

Abstract
To investigate the function of the murine cystic fibrosis transmembrane conductance regulator (CFTR), a full-length cDNA encoding wild-type murine CFTR was assembled and stably expressed in Chinese hamster ovary (CHO) cells.
Like human CFTR, murine CFTR formed Cl− channels that were regulated by cAMP-dependent phosphorylation and intracellular ATP. However, murine CFTR Cl− channels had a reduced single-channel conductance and decreased open probability (Po) compared with those of human CFTR.
Analysis of the dwell time distributions of single channels suggested that the reduced Po of murine CFTR was caused by both decreased residence in the open state and transitions to a new closed state, described by an intermediate closed time constant.
For both human and murine CFTR, ATP and ADP regulated the rate of exit from the long-lived closed state.
5′-Adenylylimidodiphosphate (AMP-PNP) and pyrophosphate, two compounds that disrupt cycles of ATP hydrolysis, stabilized the open state of human CFTR. However, neither agent locked murine CFTR Cl− channels open, although AMP-PNP increased the Po of murine CFTR.
The data indicate that although human and murine CFTR have many properties in common, some important differences in function are observed. These differences could be exploited in future studies to provide new understanding about CFTR.
The cystic fibrosis transmembrane conductance regulator (CFTR; Riordan et al. 1989) is a small conductance Cl− channel, regulated by cAMP-dependent phosphorylation and intracellular ATP, that is predominantly located in the apical membrane of epithelia (for reviews see Hanrahan, Tabcharani & Grygorczyk, 1993b; Welsh, Tsui, Boat & Beaudet, 1995). Mutations in the gene encoding CFTR cause cystic fibrosis (CF), a common lethal genetic disease in Caucasian populations (Riordan et al. 1989; Welsh et al. 1995). In CF, the loss of CFTR function disrupts the transport of fluid and electrolytes across epithelia and perturbs the quantity and composition of epithelial fluids. Defective epithelial ion transport in CF disrupts the function of a variety of organs lined by epithelia. This leads to the wide-ranging manifestations of the disease that typically include respiratory airway disease, pancreatic failure, meconium ileus, male infertility, and elevated levels of NaCl in sweat (Welsh et al. 1995).
An important goal of CF research is to understand how mutations in CFTR disrupt the function of epithelial cells and tissues, and to apply this knowledge to the development of new treatments for the disease. Towards this goal, several mouse models of CF have been developed by disrupting the murine CFTR gene in embryonic stem cells using gene targeting (for reviews see Dorin, Alton & Porteous, 1994; Grubb & Gabriel, 1997). Although CF mice exhibit several important characteristics of the human disease, including intestinal pathology and defective epithelial ion transport, they develop little or no pulmonary or pancreatic pathology (Dorin et al. 1994; Grubb & Gabriel, 1997). The failure of CF mice to develop pulmonary and pancreatic disease may reflect the presence of a Ca2+-activated apical membrane Cl− conductance in airway and pancreatic epithelia of CF mice that can compensate for the loss of CFTR function in these epithelia (Clarke, Grubb, Yankaskas, Cotton, McKenzie & Boucher, 1994). Consistent with this idea, Rozmahel et al. (1996) identified a secondary genetic factor that modulates the severity of intestinal disease in CF mice by regulating the activity of a Ca2+-activated Cl− conductance. However, other observations suggest that the failure of CF mice to develop pulmonary and pancreatic disease may reflect differences in the function of human and murine CFTR. First, human CFTR cDNAs incompletely correct the loss of CFTR function in CF mice (Zhou, Dey, Wert, DuVall, Frizzell & Whitsett, 1994; Rozmahel et al. 1997); only by using a yeast artificial chromosome with an intact human CFTR gene was the function of CFTR fully restored to CF mice (Manson et al. 1997). Second, the properties of shark and Xenopus CFTR Cl− channels differ in a number of ways from those of human CFTR (Hanrahan et al. 1993a; Price, Ishihara, Sheppard & Welsh, 1996). To investigate the function of murine CFTR, we assembled a full-length cDNA encoding wild-type murine CFTR, stably expressed this cDNA in Chinese hamster ovary (CHO) cells, and studied single channels using excised inside-out membrane patches.
METHODS
Assembly of a full-length murine CFTR cDNA and generation of stable cell lines
Overlapping portions from the 5′ end of the murine CFTR gene were amplified from adult mouse kidney (C57BL/6J mouse strain; Animal Resource Centre, Perth, Western Australia) by reverse transcriptase polymerase chain reaction (RT-PCR). These regions, denoted by the numbering of Tata et al. (1991), were amplified using the following primers: nucleotide (nt) 131 to nt 598: 5′-GAC ATCATGCAGAAGTCGCCTTTGG-3′ and 5′-TGTTCT CATCTGCATTCCAATGCGA-3′; nt 426 to nt 1308: 5′-TCC AGCCTGTCTTGCTAGGAAGAAT-3′ and 5′-CTGTGG TCATTAAGTTATACTCC-3′; nt 881 to nt 2095: 5′-GAT CAAAGAGCTGCAAAGATCAATG-3′ and 5′-AAACTG GTCAAAAGTATCATAC-3′. Amplification products were cloned into the EcoRV site of pBluescript SK II and the sequence of the inserted DNA checked for PCR errors by dideoxy sequencing with universal, primary PCR, and internal primers. Cloned inserts of these three regions, which were free of PCR errors, were assembled to form a full-length version of the murine CFTR gene. To stabilize this construct, the cryptic promoter in exon 6b was mutated as previously described (Drumm et al. 1990) with the conversion by PCR mutagenesis of the sequence starting at nt 933 from tgataat to cgacaac. In contrast to human CFTR (Drumm et al. 1990), mutating this sequence by itself did not produce a stable cDNA. A full-length murine CFTR cDNA that was stable in bacterial cells was only generated when murine intron 11 was also inserted into the construct. Full details of these assembly and mutagenesis procedures are available upon request from the authors.
The final clone, termed pFLCFTRIn, carried the complete murine CFTR coding sequence from nt 131 to nt 6298 with exon 6b mutated and exons 11 and 12 interrupted by intron 11. The sequence of the coding regions manipulated during the assembly procedure was redetermined by dideoxy sequencing. The CFTR coding sequence was isolated from this plasmid using Sma I and cloned into an end-filled Xba I site in the expression vector pEF-BOS (Mizushima & Nagata, 1990). The sequence of the entire murine CFTR protein coding region in this plasmid, termed pEFmCFTR, was again verified on both DNA strands using dideoxy sequencing with primers positioned at 300 base pair intervals. The final derived sequence encoded a protein identical to that predicted by Yorifuji et al. (1991) for murine CFTR.
CHO-K1 cells (American Type Culture Collection, Rockville, MD, USA) were co-transfected with the plasmids pEFmCFTR and ppgk-neo (Tybulewicz, Crawford, Jackson, Bronson & Mulligan, 1991) using the cationic lipid DOTAP (Boehringer Mannheim) and selected for neomycin resistance (neomycin, 600 μg ml−1). Single colonies were expanded and those expressing the murine CFTR gene were identified by RT-PCR. For the studies described below, two CHO cell lines stably expressing wild-type murine CFTR were used. As similar results were obtained with both cell lines the data have been pooled.
Cell culture
CHO cells were grown in Ham's F-12 nutrient medium, supplemented with 10% fetal calf serum, and 600 μg ml−1 neomycin (all from Life Technologies Ltd, Paisley, UK), at 37°C in a humidified atmosphere of 5% CO2. Mouse mammary epithelial cells (C127 cells) stably expressing wild-type human CFTR (Marshall et al. 1994) were cultured as previously described (Sheppard & Robinson, 1997). C127 and CHO cells expressing wild-type human CFTR were generous gifts of Drs S. H. Cheung, C. R. O'Riordan and A. E. Smith (Genzyme, Framingham, MA, USA). For experiments using excised inside-out membrane patches, cells were seeded onto glass coverslips and used within 48 h.
Electrophysiology
CFTR Cl− channels were recorded in excised inside-out membrane patches using an Axopatch 200A patch-clamp amplifier (Axon Instruments Inc.) and pCLAMP data acquisition and analysis software (version 6.03; Axon Instruments Inc.) as previously described (Hamill, Marty, Neher, Sakmann & Sigworth, 1981; Sheppard & Robinson, 1997). The established sign convention was used throughout; currents produced by positive charge moving from intra- to extracellular solutions (anions moving in the opposite direction) are shown as positive currents.
The bath (intracellular) solution contained (mM): 140 N-Methyl-d- glucamine (NMDG), 3 MgCl2, 1 CsEGTA, and 10 Tes, adjusted to pH 7.3 with HCl ([Cl−], 147 mM; free [Ca2+], < 10−8 M). The pipette (extracellular) solution contained (mM): 140 NMDG, 140 aspartic acid, 5 CaCl2, 2 MgSO4, and 10 Tes, adjusted to pH 7.3 with Tris ([Cl−], 10 mM). In some experiments, the pipette contained the 147 mM Cl− intracellular solution. CFTR Cl− channels were activated by the addition of the catalytic subunit of protein kinase A (PKA; 75 nM) and ATP (1 mM) to the intracellular solution within 5 min of excising membrane patches. There was no difference in single-channel open probability (Po) between experiments in which membrane patches were excised directly into ATP and PKA and those where ATP and PKA were added after membrane patch excision. (For murine CFTR, Po= 0.08 ± 0.01 (n = 5) both when membrane patches were excised directly into ATP and PKA and when ATP and PKA were added after patch excision; P = 0.95.) To prevent channel run-down, PKA was maintained in the intracellular solution for the duration of the experiment. Membrane patches were clamped at -50 mV, and the intracellular solution was maintained at 37°C using a temperature-controlled microscope stage (Brook Industries, Lake Villa, IL, USA).
In this study, we used membrane patches that contained five or less and four or less active channels for human and murine CFTR, respectively. The number of channels in each patch was determined from the maximum number of simultaneous channel openings observed during the course of an experiment. An experiment typically lasted 30-90 min and included multiple interventions each of 4 min duration that significantly stimulated Po (e.g. ATP (1-3 mM) + PKA (75 nM)). To examine the effect of ATP, ADP, 5′-adenylylimidodiphosphate (AMP-PNP), and pyrophosphate (PPi) on the activity of murine CFTR, interventions of 4 min duration were compared with the average of pre- and postintervention control periods each of 4 min duration, during which time the intracellular solution contained ATP (0.3 mM).
Single-channel currents were initially recorded on digital audiotape using a digital tape recorder (Biologic Scientific Instruments, model DTR-1204; Intracel Ltd, Royston, UK) at a bandwidth of 10 kHz. On playback, records were filtered with an eight-pole Bessel filter (Frequency Devices, model 902LPF2; SCENSYS Ltd, Aylesbury, UK) at a corner frequency of 500 Hz and acquired using a Digidata 1200 interface (Axon Instruments, Inc.) and pCLAMP at a sampling rate of 5 kHz.
Single-channel current amplitudes were determined from the fit of Gaussian distributions to current amplitude histograms. We have previously reported that using a Cl− concentration gradient (external [Cl−], 10 mM; internal [Cl−], 147 mM), wild-type human CFTR has a rectifying current-voltage (I-V) relationship with a reversal potential at about +60 mV, consistent with Cl− selectivity (expected chloride equilibrium potential (ECl), +71 mV; Sheppard, Rich, Ostedgaard, Gregory, Smith & Welsh, 1993). The fit of linear least-squares regression lines to single-channel I-V relationships was used to determine single-channel conductance at negative voltages, where the I-V relationship was linear.
For Po and kinetic analyses, lists of open and closed times were created using a half-amplitude crossing criterion for event detection. Transitions less than 1 ms in duration were excluded from the analyses. Single-channel open and closed time histograms were plotted with logarithmic x-axes with 10 bins decade−1, and the maximum likelihood method was used to fit a one-, two- or three-component exponential to the data. To determine whether a sum of exponential components was significantly better than a fit with fewer components, the F statistic and the log-likelihood ratio (LLR) test were used as previously described (Winter, Sheppard, Carson & Welsh, 1994). The F statistic was tested at the P < 0.05 significance level and the LLR test was considered statistically significant at a value of 2.0 or greater. Only membrane patches that contained a single active channel were used for single-channel kinetic analyses.
Reagents
The catalytic subunit of PKA was obtained from Promega Ltd (Southampton, UK). ADP, ATP (both disodium salts), AMP-PNP (lithium salt), and PPi (tetrasodium salt) were purchased from Sigma-Aldrich Company Ltd (Poole, UK). All other chemicals were of reagent grade.
Statistics
Results are expressed as means ±s.e.m. of n observations. To test for differences between groups of data, an analysis of variance (ANOVA) was used. To compare only two sets of data, we used Student's t test. Differences were considered statistically significant when P < 0.05. All tests were performed using SigmaStat (version 1.03; Jandel Scientific GmbH, Erkrath, Germany).
RESULTS
Wild-type murine CFTR forms a regulated Cl− channel
To learn whether wild-type murine CFTR forms a regulated Cl− channel, we expressed wild-type murine CFTR in CHO cells and studied channels using excised inside-out membrane patches. Figure 1 shows a typical recording from a membrane patch containing several channels. Under basal conditions no channel activity was observed. Addition of ATP (1 mM) to the intracellular solution was without effect, but the addition of PKA (75 nM) in the continued presence of ATP (1 mM) activated channels (Fig. 1). When ATP and PKA were removed channels closed (Fig. 1, Wash). However, readdition of ATP (0.3 mM) reactivated channels (Fig. 1). When added to membrane patches excised from untransfected CHO cells ATP and PKA were without effect (n = 10, data not shown). These results suggest that like wild-type human CFTR, murine CFTR is regulated by cAMP-dependent phosphorylation and intracellular ATP.
Figure 1. Wild-type murine CFTR forms a channel regulated by cAMP-dependent phosphorylation and intracellular ATP.
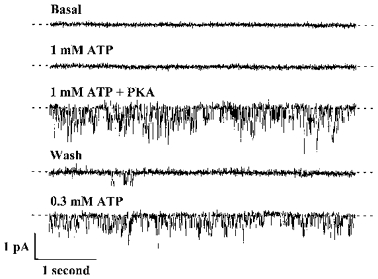
Representative records are from an excised inside-out membrane patch from a CHO cell stably expressing wild-type murine CFTR. ATP (0.3 mM or 1 mM) and the catalytic subunit of PKA (75 nM) were present in the intracellular solution during the times indicated. Each trace is 5 s long. Voltage was clamped at -50 mV, and there was a large Cl− concentration gradient across the membrane patch (external [Cl−], 10 mM; internal [Cl−], 147 mM). Dashed lines indicate the closed channel state and downwards deflections correspond to channel openings. For the purpose of illustration, records were filtered at 500 Hz and digitized at 1 kHz.
To investigate further the function of murine CFTR, we examined the single-channel properties of murine CFTR. For comparison, we studied wild-type human CFTR Cl− channels using excised inside-out membrane patches from C127 cells that stably express wild-type human CFTR (Marshall et al. 1994). We used C127 cells because they have a lower density of wild-type human CFTR Cl− channels than CHO cells (data not shown). However, the properties and regulation of wild-type human CFTR Cl− channels do not differ between the two cell types. For further information, see the legends of Figs 2 and 8.
Figure 2. Comparison of the single-channel properties of human and murine CFTR.
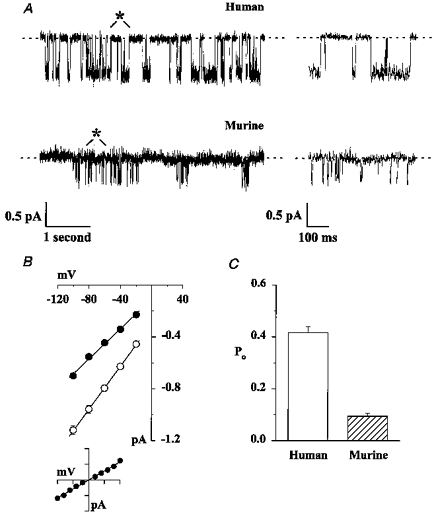
A, representative single-channel records of human and murine CFTR. ATP (1 mM) and the catalytic subunit of PKA (75 nM) were present in the intracellular solution. Voltage was clamped at -50 mV, and there was a large Cl− concentration gradient across the membrane patch (external [Cl−], 10 mM; internal [Cl−], 147 mM). Traces on the left are 5 s long, the 500 ms portions indicated by asterisks are shown on an expanded time scale to the right. B, single-channel I-V relationships of human (○) and murine (•) CFTR. Symbols and error bars indicate means ±s.e.m. of n = 5-6 and n = 7 for human and murine CFTR, respectively, determined using the conditions described in A. Where error bars are not evident the symbol has obscured them. The inset shows the I-V relationship of murine CFTR when the membrane patch was bathed in symmetrical 147 mM Cl− solutions (abscissa: -100 to +100 mV; ordinate: -1 to +1 pA). Data are means ±s.e.m. (n = 5). Under these conditions, reversal potential (Erev) was 2.0 ± 0.7 mV (n = 5) and ECl was 0 mV. Lines indicate the mean slope conductance calculated from the slope conductance of individual experiments. C, Po of human and murine CFTR. Columns and error bars indicate means +s.e.m. (n = 6). Other details as in A. In excised inside-out membrane patches from CHO cells expressing wild-type human CFTR, the single-channel conductance was 9.03 ± 0.18 pS (n = 7) and the Po was 0.43 ± 0.05 (n = 5) under the conditions described in A. These values are not significantly different from those of human CFTR Cl− channels in membrane patches from C127 cells expressing wild-type human CFTR (P > 0.1).
Figure 8. Pyrophosphate fails to stimulate murine CFTR Cl− channels.
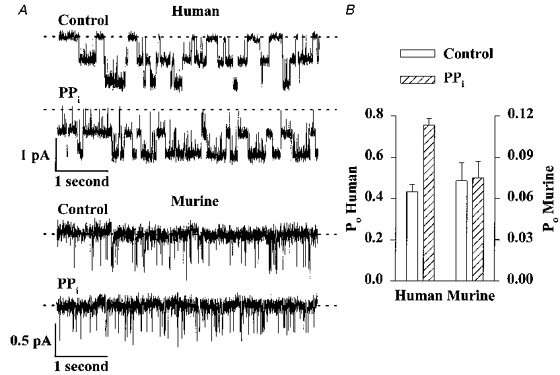
A, effect of PPi (5 mM) on the activity of two human and two murine CFTR Cl− channels. ATP (0.3 mM) and PKA (75 nM) were continuously present in the intracellular solution; voltage was -50 mV. Each trace is 5 s long. B, effect of PPi (5 mM) on the Po of human (left ordinate) and murine (right ordinate) CFTR Cl− channels. Note the change in scale. Columns and error bars indicate means +s.e.m. of n = 4 and n = 6 for human and murine CFTR, respectively. PPi (5 mM) increased the Po of human CFTR to 175 ± 8% of the control value (n = 4; P < 0.01), but was without effect on the Po of murine CFTR (n = 6; P > 0.05). Other details as in A. Using the conditions described, PPi (5 mM) increased the Po of human CFTR Cl− channels in membrane patches excised from CHO cells expressing wild-type human CFTR to 0.82 ± 0.13 (mean ±s.d.; n = 2; 189 ± 6% of the control value; n = 2).
Figure 2A shows representative single-channel traces of human and murine CFTR. Two important differences between human and murine CFTR are apparent from these traces. First, the single-channel current amplitude of murine CFTR was less than that of human CFTR. To quantify this difference, we measured single-channel conductance (Fig. 2B). The single-channel conductance of murine CFTR (5.76 ± 0.16 pS; n = 7) was significantly less than that of human CFTR (8.29 ± 0.18 pS; n = 6; P < 0.001). However, the gating behaviour of murine CFTR was characterized by many poorly resolved channel openings (Fig. 2A). Because these openings might reduce the single-channel current amplitude of murine CFTR when averaged with well-resolved channel openings, we verified that the single-channel conductance of murine CFTR was decreased. Using values of single-channel current amplitude determined by cursor measurements of individual well-resolved channel openings, the single-channel conductance of murine CFTR was 5.66 ± 0.12 pS (n = 3). This value did not differ from that calculated using current amplitude histograms to determine single-channel current amplitude (5.41 ± 0.13 pS; n = 3; P > 0.05). Like human CFTR, channels were Cl− selective (Fig. 2B, inset).
Second, the gating behaviour of murine CFTR was dramatically different from that of human CFTR (Fig. 2A). As previously described (Winter et al. 1994), the gating behaviour of human CFTR was characterized by bursts of channel openings, interrupted by brief flickery closures, separated by longer closures between bursts. In contrast, the pattern of gating of murine CFTR was characterized by an apparent shortening of the time that channels were open (Fig. 2A). To quantify the difference in gating behaviour between human and murine CFTR, we measured Po. Figure 2C shows that the Po of murine CFTR was significantly less than that of human CFTR. Because the decreased open time of murine CFTR might cause the number of channels in a membrane patch (N) to be underestimated, we verified that experiments were of sufficient length. The time required to observe at least one single all-open event is (3τo/N)/(Po)N (Venglarik, Schultz, Frizzell & Bridges, 1994). For murine CFTR using values of τo= 6.22 ms and Po= 0.094 (Table 1 and Fig. 2C), 59 s were required to detect four channels in a membrane patch and 501 s were required to exclude the possibility that five channels were present. These results suggest that experiments which typically lasted 30-90 min were of adequate length to determine that membrane patches contained four or less active murine CFTR Cl− channels.
Table 1.
Open and closed time constants of human and murine CFTR Cl− channels
| Area under curve | ||||||||
|---|---|---|---|---|---|---|---|---|
| τo (ms) | τcf (ms) | τci (ms) | τcs (ms) | τcf | τci | τcs | n | |
| Human | 66.8 ± 8.59 | 3.18 ± 0.20 | — | 169.4 ± 26.7 | 0.59 ± 0.02 | — | 0.41 ± 0.02 | 6 |
| Murine | 6.22 ± 0.83 | 5.27 ± 0.76 | 84.0 ± 6.47 | 410.1 ± 42.4 | 0.51 ± 0.03 | 0.43 ± 0.04 | 0.06 ± 0.01 | 6 |
Time constants were derived from the fitting of one-, two- or three-component exponential functions to open and closed time histograms using the maximum likelihood method as described in the Methods. τo, open time constant; τcf, fast closed time constant; τci, intermediate closed time constant; τcs, slow closed time constant. Area under curve indicates the proportion of the total closed time distribution that corresponds to the different closed time constants. Measurements were made in the presence of ATP (1 mM) and PKA (75 nM) at −50 mV. Data are means ±s.e.m. of n single-channel patches and in each patch approximately 5000 events were analysed. Although the number of events in the closed time histograms were relatively small (1000 to 3000), for murine CFTR six of six closed time histograms were fitted with a three-component exponential and all six fits were significant both by the F statistic (P < 0.05) and the LLR test (> 2). In contrast, for human CFTR only two of six closed time histograms could be fitted with a three-component exponential and of these only one fit was significant by both the F statistic (P < 0.05) and the LLR test (> 2). For murine CFTR, similar results were obtained when the number of closed time events was greater than 7600.
We speculated that the reduced Po of murine CFTR may result from the incomplete phosphorylation of murine CFTR by PKA (75 nM). However, addition of higher concentrations of PKA did not significantly increase the Po of murine CFTR Cl− channels (75 nM PKA, Po= 0.09 ± 0.01 (n = 4); 225 nM PKA, Po= 0.10 ± 0.01 (n = 4); P > 0.5). For both human and murine CFTR, Po was relatively voltage independent (data not shown).
Analysis of the open and closed time distributions of human and murine CFTR
To understand why the Po of murine CFTR was reduced, we examined the distribution of open and closed times using membrane patches that contained only a single active channel (Fig. 3 and Table 1). Consistent with previous results, the open and closed time histograms of human CFTR were best fitted with one- and two-component functions, respectively (Fig. 3 and Table 1; Winter et al. 1994). In contrast, the open and closed time histograms of murine CFTR were best fitted with one- and three-component functions, respectively (Fig. 3 and Table 1). The new population of closed times of murine CFTR was described by an intermediate closed time constant (τci; Fig. 3 and Table 1).
Figure 3. Open (A) and closed (B) time histograms of human and murine CFTR Cl− channels.
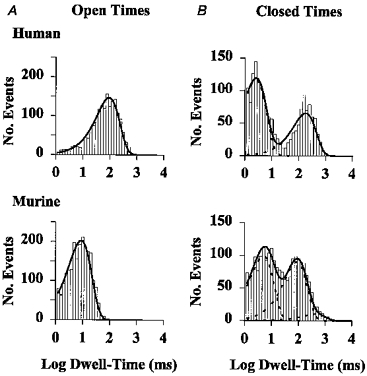
Data are from experiments in which the membrane patch contained only one active channel, studied in the presence of ATP (1 mM) and PKA (75 nM) at -50 mV. For the open time histograms, the continuous line is the fit of a one-component exponential function. For the closed time histograms, the continuous line is the fit of either a two- (human CFTR) or a three- (murine CFTR) component exponential function and the dashed lines show the individual components of the exponential functions. Logarithmic x-axes with 10 bins decade−1 were used for both open and closed time histograms. Other details as in Table 1.
The data presented in Table 1 suggest that the decreased Po of murine CFTR is a consequence of changes in both open and closed times. The main causes were first, the greater than 10-fold decrease in the open time constant of murine CFTR compared with that of human CFTR (P < 0.001) and second, the new population of closed times of murine CFTR described by τci; τci represented 43% of the total closed time distribution of murine CFTR. The slow closed time constant (τcs) of murine CFTR was more than double that of human CFTR. However, this change had only a small effect on Po because τcs represented only 6% of the total closed time distribution of murine CFTR, but 41% of that of human CFTR. The fast closed time constant (τcf) of murine CFTR was almost double that of human CFTR, but its share of the closed time distribution was little changed. These results suggest that the decreased Po of murine CFTR mainly results first from reduced residence in the open state, and second from frequent transitions to a new closed state, described by an intermediate closed time constant.
Effect of intracellular ATP and ADP on the activity of murine CFTR
Previous studies of human CFTR Cl− channels suggest that ATP regulates the rate of transition from the long-lived closed state to a burst of activity in which the channel flickers back and forth between the open state and the short-lived closed state (Winter et al. 1994). To investigate how intracellular ATP regulates murine CFTR, we examined the effect of ATP concentration on channel activity. Figure 4 shows that as the ATP concentration increased, the activity of both human and murine CFTR Cl− channels increased. For both human and murine CFTR, the relationship between Po and ATP concentration was best fitted by a Michaelis-Menten function (human CFTR: Km= 117 μM, maximum Po (Po,max) = 0.61; murine CFTR: Km= 106 μM, Po,max= 0.10; Fig. 4B). These data suggest that the ATP dependence of murine CFTR was similar to that of human CFTR, despite the large difference in Po,max values. Similar Km values for human CFTR have been reported by Gunderson & Kopito (1994). However, other studies have suggested both lower (e.g. 24 μM; Venglarik et al. 1994) and higher (e.g. 3 mM; Quinton & Reddy, 1992) Km values for human CFTR.
Figure 4. Effect of ATP concentration on the activity of human and murine CFTR Cl− channels.
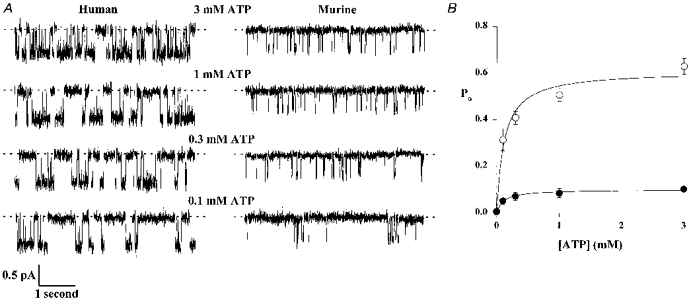
A, comparison of the effect of different ATP concentrations on the activity of human and murine CFTR Cl− channels. PKA (75 nM) was present throughout; voltage was −50 mV. Each trace is 5 s long. B, relationship between ATP concentration and Po for human (○) and murine (•) CFTR. Symbols and error bars indicate means ±s.e.m. of n = 4-5 values at each concentration. Continuous lines are Michaelis-Menten fits to the mean data.
In a small number of membrane patches that contained only a single active channel, the effect of ATP concentration on open and closed time constants was examined. For both human and murine CFTR, only τcs was ATP dependent (Fig. 5). As the ATP concentration increased, τcs decreased (P < 0.05, one-way ANOVA). None of the other open and closed time constants of human and murine CFTR were ATP dependent (P > 0.05, one-way ANOVA; Fig. 5). Using the data in Table 1, the rate of exit from the long-lived closed state (1/τcs) may be calculated for human and murine CFTR. For human CFTR, the rate of exit from the long-lived closed state was 6.03 ± 0.62 s−1 (n = 6), measured in the presence of ATP (1 mM) and PKA (75 nM) at -50 mV, in good agreement with previous reports (Winter et al. 1994; Li et al. 1996). However, under the same conditions, the rate of exit of murine CFTR from the long- lived closed state (3.00 ± 0.32 s−1; n = 6) was significantly less than that of human CFTR (P < 0.002).
Figure 5. Relationship between open and closed time constants and ATP concentration for human and murine CFTR.
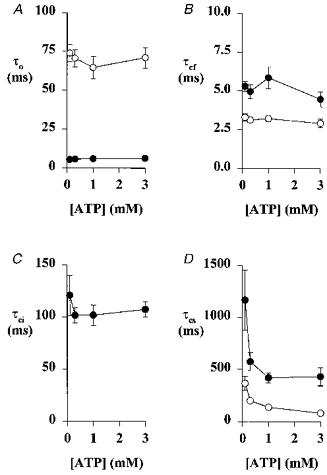
Open time constant (τo, A), fast closed time constant (τcf, B), intermediate closed time constant (τci, C), and slow closed time constant (τcs, D) for human (○) and murine (•) CFTR. For human CFTR, symbols and error bars indicate means ±s.e.m. (n = 4-6) at each concentration. For murine CFTR, symbols and error bars indicate means ±s.e.m. (n = 4-5) at each concentration, except τcs at 3 mM ATP where n = 2.
Intracellular ADP increases the interburst duration of human CFTR Cl− channels without altering the length of bursts (Winter et al. 1994). To examine the effect of ADP on the activity of murine CFTR Cl− channels, we added ADP to the intracellular solution in the continuous presence of ATP and PKA (Fig. 6). ADP significantly reduced the activity of both human and murine CFTR Cl− channels (Fig. 6). ADP also increased the residence of murine CFTR in the long-lived closed state (see Fig. 6 legend).
Figure 6. ADP inhibits human and murine CFTR Cl− channels.
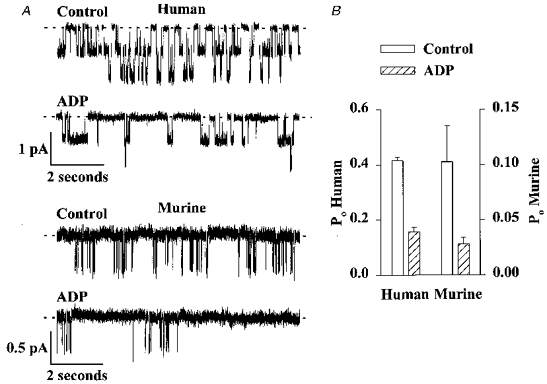
A, effect of ADP (0.3 mM) on the activity of two human CFTR Cl− channels and one murine CFTR Cl− channel. ATP (0.3 mM) and PKA (75 nM) were continuously present in the intracellular solution; voltage was -50 mV. Each trace is 10 s long. B, effect of ADP (0.3 mM) on the Po of human (left ordinate) and murine (right ordinate) CFTR Cl− channels. Note the change in scale. Columns and error bars indicate means +s.e.m. (n = 5) for each condition. ADP (0.3 mM) reduced the Po of human and murine CFTR Cl− channels to 38 ± 4 and 27 ± 6% of the control values, respectively (n = 5; P < 0.05). Other details as in A. In one membrane patch that contained only a single murine CFTR Cl− channel, ADP increased τcs, but was without effect on the other dwell time constants (Control: τo= 7.59 ms, τcf= 5.09 ms, τci= 92.8 ms, τcs= 527.7 ms; ADP: τo= 7.65 ms, τcf= 5.17 ms, τci= 88.1 ms, τcs= 952.0 ms).
AMP-PNP and PPi fail to stabilize the open state of murine CFTR
In the presence of ATP, the non-hydrolysable ATP analogue AMP-PNP prolongs the burst duration of human CFTR Cl− channels (Gunderson & Kopito, 1994; Hwang, Nagel, Nairn & Gadsby, 1994; Carson, Travis & Welsh, 1995a). Because the pattern of gating of murine CFTR differed from that of human CFTR, we speculated that the interaction of AMP-PNP with murine CFTR may differ from that of human CFTR. To investigate the effect of AMP-PNP on the activity of murine CFTR, we added AMP-PNP to the intracellular solution in the continuous presence of ATP and PKA (Fig. 7); temperature was 37°C. Consistent with previous results (Gunderson & Kopito, 1994; Hwang et al. 1994; Carson et al. 1995a), AMP-PNP greatly prolonged the duration of the occasional burst of activity of human CFTR and hence, increased Po (Fig. 7). AMP-PNP also increased the Po of murine CFTR (Fig. 7). However, it did not lock murine CFTR Cl− channels in the open state, in contrast to human CFTR (Fig. 7).
Figure 7. Effect of AMP-PNP on the activity of human and murine CFTR Cl− channels.
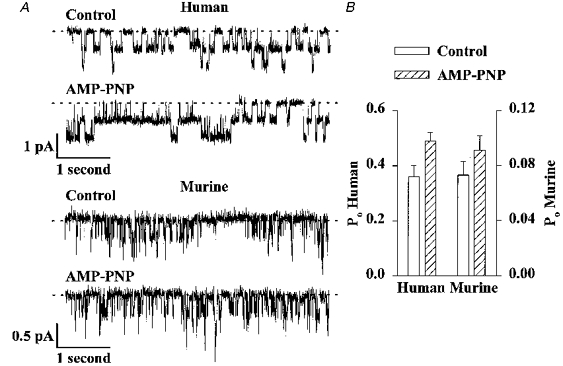
A, comparison of the effect of AMP-PNP (1 mM) on the activity of two human and three murine CFTR Cl− channels. ATP (0.3 mM) and PKA (75 nM) were continuously present in the intracellular solution; voltage was -50 mV. Each trace is 5 s long. B, effect of AMP-PNP (1 mM) on the Po of human (left ordinate) and murine (right ordinate) CFTR Cl− channels. Note the change in scale. Columns and error bars indicate means +s.e.m. (n = 6) for each condition. AMP-PNP (1 mM) increased the Po of human and murine CFTR Cl− channels to 145 ± 17 and 130 ± 11% of the control values, respectively (n = 6; P < 0.05). Other details as in A.
Previous studies have shown that at 37°C the inorganic phosphate analogue PPi more potently prolongs the burst duration of human CFTR than AMP-PNP (Gunderson & Kopito, 1994; Carson et al. 1995a; Carson, Winter, Travis & Welsh, 1995b). To learn whether PPi would lock murine CFTR Cl− channels open, we added PPi to the intracellular solution in the continuous presence of ATP and PKA (Fig. 8). Consistent with previous results (Gunderson & Kopito, 1994; Carson et al. 1995b), PPi greatly increased the burst duration of human CFTR and significantly stimulated Po (Fig. 8). In contrast, PPi did not stimulate the activity of murine CFTR and no increase in the open time of murine CFTR was apparent (Fig. 8).
DISCUSSION
When expressed in CHO cells, wild-type murine CFTR formed a regulated Cl− channel with many properties similar to those of human CFTR. However, some important differences in function were observed between human and murine CFTR Cl− channels. Murine CFTR Cl− channels had a reduced single-channel conductance, decreased Po, altered gating behaviour, and were insensitive to stimulation with PPi.
The function of human, murine, shark and Xenopus CFTR has been examined using heterologous cells (Hanrahan et al. 1993a, b; Welsh et al. 1995; Price et al. 1996; this study). In each case, expression of CFTR generated Cl−-selective channels that were regulated by cAMP-dependent phosphorylation. However, single-channel conductance varied between species, decreasing in the following order: Xenopus > human > murine ≥ shark (Hanrahan et al. 1993a; Price et al. 1996; this study). Previous studies have shown that mutation of a number of residues within the membrane-spanning domains (MSDs), including R117, R334, K335, S341, R347, and T1134, reduces the single-channel conductance of human CFTR (Sheppard et al. 1993; Tabcharani et al. 1993; McDonough, Davidson, Lester & McCarty, 1994). Because these residues are conserved across species, other residues within the MSDs probably account for the variation in the single-channel conductance of CFTR between species. These residues could be located within the transmembrane segments that contribute to the CFTR pore (Sheppard et al. 1993; Tabcharani et al. 1993; McDonough et al. 1994; Cheung & Akabas, 1997). Alternatively, they may be located within the extra- and intracellular loops that link the transmembrane segments. Consistent with this latter possibility, deletion of nineteen residues from the second intracellular loop (ICL2) promoted transitions to a subconductance state, while the I-V relationships of the mutations S945L and G970R located in ICL3 showed weak outward rectification in contrast to that of wild-type CFTR (Xie, Drumm, Ma & Davis, 1995; Seibert, Linsdell, Loo, Hanrahan, Riordan & Clarke, 1996b).
When expressed in heterologous cells, the single-channel properties of recombinant wild-type human CFTR accurately reproduce those of native human CFTR Cl− channels in airway and intestinal epithelia (Hanrahan et al. 1993b; Welsh et al. 1995). In contrast, little is known about the single-channel function of murine CFTR in these tissues. Chloride channels with properties resembling those of CFTR have been identified in epithelial cells isolated from rat pancreas and mouse gall bladder (Gray, Greenwell & Argent, 1988; French et al. 1996). The properties of these native rodent CFTR Cl− channels show some similarities to those of recombinant murine CFTR, but also some differences. The most notable difference is the reduced Po of recombinant murine CFTR compared with that of native rodent CFTR Cl− channels (Gray et al. 1988; French et al. 1996; this study). The reason for this difference is not known. It may reflect either differences in the cellular complement of kinases and phosphatases between CHO cells and rodent epithelial cells or the level of expression of CFTR in these different cells (Becq et al. 1994; Welsh et al. 1995).
Based on previous studies of human CFTR (Chang et al. 1993; Hwang et al. 1994; Winter et al. 1994; Carson et al. 1995a; Li et al. 1996; Winter & Welsh, 1997), the decreased Po of recombinant murine CFTR may be a consequence of either inadequate phosphorylation by PKA or altered regulation by ATP hydrolysis. Several lines of evidence suggest that the phosphorylation of murine CFTR by PKA was not defective. First, the major regulatory sites for cAMP-dependent phosphorylation (S660, S737, S795 and S813) are conserved between human and murine CFTR (Riordan et al. 1989; Tata et al. 1991). Second, when we increased the concentration of PKA used to activate murine CFTR, no increase in the Po of murine CFTR was observed. Third, simultaneous mutation of the major PKA regulatory sites (S660,737,795,813A) increased the residence of human CFTR in the long-lived closed state without altering burst duration (Winter & Welsh, 1997). In contrast, murine CFTR resided only briefly in the long-lived closed state. Fourth, a CF-associated mutation in ICL4 that affects the residue G1069 replaces the glycine of human CFTR with the arginine of murine CFTR (Savov, Mercier, Kalaydjieva & Férec, 1994). When expressed in CHO cells, G1069R had a Po intermediate between that of human and murine CFTR (Seibert, Linsdell, Loo, Hanrahan, Clarke & Riordan, 1996a; this study). These data suggest that the low Po of murine CFTR is not likely to result from the defective phosphorylation of murine CFTR by PKA. Instead, the low Po of murine CFTR may, in part, reflect amino acid sequence variation between human and murine CFTR. However, in addition to PKA, other kinases modulate the activity of CFTR, while phosphatases dephosphorylate and inactivate CFTR (Becq et al. 1994; Welsh et al. 1995; Jia, Mathews & Hanrahan, 1997). Therefore, the low Po of murine CFTR expressed in CHO cells may also reflect altered regulation of murine CFTR by kinases other than PKA or the activity of phosphatases. Further studies are required to test these possibilities.
A number of kinetic models which describe the regulation of human CFTR Cl− channels by ATP have been proposed (Hwang et al. 1994; Venglarik et al. 1994; Winter et al. 1994; Carson et al. 1995a; Gunderson & Kopito, 1995). Winter et al. (1994) developed a linear model containing one open (O) and two closed (C) states (C1⇌ C2⇌ O) using excised membrane patches that contained single, phosphorylated CFTR Cl− channels. In this model, ATP and ADP regulated the rate of transition from C1 to C2 without affecting other transition rates (Winter et al. 1994). Although this model has been widely used to investigate the gating behaviour of CFTR, it is a minimal model of channel gating. One reason is that each state or transition likely represents more than one physical event or conformational change. Furthermore, this model does not describe the binding and hydrolysis of ATP at one or both of the nucleotide-binding domains (NBDs).
Using data from excised membrane patches that contained multiple CFTR Cl− channels, Venglarik et al. (1994) developed a linear scheme with four closed states and one open state (C0⇌ ATP + C1⇌ ATP-C2⇌ ATP-O ⇌ ATP-C3). In this model, an ATP-bound closed state (C2) was interposed between the phosphorylated closed state (C1) and the ATP-bound open state (O) to account for the observation that the maximal Po of human CFTR is less than one (Venglarik et al. 1994). A different scheme with three closed states and two open states was proposed by Fischer & Machen (1994) to describe the gating behaviour of CFTR in cell-attached membrane patches. Interestingly, in this model, three of the states were voltage dependent (Fischer & Machen, 1994). To describe the regulation of CFTR gating behaviour by phosphorylation at the R domain and ATP binding and hydrolysis at the two NBDs, more complex models have been proposed (Hwang et al. 1994; Carson et al. 1995a; Gunderson & Kopito, 1995). Two such models involve cycles of ATP hydrolysis at each NBD and interactions between the NBDs (Hwang et al. 1994; Carson et al. 1995a). In these models, ATP hydrolysis at NBD1 is required to open the channel and ATP hydrolysis at NBD2 is required to close the channel (Hwang et al. 1994; Carson et al. 1995a). In another model, an asymmetric cycle of ATP binding and hydrolysis at NBD2 controls channel gating (Gunderson & Kopito, 1995).
Analysis of histograms of open and closed times suggest that the kinetics of phosphorylated murine CFTR Cl− channels were described by a model containing a minimum of one open state and three closed states. Because the exponential functions fitted to dwell time histograms do not have a simple relationship to the rate constants used to describe transitions between states, the connections between the open and closed states could not be determined. However, several lines of evidence suggest that the intermediate closed state of murine CFTR may represent a closed state that interrupts bursts of channel openings rather than a closed state which links the phosphorylated closed state to the open state. First, visual inspection of single-channel records (for example see Figs 2A, 4A and 6A) suggested that the gating behaviour of murine CFTR is characterized by bursts of channel activity. However, bursts of activity of murine CFTR were interrupted by comparatively long closures in contrast to the brief closures that interrupt bursts of activity of human CFTR (Figs 2A, 4A and 6A). Second, like human CFTR, bursts of activity of murine CFTR were separated by long closures (Figs 2A, 4A and 6A). Third, for both human and murine CFTR, ATP regulated the rate of exit from the long-lived closed state, although the rate of exit of murine CFTR was reduced compared with that of human CFTR; ADP also inhibited human and murine CFTR at this transition. Fourth, PPi failed to increase the Po of murine CFTR (see below). Thus, the intermediate closed state may represent closures that interrupt bursts of channel activity.
The non-hydrolysable ATP analogue AMP-PNP has been widely used to investigate the gating behaviour of CFTR (Quinton & Reddy, 1992; Gunderson & Kopito, 1994; Hwang et al. 1994; Carson et al. 1995a; Welsh et al. 1995; Mathews, Tabcharani, Chang, Riordan & Hanrahan, 1996). In the absence of ATP, AMP-PNP failed to support channel activity (Hwang et al. 1994; Welsh et al. 1995). In contrast, in the presence of ATP and cAMP agonists AMP-PNP increased apical Cl− current in sweat duct epithelia (Quinton & Reddy, 1992). Quinton & Reddy (1992) interpreted this observation to suggest that AMP-PNP interacts with CFTR by a non-hydrolytic mechanism to control channel gating. However, other studies of the effect of mixtures of ATP and AMP-PNP on the gating behaviour of CFTR Cl− channels were interpreted to suggest that the hydrolysis of ATP at NBD2 was required to close the channel (Gunderson & Kopito, 1994; Hwang et al. 1994; Carson et al. 1995a). The data also suggest that the effect of mixtures of ATP and AMP-PNP on channel gating is complex. Because AMP-PNP only prolonged the burst duration of CFTR Cl− channels in guinea-pig cardiac myocytes in the presence of ATP and PKA at 25°C, Hwang et al. (1994) suggested that AMP-PNP only locks open highly phosphorylated CFTR Cl− channels. However, mixtures of ATP and AMP-PNP locked open a poorly phosphorylated CFTR variant lacking all ten dibasic PKA consensus sequences (Mathews et al. 1996) and the ability of ATP and AMP-PNP to prolong the burst duration of CFTR was strongly temperature dependent (Hwang et al. 1994; Carson et al. 1995a; Mathews et al. 1996). In contrast, mixtures of ATP and polyphosphates, such as PPi, prolonged the burst duration of CFTR regardless of the temperature (Gunderson & Kopito, 1994; Carson et al. 1995b).
The present results demonstrate that although AMP-PNP and PPi greatly prolonged the burst duration of human CFTR, neither agent locked murine CFTR Cl− channels open. In addition, AMP-PNP increased the Po of murine CFTR, whereas PPi was without effect. The reason for this difference is not known. However, an explanation for the failure of AMP-PNP and PPi to increase the burst duration of murine CFTR is suggested by the effect of PPi on the gating kinetics of human CFTR (Carson et al. 1995b). PPi increased the activity of human CFTR in two ways: first, it increased the rate of channel opening into a burst and second, it decreased the rate of channel closure from a burst (Carson et al. 1995b). With the experimental conditions used, murine CFTR resides only briefly in the long-lived closed state. Therefore, any effect of PPi on the rate of entry to or exit from a burst would be predicted to have only a minimal effect on the Po of murine CFTR. Consistent with this idea, PPi failed to increase the Po of murine CFTR. To increase the Po of murine CFTR, it will be necessary to identify agents that decrease the rate of entry to or promote the rate of exit from the intermediate closed state.
In conclusion, our data indicate that although human and murine CFTR have many properties in common, some important differences in function are observed. These differences could be exploited in future studies to provide new understanding about the relationship between the structure and function of CFTR.
Acknowledgments
We thank Professor M. J. Welsh and colleagues at the University of Iowa, and Professor D. J. Porteous for advice and critical comments. We thank Drs S. H. Cheng, C. R. O'Riordan, and A. E. Smith (Genzyme, Framingham, MA, USA) for the generous gifts of C127 and CHO cells expressing wild-type human CFTR and Drs R. H. Ashley and K. A. Volk for assistance with curve fitting. We also thank the University of Edinburgh Biomedical Sciences Planning Unit Workshop and M. D. McGregor for their services. This work was supported by the Biotechnology and Biological Sciences Research Council, Cystic Fibrosis Trust and National Health and Medical Research Council of Australia.
References
- Becq F, Jensen T J, Chang X-B, Savoia A, Rommens J M, Tsui L-C, Buchwald M, Riordan J R, Hanrahan J W. Phosphatase inhibitors activate normal and defective CFTR chloride channels. Proceedings of the National Academy of Sciences of the USA. 1994;91:9160–9164. doi: 10.1073/pnas.91.19.9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson M R, Travis S M, Welsh M J. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. Journal of Biological Chemistry. 1995a;270:1711–1717. doi: 10.1074/jbc.270.4.1711. 10.1074/jbc.270.4.1711. [DOI] [PubMed] [Google Scholar]
- Carson M R, Winter M C, Travis S M, Welsh M J. Pyrophosphate stimulates wild-type and mutant cystic fibrosis transmembrane conductance regulator Cl− channels. Journal of Biological Chemistry. 1995b;270:20466–20472. doi: 10.1074/jbc.270.35.20466. 10.1074/jbc.270.35.20466. [DOI] [PubMed] [Google Scholar]
- Chang X-B, Tabcharani J A, Hou Y-X, Jensen T J, Kartner N, Alon N, Hanrahan J W, Riordan J R. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. Journal of Biological Chemistry. 1993;268:11304–11311. [PubMed] [Google Scholar]
- Cheung M, Akabas M H. Locating the anion-selectivity filter of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel. Journal of General Physiology. 1997;109:289–299. doi: 10.1085/jgp.109.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke L L, Grubb B R, Yankaskas J R, Cotton C U, McKenzie A, Boucher R C. Relationship of a non-cystic fibrosis transmembrane conductance regulator-mediated chloride conductance to organ-level disease in cftr(-/-) mice. Proceedings of the National Academy of Sciences of the USA. 1994;91:479–483. doi: 10.1073/pnas.91.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorin J R, Alton E W F W, Porteous D J. Mouse models for cystic fibrosis. In: Dodge J A, Brock D J H, Widdicombe J H, editors. Cystic Fibrosis - Current Topics. Vol. 2. Chichester: John Wiley & Sons Ltd; 1994. pp. 3–31. [Google Scholar]
- Drumm M L, Pope H A, Cliff W H, Rommens J M, Marvin S A, Tsui L-C, Collins F S, Frizzell R A, Wilson J M. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell. 1990;62:1227–1233. doi: 10.1016/0092-8674(90)90398-x. [DOI] [PubMed] [Google Scholar]
- Fischer H, Machen T E. CFTR displays voltage dependence and two gating modes during stimulation. Journal of General Physiology. 1994;104:541–566. doi: 10.1085/jgp.104.3.541. 10.1085/jgp.104.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French P J, van Doorninck J H, Peters R H P C, Verbeek E, Ameen N A, Marino C R, De Jonge H R, Bijman J, Scholte B J. A ΔF508 mutation in mouse cystic fibrosis transmembrane conductance regulator results in a temperature-sensitive processing defect in vivo. Journal of Clinical Investigation. 1996;98:1304–1312. doi: 10.1172/JCI118917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M A, Greenwell J R, Argent B E. Secretin-regulated chloride channel on the apical plasma membrane of pancreatic duct cells. Journal of Membrane Biology. 1988;105:131–142. doi: 10.1007/BF02009166. [DOI] [PubMed] [Google Scholar]
- Grubb B R, Gabriel S E. Intestinal physiology and pathology in gene-targeted mouse models of cystic fibrosis. American Journal of Physiology. 1997;273:G258–266. doi: 10.1152/ajpgi.1997.273.2.G258. [DOI] [PubMed] [Google Scholar]
- Gunderson K L, Kopito R R. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane regulator channel gating. Journal of Biological Chemistry. 1994;269:19349–19353. [PubMed] [Google Scholar]
- Gunderson K L, Kopito R R. Conformational states of CFTR associated with channel gating: the role of ATP binding and hydrolysis. Cell. 1995;82:231–239. doi: 10.1016/0092-8674(95)90310-0. [DOI] [PubMed] [Google Scholar]
- Hamill O P, Marty A, Neher E, Sakmann B, Sigworth F J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hanrahan J W, Duguay F, Sansom S, Alon N, Jensen T, Riordan J R, Grzelczak Z. Low-conductance chloride channel activated by cAMP in the rectal gland of the shark Squalus acanthias and in cells heterologously expressing shark CFTR. Bulletin Mount Desert Island Biological Laboratory. 1993a;32:48–49. [Google Scholar]
- Hanrahan J W, Tabcharani J A, Grygorczyk R. Patch clamp studies of apical membrane chloride channels. In: Dodge J A, Brock D J H, Widdicombe J H, editors. Cystic Fibrosis - Current Topics. Vol. 1. Chichester: John Wiley & Sons Ltd; 1993b. pp. 93–137. [Google Scholar]
- Hwang T-C, Nagel G, Nairn A C, Gadsby D C. Regulation of the gating of cystic fibrosis transmembrane conductance regulator Cl channels by phosphorylation and ATP hydrolysis. Proceedings of the National Academy of Sciences of the USA. 1994;91:4698–4702. doi: 10.1073/pnas.91.11.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y, Mathews C J, Hanrahan J W. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. Journal of Biological Chemistry. 1997;272:4978–4984. doi: 10.1074/jbc.272.8.4978. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Wang W, Garami E, Hewryk M, Lee D, Rommens J M, Galley K, Bear C E. ATPase activity of the cystic fibrosis transmembrane conductance regulator. Journal of Biological Chemistry. 1996;271:28463–28468. doi: 10.1074/jbc.271.45.28463. [DOI] [PubMed] [Google Scholar]
- McDonough S, Davidson N, Lester H A, McCarty N A. Novel pore-lining residues in CFTR that govern permeation and open-channel block. Neuron. 1994;13:623–634. doi: 10.1016/0896-6273(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Manson A L, Trezise A E O, MacVinish L J, Kasschau K D, Birchall N, Episkopou V, Vassaux G, Evans M J, Colledge W H, Cuthbert A W, Huxley C. Complementation of null CF mice with a human CFTR YAC transgene. EMBO Journal. 1997;16:4238–4249. doi: 10.1093/emboj/16.14.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall J, Fang S, Ostedgaard L S, O'Riordan C R, Ferrara D, Amara J F, Hoppe H, IV, Scheule R K, Welsh M J, Smith A E, Cheng S H. Stoichiometry of recombinant cystic fibrosis transmembrane conductance regulator in epithelial cells and its functional reconstitution into cells in vitro. Journal of Biological Chemistry. 1994;269:2987–2995. [PubMed] [Google Scholar]
- Mathews C J, Tabcharani J A, Chang X-B, Riordan J R, Hanrahan J W. Characterization of nucleotide interactions with CFTR channels. Pediatric Pulmonology Supplement. 1996;13:221. [Google Scholar]
- Mizushima S, Nagata S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Research. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M P, Ishihara H, Sheppard D N, Welsh M J. Function of Xenopus cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channels and use of human-Xenopus chimeras to investigate the pore properties of CFTR. Journal of Biological Chemistry. 1996;271:25184–25191. doi: 10.1074/jbc.271.41.25184. [DOI] [PubMed] [Google Scholar]
- Quinton P M, Reddy M M. Control of CFTR chloride conductance by ATP levels through non-hydrolytic binding. Nature. 1992;360:79–81. doi: 10.1038/360079a0. [DOI] [PubMed] [Google Scholar]
- Riordan J R, Rommens J M, Kerem B-S, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou J-L, Drumm M L, Iannuzzi M C, Collins F S, Tsui L-C. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rozmahel R, Gyömörey K, Plyte S, Nguyen V, Wilschanski M, Durie P, Bear C E, Tsui L-C. Incomplete rescue of cystic fibrosis transmembrane conductance regulator deficient mice by the human CFTR cDNA. Human Molecular Genetics. 1997;6:1153–1162. doi: 10.1093/hmg/6.7.1153. [DOI] [PubMed] [Google Scholar]
- Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, Moore A, Forstner J, Durie P, Nadeau J, Bear C, Tsui L-C. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nature Genetics. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- Savov A, Mercier B, Kalaydjieva L, Férec C. Identification of six novel mutations in the CFTR gene of patients from Bulgaria by screening the twenty seven exons and exon/intron boundaries using DGGE and direct DNA sequencing. Human Molecular Genetics. 1994;3:57–60. doi: 10.1093/hmg/3.1.57. [DOI] [PubMed] [Google Scholar]
- Seibert F S, Linsdell P, Loo T W, Hanrahan J W, Clarke D M, Riordan J R. Disease-associated mutations in the fourth cytoplasmic loop of cystic fibrosis transmembrane conductance regulator compromise biosynthetic processing and chloride channel activity. Journal of Biological Chemistry. 1996a;271:15139–15145. doi: 10.1074/jbc.271.25.15139. [DOI] [PubMed] [Google Scholar]
- Seibert F S, Linsdell P, Loo T W, Hanrahan J W, Riordan J R, Clarke D M. Cytoplasmic loop three of cystic fibrosis transmembrane conductance regulator contributes to regulation of chloride channel activity. Journal of Biological Chemistry. 1996b;271:27493–27499. doi: 10.1074/jbc.271.44.27493. [DOI] [PubMed] [Google Scholar]
- Sheppard D N, Rich D P, Ostedgaard L S, Gregory R J, Smith A E, Welsh M J. Mutations in CFTR associated with mild disease form Cl− channels with altered pore properties. Nature. 1993;362:160–164. doi: 10.1038/362160a0. [DOI] [PubMed] [Google Scholar]
- Sheppard D N, Robinson K A. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. Journal of Physiology. 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani J A, Rommens J M, Hou Y-X, Chang X-B, Tsui L-C, Riordan J R, Hanrahan J W. Multi-ion pore behaviour in the CFTR chloride channel. Nature. 1993;366:79–82. doi: 10.1038/366079a0. [DOI] [PubMed] [Google Scholar]
- Tata F, Stanier P, Wicking C, Halford S, Kruyer H, Lench N J, Scambler P J, Hansen C, Braman J C, Williamson R, Wainwright B J. Cloning the mouse homolog of the human cystic fibrosis transmembrane conductance regulator gene. Genomics. 1991;10:301–307. doi: 10.1016/0888-7543(91)90312-3. [DOI] [PubMed] [Google Scholar]
- Tybulewicz V L J, Crawford C E, Jackson P K, Bronson R T, Mulligan R C. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Venglarik C J, Schultz B D, Frizzell R A, Bridges R J. ATP alters current fluctuations of cystic fibrosis transmembrane conductance regulator: evidence for a three-state activation mechanism. Journal of General Physiology. 1994;104:123–146. doi: 10.1085/jgp.104.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh M J, Tsui L-C, Boat T F, Beaudet A L. Cystic fibrosis. In: Scriver C R, Beaudet A L, Sly W S, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York: McGraw-Hill Inc.; 1995. pp. 3799–3876. [Google Scholar]
- Winter M C, Sheppard D N, Carson M R, Welsh M J. Effect of ATP concentration on CFTR Cl− channels: a kinetic analysis of channel regulation. Biophysical Journal. 1994;66:1398–1403. doi: 10.1016/S0006-3495(94)80930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M C, Welsh M J. Stimulation of CFTR activity by its phosphorylated R domain. Nature. 1997;389:294–296. doi: 10.1038/38514. [DOI] [PubMed] [Google Scholar]
- Xie J, Drumm M L, Ma J, Davis P B. Intracellular loop between transmembrane segments IV and V of cystic fibrosis transmembrane conductance regulator is involved in regulation of chloride channel conductance state. Journal of Biological Chemistry. 1995;270:28084–28091. doi: 10.1074/jbc.270.47.28084. [DOI] [PubMed] [Google Scholar]
- Yorifuji T, Lemna W K, Ballard C F, Rosenbloom C L, Rozmahel R, Plavsic N, Tsui L-C, Beaudet A L. Molecular cloning and sequence analysis of the murine cDNA for the cystic fibrosis transmembrane conductance regulator. Genomics. 1991;10:547–550. doi: 10.1016/0888-7543(91)90434-g. [DOI] [PubMed] [Google Scholar]
- Zhou L, Dey C R, Wert S E, DuVall M D, Frizzell R A, Whitsett J A. Correction of lethal intestinal defect in a mouse model of cystic fibrosis by human CFTR. Science. 1994;266:1705–1708. doi: 10.1126/science.7527588. [DOI] [PubMed] [Google Scholar]