

Abstract
The effect of extracellularly applied divalent cations upon cytosolic Ca2+ levels ([Ca2+]) was investigated in fura-2-loaded mouse Leydig (TM3) cells.
The extracellular application of Ca2+ (2.5–15 mM) or Ni2+ (0.5–5 mM) elicited concentration-dependent elevations in cytosolic [Ca2+] that were followed by decays to baseline levels. Extracellular Mg2+ (0.8–15 mM) failed to influence cytosolic [Ca2+].
Conditioning applications of Ca2+ (2.5–10 mM), Mg2+ (2.5–15 mM) or Ni2+ (0.5–5 mM) all attenuated the cytosolic Ca2+ response to a subsequent test application of 5 mM [Ni2+].
The amplitude of Ni2+-induced cytosolic Ca2+ signals remained constant in low-Ca2+ solutions. Such findings suggest a participation of Ca2+ release from intracellular stores. In parallel, depletion of Ca2+ stores by either ionomycin (5 μM, in low-Ca2+ solutions) or thapsigargin (4 μM) abolished or attenuated Ni2+-induced Ca2+ transients.
Ionomycin (5 μM) elevated cytosolic [Ca2+] in Ca2+-free solutions even after prior Ni2+ application, indicating the presence of Ni2+-insensitive stores.
Caffeine (250 and 500 μM) elevated cytosolic [Ca2+] and attenuated Ni2+-induced Ca2+ release. Furthermore, TM3 cells stained intensely with a specific anti-ryanodine receptor antiserum, Ab34. These findings suggest that Ca2+ release is regulated by ryanodine receptors.
Both membrane depolarization and hyperpolarization, brought about by changes in extracellular [K+] ([K+]e) in the presence of valinomycin (5 μM), altered the waveform of the Ni2+-induced cytosolic Ca2+ signal. Hyperpolarization, in addition, diminished the response magnitude. Such voltage-induced response modulation localizes the regulatory events to the Leydig cell plasma membrane.
We propose the existence of a cell surface divalent cation (Ca2+) receptor in Leydig cells, the activation of which triggers Ca2+ fluxes through ryanodine receptors.
Leydig cells secrete the androgenic steroid testosterone that is vital both for male sexual development and the maintenance of skeletal integrity (Jackson, 1993). Testosterone secretion from Leydig cells in response to luteinizing hormone is modulated through changes in the intracellular levels of both Ca2+ and cyclic AMP (cAMP); the effect of cAMP is also exerted ultimately through a change in cytosolic [Ca2+] (Sullivan & Cooke, 1986). Furthermore, intracellular Ca2+ also influences steroidogenesis although the precise mechanism of this effect is unclear. It is believed that mitochondrial cholesterol transport and enzymatic side chain cleavage are both Ca2+-sensitive (Sullivan & Cooke, 1986). In addition to intracellular [Ca2+] changes, changes in extracellular [Ca2+] ([Ca2+]e) also modulate testosterone secretion. The latter doubles when extracellular [Ca2+] is increased from 1 to 10 mM (Meikle et al. 1991). Again, the mechanism through which an elevated [Ca2+]e is transduced into enhanced testosterone secretion is unknown. Additionally, we are also unclear about the altered or steady-state [Ca2+]e experienced by Leydig cells in vivo.
Previous studies have shown that certain eukaryotic cells ‘sense’ changes in their extracellular [Ca2+], a property that has been attributed to the existence of a variety of surface membrane Ca2+-sensing receptors (Brown et al. 1995). These cells include parathyroid hormone-secreting chief cells (Brown, 1991; Brown et al. 1993), calcitonin-secreting thyroid C cells (Garrett et al. 1995), Ca2+-absorbing gastric mucosal and intestinal cells (Pazianas et al. 1995; Cima et al. 1997; Gama et al. 1997), Ca2+-reabsorbing renal medullary and cortical cells (Ricardi et al. 1995), cytotrophoblasts (Lundgren et al. 1994; Bax et al. 1994), neurones (Ruat et al. 1995; Quinn et al. 1997), bone-resorbing osteoclasts (Zaidi et al. 1989; Malgaroli et al. 1989) and bone-forming osteoblasts (Honda et al. 1995). Notably, the osteoclast Ca2+ receptor, which we believe is a functional component of a surface ryanodine receptor, responds to low-millimolar [Ca2+] changes generated locally as a result of hydroxyapatite dissolution (Zaidi et al. 1995). Its activation results in the inhibition of bone resorption, possibly as part of a feedback mechanism of osteoclast control (Moonga et al. 1990).
All Ca2+-sensing receptors are thought to trigger intracellular signals in response to changed [Ca2+]e. This, in turn, regulates cell function. Most commonly, this signal is a cytosolic [Ca2+] change that results from both transmembrane Ca2+ influx and intracellular Ca2+ release (Brown, 1991; Zaidi et al. 1993a). The latter involves either ryanodine receptors or inositol trisphosphate (IP3) receptors (Brown, 1991; Shankar et al. 1995b). In the osteoclast, however, the plasma membrane ryanodine receptor itself gates Ca2+ influx (Zaidi et al. 1995; Adebanjo et al. 1996).
This study has used methods employed previously to characterize Ca2+-sensing in other cells (Brown, 1991; Zaidi et al. 1993a). Specifically, divalent cations have been used as surrogate Ca2+ agonists allowing us to distinguish Ca2+ release from Ca2+ influx (Nemeth, 1990; Shankar et al. 1993). Our results demonstrate a plasma membrane divalent cation (Ca2+) receptor in Leydig cells, the activation of which triggers Ca2+ release from ryanodine receptor-gated intracellular Ca2+ stores.
METHODS
Reagents
Fura-2, fura-2 AM and ionomycin were purchased from Molecular Probes, Inc. (Eugene, OR, USA). Tissue culture materials, including Hepes and heat-inactivated fetal calf serum (FCS) were purchased from Gibco-BRL. EDTA, EGTA, Triton X-100 and trypsin were all obtained from Sigma Chemical Co. Measurements of cytosolic [Ca2+] were carried out in either RPMI-1640 ([Ca2+], 1.25 mM) (Gibco BRL) or modified Krebs (Ca2+-free) medium. The latter comprised (mM): 130 NaCl, 5 KCl, 0.8 MgCl2, 5 glucose, 10 Hepes and 1.2 EGTA (pH 7.4) ([Ca2+] < 5 nM, by fura-2 measurements). An antibody, Ab34, raised to the consensus calmodulin-binding sequence of the ryanodine receptor was kindly provided for us by Dr F. A. Lai (National Institute for Medical Research, London, UK). The antibody has been shown not to differentiate between the three known ryanodine receptor isoforms, types I, II and III. It also does not bind to any one of the known IP3 receptor isoforms (Zaidi et al. 1995).
Culture of TM3 cells
Leydig cells (TM3, ATCC-CRL-1714, American Tissue Culture Collection, Riversville, MD, USA) derived from BALB/c mice have primary epithelial cell characteristics, are non-tumorigenic and express receptors for epidermal growth factor, luteinizing hormone, androgens, oestrogen and progesterone. The cells were grown in Hepes-buffered RPMI-1640 supplemented with FCS (10 % v/v), glutamine (1 % w/v), penicillin (50 kU l−1) and streptomycin (50 mg l−1). The cells were sub-cultured at confluence by washing in EDTA, followed by trypsin treatment (0.025 % w/v) for 2 min, addition of RPMI-1640 before centrifugation, and resuspension in medium. The cells were maintained in tissue culture flasks (Fisher Scientific) at 37°C and were harvested in their logarithmic growth phase.
Cytosolic [Ca2+] measurements
A fluorescence method employing an inverted phase-contrast microscope (Diaphot, Nikon) was used to measure cytosolic [Ca2+] in single TM3 cells using the Ca2+-sensitive fluorochrome, fura-2 (Shankar et al. 1993). Glass coverslips containing dispersed cells were incubated with 10 μM fura-2 AM in serum-free RPMI-1640 for 30 min at 37°C. They were then transferred to a Perspex bath on the microscope stage and exposed to agonists by pipetting solutions that were pre-warmed to 37°C. The temperature of the solution was kept constant by a thermostatically controlled heating device. Its volume was maintained at 2 ml using a vacuum withdrawal of fluid rising beyond a constant bath level.
Fluorochrome-loaded TM3 cells were exposed alternately to excitation wavelengths of 340 and 380 nm approximately every second. This was achieved by using a microcomputer-driven wheel to which band-pass interference filters had been fitted. The emitted fluorescence was deflected to a dichroic mirror (400 nm), filtered at 510 nm, and directed to the microscope side-port fitted with a photomultiplier tube (PM28B, Thorn EMI). The photomultiplier tube produced single photon currents that were fed into a photon counter (Newcastle Photometric Systems, Newcastle-upon-Tyne, UK). Photon counts per second (c.p.s.) were recorded on an IBM microcomputer. The ratio of emitted fluorescence intensities due to excitation at 340 and 380 nm, F340/F380, was calculated and displayed.
The fura-2 signals were calibrated using a protocol for intracellular calibration described previously by Tsien & Pozzan (1989) and adopted by us (Shankar et al. 1993). Briefly, fura-2-loaded cells were bathed in a Ca2+-free, EGTA-containing solution consisting of (mM): 130 NaCl, 5 KCl, 5 glucose, 0.8 MgCl2, 10 Hepes and 0.1 EGTA. Ionomycin (5 μM) was first applied in order to obtain the minimum ratio due to lowest cytosolic [Ca2+] (Rmin) and the maximal fluorescent intensity at 380 nm (Fmax). CaCl2 (1 mM) was then applied together with 5 μM ionomycin in order to obtain values for the maximum ratio due to elevated cytosolic [Ca2+] (Rmax) and the minimal fluorescent intensity at 380 nm (Fmin). Note that, at this concentration, ionomycin has been shown to equilibrate extracellular and intracellular Ca2+ pools in the osteoclast (Shankar et al. 1994). The dissociation constant (Kd) for Ca2+ and fura-2 at a temperature of 20°C, an ionic strength of 0.1 M, and a pH of 6.85, is 224 nM (Tsien & Pozzan, 1989). The values, together with the experimental signal, R, were substituted into the equation:
The resulting cytosolic [Ca2+] levels between treatment groups were compared by Analysis of Variance (ANOVA) with Bonferroni's correction for inequality.
Immunocytochemistry
Coverslips containing TM3 cells were fixed with glutaraldehyde (10 % v/v) and permeabilized gently with Triton X-100 (0.1 % v/v). They were then incubated with normal goat serum diluted in 10 mM phosphate-buffered saline (PBS; 1 in 10, pH 7.4) in multiwell dishes for 15 min. Excess serum was removed and replaced with Hanks’ Balanced Salt Solution (HBSS). The cells were then incubated with non-immune rabbit serum (control) or Ab34 (both diluted in HBSS, 1 in 100, v/v). After 1 h of incubation, the coverslips were rinsed gently with HBSS, drained, and re-incubated for a further hour with goat anti-rabbit FITC (1 in 20, diluted in HBSS). Finally, the coverslips were washed gently and drained. Cells were visualized on an epifluorescence microscope (Diaphot).
RESULTS
Effect of extracellular Ca2+ and Ni2+ on cytosolic [Ca2+] in TM3 cells
Application of RPMI-1640 containing added CaCl2 extracellularly to TM3 cells at a [Ca2+]e between 2.5 and 15 mM ([Mg2+], 0.8 mM) produced transient elevations of cytosolic [Ca2+] (Fig. 1). Each cytosolic Ca2+ response consisted of a rise over ∼50 s followed by a decline towards basal levels (Fig. 1A). Figure 1C plots values of the magnitude (Δ) of the cytosolic [Ca2+] change (peak minus basal cytosolic [Ca2+]: means +s.e.m.) versus the concentration of applied extracellular Ca2+. An ANOVA with Bonferroni's correction for inequality revealed a significant difference in Δ cytosolic [Ca2+] when the response to an application of 2.5 mM [Ca2+]e was compared with that to 10 mM [Ca2+]e (P = 0.018), but not when a similar comparison was made between the response to 2.5 mM [Ca2+]e and that to either 5 or 15 mM [Ca2+]e (P = 0.702 and 0.490, respectively). In contrast, application of the related alkaline earth metal Mg2+ failed to elicit cytosolic Ca2+ signals at any concentration between 0.8 and 15 mM ([Ca2+]e, 1.25 mM).
Figure 1. TM3 cell responses to elevations of extracellular [Ca2+].
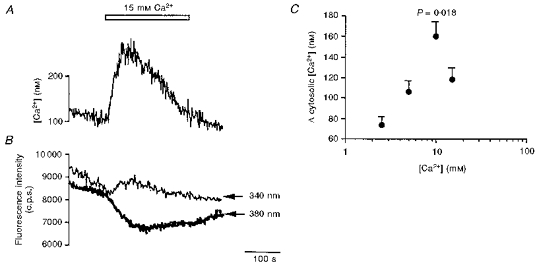
A and B, representative traces showing the effects of elevating extracellular [Ca2+] to 15 mM on cytosolic [Ca2+] (nM) (A) and fluorescence intensities (counts per second (c.p.s.)) at excitation wavelengths of 340 and 380 nm (B) in cultured TM3 cells (for details see Methods). C, effect of a range of extracellular [Ca2+] on the mean peak change (Δ) in cytosolic [Ca2+] (nM). The latter data points were derived by subtracting the basal from peak cytosolic [Ca2+]. Each data point (mean +s.e.m.) was then compared with the response to 2.5 mM [Ca2+] (regarded as the control) by ANOVA with Bonferroni's correction for inequality. Except for 10 mM [Ca2+], the rest of the points were not significantly different (P > 0.05) from control (n = 4–6 for each point).
In previous studies with osteoclasts, the transition metal cation Ni2+, when used instead of Ca2+, has allowed a clear distinction between processes attributable to intracellular Ca2+ release and those resulting from extracellular Ca2+ influx (Shankar et al. 1993). These studies went on to explore the effect of a range of divalent and trivalent metal ions and emerged with a rank order of potency of action, Cd2+ > Ni2+ = La3+ > Al3+ > Ca2+ > Ba2+ = Sr2+ > Mg2+ (Zaidi et al. 1991; Shankar et al. 1992). The present study similarly assessed the effect of the transition metal cation Ni2+ on cytosolic [Ca2+] in cultured Leydig cells. Figure 2A demonstrates that Ni2+, when applied to cells bathed in 1.25 mM [Ca2+]e and 0.8 mM [Mg2+]e, typically triggered a transient elevation in cytosolic [Ca2+] at concentrations > 1.5 mM. The responses to Ni2+, obtained over 60 and 120 s, typically consisted of a rapid elevation of cytosolic [Ca2+] to a peak followed by a decay to baseline that was more complex in waveform than that expected from a single exponential decline in some traces. Their maximum amplitude increased with [Ni2+] up to 3 mM (P = 0.028), but fell at 5 mM [Ni2+], nevertheless remaining significantly higher (P = 0.009) than the corresponding response to 0.5 mM [Ni2+] (Fig. 2B). The larger responses to Ni2+ as compared with Ca2+ were consistent with the order of potency demonstrated previously with the osteoclast system (Zaidi et al. 1991; Shankar et al. 1992).
Figure 2. Cytosolic [Ca2+] responses to extracellularly applied [Ni2+].
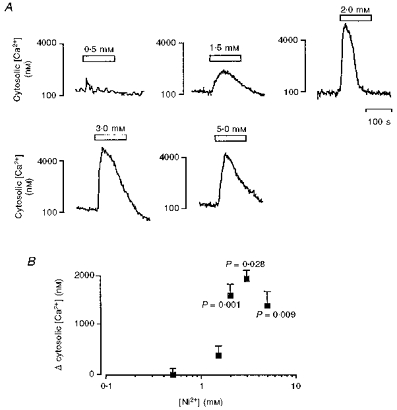
A, representative traces showing the effect of extracellularly applied [Ni2+] (0.5-5 mM) on cytosolic [Ca2+] in cultured TM3 cells (for details, see Methods). B, effect of a range of extracellular [Ni2+] (0.5-5 mM) on the mean peak change (Δ) in cytosolic [Ca2+] (nM). The latter data points were derived by subtracting the basal from the peak cytosolic [Ca2+]. Each data point was then compared with the response to 0.5 mM [Ni2+] (regarded as the control) by ANOVA with Bonferroni's correction for inequality. P values are shown, n = 4–6 for each point.
Conditioning cation applications inactivate the response to 5 mM [Ni2+]
The effect of Ni2+ on cytosolic [Ca2+] was inactivated by prior exposures to Ni2+, Ca2+ or Mg2+. In the first set of experiments, further cells were exposed to a range of Ni2+ concentrations (0.5-5 mM) that were themselves effective in triggering cytosolic Ca2+ transients (see above). When the cytosolic [Ca2+] had returned to baseline, the cells were washed with serum-free medium, and then exposed to a second pulse of 5 mM [Ni2+] within 1 or 2 min. Figure 3A and B displays traces of emitted fluorescence (F340 and F380; photon c.p.s.) due to excitation at wavelengths of 340 and 380 nm, respectively (F340 and F380; photon c.p.s.) below the resulting ratiometric (F340/F380) signals under two sets of experimental conditions. Figure 3A shows the results from a dye-loaded TM3 cell bathed in RPMI-1640 that was exposed to a conditioning application of 3 mM Ni2+ followed by a test application of 5 mM Ni2+ (horizontal open bars). Figure 3B displays the contrasting results of adding the 5.0 mM Ni2+directly to free fura-2 (10 micromolar) in the bath solution.
Figure 3. Fluorescence intensities at excitation wavelengths of 340 and 380 nm and the ratio F340/F380 under two sets of experimental conditions.
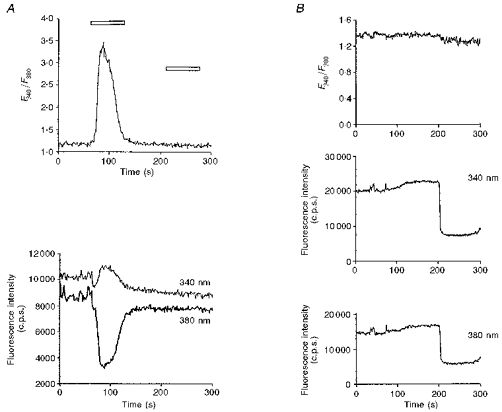
A, typical results from a dye-loaded cell bathed in RPMI-1640, exposed to a conditioning application of 3 mM Ni2+ followed by a test application of 5 mM Ni2+ (horizontal open bars). B, results of adding 5.0 mM Ni2+ directly to free fura-2 (10 micromolar) in the bath solution containing (mM): 130 NaCl, 5.0 KCl, 5 glucose and 10 Hepes. Note the downward deflections in both the F340 and the F380 signals after adding Ni2+ in B.
A comparison of these results makes it unlikely that the traces in Fig. 3A primarily reflect a fura-2 reaction with Ni2+ that had permeated into the cytosol rather than a reflection of changes in cytoplasmic [Ca2+]. First, Fig. 3A shows that the initial agonist application to the TM3 cells produced upward deflections in the F340 trace but downward deflections in the F380 traces. This is in contrast to the direct reaction between fura-2 and Ni2+ that produced downward deflections in both the F340 and the F380 signals (Fig. 3B). The relative deflections in the F340 and F380 traces in Fig. 3A thus do not fulfil the predictions of a direct reaction of dye with Ni2+. Secondly, both (F340 and F380) fluorescence traces from the TM3 cells eventually returned towards their previous stable baseline values (Fig. 3A). In contrast, the direct reaction between Ni2+ and free fura-2 produced a sustained deflection in both traces with no recovery whatsoever to the initial baseline (Fig. 3B). The latter finding would require a specific cellular mechanism for altering cytosolic Ni2+ in order to reproduce a trace of the form of Fig. 3A. Thirdly, the ratiometric (F340/F380) signal from the TM3 cells showed a corresponding deflection followed by return to baseline consistent with a net flux of Ca2+ into the cytosolic compartment followed by its net withdrawal. In contrast, Fig. 3B shows little significant alteration in the F340/F380 ratio with the direct addition of Ni2+. Finally, Fig. 3A shows that the subsequent test applications of Ni2+ to TM3 cells influenced neither the F340 and F380 traces nor their ratio, suggesting an inactivation of a process modifying cytosolic Ca2+ rather than a capacity for passive Ni2+ entry.
Figure 4 displays cytosolic Ca2+ responses resulting from a test application of 5 mM Ni2+ following conditioning applications of Ni2+ over a range of Ni2+ concentrations. Responses to the second Ni2+ pulse were diminished to an extent that depended upon the conditioning [Ni2+] (Fig. 4A and B). This reduction was significant at conditioning [Ni2+] of 4 and 5 mM (P = 0.009 and 0.027, respectively).
Figure 4. Conditioning applications of extracellular [Ni2+] inactivate the cytosolic [Ca2+] response to subsequent [Ni2+] applications.
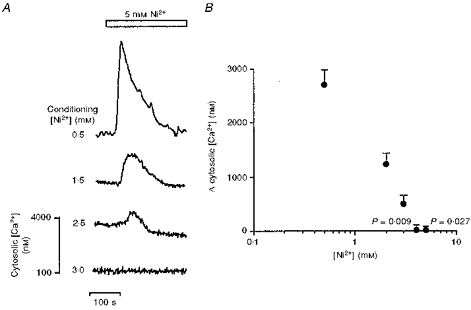
A, representative traces showing the effect of conditioning with extracellular [Ni2+] (0.5-3 mM) on the cytosolic [Ca2+] change induced by the subsequent application of 5 mM [Ni2+] (open bar) to cultured TM3 cells. The scale bar refers to changes in the levels of the cytosolic [Ca2+] (nM) in the bottom trace. B, effect of a range of conditioning extracellular [Ni2+] (0.5-5 mM) on the mean peak change (Δ) in cytosolic [Ca2+] (nM) elicited by the subsequent application of 5 mM [Ni2+]. The latter data points were derived by subtracting the basal from peak cytosolic [Ca2+]. Each data point was then compared with the response to a conditioning 0.5 mM [Ni2+] (regarded as control) by ANOVA with Bonferroni's correction for inequality. P values shown, n = 4–6 for each point.
Figure 5 summarizes typical results from experiments that investigated whether extracellular Ca2+ or Mg2+ could inactivate the cytosolic Ca2+ response to 5 mM [Ni2+]. In these experiments, the cells were exposed to a range of [Ca2+]e (2.5–10 mM) or [Mg2+]e (0.8-15 mM). Following recovery from any resulting cytosolic [Ca2+] change, as in the case of [Ca2+]e elevation, the cells were washed with serum-free medium and a pulse of 5 mM [Ni2+] was applied within 1–2 min. Prior application of Ca2+ (panel A) or Mg2+ (panel B) resulted in a progressive concentration-dependent diminution of the cytosolic Ca2+ responses to Ni2+. Each resulting transient increase in cytosolic [Ca2+] was again followed by a decay that was often more complex in waveform than that which might be expected from a simple exponential decay. The attenuation was maximal at 10 mM [Ca2+] or 10 mM [Mg2+]; thus, at these concentrations, peak versus baseline cytosolic [Ca2+] was not significantly different (P = 0.1 and 0.765, respectively). Note that when solutions without added Ca2+ and Mg2+ were used, the Ni2+-induced peak cytosolic [Ca2+] was not significantly different from control (cf. Fig. 2).
Figure 5. Conditioning applications of Ca2+ or Mg2+ inactivate the cytosolic [Ca2+] response to extracellularly applied Ni2+.
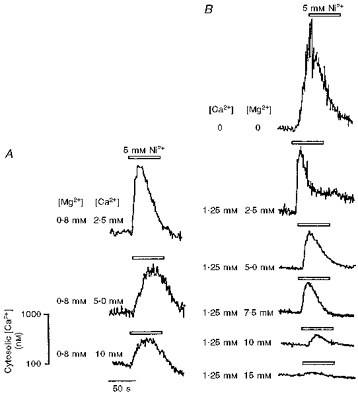
Representative traces showing the effect of extracellular [Ni2+] (5 mM) (open bars) on the cytosolic [Ca2+] (nM) of cultured TM3 cells, in various protocols wherein the cells were bathed in media with different [Ca2+] and [Mg2+] values, as indicated. Prior application of Ca2+ (A) or Mg2+ (B) resulted in a progressive concentration-dependent diminution of the cytosolic Ca2+ responses to Ni2+. The vertical scale bar refers to the bottom trace, which was obtained under conditions of 0.8 mM Mg2+ and 10 mM Ca2+.
Ni2+-induced Ca2+ release persists despite reductions in extracellular [Ca2+]
We next examined the extent to which the cytosolic Ca2+ signals observed following application of divalent cations might be attributed to the release of intracellularly stored Ca2+ as opposed to, but not excluding, transmembrane Ca2+ influx. The initial experiments investigated the effect of reducing the net inward electrochemical gradient on the movement of Ca2+ by employing Ni2+ as a surrogate Ca2+ agonist in cells bathed in modified Krebs solution containing 1.2 mM EGTA (see Methods). When added to non-esterified fura-2, the latter solutions caused a shift in baseline fluorescence ratio, F340/F380, consistent with a [Ca2+] < 5 nM. In order to correct for the binding of Ni2+ to EGTA that would displace Ca2+ from the Ca2+-EGTA complex, we used a calculated [Ni2+] of 6 mM. This corresponded to an effective [Ni2+] of 5 mM (Caputo, 1981) in view of the greater binding affinity of EGTA for Ni2+ as compared with Ca2+. Under these conditions any contaminating Ca2+ would become free in solution. This would be at concentrations of the order of 1–10 μM that are considerably smaller (by around three orders of magnitude) than the Ca2+-containing solution ([Ca2+], 1.25 mM) used in the preceding controls. Finally, there was also no evidence for an effect of Ni2+ upon the fura-2 ratiometric signal. Thus Fig. 3B illustrates that the direct application of Ni2+ to non-esterified fura-2 failed significantly to shift the baseline fluorescence ratio, F340/F380. This indicated that fura-2 signals remained mostly unaffected by Ni2+, although this does not exclude binding between the fluorochrome and Ni2+.
Figure 6A-D illustrates further protocols wherein TM3 cells were exposed to Ni2+ either in the presence of Ca2+ (1.25 mM, panel A) or in Ca2+-free, EGTA-containing medium ([Ca2+] < 5 nM, panel B). Comparison of Fig. 6A and B confirms that exposure of TM3 cells to Ni2+ in EGTA-containing solution as opposed to a 1.25 mM [Ca2+]e did not result in a measurable difference in the overall magnitude of the resulting cytosolic [Ca2+] signal. The decay phases of the cytosolic [Ca2+] responses in the cells that were exposed to normal levels of extracellular [Ca2+] did appear in some cases to be slightly more prolonged than responses from cells where extracellular [Ca2+] was reduced. Table 1 shows that the Ni2+-induced peak Δ cytosolic [Ca2+] in the two situations was not significantly different (P = 0.418). When the cytosolic Ca2+ transient returned to baseline, the cells bathed in Ca2+-free, EGTA-containing medium were exposed to 5 μM ionomycin to deplete any remaining intracellular Ca2+ stores. There was still some rise (P = 0.094) in cytosolic [Ca2+], suggesting that a single application of Ni2+ did not deplete all the intracellular Ca2+ stores.
Figure 6. Ni2+-induced cytosolic [Ca2+] elevations utilize intracellular Ca2+ stores.
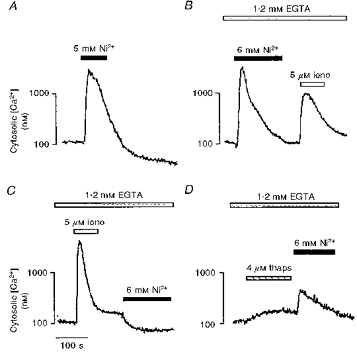
Representative traces showing the effects of extracellular [Ni2+] (5 mM; filled bars), ionomycin (iono, 5 μM; open bars) and thapsigargin (thaps, 4 μM; shaded bars), in various protocols, on the cytosolic [Ca2+] of cultured TM3 cells bathed either in medium containing 1.25 mM Ca2+ and 0.8 mM Mg2+ (A) or in modified Krebs solution containing 1.2 mM EGTA (B-D). The statistical analysis is given in Table 1.
Table 1.
Basal (pretreatment) and peak cytosolic [Ca2+] from cytosolic [Ca2+] responses of TM3 cells under different experimental conditions
| External solution | Application | Pretreatment [Ca2+]1 (nm) | Peak [Ca2+]1(nm) |
|---|---|---|---|
| [Ca2+]e = 1.25 mm | 5 mm[Ni2+] | 111 ± 46.4 | 1482 ± 439a |
| 1.2 mm EGTA, [Ca2+]e < 5 nm | 6 mm[Ni2+] | 78.9 ± 11.9 | 2216 ± 581a1 |
| then 5 μm ionomycin | 955 ± 23.1b | 1373 ± 39.0b1 | |
| 1.2 mm EGTA, [Ca2+]e< 5 nm | 5 μm ionomycin | 119 ± 18.1c | 6091 ± 690c1 |
| then 6 mm[Ni2+] | 169 ± 4.06d | 61.8 ± 3.90d1 | |
| 1.2 mm EGTA, [Ca2+]e < 5 nm | 4 μm thapsigargin | 113 ± 26.0e | 206 ± 66.6e1 |
| then 6 mm[Ni2+] | 145 ± 4.77 | 288 ± 122a2 |
Statistics by ANOVA with Bonferroni's correction for inequality.
a vs. a1P = 0.418
b vs. b1P = 0.094
c vs. c1 and d vs. d1P = 0.001
e vs. e1P = 0.222
a vs. a2P = 0.033 (n = 3–6 cells for each variable).
Ni2+-induced Ca2+ release depends on intracellular Ca2+ stores
Figure 6C and D represents typical results from two complementary experiments that sought to investigate the effect of depleting intracellular Ca2+ stores on the Ni2+ effect. First, cells were treated with 5 μM ionomycin in Ca2+-free medium. As expected, this produced a highly significant (P < 0.001), transient rise in cytosolic [Ca2+]; a subsequent Ni2+ application then failed to elevate cytosolic [Ca2+]. Instead there was a small, but significant (P < 0.001) decrement in basal cytosolic [Ca2+] (Fig. 6C and Table 1). Parallel experiments used thapsigargin, a microsomal Ca2+-ATPase inhibitor. In Ca2+-free medium, thapsigargin elicited a small rise in cytosolic [Ca2+], indicating store depletion (Table 1). The subsequent application of Ni2+ triggered a cytosolic Ca2+ signal that was significantly attenuated compared with that elicited in the absence of thapsigargin, either in the presence (cf. Fig. 6A, P = 0.041) or absence (cf. Fig. 6B, P = 0.033) of extracellular Ca2+. These results confirmed a participation of Ca2+ release from intracellular stores in the Leydig cell response to extracellular applications of divalent cations.
Ni2+-induced Ca2+ release may involve ryanodine receptor-gated Ca2+ stores
We next investigated the sensitivity of the intracellular Ca2+ stores to caffeine, a known ryanodine receptor agonist. Caffeine itself elevated cytosolic [Ca2+] when applied at concentrations of 250 and 500 μM (P = 0.004 and 0.01, respectively). More importantly, at both concentrations caffeine also inhibited the cytosolic Ca2+ response to Ni2+ significantly (P = 0.006 and 0.06, respectively) (Table 2). That ryanodine receptors were present in TM3 cells was next confirmed immunocytochemically. Notably, permeabilized TM3 cells stained strongly with the antiserum Ab34. Cells incubated with non-immune rabbit serum, instead of the antiserum, did not stain (Fig. 7). Taken together, the data suggest that (a) caffeine-sensitive, ryanodine receptor-gated Ca2+ stores are present in TM3 cells, and (b) these stores appear to be involved in Ni2+-induced cytosolic Ca2+ release. Note that caffeine is also a phosphodiesterase inhibitor at the concentrations tested and hence is expected to increase cellular cAMP levels. To exclude the latter as a mechanism of caffeine action, we tested the effect of a cell-permeant cAMP analogue, dibutyryl cAMP, on Ni2+-induced Ca2+ release. Dibutyryl cAMP (200 μM) neither elevated cytosolic [Ca2+] nor inhibited Ni2+-induced Ca2+ release (P = 0.274) (Table 2).
Table 2.
Peak change (Δ) cytosolic [Ca2+] of TM3 cells under different experimental conditions
| Protocol | Concentration | Δ[Ca2+]1 (nm) |
|---|---|---|
| Ni2+ (control) | 5 mm | 1371 ± 393 |
| Caffeine | 250 μm | 66.8 ± 13.61 |
| 500 μm | 46.4 ± 10.52 | |
| Ni2+ after caffeine | 250 μm | 136 ± 44.03 |
| 500 μm | 401 ± 2164 | |
| db-cAMP | 200 μm | 0 |
| Ni2+ after db-cAMP | 200 μm | 2443 ± 6855 |
Statistics by ANOVA with Bonferroni's correction for inequality. Basal compared with peak cytosolic [Ca2+]:
P = 0.004
P = 0.010. Peak δ[Ca2+]1 of treatment compared with Ni2+ alone (control):
P = 0.006
P = 0.057
P = 0.274 (n = 3–6 cells for each variable). db-cAMP, dibutyryl cyclic AMP.
Figure 7. Presence of ryanodine receptors in TM3 cells.

Immunofluorescent micrographs of TM3 cells incubated with either antiserum Ab34 (a) or with non-immune rabbit serum (b; negative control). Field of view, 375 μm × 255 μm.
Membrane potential modulates Ni2+-induced cytosolic Ca2+ transients
We finally sought to investigate the effect of changing the cell membrane potential on Ni2+-induced Ca2+ release. This was achieved by using 5 μM valinomycin, a K+ ionophore, in the presence of either 5 or 100 mM [K+]e. In the presence of valinomycin, 5 mM [K+]e is known to shift the membrane potential in the negative direction, whereas 100 mM [K+]e causes membrane depolarization (Shankar et al. 1995a). The latter manoeuvre slowed the decline of the Ni2+-induced cytosolic Ca2+ signal in the absence of any effect on peak Δ cytosolic [Ca2+] (Fig. 8). Conversely, hyperpolarization attenuated the magnitude of the cytosolic Ca2+ signal in addition to slowing its decay phase (Fig. 8). These effects of membrane voltage change are consistent with regulatory events that are localized to the Leydig cell membrane.
Figure 8. Membrane potential modulates Ni2+-induced cytosolic Ca2+ transients.
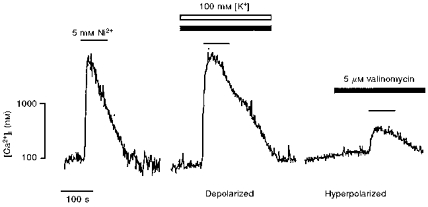
Representative traces showing the effect of extracellular [Ni2+] (5 mM; black lines) on the cytosolic [Ca2+] of cultured TM3 cells, in various protocols whereby the membrane potential was altered by using the K+ ionophore, valinomycin (5 μM; filled bars), in the presence of either 5 mM (hyperpolarized) or 100 mM [K+]e (open bar) (depolarized).
DISCUSSION
The present study was prompted by the observation that extracellular [Ca2+], when elevated from 1 to 10 mM, doubles Leydig cell testosterone secretion (Meikle et al. 1991). Here we show, for the first time, that such elevated extracellular Ca2+ levels elicit sharp increases in cytosolic [Ca2+]. In addition, by employing experimental strategies used to study extracellular Ca2+ sensing in other cells (for reviews see Brown, 1991; Zaidi et al. 1993a; Brown et al. 1995), we have obtained new insights into transduction mechanisms that Leydig cells use to trigger Ca2+ release in response to changes in [Ca2+]e.
The transition metal cation, Ni2+, was used in our earlier studies to demonstrate and to characterize a Ca2+-sensing receptor on the osteoclast (Bax et al. 1993; Shankar et al. 1993; Zaidi et al. 1993a). Here we provide evidence that Ni2+ acts at the Leydig cell surface consistent with the existence of a specific receptor activated by Ca2+ and Ni2+. Firstly, Ni2+ is thought not to permeate cells; instead it blocks plasma membrane Ca2+ channels (Caputo, 1981; Huang, 1988). Secondly, by monitoring the fluorescence separately at each excitation wavelength, 340 and 380 nm, we did not observe classical quenching of the fura-2 signals at any Ni2+ concentration tested. Thirdly, it is unlikely that the observed fluorescence changes would have resulted from a fura-Ni2+ interaction intracellularly. If so, we should have seen a non-decaying signal as there is no known mechanism of Ni2+ efflux from cells. Furthermore, we have confirmed that in the same experimental system, the direct exposure of non-esterified (free) fura-2 to Ni2+ did not appreciably alter the ratio of the emission, F340/F380. Thus, it is unlikely that the Ni2+-induced changes in the fluorescence ratio F340/F380 could have resulted primarily from the binding of permeated Ni2+ to fura-2 within the cells.
Further evidence implicates a cell surface site for Ni2+ action more directly. Such an activation site should extend across the cell surface membrane, and hence be exposed to the transmembrane electric field. Thus, we find that a change in cell membrane voltage alters the magnitude and waveform of the Ni2+-induced Ca2+ signal (Shankar et al. 1995a). In the presence of the K+ ionophore valinomycin, 5 mM [K+]e was found to attenuate and prolong the Ni2+-induced cytosolic Ca2+ signal. Likewise, 100 mM [K+]e prolonged inactivation. The receptor for Ni2+ may therefore well be an integral protein that is either itself sensitive to the transmembrane electric field, or whose binding with charged ligands is voltage dependent.
In previous studies with the osteoclast, we have used Ni2+ as a substitute for Ca2+ to examine changes in cytosolic [Ca2+] in the absence of transmembrane Ca2+ influx (Bax et al. 1993; Shankar et al. 1993; Zaidi et al. 1993a). Here, we demonstrate a major role for intracellular Ca2+ release in the generation of the Ni2+-induced cytosolic Ca2+ signal. Thus, a marked reduction of Ca2+ in the extracellular solution was found to conserve the magnitude of the Ca2+ signal. These results should be treated with caution, as the binding of Ni2+ to EGTA and the consequent displacement of Ca2+ into the solution would not reverse the cellular electrochemical gradient of Ca2+. There is therefore the possibility that even in EGTA-containing solutions, Ni2+ may trigger Ca2+ influx; the latter may, in turn, contribute to the Ni2+-induced cytosolic Ca2+ signal. However, depletion of releasable Ca2+ stores using either a Ca2+ ionophore, ionomycin (in Ca2+-free medium) or, more specifically, a microsomal membrane Ca2+-ATPase inhibitor, thapsigargin (in Ca2+-free medium), abolished the Ni2+ response (Zaidi et al. 1993b; Shankar et al. 1994).These results with thapsigargin provide more direct evidence for the release of Ca2+ from intracellular stores and are reminiscent of hormone effects on membrane receptors or, indeed, cation effects on cell surface Ca2+-sensing receptors (Brown, 1991; Berridge, 1993; Zaidi et al. 1993a; Brown et al. 1995).
The latter studies prompted us to examine whether such Ni2+-induced Ca2+ release involved a participation of caffeine-sensitive ryanodine receptors. Note that apart from their classical location in microsomal membranes, ryanodine receptors are also present on the plasma membranes of osteoclasts (Zaidi et al. 1995). We thus tested the effect of caffeine on basal cytosolic Ca2+ levels and on Ni2+-induced Ca2+ release. Caffeine, applied at 250 and 500 μM, itself triggered cytosolic Ca2+ signals. At the same concentrations, caffeine significantly inhibited Ni2+-induced cytosolic Ca2+ release. The concentrations appear somewhat lower than those used in skeletal muscle (1-10 mM), but are similar to those effective in the osteoclast (50-500 μM) (Shankar et al. 1995b). In contrast, dibutyryl cAMP did not elevate cytosolic [Ca2+] or attenuate Ni2+-induced Ca2+ release. This excludes an effect of caffeine through its inhibition of phosphodiesterase, and a consequent elevation in cellular cAMP. In parallel experiments, an anti-ryanodine receptor antiserum, Ab34, stained Leydig cells strongly and specifically, confirming the expression of ryanodine receptors. Taken together, the results argue strongly for the involvement of ryanodine receptors in Ni2+ action on Leydig cells, without ruling out the participation of IP3 receptors.
Finally, we investigated the interaction between the three cations, Ca2+, Mg2+ and Ni2+. Mg2+ itself was found not to elevate cytosolic [Ca2+]. This is similar to the cation's action on the osteoclast (Zaidi et al. 1991), but contrasts with its potent activating action in parathyroid cells (Brown, 1991). However, both Mg2+ and Ca2+ inhibited Ni2+-induced Ca2+ release in a concentration-dependent manner. This inhibition is unlikely to be due to empty Ca2+ stores, as Mg2+ itself did not trigger Ca2+ release from these stores. It is likely that Mg2+ and Ca2+ compete with, or else displace Ni2+ from its cell surface binding site. Hypothetically, this could result from differences in the physicochemical properties of the cations, such as their crystal ionic radii (0.099 nm for Ca2+versus 0.069 nm for Ni2+).
In conclusion, the results provide strong evidence that a divalent cation (Ca2+) receptor is present on the Leydig cell surface. The receptor appears to be coupled to Ca2+ release from ryanodine receptor-gated intracellular Ca2+ stores. Currently, we have no structural information on this putative entity. Its molecular characterization may nevertheless have significant therapeutic implications. Notably, testosterone and its analogues are being currently investigated for use in preventing muscle and bone loss in ageing men. Furthermore, in men with prostate cancer, testosterone levels must be reduced. Hence it is of interest to modulate endogenous testosterone secretion in vivo, potentially by a molecule that could activate or inhibit the Leydig cell Ca2+-sensing receptor. The latter strategy has been used to develop a novel ‘calcimimetic’, a potent inhibitor of parathyroid hormone secretion for use in humans (Silverberg et al. 1997).
Acknowledgments
This study was supported by grants to M. Z. from the National Institute on Aging (NIH RO1 AG 14917-02), the Department of Veterans Affairs (Merit Review Award) and the Amgen Corporation, Inc., Thousand Oaks, CA, USA. C. L.-H. H. acknowledges the support of the Leverhulme Trust (UK) and the Biotechnology and Biological Research Council (BBSRC) of the UK. J. I. acknowledges the support of grants AI41231 and RR03034 from the NIH.
References
- Adebanjo OA, Shankar VS, Pazianas M, Simon B, Lai FA, Huang CL-H, Zaidi M. Extracellularly applied ruthenium red and cyclic ADP-ribose elevate cytosolic Ca2+ in isolated rat osteoclasts. American Journal of Physiology. 1996;270:F469–475. doi: 10.1152/ajprenal.1996.270.3.F469. [DOI] [PubMed] [Google Scholar]
- Bax BE, Shankar VS, Bax CMR, Alam ASMT, Zara SJ, Pazianas M, Huang CL-H, Zaidi M. Functional consequences of the interaction of Ni2+ with the osteoclast Ca2+ receptor. Experimental Physiology. 1993;78:517–529. doi: 10.1113/expphysiol.1993.sp003703. [DOI] [PubMed] [Google Scholar]
- Bax CMR, Bax BE, Bain M, Zaidi M. Ca 2+ channels in human term trophoblastic cells. A study using the Ca2+-sensitive dye, fura-2. Trophoblast Research. 1994;8:573–580. [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signaling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Brown EM. Extracellular Ca2+ sensing, regulation of parathyroid cell function, and role of Ca2+ and other ions as extracellular (first) messengers. Physiological Reviews. 1991;71:371–411. doi: 10.1152/physrev.1991.71.2.371. [DOI] [PubMed] [Google Scholar]
- Brown EM, Gamba G, Ricardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular calcium sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. doi: 10.1038/366575a0. [DOI] [PubMed] [Google Scholar]
- Brown EM, Pollak M, Seidman CE, Seidman JG, Chou Y-HW, Ricardi D, Hebert SC. Calcium-ion-sensing cell surface receptors. New England Journal of Medicine. 1995;333:234–240. doi: 10.1056/NEJM199507273330407. [DOI] [PubMed] [Google Scholar]
- Caputo C. Nickel substitution for calcium and the timecourse of potassium conductances for single muscle fibres. Journal of Muscle Research and Cell Motility. 1981;2:167–182. doi: 10.1007/BF00711867. [DOI] [PubMed] [Google Scholar]
- Cima RR, Cheng I, Klingensmith ME, Chattopadhyay N, Kifor O, Hebert SC, Brown EM, Soybel D. Identification and functional assay of an extracellular Ca2+ receptor in Necturus gastric mucosa. American Journal of Physiology. 1997;273:G1051–1060. doi: 10.1152/ajpgi.1997.273.5.G1051. [DOI] [PubMed] [Google Scholar]
- Gama L, Baxendale-Cox LM, Breitwieser GE. Ca2+ sensing receptors in intestinal epithelium. American Journal of Physiology. 1997;273:C1168–1175. doi: 10.1152/ajpcell.1997.273.4.C1168. [DOI] [PubMed] [Google Scholar]
- Garrett JE, Tamir H, Kifor O, Simin RT, Rogers KV, Mithal A, Gagel RF, Brown EM. Calcitonin-secreting cells of the thyroid express an extracellular calcium receptor gene. Endocrinology. 1995;136:5202–5211. doi: 10.1210/endo.136.11.7588259. 10.1210/en.136.11.5202. [DOI] [PubMed] [Google Scholar]
- Honda Y, Fitzsimmons RJ, Baylink DJ, Mohan S. Effects of extracellular calcium on insulin-like growth factor II in human bone cells. Journal of Bone and Mineral Research. 1995;10:1660–1665. doi: 10.1002/jbmr.5650101108. [DOI] [PubMed] [Google Scholar]
- Huang CL-H. Intramembrane charge movements in skeletal muscle. Physiological Reviews. 1988;68:1197–1247. doi: 10.1152/physrev.1988.68.4.1197. [DOI] [PubMed] [Google Scholar]
- Jackson JA. Osteoporosis in men. In: Favus MJ, editor. Primer on Metabolic Bone Diseases and Disorders of Mineral Metabolism. Philadelphia: Lippincott Raven; 1993. pp. 255–257. [Google Scholar]
- Lundgren S, Hjalm G, Hellman P. A protein involved in calcium sensing of the human parathyroid and placental cytotrophoblast cells belongs to the LDL-receptor protein superfamily. Experimental Cell Research. 1994;212:344–350. doi: 10.1006/excr.1994.1153. 10.1006/excr.1994.1153. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Meldolesi J, Zambonin-Zallone A, Teti A. Control of cytosolic free calcium in rat and chicken osteoclasts. The role of extracellular calcium and calcitonin. Journal of Biological Chemistry. 1989;264:14342–14347. [PubMed] [Google Scholar]
- Meikle AW, Liu X-A, Stringham JD. Extracellular calcium and luteinizing hormone effects on 22-hydroxycholesterol used for testosterone production in mouse Leydig cells. Journal of Andrology. 1991;12:148–151. [PubMed] [Google Scholar]
- Moonga BS, Moss DW, Patchell A, Zaidi M. Intracellular regulation of enzyme secretion from rat osteoclasts and evidence for a functional role in bone resorption. The Journal of Physiology. 1990;429:29–45. doi: 10.1113/jphysiol.1990.sp018242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeth EF. Regulation of cytosolic calcium by extracellular divalent cations in C-cells and parathyroid cells. Cell Calcium. 1990;11:323–327. doi: 10.1016/0143-4160(90)90033-q. 10.1016/0143-4160(90)90033-Q. [DOI] [PubMed] [Google Scholar]
- Pazianas M, Adebanjo OA, Shankar VS, James SV, Colston KW, Maxwell JD, Zaidi M. Extracellular Ca 2+ sensing by the enterocyte. Prediction of a novel divalent cation sensor. Biochemical and Biophysical Research Communications. 1995;210:448–453. doi: 10.1006/bbrc.1995.1748. [DOI] [PubMed] [Google Scholar]
- Quinn SJ, Ye C-P, Diaz R, Kifor O, Bai M, Vassilev P, Brown E. The Ca2+-sensing receptor: a target for polyamines. American Journal of Physiology. 1997;273:C1315–1323. doi: 10.1152/ajpcell.1997.273.4.C1315. [DOI] [PubMed] [Google Scholar]
- Ricardi D, Pak J, Lee W-S, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proceedings of the National Academy of Sciences of the USA. 1995;9:131–135. doi: 10.1073/pnas.92.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruat M, Molliver ME, Snowman AM, Snyder SH. Calcium sensing receptor: molecular cloning and localization to nerve terminals. Proceedings of the National Academy of Sciences of the USA. 1995;92:3161–3165. doi: 10.1073/pnas.92.8.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar VS, Bax CMR, Alam ASMT, Bax BE, Huang CL-H, Zaidi M. The osteoclast Ca 2+ receptor is highly sensitive to activation by transition metal cations. Biophysical and Biochemical Research Communications. 1992;187:913–918. doi: 10.1016/0006-291x(92)91284-w. [DOI] [PubMed] [Google Scholar]
- Shankar VS, Bax CMR, Bax BE, Alam ASMT, Simon B, Pazianas M, Moonga BS, Huang CL-H, Zaidi M. Activation of the Ca 2+‘receptor’ on the osteoclast by Ni2+ elicits cytosolic Ca2+ signals: evidence for receptor activation and inactivation, intracellular Ca2+ redistribution and divalent cation modulation. Journal of Cellular Physiology. 1993;155:120–129. doi: 10.1002/jcp.1041550116. [DOI] [PubMed] [Google Scholar]
- Shankar VS, Huang CL-H, Adebanjo OA, Pazianas M, Zaidi M. Calcium influx and redistribution in isolated rat osteoclasts. Experimental Physiology. 1994;79:537–545. doi: 10.1113/expphysiol.1994.sp003786. [DOI] [PubMed] [Google Scholar]
- Shankar VS, Huang CL-H, Adebanjo OA, Simon BJ, Alam ASMT, Moonga BS, Pazianas M, Scott RH, Zaidi M. The effect of membrane potential on surface Ca2+ receptor activation in rat osteoclasts. Journal of Cellular Physiology. 1995a;162:1–8. doi: 10.1002/jcp.1041620102. [DOI] [PubMed] [Google Scholar]
- Shankar VS, Pazianas M, Huang CL-H, Simon B, Adebanjo O, Zaidi M. Caffeine modulates Ca2+ receptor activation in isolated rat osteoclasts and induces intracellular Ca2+ release. American Journal of Physiology. 1995b;268:F447–454. doi: 10.1152/ajprenal.1995.268.3.F447. [DOI] [PubMed] [Google Scholar]
- Silverberg SJ, Bone HGIII, Marriott TB, Locker FG, Thys-Jacobs S, Dziem G, Kaatz S, Sanguinetti EL, Bilezikian JP. Short-term inhibition of parathyroid hormone secretion with calcium receptor agonist in patients with primary hyperparathyroidism. New England Journal of Medicine. 1997;337:1506–1510. doi: 10.1056/NEJM199711203372104. 10.1056/NEJM199711203372104. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Cooke BA. The role for Ca2+ in steroidogenesis in Leydig cells. Biochemical Journal. 1986;236:45–51. doi: 10.1042/bj2360045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY, Pozzan T. Measurement of cytosolic free Ca2+ with quin 2. Methods in Enzymology. 1989;172:232–262. doi: 10.1016/s0076-6879(89)72017-6. [DOI] [PubMed] [Google Scholar]
- Zaidi M, Alam ASMT, Huang CL-H, Pazianas M, Bax CMR, Bax BE, Moonga BS, Bevis PJR, Shankar VS. Extracellular Ca2+ sensing by the osteoclast. Cell Calcium. 1993a;14:271–277. doi: 10.1016/0143-4160(93)90048-b. 10.1016/0143-4160(93)90048-B. [DOI] [PubMed] [Google Scholar]
- Zaidi M, Datta HK, Patchell A, Moonga BS, MacIntyre I. ‘Calcium-activated’ intracellular calcium elevation: a novel mechanism of osteoclast regulation. Biochemical and Biophysical Research Communications. 1989;163:1461–1465. doi: 10.1016/0006-291x(89)91143-1. [DOI] [PubMed] [Google Scholar]
- Zaidi M, Kerby J, Huang CL-H, Alam ASMT, Rathod H, Chambers TJ, Moonga BS. Divalent cations mimic the inhibitory effects of extracellular ionized calcium on bone resorption by isolated rat osteoclasts: further evidence for a ‘calcium receptor’. Journal of Cellular Physiology. 1991;149:422–427. doi: 10.1002/jcp.1041490310. [DOI] [PubMed] [Google Scholar]
- Zaidi M, Shankar VS, Bax CMR, Bax BE, Bevis PJR, Pazianas M, Alam ASMT, Huang CL-H. Linkage of extracellular and intracellular control of cytosolic Ca 2+ in rat osteoclasts in the presence of thapsigargin. Journal of Bone and Mineral Research. 1993b;8:961–967. doi: 10.1002/jbmr.5650080809. [DOI] [PubMed] [Google Scholar]
- Zaidi M, Shankar VS, Tunwell RE, Adebanjo OA, McKrill J, Pazianas M, O'Connell D, Simon B, Rifkin BR, Venkitaraman A, Huang CL-H, Lai FA. A ryanodine receptor-like molecule expressed in the osteoclast plasma membrane functions in extracellular Ca2+ sensing. Journal of Clinical Investigation. 1995;96:1582–1590. doi: 10.1172/JCI118197. [DOI] [PMC free article] [PubMed] [Google Scholar]