

Abstract
The mechanism of the protochlorophyllide (PChlide) photoreduction reaction operating in light-adapted plants and catalyzed by NADPH:protochlorophyllide oxidoreductase B (PORb) has been analyzed by low-temperature fluorescence spectroscopy by using purified barley PORb overexpressed heterologously in Escherichia coli as a fusion protein with the maltose-binding protein. We show that the PORb-catalyzed PChlide reduction reaction consists of two steps, one photochemical and the other nonphotochemical. The initial photochemical reaction follows a single quantum mechanism and leads to the formation of an unstable intermediate with mixed pigment electronic structure and an EPR spectrum that suggests the presence of a free electron. The second step involves the spontaneous conversion of the unstable intermediate into chlorophyllide as defined by its spectroscopic characteristics and migration on an HPLC column. Both steps of the reaction can be performed at subzero temperatures in frozen samples, suggesting that they do not include major changes in enzyme conformation or pigment rearrangement within the active site. The rate of the reaction at room temperature depends linearly on enzyme and substrate (PChlide) concentration, and the kinetic parameters are consistent with one molecule of substrate bound per active monomer in solution. The PORb-catalyzed reaction in vitro is spectroscopically similar to that identified in leaves of light-adapted plants, suggesting that the same reaction sequence observed operates in planta.
In cyanobacteria, green algae, and most vascular plants, chlorophyll formation is regulated, at least in part, by the light-dependent reduction of protochlorophyllide (PChlide) to chlorophyllide (Chlide) catalyzed by the enzyme NADPH:protochlorophyllide oxidoreductase (POR; EC 1.6.99.1) (1). POR is one of only two enzymes known to exist in nature that require light for their catalytic activity (1, 2), making it one of the more intriguing reactions known in plant metabolism.
Previously, POR-catalyzed PChlide photoreduction was studied mainly in etiolated (or dark-grown) plants, where, because of the high levels of enzyme (POR) and substrate (PChlide) accumulated, the process was relatively easy to monitor (3, 4). At least three different spectral forms of PChlide are recognized in intact tissues based on their fluorescence emission maximum (in nm): PChlide F632, PChlide F644, and PChlide F655 (5). The main photoactive form present in etiolated plants is PChlide F655, which, after illumination, is converted to Chlide and, subsequently, to chlorophyll (F682) through the formation of several, long wavelength intermediates (5–9). In most angiosperms, POR and its substrate localize to specific structures termed prolamellar bodies that are not detected in light-adapted plants (10). Some or all of the variability in the spectral composition of PChlide in vivo may be a consequence of enzyme ternary complex formation and aggregation within the etioplast and prolamellar bodies (8–10).
Several years ago, we developed a method to study chlorophyll formation in the tissues of light-grown plants (11) and demonstrated that a difference exists in the mechanism of PChlide photoreduction in etioplasts and chloroplasts. Only two PChlide species, PChlide F632 and PChlide F644, were found in light-grown plants and the green alga Chlamydomonas reinhardtii after redarkening (11, 12). After illumination, both disappeared concomitant with the appearance of Chlide F675 (12, 13). The same reaction has been reported to be a minor pathway in etiolated plants (6, 7, 9) and was the only pathway detected in DET340, a photomorphogenetic mutant of Arabidopsis thaliana that exhibits many of the gene-expression patterns and morphological features of light-adapted plants when grown in darkness (14).
Two Por genes, designated PorA and PorB, have been identified in angiosperm species. Although both genes are expressed in etiolated plants, only PorB is expressed in light-adapted plants and in the DET340 mutant (14–16). Because only PorB is expressed in light-adapted plants, the encoded PORb protein must be the only enzyme responsible for PChlide photoreduction in chloroplasts.
The use of heterologously expressed POR proteins has proven to be extremely useful in analyzing the general requirements for POR catalysis (17–19). To better understand the mechanism of PChlide photoreduction in light-adapted plants, in the present study we constructed and heterologously expressed in E. coli a fusion protein consisting of the barley PORb amino-terminally linked to the bacterial maltose-binding protein (MBP). The reaction catalyzed by the MBP-PORb fusion protein then was analyzed in vitro. Our data show that PORb-catalyzed PChlide photoreduction occurs in two steps, involving first the light-induced formation of an unstable intermediate and followed by the spontaneous conversion of this intermediate into the final product, Chlide. The reaction is performed by a monomeric enzyme that binds a single pigment molecule. Both steps of PChlide reduction can be completed in frozen samples and, thus, they do not include major protein conformational changes or pigment movements. The photoactive enzyme complex formed by PORb and the final product of the reaction in vitro have spectral properties similar to those found in light-adapted plants. No long-wavelength PChlide F655 or long-wavelength spectroscopic intermediates found in etiolated plants were detected in the course of PORb-catalyzed PChlide photoreduction in vitro.
MATERIALS AND METHODS
Construction of MBP-PORb Fusions, Bacterial Expression, and Enzyme Purification.
Nucleotide sequences encoding the mature PORb protein of barley (15) were amplified by a TAQ polymerase-catalyzed PCR from pBluescript SK (Stratagene) plasmids encoding the full-length PORb cDNA (kindly provided by Klaus Apel, ETH-Zentrum, Zurich) by using synthetic oligonucleotide primers specific to the 5′ and 3′ ends of the coding sequence of mature protein. The forward primer (5′-aatttcaggatccatgatcgacggcgcggagttc) introduced a BamHI site (underlined) whereas the reverse primer (5′-tttaaaactgcagtcatgcgagcccgacgagcttc-3′) included a PstI recognition site (underlined). The PCR consisted of 20 ng of template DNA, 0.5 mg of each primer, and 4 units of Taq polymerase in 50 ml of buffer. Amplification reactions were performed in a Perkin–Elmer thermocycler as described in ref. 17 except that 30 cycles of amplification were used. The resulting amplification products were digested with BamHI and PstI and purified by electrophoresis through 1% low-melting agarose gels, and the resulting fragments were ligated into the pMAL-c2 expression vector digested previously with the same restriction enzymes (20). The recombinant plasmids (pMAL-PORb) were transformed into competent E. coli ER2508 cells, plasmid DNA was prepared, and the nucleotide sequence of the MBP-PORb fusions was confirmed by sequencing using standard procedures (20). For enzyme isolation, E. coli cells were grown overnight in yeast-tryptone (YT) medium containing 100 mg/ml ampicillin and 15 mg/ml tetracycline (17). Exponentially growing cultures were treated with 0.1 mM isopropyl β-d-thiogalactoside for 4 hr to induce MBP-PORb expression. The cells were collected by centrifugation, washed two times with extraction buffer (50 mM Tris⋅HCl, pH 7.5/500 mM NaCl/0.1% Triton X-100/5 mM EDTA/1 mM DTT), and sonicated on ice for 15 sec in VirSonic 60/VirTis at output power 2 W. The cell debris was removed by centrifugation at 9,000 × g for 20 min. The resulted cell extracts were diluted to 50 mM Tris⋅HCl, pH 7.5/250 mM NaCl/0.05% Triton X-100/2.5 mM EDTA/1 mM DTT (column buffer) and applied to an amylose column equilibrated with the same buffer. The column was washed with 10 vol of the column buffer, and the proteins were eluted with 50 mM Tris⋅HCl/50 mM NaCl/0.1% Triton X-100/1 mM DTT/10 mM maltose (elution buffer). The eluted fusion protein had the predicted molecular mass (78 kDa) and possesses PChlide-photoreducing activity.
SDS/PAGE Analysis, Immunoblotting, and Protein Assay.
The protein concentration of samples was determined as described (21). For SDS/PAGE, the samples were diluted with electrophoresis sample buffer [final concentration, 70 mM Tris⋅HCl, pH 6.8/1% (wt/vol) SDS/7% (vol/vol) glycerol/1.5% (vol/vol) 2-mercaptoethanol/0.03% (wt/vol) bromphenol blue], aliquots were heat-denatured (95°C for 3 min), and the proteins were fractionated on 8% (wt/vol) polyacrylamide gels (22). After electrophoresis, the gels were either stained with Coomassie Brilliant Blue R-250 to visualize the proteins or the proteins were electrophoretically blotted to poly(vinylidene difluoride) membranes (Bio-Rad) for immunological analysis. Immunological detection of POR was performed by using polyclonal antiserum raised against purified wheat POR (kindly provided by W. T. Griffiths, University of Bristol, U.K.) in conjunction with the Enhanced Chemiluminescence Detection Kit (Amersham Pharmacia) (14, 23).
Assay of POR Enzymatic Activity, Low-Temperature Fluorescence Analysis, and EPR Spectroscopy.
POR activity was assayed in reaction buffer [50 mM Tris⋅HCl, pH 7.5/0.1% (vol/vol) Triton X-100] containing 10 mg/ml protein, 20 nM PChlide, and 4 mM NADPH. The reaction mixture was assembled and allowed to equilibrate at room temperature (22°C) for 10 min in the dark. The mixture then was illuminated for 10 min with white light of a luminescence tube (33 mE⋅m−2⋅sec−1). The pigments were extracted immediately with 90% acetone (final concentration) and assayed spectrofluorometrically under excitation at 433 nm unless noted otherwise in the text. Low-temperature fluorescence experiments were performed in the same reaction mix, except that the concentration of POR protein was increased to 100 mg/ml. For analysis of fluorescence spectra, samples were placed on a sample holder and quick-frozen in liquid nitrogen. As indicated in the text, the samples were warmed to the desired temperature and then illuminated in a Dewar flask with continuous white, red (>600 nm), or blue (360–480 nm) light emitted from a tungsten lamp or with light of a 1-msec photoflash. The light intensity was attenuated with neutral-density glass filters. Fluorescence spectra were recorded with a SPEX Fluorolog-2 fluorometer. Room-temperature spectra were taken of samples held in a 0.5-cm rectangular cuvette, and low-temperature spectra were recorded by using specially designed, low-temperature sample holders maintained in or over the surface of liquid nitrogen. The temperature of the sample was controlled with a thermocouple sensor. Both excitation and emission monochromator slit widths were about 4 nm. All spectra were corrected for intensity of the excitation light and sensitivity of the photomultiplier. Difference spectra and second derivatives of the spectra, as well as Gaussian deconvolution by nonlinear regression analysis using the Quasi-Newton method, were performed with the statistica 5 package. EPR spectra were recorded with a Varian E9 spectrometer at a frequency of 9.463 GHz and microwave power of 10 mW. The temperature of samples was maintained by a steam of liquid nitrogen (−170°C). The spectra were recorded after 25 accumulations.
Fluorescence Quantum Yields and Activation Energy of the Reaction Steps.
To analyze the effect of temperature on fluorescence quantum yield, fluorescence intensities were recorded by using a multiwavelength program with the following excitation/emission wavelength pairs: 416 nm/635 nm for nonactive PChlide, 466 nm/644 nm for active PChlide, 452 nm/484 nm for the unstable intermediate, and 416 nm/675 nm for Chlide. The slit widths of excitation and emission monochromators were set to 4 nm, and integration time was 5 sec. Activation energy of the reaction steps was calculated based on the initial rates of the intermediate and Chlide formation under illumination with weak monochromatic light (1.5 mE⋅m−2⋅sec−1) at different temperatures.
HPLC Analysis of Pigments and Reaction Intermediates.
HPLC separation of PChlide and Chlide was performed on a Beckman System Gold apparatus by using a Vydac reverse-phase C18 column (4.6 mm × 250 mm) with methanol/ethyl acetate/water (55:36:9) as eluant and a flow rate of 1 ml/min. Pigments were identified by fluorescence with a Jasco FP920 Intelligent Fluorescence Detector by using an excitation light at 435 nm and collecting emission at 635 nm (PChlide) or 672 nm (Chlide).
RESULTS
Purification of MBP-PORb and Spectral Analysis of Photoactive Pigment-PORb Complexes in Vitro.
We and others have shown that enzymatically active POR fusion proteins can be prepared from the soluble fraction of E. coli cells overexpressing chimeric POR genes (17, 18). In the present study, the coding region of the mature PORb of barley was amino-terminally linked to sequences encoding the bacterial MBP, the chimeric MBP-PorB gene was expressed in E. coli ER2508 cells, and affinity chromatography was used to prepare highly purified, enzymatically active fusion protein. The 78-kDa MBP-PORb fusion protein is the predominate protein species present after affinity purification on maltose resin and, as determined by SDS/PAGE and immunoblot analysis, is greater than 95% pure.
Incubation of the purified MBP-PORb with NADPH and PChlide in the dark at room temperature resulted in the formation of active (POR-PChlide-NADPH) ternary complex. This can be demonstrated by the capacity of the enzyme to convert PChlide into Chlide upon continuous or flash illumination of the reaction mix. Illumination of the reaction mixtures containing reconstituted POR ternary complex results in a decrease in the amounts of PChlide and accumulation of Chlide with an emission maximum at 672 nm in acetone at room temperature (Fig. 1A). After approximately 10 min of illumination, the reaction saturates. At this point, about 50% of initial PChlide molecules available in the reaction mix are photoreduced. Prolonged illumination of the reaction resulted in a decrease in the detectable amounts of both PChlide and the newly formed Chlide (Fig. 1A). The magnitude of the decrease depended on light intensity and could be eliminated to a large extent by saturation of the reaction mix with argon, indicating that some or all of the decrease was the result of pigment photooxidation. To exclude interference of photooxidation with PChlide photoreduction in our assays, subsequent experiments were performed in an argon atmosphere.
Figure 1.

PORb activity measured in vitro at room temperature (+22°C). Purified MBP-PORb fusion protein was equilibrated in reaction mix in the dark for 10 min and then illuminated as indicated. (A) Pigment fluorescence in acetone extracts before and after illumination of the reaction mix with continuous white light (33 mE⋅m−2⋅s−1) for various lengths of time. Kinetics of Chlide accumulation is shown in Inset. (B) Effect of MBP-PORb protein concentration on the enzyme activity after illumination with continuous white light for 3 min. Chlide accumulation is shown by a dashed line, and PChlide disappearance is shown by a dotted line. (C) Effect of PChlide concentration on the yield of Chlide formation after 1-ms light flash. Pigment concentrations were calculated from the fluorescence spectra after corrections for band overlap and differences in the molar extinction coefficients and quantum yields. (D) Chlide accumulation after a series of 1-ms flashes of different light intensity, separated by 10-sec dark intervals. The intensity (in joules) of the flashes is shown near the lines. Light-intensity dependence of the amounts of Chlide accumulated after six flashes is shown in Inset.
PChlide Photoreduction at Room Temperature: Concentration and Light Intensity Dependence.
The rate of the PChlide photoreduction reaction at room temperature depended on the concentration of enzyme (Fig. 1B) and substrate (Fig. 1C) present in the reaction mixture and light intensity (Fig. 1D). To determine the quantum requirements for PChlide reduction, we assayed the yield of Chlide formation after illumination of POR complexes with one or more light flashes of different intensities. As shown in Fig. 1 D and Inset, Chlide amounts increased linearly with the number of flashes given and with flash intensity, saturating at bright flashes. The linear dependence of Chlide formation on light intensity is consistent with a single quantum-reaction mechanism. A similar dependence on light intensity was observed for POR-catalyzed PChlide reduction by using etioplast membrane preparations (24).
To determine whether the MBP-PORb fusion is capable of reloading substrate after the initial Chlide-formation event, samples containing excess amounts of NADPH (4 mM) and PChlide (0.2 mM) were illuminated with a series of saturating 1-ms flashes separated by various time intervals (data not shown). Little to no Chlide formation was detected after the second flash, when the interval between the flashes was less than 1 ms. When the interval between flashes reached 10 sec, the next saturating flash gave the yield of Chlide nearly the same as the first one (Fig. 1D). Thus, under our experimental conditions the enzyme can be reloaded and not more than 10 sec is required to liberate Chlide and reload a new PChlide molecule.
To determine the amount of pigment required for a single photochemical reaction, we examined the dependence of the reaction yield on PChlide concentration (Fig. 1C). No Chlide formation was detected at very low PChlide concentration, even under saturating light flash. At PChlide concentrations between 0.1 and 3 nM, the yield of the reaction increased linearly with the pigment concentration. At high amounts of pigment, the reaction saturates. Because the amount of Chlide formed after a single, saturating flash in the presence of excess enzyme is about 5% of the total amount of PChlide in the system, this indicates that not less than about 5% of the pigment is in the state of photoactive complex. This corresponds to the amounts determined for MBP-PORa from Michaelis–Menten parameters (17).
The rather high protein concentrations in the reaction system do not exclude possibility of the formation of enzyme dimers. To identify which state of POR is photochemically active, we assayed the yield of the reaction as a function of POR concentration (Fig. 1B). No square dependence was detected, even at very low POR concentration, suggesting participation of monomeric enzyme in the reaction.
PChlide Photoreduction at Low Temperature: Separation of the Reaction Steps.
When frozen solutions of MBP-PORb ternary complexes were illuminated with continuous or flashed light of moderate intensity (33 mE⋅m−2⋅sec−1) at temperatures below −90°C, no changes in their spectra were detected. Above −90°C, however, evidence for a photochemical reaction was apparent by the appearance of a new fluorescence band at 682 nm (F682) (Fig. 2A). The efficiency of the formation of this band per 1-ms flash increases with temperature, reaching a maximum at −50°C. This suggests that activation energy is required for the initiation of the reaction. Formation of the F682 band is strictly light-dependent and does not occur in the dark at any temperature. No formation of the F682 band is observed in the absence of NADPH in the reaction mixture.
Figure 2.

Steps of PChlide photoreduction resolved by illumination at low temperatures. (A) Fluorescence spectrum of initial sample frozen in the dark to −196°C (INIT. SAMPLE). The same sample was heated in the dark to −50°C, illuminated with a 1-ms flash of white light (1×F), and then cooled to −196°C, where the fluorescence spectrum was recorded. In control experiments, samples were heated in the dark to −25°C, maintained at this temperature for 10 min, and then cooled to −196°C (DA10′). Note that the difference spectrum (LIGHT–DARK, +NADPH) shows that photoactive PChlide has emission maximum at 644 nm, and its photoreduction leads to the formation of an intermediate with fluorescence maximum at 682 nm. No such change was detected in the absence of NADPH (LIGHT–DARK, −NADPH). No changes in PChlide fluorescence around 655 nm were detected under any of the conditions tested. (B) Initial sample was heated in the dark to −50°C, illuminated at this temperature with a 1-ms flash of white light, and then either cooled to −196°C, where its fluorescence was recorded (1×F), or heated in the dark to −25°C, kept at this temperature for 4 min, and then cooled to −196°C (1×F+DA4′). Note that heating of the illuminated sample in the dark without thawing leads to the shift of the emission band of the intermediate product from 682 to 676 nm without any changes in the main PChlide fluorescence band at about 633 nm. All spectra were recorded at −196°C, with an excitation wavelength of 416 nm.
Once the F682 band is formed, elevation of the temperature of the sample in the dark to −25°C leads to a spontaneous shift in the maximum of this band to 675 nm (F675) (Fig. 2B). This F675 band is stable even after melting the sample and was spectroscopically identical to the spectral band that formed under illumination at room temperature. Illumination of the frozen samples at −25°C with a second or third light flash did not induce any further changes in the pigment fluorescence spectra.
Of several spectral bands of PChlide that can be detected in the reaction mixture at low temperature in the dark, only the F644 band disappears after illumination (Fig. 2A). This result is in line with the formation of this band only in the presence of POR (data not shown) and indicates that this is the PChlide form responsible for photochemical activity of POR in solution. Interestingly, the disappearance of F644 after illumination induces not only the formation of the intermediate F682 but also increases the intensity of monomeric PChlide at 628 nm (Fig. 2A). This indicates either that light induces decomposition of the photoactive complex or that the primary photoreaction is partially reversible.
Spectroscopic Identification of the Low-Temperature Intermediate.
It is known that absorption and fluorescence excitation spectra of PChlide have a specific split structure in the Soret region because of the asymmetry of Bx and By transitions (25). Reduction of ring D induces an increase in molecular symmetry and disappearance of the splitting in the spectra. Similar to PChlide, the excitation spectrum of the F682 band shows a split spectral structure (Fig. 3A). Thus, this fluorescence belongs to a pigment in which the C17—C18 carbon–carbon double bond in ring D is not yet reduced. On the other hand, the next step of the reaction, namely, the formation of the F675 band, is accompanied by the disappearance of the splitting and appearance of fluorescence excitation spectrum typical for Chlide (Fig. 3A).
Figure 3.

Identification of the intermediate and final product of PChlide photoreduction reaction. (A) Fluorescence excitation spectra (top lines) and second derivatives (bottom lines) of the initial PChlide (DA,F675 and DA,F685), the intermediate F685, formed after illu mination of the sample with white light (33 mE⋅m−2⋅s−1) for 10 min at −50°C (WL10′ at −50C, F685), and the Chlide F675, formed after illumination of the sample with a light flash given at −50°C and subsequent heating of the sample in the dark to −25°C (1×F at50C+DA-25C). All fluorescence spectra were recorded at −196°C. Note that the formation of the intermediate F682 does not lead to degeneration of Bx and By bands in the excitation spectrum whereas formation of the final product (F675) does. (B) HPLC identification of Chlide formed after a single light flash at −50°C and subsequent heating to −25°C (1×F at-50C+DA5′ at−25C) and after illumination with continuous white light at +22°C for 10 min (WL10′ at +22°C). The pigments were extracted from the frozen samples with acetone in complete darkness. The arrows indicate retention times of pure PChlide and Chlide. The pigments were detected in the column eluant fluorometrically under excitation at 435 and emission at 675 nm. (C) EPR signal that appears after illumination of the reaction mix with white light for 10 min at −50°C (WL10′ at−50C). No such signal was detected when sample was heated to −50°C and kept at this temperature for 10 min without illumination (DA10′ at −50C). For EPR spectral recording, the frozen samples were cooled to −170°C and assayed at a frequency of 9.463 GHz and microwave power = 10 mW.
HPLC analysis of the reaction products from samples of ternary complex flash illuminated at −50°C and warmed to −25°C showed that the product of the reaction chromatographs with the same mobility as Chlide, confirming that the reaction can proceed to completion under these conditions (Fig. 3C). Because formation of the F675 band occurs at temperatures at which the reaction solution is frozen and pigment movement, therefore, is unlikely, the F675 band must belong to a pigment bound to the enzyme.
In an attempt to identify the nature of the unstable fluorescence intermediate formed at low temperatures, an EPR analysis of the system was carried out. MBP-PORb ternary complexes were formed at room temperature in the dark, frozen at −196°C, and then warmed to −50°C and either flash-illuminated as described above or maintained in the dark. The samples then were refrozen (−170°C) and EPR spectra were taken. Control samples showed no detectable peak in their spectra, whereas samples flash-illuminated at −50°C yielded an EPR signal with a g factor of 2.002 and width of about 1 mT (Fig. 3B). Such a spectrum suggests the presence of free electrons. A similar EPR signal was detected in etiolated plants (26). Unfortunately, the intensity of the signal was consistently too low to permit precise determination of its spectral characteristics at this time. Therefore, origin of the paramagnetic species remains to be determined.
DISCUSSION
In the present study the mechanism of PChlide photoreduction that operates in light-adapted plants has been analyzed in vitro by using a heterologously expressed MBP-PORB fusion protein and low-temperature fluorescence spectroscopy. Our studies have led to two significant observations.
First, we have shown that PORb-catalyzed PChlide photoreduction in vitro occurs in two steps involving the formation of an unstable intermediate. By working at low temperatures, we were able to control the progress of the PORb-catalyzed reduction reaction and separate steps in the reduction process:
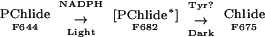 |
The first step leads to the production of an intermediate that is stable at temperatures below −50°C. Formation of this intermediate requires a single quantum of light energy. The second step is nonphotochemical and leads to the formation of Chlide. This step proceeds spontaneously at temperatures above −50°C, even in the dark. Because the overall reaction can take place in frozen solutions, gross enzyme conformational changes or pigment diffusion is unlikely to be significant.
The temperature dependence of the first step of the reaction indicates that it is not a simple photochemical act. This conclusion is confirmed by analysis of temperature dependence of PChlide fluorescence quantum yield showing specific quenching of the photoactive PChlide (F644) before formation of the intermediate (unpublished data). Most likely, the pigment singlet-excited state participates in the reaction and the reaction proceeds through an ionic mechanism. The low intensity of EPR signal would agree with this possibility. The activation energy of the second step of the reaction (about 0.20 eV) indicates that proton translocation might be the rate-limiting factor in this step.
Although the exact molecular nature of the unstable intermediate is not known at this time, one possibility is that it is a semireduced PChlide radical, formed by hydrogen transfer from NADPH (4, 17). Such a conclusion would be consistent with our fluorescence and EPR spectral data indicating that an intermediate is not formed in the absence of NADPH cofactor and with our previous data suggesting that NADPH directly participates in the first step of the PChlide reduction reaction in isolated, photoactive POR preparations from etiolated plants (27). Under this scenario, the second, light-independent step of the reaction would be the transfer of hydrogen from an active-site amino acid residue or solvent. Mutagenesis studies of the pea POR identified a highly conserved TYR residue within the active site essential for activity that could be the source of the reducing hydrogen (28).
We cannot rule out the possibility that the unstable intermediate represents a product of a photoisomerization event similar to those described in other photoactive proteins, such as the photoactive yellow protein, rhodopsin, and bacteriorhodopsin (29, 30). Under this scenario, photon absorption by the POR-bound PChlide results in the formation of a electronically excited state and an isomerization of the pigment. Pigment isomerization then would trigger changes in the bonding network with active-site residues that stabilized the pigment in the dark-state structure. The combination of an excited electronic state in the isomerized pigment and subtle changes in the active-site bonding easily could lead to a new energy minimum that would favor hydride transfer from bound NADPH cofactor and subsequent protonation from an active-site TYR or solvent molecule. Once transfer were initiated, reisomerization and return to the dark-state active-site environment would take place near or at the site of isomerization. The speed at which pigment isomerization and return to the ground state could occur would preclude a requirement for any substantive protein conformational changes. Thus, the initial stages could take place even under low temperatures as observed in the present study. If this were the case, it should be possible to identify active-site residues critical to forming the bonding patterns necessary to allow isomerization upon photon absorption that separate isomerization from subsequent photoreduction. In this regard, we recently have found that mutations within conserved TYR and LYS residues in the active site, known to block photoreduction of PChlide, do not prevent the formation of the unstable intermediate (unpublished data). Further studies clearly are necessary to identify the nature of the PChlide side groups and POR active-site residues that might be involved in this process.
In our study, we demonstrated that the reaction catalyzed by PORb in vitro is similar to the reaction described in light-adapted plants. Because PORb is the only form of POR expressed in light-adapted vascular plants, this must be the enzyme responsible for chlorophyll biosynthesis in mature greened tissues (14–16). Both the initial photoactive PChlide complex and the final product of the reaction catalyzed by PORb in vitro are spectroscopically similar to that detected in light-adapted plants (11, 13) and etiolated DET340 (14). No long-wavelength PChlide spectral forms and long-wavelength reaction intermediates typical for etiolated plants were detected in the course of PChlide photoreduction in vitro. A similar reaction was reported as a minor component in etiolated plants (6, 8, 9) This spectroscopic identity implies that the catalytic mechanisms must be the same in vivo as determined in vitro.
Previously published experiments performed with etiolated plants or etioplast membrane preparations have shown that photoactive PChlide has fluorescence emission maximum at 655 nm (4, 6, 7, 9). After illumination, this PChlide species subsequently converts into intermediates with fluorescence maxima at 684, 690, 695, and 682 nm. Neither photoactive PChlide F655 nor the final product, F682, or long-wavelength intermediates (F690 and F695) were detected in the course of PChlide photoreduction by MBP-PORb in vitro. Several processes can complicate the PChlide photoreduction in etiolated plants and etioplast membrane preparations: POR aggregation and prolamellar body formation (10), energy migration among PChlide molecules and between PChlide and newly formed Chlide (1, 32), and/or the presence of two different POR enzymes (14–16). The individual contribution of these factors remains to be determined.
Preliminary experiments indicate that monomeric MBP-PORa and MBP-PORb have similar steps and intermediates of PChlide photoreduction in vitro (unpublished data). Thus, the specific role of PORa in PChlide photoreduction in etiolated plants likely is not the result of a difference in the catalytic mechanism between the PORa and PORb proteins, but is due to specificity of pigment to enzyme binding and an association of the enzymes (31, 32).
In conclusion, the mechanism of PChlide photoreduction that operates in light-adapted plants can be modeled by monomeric PORb activity in the enzyme solutions. The reaction runs both at room and subzero temperatures. It consists of two steps, one photochemical and the other nonphotochemical, with the formation of an unstable reaction intermediate between them.
Acknowledgments
We thank Klaus Apel for providing the barley cDNA clones, Trevor Griffiths for the antiserum to wheat POR, Jeff Elena for his help with low-temperature EPR experiments, Wilson McIvor for technical assistance with HPLC, and Samuel Beale for comments about HPLC of PChlide. This work was supported by a grant from the U.S. Department of Energy (DEFGO5-94ER20144).
ABBREVIATIONS
- Chlide
chlorophyllide
- MBP
maltose-binding protein
- PChlide
protochlorophyllide
- POR
NADPH:protochlorophyllide oxidoreductase
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Lebedev N, Timko M P. Photosynth Res. 1998;58:5–23. doi: 10.1023/A:1020999325065. [DOI] [PubMed] [Google Scholar]
- 2.Begley T P. Acc Chem Res. 1994;27:394–401. [Google Scholar]
- 3.Apel K, Santel H J, Redlinger T E, Falk H. Eur J Biochem. 1980;111:251–258. doi: 10.1111/j.1432-1033.1980.tb06100.x. [DOI] [PubMed] [Google Scholar]
- 4.Griffiths W T. In: Chlorophylls. Scheer H, editor. Boca Raton, FL: CRC; 1991. pp. 433–449. [Google Scholar]
- 5.Litvin F F, Krasnovsky A A. Dokl Akad Nauk SSSR. 1957;117:106–109. [Google Scholar]
- 6.Litvin F F, Belyaeva O B. Photosynthetica. 1971;5:200–209. [Google Scholar]
- 7.Franck F, Mathis P. Photochem Photobiol. 1980;32:799–803. [Google Scholar]
- 8.Boddi B, Ryberg M, Sundqvist C. J Photochem Photobiol B. 1993;21:125–133. [Google Scholar]
- 9.Boddi B, Franck F. J Photochem Photobiol B. 1998;41:73–82. doi: 10.1016/s1011-1344(02)00389-5. [DOI] [PubMed] [Google Scholar]
- 10.Sundqvist C, Dahlin C. Physiol Plant. 1997;100:748–759. [Google Scholar]
- 11.Lebedev N N, Siffel P, Krasnovskii A A. Photosynthetica. 1985;19:183–187. [Google Scholar]
- 12.Lebedev N N. Photosynthetica. 1996;32:569–585. [Google Scholar]
- 13.Siffel P, Lebedev N N, Krasnovskii A A. Photosynthetica. 1987;21:23–28. [Google Scholar]
- 14.Lebedev N, VanCleve B, Armstrong G, Apel K. Plant Cell. 1995;7:2081–2090. doi: 10.1105/tpc.7.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holtorf H, Reinbothe S, Reinbothe C, Bereza B, Apel K. Proc Natl Acad Sci USA. 1995;92:3254–3258. doi: 10.1073/pnas.92.8.3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armstrong G A, Runge S, Frick G, Sperling U, Apel K. Plant Physiol. 1995;108:1505–1517. doi: 10.1104/pp.108.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin G E M, Timko M P, Wilks H M. Biochem J. 1997;325:139–145. doi: 10.1042/bj3250139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Townley H E, Griffiths W T, Nugent J P. FEBS Lett. 1998;422:19–22. doi: 10.1016/s0014-5793(97)01589-5. [DOI] [PubMed] [Google Scholar]
- 19.Dahlin C, Aronsson H, Wilks H M, Lebedev N, Sundqvist C, Timko M P. Plant Mol Biol. 1999;39:309–323. doi: 10.1023/a:1006135100760. [DOI] [PubMed] [Google Scholar]
- 20.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 21.Esen A. Anal Biochem. 1978;89:264–273. doi: 10.1016/0003-2697(78)90749-2. [DOI] [PubMed] [Google Scholar]
- 22.Oliver R P, Griffiths W T. Biochem J. 1980;191:277–280. doi: 10.1042/bj1910277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spano A J, He Z, Michel H, Hunt D F, Timko M P. Plant Mol Biol. 1992;18:967–997. doi: 10.1007/BF00019210. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths W T, McHugh T, Blankenship R E. FEBS Lett. 1996;398:235–238. doi: 10.1016/s0014-5793(96)01249-5. [DOI] [PubMed] [Google Scholar]
- 25.Hanson L K. In: Chlorophylls. Scheer H, editor. Boca Raton, FL: CRC; 1991. pp. 993–1014. [Google Scholar]
- 26.Belyaeva O B, Timofeev K N, Litvin F F. Photosynth Res. 1988;15:247–256. doi: 10.1007/BF00047356. [DOI] [PubMed] [Google Scholar]
- 27.Lebedev N N, Dujardin E. Z Naturforsch. 1993;48:402–405. [Google Scholar]
- 28.Wilks H M, Timko M P. Proc Natl Acad Sci USA. 1995;92:724–728. doi: 10.1073/pnas.92.3.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Genick U K, Borgstahl G E O, Ng K, Ren Z, Pradervand C, Burke P M, Srajer V, Teng T Y, Schildkamp W, Mcree D E, et al. Science. 1997;275:1471–1475. doi: 10.1126/science.275.5305.1471. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Schoenlein R W, Peteanu L A, Mathies R A, Shank C V. Science. 1994;266:422–424. doi: 10.1126/science.7939680. [DOI] [PubMed] [Google Scholar]
- 31.Sperling U, Frank F, van Cleve B, Frick G, Apel K, Armstrong G. Plant Cell. 1998;10:283–296. doi: 10.1105/tpc.10.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reinbothe C, Lebedev N, Reinbothe S. Nature (London) 1999;397:80–84. [Google Scholar]