explanation for this decrease may be based on the capacity of ATX II to affect the properties of Na+ channels leading to the stabilization of a low-conductance state, as observed in single-channel recordings made by Castillo, Piernavieja & Recio-Pinto (1996). The early increase in peak amplitude following the addition of ATX II (see Fig. 1D) may be progressively masked by the changes in the conductive properties of the channel which, in our experiments, seemed to become the preponderant effect of the toxin after a few minutes. The limited recovery in INa,f amplitude during toxin wash-out is more difficult to account for, as it evidently contrasts with the virtually complete recovery of the INa,p to control values; we did not try to clarify this phenomenon, which was beyond the scope of our study, but it appeared to add to the slight INa,f run-down often observed during long-lasting whole-cell recordings.
Effects of ATX II-induced changes in INa on firing behaviour of neocortical pyramidal neurones
In layer V pyramidal neurones, ATX II was found to activate rhythmic burst discharges not only in IB, but also in RSNA neurones, suggesting that both of these neuronal classes are endowed with similar membrane characteristics. This hypothesis is supported by the evidence that, even under control conditions, RSNA neurones share a number of physiological characteristics with IB neurones that differentiate them from RSAD neurones: both IB and RSNA neurones were found to generate faster individual APs followed by a consistent DAP (Franceschetti et al. 1995), and both respond with a non-adapting rhythmic firing to depolarizing current injections, depolarizing the Vm slightly in comparison with Vrest. The physiological characteristics distinguishing the neurones that we classified as RSNA neurones are very similar to those found by Chagnac-Amitai & Connors (1989) in a subclass of RS neurones, and to the ‘simple spiking’ pyramidal neurone described by Silva et al. (1991). The firing properties of the RSNA could simply be due to the fact that, under control conditions, they are endowed with a persistent fraction of INa that is sufficient for DAP generation, but not strong enough to sustain burst discharges.
ATX II perfusion never induced burst firing in RSAD neurones, but it did significantly increase firing frequency and reduce spike frequency adaptation. We did not evaluate the effect of the INa,p in these neurones in any detail, but the observed increase in firing frequency substantially agrees with previous evidence (Stafstrom et al. 1985; Fledervish & Gutnick, 1996), suggesting that INa,p can substantially affect the AP threshold and firing rate. When low-frequency discharges were induced by a just-suprathreshold depolarizing current in IB and RSNA neurones, ATX II converted individual APs to recurrent bursts without affecting the firing frequency, probably because of the powerful counteracting effects of post-burst hyperpolarizing currents (i.e. K+- and Ca2+-mediated currents). In these neurones, unlike RSAD neurones, an increase in firing frequency could be detected only by injecting large current pulses, which caused the INa-dependent depolarization to overcome the action of hyperpolarizing currents.
During ATX II perfusion, low-frequency repetitive burst discharges in both IB and RSNA neurones, were often reversibly replaced by individual APs, each followed by a shoulder lasting a few milliseconds. We assume that these shoulders strictly correlate with the burst firing enhancement, and only depend on a further slight increase in toxin-induced INa inactivation, which leads to a short depolarized plateau rather than closely spaced APs. In accordance with this view is the fact that this type of repetitive firing was never found in RSAD neurones.
The more dramatic ATX II-induced effect is the long-lasting depolarizing shifts that were observed in all of the neuronal subclasses and seem to be unrelated to their basal firing properties. We assume that this ‘metastable’ depolarized status of Vm can be attributed to a transient peak in the toxin-induced inhibition of INa inactivation.
The concurrent decrease in both the depolarizing slope and the amplitude of the APs can be explained by the toxin-induced decrease in the INa,f peak amplitude, as found in the voltage-clamp experiments.
Our results suggest that IB and RSNA neurones may be grouped together on the basis of their similar behaviour in the presence of ATX II, whereas RSAD neurones should be considered separately. Outward currents may contribute to the control of burst duration and the regulation of the typical rhythmic firing characterizing both IB and RSNA neurones. Our observations of differences in behaviour between IB, RSNA and RSAD neurones following the increase in INa,p are consistent with the hypothesis that IB and RSNA neurones have a lower expression of fast repolarizing currents than RSAD neurones.
Effects of other agents acting on Na+ current inactivation
In our experiments, veratridine (like ATX II) was found to enhance burst discharges in IB neurones and induce them in RSNA neurones; however, the most conspicuous change observed during alkaloid perfusion was an extreme increase in firing frequency and, particularly, a protracted post-stimulus discharge. Similar long-lasting discharges have been previously described in hippocampal CA1 pyramidal neurones, and have been attributed to a veratridine-induced increase in the INa,p (Alkadhi & Tian, 1996). In our opinion, this change should be principally ascribed to the leftward shift induced by veratridine in the voltage dependence of INa activation, since we never observed the same firing behaviour during ATX II administration. Because of this leftward shift in INa activation, the veratridine-modified channels did not close when Vm returned to its resting value, and could therefore have sustained the high frequency discharge persisting at the end of the depolarizing current stimulus. According to this hypothesis, the veratridine-induced increase in the INa,p fraction sustains the enhanced tendency to burst which, however, appears to be embedded in high frequency protracted discharges due to the change in INa activation. This hypothesis is further supported by the greater amplitude of the Na+ tail conductance in comparison with the conductance of the persistent component during veratridine perfusion. The decrease in the amplitude of APs in the presence of veratridine, in comparison with the control situation, can be explained by the drug-induced decrease in INa,f amplitude demonstrated by voltage-clamp experiments.
IAA is an alkylating agent used to modify proteins in biochemical studies. Its effects on ionic channels have not been widely explored, but it is known that similar agents are capable of slowing down the inactivation of the Na+ current (Ulbricht, 1990). We chose IAA because it did not affect the inactivation properties of K+ currents in the experiments performed by Oxford & Wagoner (1989). When added to the pipette solution, we found that IAA increased the INa,p in neocortical neurones, without inducing a large shift in the voltage dependence of current activation. Our data cannot support a selective IAA effect on Na+ channels, although the IAA-induced increase in the INa,p, (like that induced by ATX II) was able to enhance burst discharges and induce repetitive bursts in RSNA neurones in neocortical slices. The typical ATX II-induced long-lasting depolarizing shifts were never found during intracellular perfusion with IAA, which is in line with their ability to induce smaller increases in INa,p amplitude.
Conclusions
The present data concur with those previously published (Franceschetti et al. 1995; Guatteo et al. 1996) in supporting the hypothesis that INa,p actually sustains burst discharge in neocortical pyramidal neurones. We suggest that IB and, to a less extent, RSNA neurones are endowed with a relatively large persistent fraction of INa, which, in physiological conditions, is sufficient to sustain bursts in IB but not in RSNA neurones.
Since both IB and RSNA neurones are thought to regulate the generation of neocortical rhythms (Silva et al. 1991), any physiological or pathological mechanism able to modulate INa,p could significantly affect cortical synchronization.
Physiological modulations able to increase INa,p in a manner similar to that shown here for ATX II have been observed. In rat brain neurones, Na+ channel phosphorylation by protein kinase C has been reported to reduce the INa,f and to slow down inactivation of the current (Numann, Catterall & Scheuer, 1991). More recently, it has been shown that G protein βγ-subunits, overexpressed in the cell line tsA-201, may act directly on the Na+ channel, to generate or increase the INa,p (Ma, Catterall & Scheuer, 1997); however, the functional consequences of these changes remain unknown. A significant INa,p increase by dopamine modulation has been recently reported in rat prefrontal cortex. This effect has been suggested to significantly enhance the influences of local inputs on pyramidal neurones, and to be putatively relevant to cognitive processes (Yang & Seamans, 1996).
The possibility that changes in INa,p may be the pathophysiological basis for some inherited human disorders, is supported by the demonstration that one of the forms of the LQT cardiac syndrome and some forms of muscular myotonia and periodic paralysis are sustained by Na+ channel mutations that reduce INa fast inactivation (Bennett, Yazawa, Makita & George, 1995; Cannon, 1996). Similar mutations in brain Na+ channels may represent the pathophysiological mechanism of some forms of genetic disorders, leading to widespread and aberrant cortical synchronization; interestingly, Na+ channels have been found to be implicated in a genetic model of epilepsy (Sashihara et al. 1992). Moreover, it has been found that one of the most widely used antiepileptic drugs, phenytoin, selectively decreases INa,p at therapeutic concentrations (Chao & Alzheimer, 1995) and can preferentially diminish late sodium channel openings underlying epileptiform activity (Segal & Douglas, 1997), thus suggesting a possible antiepileptic mechanism based on a downregulation of the ability of the INa,p to sustain neocortical burst firing.
Acknowledgments
We thank E. Wanke for his support and S. Taverna for help in some experiments. This work was supported by grants from the Paolo Zorzi Foundation for Neurosciences.
References
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Alkadhi KA, Tian LM. Veratridine-enhanced persistent sodium current induces bursting in CA1 pyramidal neurons. Neuroscience. 1996;71:625–632. doi: 10.1016/0306-4522(95)00488-2. 10.1016/0306-4522(95)00488-2. [DOI] [PubMed] [Google Scholar]
- Alzheimer C. A novel voltage-dependent cation current in rat neocortical neurones. The Journal of Physiology. 1994;479:199–205. doi: 10.1113/jphysiol.1994.sp020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. The Journal of Physiology. 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AL. Molecular mechanism for an inherited cardiac arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Bergman C, Dubois JM, Rojas E, Rathmayer W. Decreased rate of sodium conductance inactivation in the node of Ranvier induced by a polypeptide toxin from sea anemone. Biochimica et Biophysica Acta. 1976;455:173–184. doi: 10.1016/0005-2736(76)90162-0. [DOI] [PubMed] [Google Scholar]
- Brown AM, Schwindt PC, Crill WE. Different voltage dependence of transient and persistent Na+ current is compatible with modal-gating hypothesis for sodium channels. Journal of Neurophysiology. 1994;71:2562–2565. doi: 10.1152/jn.1994.71.6.2562. [DOI] [PubMed] [Google Scholar]
- Cannon SC. Ion-channel defects and aberrant excitability in myotonia and periodic paralysis. Trends in Neurosciences. 1996;19:3–10. doi: 10.1016/0166-2236(96)81859-5. [DOI] [PubMed] [Google Scholar]
- Cannon SC, Corey DP. Loss of Na+ channel inactivation by anemone toxin (ATX II) mimics the myotonic state in hyperkalaemic periodic paralysis. The Journal of Physiology. 1993;466:501–520. [PMC free article] [PubMed] [Google Scholar]
- Castillo C, Piernavieja C, Recio-Pinto E. Anemone toxin II unmasks two conductance states in neuronal sodium channels. Brain Research. 1996;733:231–242. doi: 10.1016/0006-8993(96)00566-5. [DOI] [PubMed] [Google Scholar]
- Chagnac-Amitai Y, Connors BW. Synchronized excitation and inhibition driven by intrinsically bursting neurones in neocortex. Journal of Neurophysiology. 1989;62:1149–1162. doi: 10.1152/jn.1989.62.5.1149. [DOI] [PubMed] [Google Scholar]
- Chao TI, Alzheimer C. Effects of phenytoine on the persistent Na+ current of mammalian CNS neurones. NeuroReport. 1995;6:1778–1780. doi: 10.1097/00001756-199509000-00017. [DOI] [PubMed] [Google Scholar]
- Connors BW. Initiation of synchronized neuronal bursting in neocortex. Nature. 1984;310:685–687. doi: 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurones. Trends in Neurosciences. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ, Prince DA. Electrophysiological properties of neocortical neurons in vitro. Journal of Neurophysiology. 1982;48:1302–1320. doi: 10.1152/jn.1982.48.6.1302. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annual Review of Physiology. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Fleidervish IA, Gutnick MJ. Kinetics of slow inactivation of persistent sodium current in layer V neurons of mouse neocortical slices. Journal of Neurophysiology. 1996;76:2125–2130. doi: 10.1152/jn.1996.76.3.2125. [DOI] [PubMed] [Google Scholar]
- Franceschetti S, Guatteo E, Panzica F, Sancini G, Wanke E, Avanzini G. Ionic mechanisms underlying burst firing in pyramidal neurons: intracellular study in rat sensorimotor cortex. Brain Research. 1995;69:127–139. doi: 10.1016/0006-8993(95)00807-3. 10.1016/0006-8993(95)00807-3. [DOI] [PubMed] [Google Scholar]
- Guatteo E, Franceschetti S, Bacci A, Avanzini G, Wanke E. A TTX-sensitive conductance underlying burst firing in isolated pyramidal neurones from rat neocortex. Brain Research. 1996;741:1–12. doi: 10.1016/s0006-8993(96)00866-9. 10.1016/S0006-8993(96)00866-9. [DOI] [PubMed] [Google Scholar]
- Haas HL, Schaerer B, Vosmansky M. A simple perfusion chamber for the study of nervous tissue slices in vitro. Journal of Neuroscience Methods. 1979;1:323–325. doi: 10.1016/0165-0270(79)90021-9. 10.1016/0165-0270(79)90021-9. [DOI] [PubMed] [Google Scholar]
- Hartung K, Rathmayer W. Anemonia sulcata toxins modify activation and inactivation of Na+ currents in a crayfish neurone. Pflügers Archiv. 1985;404:119–125. doi: 10.1007/BF00585406. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Currents of Excitables Membranes. Sunderland, MA, USA: Sinauer Associates Inc.; 1992. p. 607. [Google Scholar]
- Hoehn K, Watson TWJ, MacVicar BA. A novel tetrodotoxin-insensitive, slow sodium current in striatal and hippocampal neurons. Neuron. 1993;30:543–552. doi: 10.1016/0896-6273(93)90341-n. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends in Neurosciences. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein βγ subunits. Neuron. 1997;19:443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends in Neurosciences. 1996;19:530–534. doi: 10.1016/s0166-2236(96)40004-2. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Avanzini G, Franceschetti S. An increment in the non inactivating fraction of the sodium current (INa) is sufficient to induce burst firing in neocortical pyramidal neurons. European Journal of Neuroscience. 1996;(suppl. 9):9.34. [Google Scholar]
- Mattia D, Hwa GGC, Avoli M. Membrane properties of rat subicular neurones in vitro. Journal of Neurophysiology. 1993;70:1244–1248. doi: 10.1152/jn.1993.70.3.1244. [DOI] [PubMed] [Google Scholar]
- Neumcke B, Schwarz JR, Stämpfli R. A comparison of sodium currents in rat and frog myelinated nerve: normal and modified sodium inactivation. The Journal of Physiology. 1987;382:175–191. doi: 10.1113/jphysiol.1987.sp016362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numann R, Catterall WA, Scheuer T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science. 1991;254:115–118. doi: 10.1126/science.1656525. [DOI] [PubMed] [Google Scholar]
- Oxford GS, Wagoner PK. The inactivating K+ current in GH3 pituitary cells and its modification by chemical reagents. The Journal of Physiology. 1989;410:587–612. doi: 10.1113/jphysiol.1989.sp017550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel α subunit. Journal of Biological Chemistry. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Romey G, Abita JP, Schweitz H, Wunderer G, Lazdunski M. Sea anemone toxin: a tool to study molecular mechanisms of nerve conduction and excitation-secretion coupling. Proceedings of the National Academy of Sciences of the USA. 1976;73:4055–4059. doi: 10.1073/pnas.73.11.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sashihara S, Yanagihara N, Kobayashi H, Izumi F, Tsuji S, Murai Y, Mita T. Overproduction of voltage-dependent Na+ channels in the developing brain of genetically seizure-susceptible E1 mice. Neuroscience. 1992;48:285–291. doi: 10.1016/0306-4522(92)90490-s. [DOI] [PubMed] [Google Scholar]
- Segal MM, Douglas AF. Late sodium channel openings underlying epileptiform activity are preferentially diminished by the anticonvulsant phenytoin. Journal of Neurophysiology. 1997;77:3021–3034. doi: 10.1152/jn.1997.77.6.3021. [DOI] [PubMed] [Google Scholar]
- Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurones. Science. 1991;251:432–436. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Schwindt PC, Chubb MC, Crill WE. Properties of persistent sodium conductance and calcium conductance of layer 5 neurones from cat sensorimotor cortex in vitro. Journal of Neurophysiology. 1985;53:153–170. doi: 10.1152/jn.1985.53.1.153. [DOI] [PubMed] [Google Scholar]
- Taylor CP. Na+ currents that fail to inactivate. Trends in Neurosciences. 1993;16:455–460. doi: 10.1016/0166-2236(93)90077-y. [DOI] [PubMed] [Google Scholar]
- Tseng GF, Prince DA. Heterogeneity of rat corticospinal neurons. Journal of Comparative Neurology. 1993;335:92–108. doi: 10.1002/cne.903350107. [DOI] [PubMed] [Google Scholar]
- Ulbricht W. The inactivation of sodium channels in the node of Ranvier and its chemical modification. In: Narahashi T, editor. Ion Channels. Vol. 2. New York: Plenum Press; 1990. pp. 123–168. [DOI] [PubMed] [Google Scholar]
- Yang CR, Seamans JK. Dopamine D1 receptor actions in layer V-VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. Journal of Neuroscience. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]