

Abstract
Sodium channels mediate fast depolarization and conduct electrical impulses throughout nerve, muscle and heart. This paper reviews the links between sodium channel structure and function.
Sodium channels have a modular architecture, with distinct regions for the pore and the gates. The separation is far from absolute, however, with extensive interaction among the various parts of the channel.
At a molecular level, sodium channels are not static: they move extensively in the course of gating and ion translocation.
Sodium channels bind local anaesthetics and various toxins. In some cases, the relevant sites have been partially identified.
Sodium channels are subject to regulation at the levels of transcription, subunit interaction and post-translational modification (notably glycosylation and phosphorylation).
Sodium channels play a central role in physiology: they transmit depolarizing impulses rapidly throughout cells and cell networks, thereby enabling co-ordination of higher processes ranging from locomotion to cognition. These channels are also of special importance for the history of physiology. Elucidation of their fundamental properties in the squid axon launched modern channel theory. In particular, the work of Hodgkin and Huxley on sodium channels, published in this Journal, revolutionized electrophysiology by elegantly dissecting the elementary processes of gating and permeation (Hodgkin & Huxley, 1952). More recently, sodium channels were the first voltage-dependent ion channels to be cloned (Noda et al. 1984), ushering in the era of heterologous expression and molecular manipulation. The cloning happily coincided with the development of patch-clamp techniques, which enabled single-channel recordings. This paper reviews the general concepts of sodium channel structure and function that have emerged over the past half-century. Because the goal of this series is to be brief, citations to the literature are selective. The reader is referred to other reviews (e.g. Fozzard & Hanck, 1996) for more encyclopaedic treatments.
Evolution
Sodium channels first appear phylogenetically in the jellyfish, where they enable the organism to transmit electrical signals efficiently throughout a dispersed neural net. Invertebrate Na+ channel expression is generally restricted to the nervous system, although in chordates Na+ channels are also present in striated muscle. The central selective pressure for Na+ channels has remained the same throughout evolution: these molecules are nature's solution to the conundrum of co-ordination and communication within large organisms, particularly when speed is of the essence. Thus, Na+ channels are richly concentrated in axons and in muscle, where they are often the most plentiful ion channels. Mammalian heart cells, for example, typically express more than 100 000 Na+ channels (Makielski, Sheets, Hanck, January & Fozzard, 1987), but only 20 000 or so L-type Ca2+ channels (Rose, Balke, Wier & Marban, 1992) and fewer copies of each family of voltage-dependent K+ channels.
Na+ channels consist of various subunits, but only the principal (α) subunit is required for function. Figure 1A shows that the α subunit has a modular architecture: it consists of four internally homologous domains (labelled I-IV), each of which contains six transmembrane segments and resembles a single α subunit of a voltage-dependent K+ channel. The four domains fold together so as to create a central pore whose structural constituents determine the selectivity and conductance properties of the channel. It is noteworthy that Ca2+ channels have a similar overall architecture, with important differences in various regions (including the pore). Because unicellular organisms express K+ and Ca2+ channels, it is plausible that the simpler K+ channels were primordial, with the subsequent evolution of Ca2+ channels by gene duplication. Na+ channels might have arisen in an analogous manner or, more likely, from mutations in a primitive Ca2+ channel.
Figure 1. Schematic depictions of the Na+ channel α subunit.
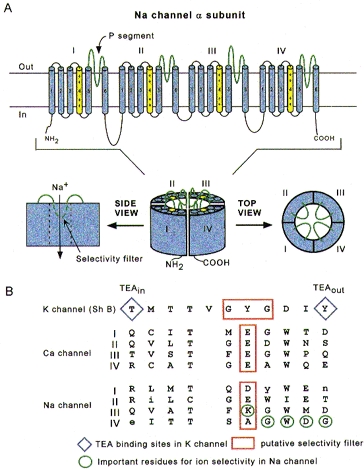
A, putative transmembrane folding. The charged S4 segments are shown in yellow, and the pore-lining P segments in green. B, aligned primary amino acid sequences in single-letter code of the P segments in a K+ channel (Shaker B), the four domains of the cardiac L-type Ca2+ channel, and the four domains of the Na+ channel. Residues shown in upper case are highly conserved among voltage-dependent Na+ channels. The diamonds indicate the external and internal binding sites for tetraethylammonium (TEA) ion in the K+ channel and the red boxes outline the putative selectivity filters, although, in the case of the Na+ channel, the residues which are most important for selectivity (circled in green) are mostly outside the box.
These evolutionary considerations serve to point out various themes that are general to voltage-dependent ion channels: first, the architecture is modular, consisting either of four homologous subunits (in K+ channels) or of four internally homologous domains (in Na+ and Ca2+ channels). Secondly, as depicted in Fig. 1A, the proteins wrap around a central pore. The pore-lining (‘P segment’) regions exhibit exquisite conservation within a given channel family of like selectivity (jellyfish, eel, fruit-fly and human Na+ channels have very similar P segments), but not among families with different selectivities (Fig. 1B). Third, the general strategy for activation gating is highly conserved: the fourth transmembrane segment (S4), stereotypically studded with positively charged residues, lies within the membrane field and moves in response to depolarization, somehow opening the channel (Stühmer et al. 1989).
Design motifs: permeation
The S5-S6 linkers or P segments of each domain come together to form the pore (Yellen, Jurman, Abramson & MacKinnon, 1991). Inspection of the primary structures of the linkers in each domain (Fig. 1B) reveals that each is unique. The structural basis of permeation thus differs fundamentally from that of K+ channels, in which four identical P segments can come together to form a K+- selective pore. Indeed, accessibility mapping studies in Na+ channels have revealed marked asymmetries in the contributions of each domain to the permeation pathway (Chiamvimonvat, Pérez-García, Ranjan, Marban & Tomaselli, 1996a;Pérez-García, Chiamvimonvat, Marban & Tomaselli, 1996; Yamagishi, Janecki, Marban & Tomaselli, 1997). Figure 2 (top) shows a top view of a space-filling model of the Na+ channel pore which is consistent with the available mutagenesis data. The asymmetry is apparent not only here but also in side views (Fig. 2, central panel). Two domains play a particularly prominent role in determining Na+ selectivity: III, in which a lysine (K1237 in the μ1 sequence) is critical for discrimination for Na+ over Ca2+ (Heinemann, Terlau, Stühmer, Imoto & Numa, 1992; Chiamvimonvat et al. 1996a; Pérez-García, Chiamvimonvat, Ranjan, Balser, Tomaselli & Marban, 1997) and IV, in which mutations of various contiguous residues (1531–1534) render the channel non-selective among monovalent cations (Chiamvimonvat et al. 1996a).
Figure 2. Structure for the Na+ channel pore proposed by Bénitah et al. (1997).
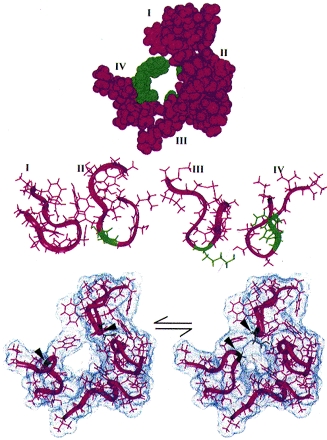
Residues which play a particularly prominent role in determining Na+ selectivity are shown in green. Top: space-filling model of the Na+ channel pore consistent with the available mutagenesis data. Centre: side views of the P segments as they might appear to permeant ions, in ribbon format. Bottom: Na+ channel pore motions illustrated by the pairing of residues D400 and G1530 (arrowheads), which can form an internal disulphide in the pore when substituted with cysteines (Bénitah et al. 1997; Tsushima et al. 1997).
It is not yet clear how these particular residues interact with the bulk solution to favour the specific flux of Na+ by factors of 100:1 or more over other cations, a feat which is particularly remarkable given the high throughput rate of each individual channel (> 107 ions s−1). In thinking about this process, it may be relevant to consider recent evidence that Na+ channels, like many enzymes, exhibit a high degree of conformational flexibility. Pairs of cysteine residues engineered into the P regions can form internal disulphides, in specific patterns that could not arise if there were not substantial mobility within the molecule (Bénitah, Ranjan, Yamagishi, Janecki, Tomaselli & Marban, 1997; Tsushima, Li & Backx, 1997). Figure 2 (lower panel) depicts one example of such motions: residues in domains I and IV which are quite distant from each other in the equilibrium structure can come sufficiently close together to occlude the pore. The motions occur over millisecond time scales and may span several nanometres in extreme cases. Interestingly, an internal disulphide crosslink renders channels less selective than those with two reduced sulfhydryls (Tsushima et al. 1997), hinting that flexibility plays an important (albeit as yet undetermined) role in selective ion translocation.
Design motifs: gating
One of the seminal contributions of Hodgkin and Huxley was the notion that Na+ channels transit among various conformational states in the process of opening (‘activation’); yet another set of conformations is entered when the channels shut during maintained depolarization (‘inactivation’). The m gates that underlie activation, and the h gate that mediates inactivation, were postulated to have intrinsic voltage dependence and to function independently (Hodgkin & Huxley, 1952). While some of the implicit structural predictions of that formulation have withstood the test of time, others have not. The four S4 segments are now widely acknowledged to serve as the activation sensors. In the process of activation, several charged residues in each S4 segment physically traverse the membrane through a narrow cuff formed by other, as yet unidentified regions of the channel (Fig. 3) (Yang & Horn, 1995; Yang, George & Horn, 1996, 1997). The S4 segment in domain IV undergoes a minimal translocation of 0.5 nm (5 Å) in response to a voltage step, emphasizing once again the importance of internal motions for the function of these proteins. The idea that the sensors are equivalent and independent turns out to be incorrect. The contributions of each S4 segment to activation are markedly asymmetrical; some of the charged residues play a much more prominent role than others in ‘homologous’ positions (Stühmer et al. 1989; Chen, Santarelli, Horn & Kallen, 1996; Kontis & Goldin, 1997). Furthermore, charge-altering mutations in multiple S4 segments do not exert simply additive effects on gating; there is some co-operativity, the extent of which varies from site to site. Gating current and mutagenesis studies have additionally revealed that activation is coupled to inactivation (Stühmer et al. 1989; O'Leary, Chen, Kallen & Horn, 1995; Chen et al. 1996; Kontis & Goldin, 1997; Kontis, Rounaghi & Goldin, 1997). Indeed, the time course of current decay predominantly reflects the voltage dependence of activation (Aldrich, Corey & Stevens, 1983), although microscopic inactivation itself does vary with voltage (particularly in cardiac channels) (Yue, Lawrence & Marban, 1989; Lawrence, Yue, Rose & Marban, 1991).
Figure 3. Schematic depictions of the Na+ channel α subunit illustrating the S4 activation sensors (top) and the III-IV linker which contributes to fast inactivation (bottom).
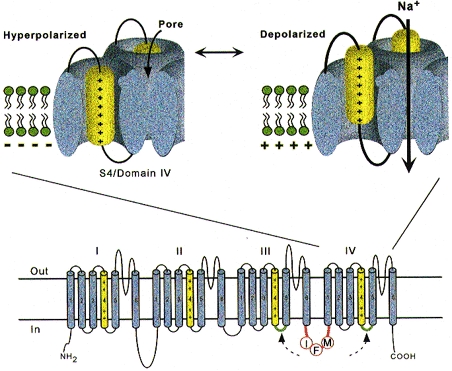
The putative receptors of the III-IV linker are shown in green. Modified from Yang et al. (1996).
If the S4s are the sensors, where are the activation gates themselves? This crucial question remains unresolved. The gates must be on the internal aspect of the permeation pathway, since pore-lining residues remain accessible to externally applied reagents regardless of whether the channels are open, closed or inactivated (Bénitah, Tomaselli & Marban, 1996; Bénitah et al. 1997). It is possible that motion of S4 distorts the S4-S5 linker, which may contribute to the inner pore (by analogy to K+ channels) and serve as a gate. Alternatively, S4 motion may displace other parts of the channel. Recent studies indicate that mutations in S6 alter Na+ channel gating as well as block by local anaesthetics (McPhee, Ragsdale, Scheuer & Catterall, 1994, 1995; Ragsdale, McPhee, Scheuer & Catterall, 1994). Taken together with the finding in K+ channels that S6 can physically occlude the inner channel mouth in response to voltage, (Holmgren, Smith & Yellen, 1997), S6 emerges as the leading contender for the physical activation gate.
Inactivation turns out to be a much more arcane process than originally envisioned by Hodgkin and Huxley. Not only is there loose coupling to activation, as discussed above, but there are also multiple inactivation processes. These are distinguishable by their recovery kinetics at strongly negative potentials: repriming from the traditional ‘fast’ inactivation occurs over tens of milliseconds, while recovery from ‘slow’ inactivation can require tens of seconds or longer (Adelman & Palti, 1969; Chandler & Meves, 1970). Fast inactivation is at least partly mediated by the cytoplasmic linker between domains III and IV (the crucial residues are labelled IFM in Fig. 3) (Stühmer et al. 1989; Moorman, Kirsch, Brown & Joho, 1990; Patton, West, Catterall & Goldin, 1992; West, Patton, Scheuer, Wang, Goldin & Catterall, 1992b), which may function as a hinged lid (West et al. 1992b) docking onto a receptor formed by the S4-S5 linkers of domains III and IV (McPhee, Ragsdale, Scheuer & Catterall, 1996). This notion fits nicely with venerable observations that fast inactivation can be disrupted by internal proteases. Nevertheless, it is increasingly clear that mutations scattered widely throughout the channel affect inactivation gating, undermining somewhat the primacy of the III-IV linker. The structural determinants of slow inactivation are even less localized than those of fast inactivation. Mutations in the P region of domain I affect both activation gating and slow inactivation (Tomaselli et al. 1995; Balser, Nuss, Chiamvimonvat, Pérez-García, Marban & Tomaselli, 1996a), while various widely scattered disease mutations as identified in paramyotonia congenita and other skeletal myopathies suppress slow inactivation (Hayward, Brown & Cannon, 1997).
Drug binding
The S6 segment of domain IV has been proposed to contain the receptor for local anaesthetics, which block Na+ channels in a voltage-dependent manner (Ragsdale et al. 1994). Block is enhanced at depolarized potentials and/or with repetitive pulsing. These observations are consistent with the idea that local anaesthetics act as allosteric effectors of inactivation gating: when they bind to the channel, they facilitate inactivation (Balser et al. 1996b). Whether or not this particular model of drug action turns out to be correct, it is clear that gating interacts with local anaesthetic block so profoundly that it is difficult to interpret at face value the localization of a ‘receptor’ to S6. As discussed above, mutations in S6, at the putative receptor sites, alter gating independent of superimposed drug effects (McPhee et al. 1994, 1995; Ragsdale et al. 1994). Mutations in distant parts of the molecule can also dramatically alter the phenotype of local anaesthetic block (Kambouris, Nuss, Johns, Tomaselli, Marban & Balser, 1998). Despite these caveats, S6 segments appear to play a special role in drug effects in a variety of channels, at least some of which appear to be independent of changes in gating.
Neurotoxins
Pharmacological competition studies and mutagenesis have defined a number of neurotoxin binding sites on the Na+ channel. Among these, tetrodotoxin (TTX), a guanidinium-containing blocker, has contributed the most to our understanding of Na+ channel structure and function. Externally applied TTX blocks Na+ channels potently (in the nanomole range) in neural and skeletal muscle isoforms, but block of cardiac channels requires much higher concentrations (∼10−5 M). The identity of one particular residue in the P region of domain I accounts for most of the isoform-specific TTX sensitivity: an aromatic residue at this position (401 in μ1) confers high affinity, while its absence renders the channel TTX resistant (Backx, Yue, Lawrence, Marban & Tomaselli, 1992; Satin et al. 1992). Many other residues in the outer mouth of the channel contribute to the binding of TTX and the related divalent guanidinium toxin, saxitoxin (STX), suggesting that the toxin has a large footprint on the external surface of the channel (Terlau et al. 1991; Lipkind & Fozzard, 1994; Chiamvimonvat et al. 1996a;Pérez-García et al. 1996).
Other natural toxins which bind with high affinity to selected Na+ channels include the μ-conotoxins, a class of inhibitory peptide toxins that specifically block the skeletal muscle isoform at a site which overlaps partially with that of TTX/STX (Catterall & Beneski, 1980; Cruz et al. 1985). These toxins have backbones which are rendered quite rigid by the three internal disulphide bonds. They are particularly useful probes of the outer vestibule of the Na+ channel (Stephan, Potts & Agnew, 1994; Chahine et al. 1995; Dudley, Todt, Lipkind & Fozzard, 1995; French, Prusak-Sochaczewski, Zamponi, Becker, Kularatna & Horn, 1996) because, unlike the channels themselves (at least to date), the toxins are amenable to structural characterization by established physical methods such as X-ray crystallography (Hill, Alewood & Craik, 1996). The fact that they can be grown in bacteria and mutated at specific sites adds considerable versatility. Although μ-conotoxins are the best characterized, other Na+ channel toxins from various species of cone snails have been described.
Sea anemone (e.g. anthopleurin A and B, ATX II) and scorpion toxins inhibit Na+ channel inactivation by binding to sites that include the S3-S4 extracellular loop of domain IV (Rogers, Qu, Tanada, Scheuer & Catterall, 1996). The fact that these toxins slow current decay by binding to an external site remote from the III-IV linker highlights the importance of disparate regions of the channel in inactivation gating.
Additional determinants of function
While this review has emphasized the importance of specific residues in the primary structure of the α subunit, many other factors contribute to the functional diversity of Na+ channels. These include variable expression of tissue-specific α subunits, differential susceptibility to phosphorylation and glycosylation, and the presence or absence of ancillary β1 and β2 subunits. These factors are considered below.
Transcriptional regulation
Control of excitability can occur at the genomic level by the regulation of transcription of channel genes. The expression of Na+ channels is developmentally regulated and tissue restricted. Patterns of electrical activity can also feed back upon and influence transcription: for example, seizures alter Na+ channel gene expression in the brain. Denervation induces the expression of the cardiac isoform of the channel in skeletal muscle, while transiently suppressing expression of the mature skeletal muscle isoform (Kallen, Sheng, Yang, Chen, Rogart & Barchi, 1990; Yang, Sladky, Kallen & Barchi, 1991). Chronic exposure to antiarrhythmic drugs which block Na+ channels can increase the steady-state levels of Na+ channel mRNA, in a manner that would tend to counteract the effects of channel blockade (Duff, Offord, West & Catterall, 1992).
The mechanisms controlling Na+ channel gene expression are only just beginning to be understood. Expression of the brain type II Na+ channel is restricted to neurons by a transcription silencer known as REST (Chong et al. 1995; Eggen & Mandel, 1997; Tapia-Ramirez, Eggen, Peral-Rubio, Toledo-Aral & Mandel, 1997). REST is a transcription factor with C2H2 zinc finger motifs homologous to the Drosophila repressor Krüppel that binds to a specific silencer element (RE-1) in the promoter of the brain II channel. REST is found in most tissues; its absence in neurons is what permits expression of the brain II isoform.
Subunits
Auxiliary (β) subunits are important modulators of Na+ channel function. Biochemical studies reveal the existence of two distinct β subunits (β1 and β2) associated with the brain Na+ channel (Hartshorne, Messner, Coppersmith & Catterall, 1982). Antibodies directed to the β1 or β2 subunit will immunoprecipitate the entire brain Na+ channel complex with a subunit stoichiometry of 1α:1β1:1β2. The β1 subunit is non-covalently associated, while β2 is linked by a disulphide bond to the α subunit (Messner & Catterall, 1985; Roberts & Barchi, 1987).
The β1 and β2 subunits have been cloned and the deduced primary structures indicate that they are unrelated proteins of molecular weights 23 and 21 kDa, respectively (Isom et al. 1992, 1995). The predicted transmembrane topology of the β subunits is similar: each contains a small carboxy-terminal cytoplasmic domain, a single membrane-spanning segment, and a large amino-terminal extracellular domain with several consensus sites for N-linked glycosylation (Fig. 4). The β2 subunit has several unique features, including an extracellular immunoglobulin-like fold with similarity to the neural cell adhesion molecule contactin. Expression of β2 with neuronal α subunits in Xenopus oocytes increases the current amplitude, modulates gating and increases the membrane capacitance (Isom et al. 1995). Co-expression of β1 subunits with either neuronal or skeletal muscle α subunits in oocytes also produces clear-cut effects on channel function. The current density increases, activation and inactivation gating are accelerated, and the steady-state inactivation curves are shifted in the hyperpolarizing direction (Isom et al. 1992; Bennett, Makita & George, 1993; Cannon, McClatchey & Gusella, 1993; Patton, Isom, Catterall & Goldin, 1994). The mRNA encoding the β1 subunit appears to be widely expressed and clearly forms an important component of neuronal and skeletal muscle Na+ channels. However, the functional role of this subunit in the heart is uncertain (Cohen & Levitt, 1993; Makita, Bennett & George, 1994; Nuss, Chiamvimonvat, Pérez-García, Tomaselli & Marban, 1995; Qu et al. 1995; Makielski, Limberis, Chang, Fan & Kyle, 1996).
Figure 4. Na+ channel phosphorylation sites and subunits.
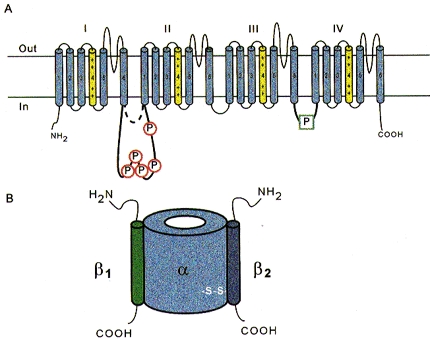
A, schematic diagram highlighting the prominent phosphorylation sites of the α subunit. These are in the III-IV linker, and in the long variant of the I-II linkers; the length of this linker is isoform specific. B, maximal subunit composition of the Na+ channel. Note that not all Na+ channels include either or both β subunits, but the α subunit is obligatory for function.
Post-translational modification: phosphorylation
Regulation of the Na+ channel by phosphorylation is complex. Isoforms of the Na+ channel α subunit fall into one of two groups, long (neuronal and cardiac) and short (skeletal muscle and eel). The neuronal isoforms have a substantially larger intracellular linker between domains I and II, which contains five consensus sites for cyclic AMP-dependent protein kinase (PKA) phosphorylation (Fig. 4). PKA modulates the function of expressed neuronal and cardiac Na+ channels. Phosphorylation of sites in the I-II linker of the brain channel reduces current amplitude without significantly affecting gating (Gershon, Weigl, Lotan, Schreibmayer & Dascal, 1992; Li, West, Numann, Murphy, Scheuer & Catterall, 1993; Murphy, Rossie, De Jongh & Catterall, 1993; Smith & Goldin, 1995, 1997; Cantrell, Smith, Goldin, Scheuer & Catterall, 1997).
The cardiac channel has eight candidate consensus PKA phosphorylation sites of the form KRXXS*, RXXS* or RXS* in the I-II linker, all of which are distinct from the neuronal channels. In vitro studies of the expressed cardiac channel demonstrate cyclic AMP-dependent phosphorylation on only two of these serines (Murphy, Rogers, Perdichizzi, Colvin & Catterall, 1996). Interestingly, when the cardiac channel is phosphorylated by PKA, the whole-cell conductance increases, suggesting the specific pattern of phosphorylation is responsible for the functional effect (Schreibmayer et al. 1994; Frohnwieser, Weigl & Schreibmayer, 1995; Murphy et al. 1996; Frohnwieser, Chen, Schreibmayer & Kallen, 1997). The function of the skeletal muscle isoform of the Na+ channel is not affected by PKA (Smith & Goldin, 1992, 1995), despite the fact that this channel is an excellent substrate for PKA-mediated phosphorylation. (Yang & Barchi, 1990) Indeed, the importance of Na+ channel phosphorylation is not always clear, and caution should be exercised when attempting to relate phenomena in heterologous expression systems to more physiological settings (and vice versa).
In contrast to PKA, protein kinase C (PKC) alters the function of all of the mammalian Na+ channel isoforms (West, Numann, Murphy, Scheuer & Catterall, 1991, 1992a; Numann, Catterall & Scheuer, 1991; Li et al. 1993; Numann, Hauschka, Catterall & Scheuer, 1994; Bendahhou, Cummins, Potts, Tong & Agnew, 1995; Qu, Rogers, Tanada, Catterall & Scheuer, 1996; Murray et al. 1997). The PKC effect is largely attributable to phosphorylation of a highly conserved serine in the III-IV linker (Fig. 4). PKC reduces the maximal conductance of the channels and alters gating in an isoform-specific fashion. The macroscopic current decay of neuronal channels is uniformly slowed by PKC (Numann et al. 1991; West et al. 1991). Disparate effects have been described in skeletal muscle and cardiac channels including acceleration of the current decay (Bendahhou et al. 1995) and a hyperpolarizing shift in the steady-state availability curve (Qu et al. 1996). The skeletal muscle Na+ channel is selectively phosphorylated by human myotonin protein kinase (HMPK) (Mounsey et al. 1995; Chahine & George, 1997). Absent or reduced phosphorylation of skeletal muscle Na+ channels by mutant HMPK may underlie the altered excitability of muscle in myotonic dystrophy, but, curiously, the genetic alterations do not appear to alter kinase function (Mounsey et al. 1995).
Post-translational modification: glycosylation
All of the subunits of the Na+ channel are modified by glycosylation. The β1, β2 and brain and muscle α subunits are heavily glycosylated, with up to 40 % (eel electroplax α subunit) (James & Agnew, 1989) of the mass being carbohydrate. In contrast, the cardiac α subunit is only 5 % sugar by weight (Cohen & Levitt, 1993). Sialic acid is a prominent component of the N-linked carbohydrate of the Na+ channel. The addition of such a highly charged carbohydrate has predictable effects on the voltage dependence of gating through alteration of the surface charge of the channel protein. Neuraminidase treatment to remove sialic acid from expressed skeletal muscle channels produces a depolarizing shift of steady-state inactivation (Bennett, Urcan, Tinkle, Koszowski & Levinson, 1997). Local surface charge is also importantly influenced by charged amino acid residues which stud the outer mouth of the pore, although the predominant effects in this case are on permeation rather than gating (Terlau et al. 1991; Chiamvimonvat, Pérez-García, Tomaselli & Marban, 1996b).
Co-translational glycosylation is essential for the maintenance of cell surface expression of the Na+ channel in neurons and Schwann cells (Ritchie, 1988; Zona, Eusebi & Miledi, 1990). Inhibition of glycosylation by tunicamycin reversibly decreases the number of STX binding sites on neuroblastoma cells (Waechter, Schmidt & Catterall, 1983). Tunicamycin also inhibits palmitation, sulphation and disulphide attachment of the β2 subunit, preventing the assembly of functional Na+ channels (Schmidt & Catterall, 1987).
Channelopathies
Alteration of ion channel function is an important pathophysiological mechanism of various familial diseases of muscle (Cannon, 1996). Na+ channel mutations underlie the aberrant excitability characteristic of some skeletal muscle myotonias and paralysis, as well as the chromosome 3-linked long-QT syndrome, an inherited cardiac arrhythmia (Wang et al. 1995). In general, these mutations disable inactivation of the Na+ channel, producing either repetitive action potential firing (myotonia) or electrical silence (flaccid paralysis) in skeletal muscle. A similar defect in the cardiac Na+ channel produces action potential prolongation and a predisposition to repetitive electrical activity (polymorphic ventricular tachycardia) in the heart. The reader is referred to several other reviews for a more detailed description of the molecular genetics and physiology of skeletal muscle (Cannon, 1996) and cardiac Na+ channelopathies (Roden, Lazzara, Rosen, Schwartz, Towbin & Vincent, 1996; see also Kambouris et al. 1998 and references therein).
Conclusions
Physiologists have many reasons to admire the Na+ channel. The study of its properties in axons laid the groundwork for all subsequent work on excitability. Its abundance in electric eel electroplax enabled the first cloning of a voltage-dependent ion channel. At the same time, the development of patch-clamp methodology left us well poised for the characterization of molecular physiology in structurally defined systems. The result has been an explosion of knowledge regarding the interrelationships between Na+ channel structure and function.
Acknowledgments
The authors thank the National Heart, Lung and Blood Institute for their support (P50 HL52307, R01 HL52768 and R01 HL50411). The authors thank Ravi Ranjan for Fig. 2.
References
- Adelman WJ, Palti Y. The effects of external potassium and long duration voltage conditioning on the amplitude of sodium currents in the giant axon of the squid, Loligo pealei. Journal of General Physiology. 1969;54:589–606. doi: 10.1085/jgp.54.5.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Backx P, Yue D, Lawrence J, Marban E, Tomaselli G. Molecular localization of an ion-binding site within the pore of mammalian sodium channels. Science. 1992;257:248–251. doi: 10.1126/science.1321496. [DOI] [PubMed] [Google Scholar]
- Balser J, Nuss H, Chiamvimonvat N, Pérez-García M, Marban E, Tomaselli G. External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. The Journal of Physiology. 1996a;494:431–442. doi: 10.1113/jphysiol.1996.sp021503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balser J, Nuss H, Orias D, Johns D, Marban E, Tomaselli G, Lawrence J. Local anesthetics as effectors of allosteric gating. Lidocaine effects on inactivation-deficient rat skeletal muscle Na+ channels. Journal of Clinical Investigation. 1996b;98:2874–2886. doi: 10.1172/JCI119116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahhou S, Cummins T, Potts J, Tong J, Agnew W. Serine-1321-independent regulation of the μ1 adult skeletal muscle Na+ channel by protein kinase C. Proceedings of the National Academy of Sciences of the USA. 1995;92:12003–12007. doi: 10.1073/pnas.92.26.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénitah J, Ranjan R, Yamagishi T, Janecki M, Tomaselli G, Marban E. Molecular motions within the pore of voltage-dependent sodium channels. Biophysical Journal. 1997;73:603–613. doi: 10.1016/S0006-3495(97)78096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénitah J, Tomaselli G, Marban E. Adjacent pore-lining residues within sodium channels identified by paired cysteine mutagenesis. Proceedings of the National Academy of Sciences of the USA. 1996;93:7392–7396. doi: 10.1073/pnas.93.14.7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett E, Urcan M, Tinkle S, Koszowski A, Levinson S. Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism. Journal of General Physiology. 1997;109:327–343. doi: 10.1085/jgp.109.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett P, Jr, Makita N, George A., Jr A molecular basis for gating mode transitions in human skeletal muscle Na+ channels. FEBS Letters. 1993;326:21–24. doi: 10.1016/0014-5793(93)81752-l. [DOI] [PubMed] [Google Scholar]
- Cannon S. Sodium channel defects in myotonia and periodic paralysis. Annual Review of Neuroscience. 1996;19:141–164. doi: 10.1146/annurev.ne.19.030196.001041. [DOI] [PubMed] [Google Scholar]
- Cannon S, McClatchey A, Gusella J. Modification of the Na+ current conducted by the rat skeletal muscle alpha subunit by coexpression with a human brain β subunit. Pflügers Archiv. 1993;423:155–157. doi: 10.1007/BF00374974. [DOI] [PubMed] [Google Scholar]
- Cantrell A, Smith R, Goldin A, Scheuer T, Catterall W. Dopaminergic modulation of sodium current in hippocampal neurons via cAMP-dependent phosphorylation of specific sites in the sodium channel α subunit. Journal of Neuroscience. 1997;17:7330–7338. doi: 10.1523/JNEUROSCI.17-19-07330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W, Beneski D. Interaction of polypeptide neurotoxins with a receptor site associated with voltage-sensitive sodium channels. Journal of Supramolecular Structure. 1980;14:295–303. doi: 10.1002/jss.400140304. [DOI] [PubMed] [Google Scholar]
- Chahine M, Chen L, Fotouhi N, Walsky R, Fry D, Santarelli V, Horn R, Kallen R. Characterizing the μ-conotoxin binding site on voltage-sensitive sodium channels with toxin analogs and channel mutations. Receptors and Channels. 1995;3:161–174. [PubMed] [Google Scholar]
- Chahine M, George A., Jr Myotonic dystrophy kinase modulates skeletal muscle but not cardiac voltage-gated sodium channels. FEBS Letters. 1997;412:621–624. doi: 10.1016/s0014-5793(97)00869-7. [DOI] [PubMed] [Google Scholar]
- Chandler WK, Meves H. Slow changes in membrane permeability and long lasting action potentials in axons perfused with fluoride solutions. The Journal of Physiology. 1970;211:707–728. doi: 10.1113/jphysiol.1970.sp009300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Santarelli V, Horn R, Kallen R. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. Journal of General Physiology. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamvimonvat N, Pérez-García M, Ranjan R, Marban E, Tomaselli G. Depth asymmetries of the pore-lining segments of the Na+ channel revealed by cysteine mutagenesis. Neuron. 1996a;16:1037–1047. doi: 10.1016/s0896-6273(00)80127-0. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat N, Pérez-García M, Tomaselli G, Marban E. Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels. The Journal of Physiology. 1996b;491:51–59. doi: 10.1113/jphysiol.1996.sp021195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Tapia-Ramirez J, Kim S, Toledo-Aral J, Zheng Y, Boutros M, Altshuller Y, Frohman M, Kraner S, Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Cohen S, Levitt L. Partial characterization of the rH1 sodium channel protein from rat heart using subtype-specific antibodies. Circulation Research. 1993;73:735–742. doi: 10.1161/01.res.73.4.735. [DOI] [PubMed] [Google Scholar]
- Cruz L, Gray W, Olivera B, Zeikus R, Kerr L, Yoshikami D, Moczydlowski E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. Journal of Biological Chemistry. 1985;260:9280–9288. [PubMed] [Google Scholar]
- Dudley S, Jr, Todt H, Lipkind G, Fozzard H. A μ-conotoxin-insensitive Na+ channel mutant: possible localization of a binding site at the outer vestibule. Biophysical Journal. 1995;69:1657–1665. doi: 10.1016/S0006-3495(95)80045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff H, Offord J, West J, Catterall W. Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel α subunit in rat cardiac muscle. Molecular Pharmacology. 1992;42:570–574. [PubMed] [Google Scholar]
- Eggen B, Mandel G. Regulation of sodium channel gene expression by transcriptional silencing. Developmental Neuroscience. 1997;19:25–26. doi: 10.1159/000111181. [DOI] [PubMed] [Google Scholar]
- Fozzard H, Hanck D. Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiological Reviews. 1996;76:887–926. doi: 10.1152/physrev.1996.76.3.887. [DOI] [PubMed] [Google Scholar]
- French R, Prusak-Sochaczewski E, Zamponi G, Becker S, Kularatna A, Horn R. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron. 1996;16:407–413. doi: 10.1016/s0896-6273(00)80058-6. [DOI] [PubMed] [Google Scholar]
- Frohnwieser B, Chen L, Schreibmayer W, Kallen R. Modulation of the human cardiac sodium channel α-subunit by cAMP-dependent protein kinase and the responsible sequence domain. The Journal of Physiology. 1997;498:309–318. doi: 10.1113/jphysiol.1997.sp021859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohnwieser B, Weigl L, Schreibmayer W. Modulation of cardiac sodium channel isoform by cyclic AMP dependent protein kinase does not depend on phosphorylation of serine 1504 in the cytosolic loop interconnecting transmembrane domains III and IV. Pflügers Archiv. 1995;430:751–753. doi: 10.1007/BF00386171. [DOI] [PubMed] [Google Scholar]
- Gershon E, Weigl L, Lotan I, Schreibmayer W, Dascal N. Protein kinase A reduces voltage-dependent Na+ current in Xenopus oocytes. Journal of Neuroscience. 1992;12:3743–3752. doi: 10.1523/JNEUROSCI.12-10-03743.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne R, Messner D, Coppersmith J, Catterall W. The saxitoxin receptor of the sodium channel from rat brain. Evidence for two nonidentical β subunits. Journal of Biological Chemistry. 1982;257:13888–13891. [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophysical Journal. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann S, Terlau H, Stühmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356:441–443. doi: 10.1038/356441a0. [DOI] [PubMed] [Google Scholar]
- Hill J, Alewood P, Craik D. Three-dimensional solution structure of μ-conotoxin GIIIB, a specific blocker of skeletal muscle sodium channels. Biochemistry. 1996;35:8824–8835. doi: 10.1021/bi960073o. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of Physiology. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren M, Smith P, Yellen G. Trapping of organic blockers by closing of voltage-dependent K+ channels - evidence for a trap door mechanism of activation gating. Journal of General Physiology. 1997;109:527–535. doi: 10.1085/jgp.109.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom L, De Jongh K, Patton D, Reber B, Offord J, Charbonneau H, Walsh K, Goldin A, Catterall W. Primary structure and functional expression of the β1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Isom L, Ragsdale D, De Jongh K, Westenbroek R, Reber B, Scheuer T, Catterall W. Structure and function of the β2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell. 1995;83:433–442. doi: 10.1016/0092-8674(95)90121-3. [DOI] [PubMed] [Google Scholar]
- James W, Agnew W. Alpha-(2–8)-polysialic acid immunoreactivity in voltage-sensitive sodium channel of eel electric organ. Proceedings of the Royal Society. 1989;B 237:233–245. doi: 10.1098/rspb.1989.0046. [DOI] [PubMed] [Google Scholar]
- Kallen R, Sheng Z, Yang J, Chen L, Rogart R, Barchi R. Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle. Neuron. 1990;4:233–242. doi: 10.1016/0896-6273(90)90098-z. [DOI] [PubMed] [Google Scholar]
- Kambouris N, Nuss H, Johns D, Tomaselli G, Marban E, Balser J. Phenotypic characterization of a novel long-QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation. 1998;97:640–644. doi: 10.1161/01.cir.97.7.640. [DOI] [PubMed] [Google Scholar]
- Kontis K, Goldin A. Sodium channel inactivation is altered by substitution of voltage sensor positive charges. Journal of General Physiology. 1997;110:403–413. doi: 10.1085/jgp.110.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontis K, Rounaghi A, Goldin A. Sodium channel activation gating is affected by substitutions of voltage sensor positive charges in all four domains. Journal of General Physiology. 1997;110:391–401. doi: 10.1085/jgp.110.4.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence J, Yue D, Rose W, Marban E. Sodium channel inactivation from resting states in guinea-pig ventricular myocytes. The Journal of Physiology. 1991;443:629–650. doi: 10.1113/jphysiol.1991.sp018855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, West J, Numann R, Murphy B, Scheuer T, Catterall W. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- Lipkind G, Fozzard H. A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophysical Journal. 1994;66:1–13. doi: 10.1016/S0006-3495(94)80746-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee J, Ragsdale D, Scheuer T, Catterall W. A mutation in segment IVS6 disrupts fast inactivation of sodium channels. Proceedings of the National Academy of Sciences of the USA. 1994;91:12346–12350. doi: 10.1073/pnas.91.25.12346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee J, Ragsdale D, Scheuer T, Catterall W. A critical role for transmembrane segment IVS6 of the sodium channel α subunit in fast inactivation. Journal of Biological Chemistry. 1995;270:12025–12034. doi: 10.1074/jbc.270.20.12025. [DOI] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A role for intracellular loop IVS4-S5 of the Na+ channel α subunit in fast inactivation. Biophysical Journal. 1996;70:318. a. [Google Scholar]
- Makielski J, Limberis J, Chang S, Fan Z, Kyle J. Coexpression of β1 with cardiac sodium channel α subunits in oocytes decreases lidocaine block. Molecular Pharmacology. 1996;49:30–39. [PubMed] [Google Scholar]
- Makielski J, Sheets M, Hanck D, January C, Fozzard H. Sodium current in voltage clamped internally perfused canine cardiac Purkinje cells. Biophysical Journal. 1987;52:1–11. doi: 10.1016/S0006-3495(87)83182-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita N, Bennett P, Jr, George A., Jr Voltage-gated Na+ channel β1 subunit mRNA expressed in adult human skeletal muscle, heart, and brain is encoded by a single gene. Journal of Biological Chemistry. 1994;269:7571–7578. [PubMed] [Google Scholar]
- Messner D, Catterall W. The sodium channel from rat brain. Separation and characterization of subunits. Journal of Biological Chemistry. 1985;260:10597–10604. [PubMed] [Google Scholar]
- Moorman JR, Kirsch GE, Brown AM, Joho RH. Changes in sodium channel gating produced by point mutations in a cytoplasmic linker. Science. 1990;250:688–691. doi: 10.1126/science.2173138. [DOI] [PubMed] [Google Scholar]
- Mounsey J, Xu P, John J, 3rd, Horne L, Gilbert J, Roses A, Moorman J. Modulation of skeletal muscle sodium channels by human myotonin protein kinase. Journal of Clinical Investigation. 1995;95:2379–2384. doi: 10.1172/JCI117931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy B, Rogers J, Perdichizzi A, Colvin A, Catterall W. cAMP-dependent phosphorylation of two sites in the α subunit of the cardiac sodium channel. Journal of Biological Chemistry. 1996;271:28837–28843. doi: 10.1074/jbc.271.46.28837. [DOI] [PubMed] [Google Scholar]
- Murphy B, Rossie S, De Jongh K, Catterall W. Identification of the sites of selective phosphorylation and dephosphorylation of the rat brain Na+ channel α subunit by cAMP-dependent protein kinase and phosphoprotein phosphatases. Journal of Biological Chemistry. 1993;268:27355–27362. [PubMed] [Google Scholar]
- Murray K, Hu N, Daw J, Shin H, Watson M, Mashburn A, George A., Jr Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circulation Research. 1997;80:370–376. doi: 10.1161/01.res.80.3.370. [DOI] [PubMed] [Google Scholar]
- Noda M, Shimizu S, Tanabe T, Takai T, Kayano T, Ikeda T, Takahashi H, Nakayama H, Kanaoka Y, Minamino N, Kanagawa K, Matsuo H, Raftery M, Hirose T, Inayama S, Hayashida H, Miyata T, Numa S. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- Numann R, Catterall W, Scheuer T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science. 1991;254:115–118. doi: 10.1126/science.1656525. [DOI] [PubMed] [Google Scholar]
- Numann R, Hauschka S, Catterall W, Scheuer T. Modulation of skeletal muscle sodium channels in a satellite cell line by protein kinase C. Journal of Neuroscience. 1994;14:4226–4236. doi: 10.1523/JNEUROSCI.14-07-04226.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuss H, Chiamvimonvat N, Pérez-García M, Tomaselli G, Marban E. Functional association of the β1 subunit with human cardiac (hH1) and rat skeletal muscle (μ1) sodium channel α subunits expressed in Xenopus oocytes. Journal of General Physiology. 1995;106:1171–1191. doi: 10.1085/jgp.106.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary M, Chen L, Kallen R, Horn R. A molecular link between activation and inactivation of sodium channels. Journal of General Physiology. 1995;106:641–658. doi: 10.1085/jgp.106.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton D, Isom L, Catterall W, Goldin A. The adult rat brain β1 subunit modifies activation and inactivation gating of multiple sodium channel α subunits. Journal of Biological Chemistry. 1994;269:17649–17655. [PubMed] [Google Scholar]
- Patton D, West J, Catterall W, Goldin A. Amino acid residues required for fast Na+-channel inactivation: charge neutralizations and deletions in the III-IV linker. Proceedings of the National Academy of Sciences of the USA. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-García M, Chiamvimonvat N, Marban E, Tomaselli G. Structure of the sodium channel pore revealed by serial cysteine mutagenesis. Proceedings of the National Academy of Sciences of the USA. 1996;93:300–304. doi: 10.1073/pnas.93.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-García M, Chiamvimonvat N, Ranjan R, Balser J, Tomaselli G, Marban E. Mechanisms of sodium/calcium selectivity in sodium channels probed by cysteine mutagenesis and sulfhydryl modification. Biophysical Journal. 1997;72:989–996. doi: 10.1016/S0006-3495(97)78751-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Isom L, Westenbroek R, Rogers J, Tanada T, McCormick K, Scheuer T, Catterall W. Modulation of cardiac Na+ channel expression in Xenopus oocytes by β1 subunits. Journal of Biological Chemistry. 1995;270:25696–25701. doi: 10.1074/jbc.270.43.25696. [DOI] [PubMed] [Google Scholar]
- Qu Y, Rogers J, Tanada T, Catterall W, Scheuer T. Phosphorylation of S1505 in the cardiac Na+ channel inactivation gate is required for modulation by protein kinase C. Journal of General Physiology. 1996;108:375–379. doi: 10.1085/jgp.108.5.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale D, McPhee J, Scheuer T, Catterall W. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Ritchie J. Sodium-channel turnover in rabbit cultured Schwann cells. Proceedings of the Royal Society. 1988;B 233:423–430. doi: 10.1098/rspb.1988.0031. [DOI] [PubMed] [Google Scholar]
- Roberts R, Barchi R. The voltage-sensitive sodium channel from rabbit skeletal muscle. Chemical characterization of subunits. Journal of Biological Chemistry. 1987;262:2298–2303. [PubMed] [Google Scholar]
- Roden D, Lazzara R, Rosen M, Schwartz P, Towbin J, Vincent G. Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. Circulation. 1996;94:1996–1202. doi: 10.1161/01.cir.94.8.1996. [DOI] [PubMed] [Google Scholar]
- Rogers J, Qu Y, Tanada T, Scheuer T, Catterall W. Molecular determinants of high affinity binding of α-scorpion toxin and sea anemone toxin in the S3–S4 extracellular loop in domain IV of the Na+ channel α subunit. Journal of Biological Chemistry. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Rose W, Balke C, Wier W, Marban E. Macroscopic and unitary properties of physiological ion flux through L-type Ca2+ channels in guinea-pig heart cells. The Journal of Physiology. 1992;456:267–284. doi: 10.1113/jphysiol.1992.sp019336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin J, Kyle J, Chen M, Bell P, Cribbs L, Fozzard H, Rogart R. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992;256:1202–1205. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Catterall W. Palmitylation, sulfation, and glycosylation of the α subunit of the sodium channel. Role of post-translational modifications in channel assembly. Journal of Biological Chemistry. 1987;262:13713–13723. [PubMed] [Google Scholar]
- Schreibmayer W, Frohnwieser B, Dascal N, Platzer D, Spreitzer B, Zechner R, Kallen R, Lester H. β-Adrenergic modulation of currents produced by rat cardiac Na+ channels expressed in Xenopus laevis oocytes. Receptors and Channels. 1994;2:339–350. [PubMed] [Google Scholar]
- Smith R, Goldin A. Protein kinase A phosphorylation enhances sodium channel currents in Xenopus oocytes. American Journal of Physiology. 1992;263:C660–666. doi: 10.1152/ajpcell.1992.263.3.C660. [DOI] [PubMed] [Google Scholar]
- Smith R, Goldin A. Phosphorylation of brain sodium channels in the I-II linker modulates channel function in Xenopus oocytes. Journal of Neuroscience. 1995;16:1965–1974. doi: 10.1523/JNEUROSCI.16-06-01965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R, Goldin A. Phosphorylation at a single site in the rat brain sodium channel is necessary and sufficient for current reduction by protein kinase A. Journal of Neuroscience. 1997;17:6086–6093. doi: 10.1523/JNEUROSCI.17-16-06086.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan M, Potts J, Agnew W. The μI skeletal muscle sodium channel: mutation E403Q eliminates sensitivity to tetrodotoxin but not to μ-conotoxins GIIIA and GIIIB. Journal of Membrane Biology. 1994;137:1–8. doi: 10.1007/BF00234993. [DOI] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Numa S. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339:597–603. doi: 10.1038/339597a0. [DOI] [PubMed] [Google Scholar]
- Tapia-Ramirez J, Eggen B, Peral-Rubio M, Toledo-Aral J, Mandel G. A single zinc finger motif in the silencing factor REST represses the neural-specific type II sodium channel promoter. Proceedings of the National Academy of Sciences of the USA. 1997;94:1177–1182. doi: 10.1073/pnas.94.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H, Heinemann S, Stühmer W, Pusch M, Conti F, Imoto K, Numa S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Letters. 1991;293:93–96. doi: 10.1016/0014-5793(91)81159-6. [DOI] [PubMed] [Google Scholar]
- Tomaselli G, Chiamvimonvat N, Nuss H, Balser J, Pérez-García M, Xu R, Orias D, Backx P, Marban E. A mutation in the pore of the sodium channel alters gating. Biophysical Journal. 1995;68:1814–1827. doi: 10.1016/S0006-3495(95)80358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsushima R, Li R, Backx P. P-loop flexibility in Na+ channel pores revealed by single- and double-cysteine replacements. Journal of General Physiology. 1997;110:59–72. doi: 10.1085/jgp.110.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waechter C, Schmidt J, Catterall W. Glycosylation is required for maintenance of functional sodium channels in neuroblastoma cells. Journal of Biological Chemistry. 1983;258:5117–5123. [PubMed] [Google Scholar]
- Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- West J, Numann R, Murphy B, Scheuer T, Catterall W. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science. 1991;254:866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- West J, Numann R, Murphy B, Scheuer T, Catterall W. Phosphorylation of a conserved protein kinase C site is required for modulation of Na+ currents in transfected Chinese hamster ovary cells. Biophysical Journal. 1992a;62:31–33. doi: 10.1016/S0006-3495(92)81769-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J, Patton D, Scheuer T, Wang Y, Goldin A, Catterall W. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proceedings of the National Academy of Sciences of the USA. 1992b;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi T, Janecki M, Marban E, Tomaselli G. Topology of the P segments in the sodium channel pore revealed by cysteine mutagenesis. Biophysical Journal. 1997;73:195–204. doi: 10.1016/S0006-3495(97)78060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Barchi R. Phosphorylation of the rat skeletal muscle sodium channel by cyclic AMP-dependent protein kinase. Journal of Neurochemistry. 1990;54:954–962. doi: 10.1111/j.1471-4159.1990.tb02343.x. [DOI] [PubMed] [Google Scholar]
- Yang J, Sladky J, Kallen R, Barchi R. TTX-sensitive and TTX-insensitive sodium channel mRNA transcripts are independently regulated in adult skeletal muscle after denervation. Neuron. 1991;7:421–427. doi: 10.1016/0896-6273(91)90294-a. [DOI] [PubMed] [Google Scholar]
- Yang N, George A, Jr, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Yang N, George A, Horn R. Probing the outer vestibule fo a sodium channel voltage sensor. Biophysical Journal. 1997;73:2260–2268. doi: 10.1016/S0006-3495(97)78258-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Horn R. Evidence for voltage-dependent S4 movement in sodium channels. Neuron. 1995;15:213–218. doi: 10.1016/0896-6273(95)90078-0. [DOI] [PubMed] [Google Scholar]
- Yellen G, Jurman ME, Abramson T, MacKinnon R. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science. 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]
- Yue D, Lawrence J, Marban E. Two molecular transitions influence cardiac sodium channel gating. Science. 1989;244:349–352. doi: 10.1126/science.2540529. [DOI] [PubMed] [Google Scholar]
- Zona C, Eusebi F, Miledi R. Glycosylation is required for maintenance of functional voltage-activated channels in growing neocortical neurons of the rat. Proceedings of the Royal Society. 1990;B 239:119–127. doi: 10.1098/rspb.1990.0011. [DOI] [PubMed] [Google Scholar]