

Abstract
The NR1I subfamily of nuclear hormone receptors includes the 1,25-(OH)2-vitamin D3 receptor (VDR; NR1I1), pregnane X receptor (PXR; NR1I2), and constitutive androstane receptor (CAR; NR1I3). PXR and VDR are found in diverse vertebrates from fish to mammals while CAR is restricted to mammals. Current evidence suggests that the CAR gene arose from a duplication of an ancestral PXR gene, and that PXR and VDR arose from duplication of an ancestral gene, represented now by a single gene in the invertebrate Ciona intestinalis. Aside from the high-affinity effects of 1,25-(OH)2-vitamin D3 on VDRs, the NR1I subfamily members are functionally united by the ability to bind potentially toxic endogenous compounds with low affinity and initiate changes in gene expression that lead to enhanced metabolism and elimination (e.g., induction of cytochrome P450 3A4 expression in humans). The detoxification role of VDR seems limited to sensing high concentrations of certain toxic bile salts, such as lithocholic acid, whereas PXR and CAR have the ability to recognize structurally diverse compounds. PXR and CAR show the highest degree of cross-species variation in the ligand-binding domain of the entire vertebrate nuclear hormone receptor superfamily, suggesting adaptation to species-specific ligands. This review examines the insights that phylogenetic and experimental studies provide into the function of VDR, PXR, and CAR, and how the functions of these receptors have expanded to evolutionary advantage in humans and other animals.
Keywords: Nuclear hormone receptor, xenobiotics, cholestasis, positive selection, metabolism, molecular evolution, bile salts, steroids
INTRODUCTION
Nuclear hormone receptors (NRs) form a superfamily of transcription factors found throughout the animal kingdom. Activity of most NRs is controlled by small molecule ligands ranging from endogenous compounds (e.g., steroid hormones, thyroid hormone, bile salts) to xenobiotics. Genome sequencing projects have revealed 48 NRs in humans [1, 2], 49 in mice [1], and 47 in rats [1]. Telost fish have a somewhat larger complement of NR genes due to gene duplication [3–5], illustrated by the 68 NR genes in the pufferfish Fugu rubripes [6]. Currently, NRs are classified into 6 families with the most intensively studied NRs falling into the NR1, NR2, and NR3 families [2, 7, 8].
NRs share a modular domain structure, which includes, from N-terminus to C-terminus, a modulatory A/B domain, the DNA-binding domain (DBD; C domain), the hinge D domain, the ligand-binding domain (LBD; E domain) and a variable F domain. Sequence-specific binding to ‘responsive elements’ in target genes is mediated by the DBD. The LBD mediates ligand activation, ligand-independent repression, and dimerization. Most of the vertebrate NR1 subfamily members heterodimerize with retinoid X receptors (RXRα, β, γ; NR2B1, 2B2, and 2B3) [9, 10].
This review focuses on the NR1I subfamily, which includes the 1,25-(OH)2-vitamin D3 receptor (VDR; NR1I1), pregnane X receptor (PXR; NR1I2), and the constitutive androstane receptor (CAR; NR1I3). This article reviews the insights that experimental and evolutionary analyses provide into the functions and interrelationships of these three receptors. For PXR and CAR, a central theme of comparative evolutionary analysis is to explain why these receptors show such extensive sequence divergence across animal species but very little sequence variation between humans. Emerging evidence suggests that PXR has adapted to cross-species differences in endogenous ligands, an unusual finding in the NR superfamily. Little is known about the functions of PXR and VDR outside mammals, hinting that there is still much to be learned about the evolution and function of the NR1I subfamily in vertebrates.
Discovery of NR1I subfamily members
VDR was the first NR1I subfamily member to be cloned, with reports of the cloning of chicken, human, and rat VDR cDNAs appearing in 1987 and 1988 [11–14]. Functional expression of this gene yields a product which binds 1,25-(OH)2-vitamin D3 (calcitriol) with high affinity and mediates classic calcitriol effects such as regulation of calcium and phosphate homeostasis. Subsequently, VDRs have been shown to influence a variety of physiological functions, affecting nearly every organ and tissue [15–19]. VDR genes have been partially or fully sequenced in 16 vertebrates including fish, amphibians, reptiles, birds, and mammals (Table 1); recently, a VDR was cloned and functionally expressed from the sea lamprey (Petromyzon marinus), a jawless vertebrate that lacks calcified tissue [20]. Although little pharmacological characterization of non-mammalian VDRs has been reported, human, mouse, and sea lamprey VDRs have high affinities (nanomolar to subnanomolar range) for binding calcitriol [14, 20–22].
Table 1.
Sequences of NR1I Subfamily Members (all accession numbers from Genbank unless otherwise specified)
| Receptor | Species | Accession number(s) | Reference(s) |
|---|---|---|---|
|
| |||
| VDR | Human | NM_000376 | [14] |
| VDR | Chimpanzee | ENSPTRT00000009010 (Ensembl, partial) | Unpublished |
| VDR | Cotton-top tamarin monkey (Saguinus oedipus) | AF354232 | [93] |
| VDR | Mouse | NM_009504 | [200] |
| VDR | Rat | NM_017058 | [12, 13] |
| VDR | Chicken | AF011356 | [201] |
| VDR | Japanese quail | U12641 | [63] |
| VDR | African clawed frog (Xenopus laevis) | U91846 | [64] |
| VDR | Nile crocodile | AJ011391 (partial) | [202] |
| VDR | Snake (Elaphe sp.) | AJ286866 (partial) | Unpublished |
| VDR | Red-eared slider turtle (Trachemys scripta elegans) | AJ286870 (partial) | Unpublished |
| VDR | Giant toad (Bufo marinus) | AY268062 (partial) | [20] |
| VDR | Tokay (Gekko gecko) | AY254096 (partial) | [20] |
| VDRα | Japanese flounder (Paralichthys olivaceus) | AB037674 (expressed in all tissues) | [184] |
| VDRβ | Japanese flounder | AB037673 (not expressed in liver or muscle) | [184] |
| VDRα | Pufferfish (Fugu rubripes) | SINFRUT00000174563 (Ensembl) | [6] |
| VDRβ | Pufferfish | SINFRUT00000165525 (Ensembl) | [6] |
| VDR | Zebrafish (Danio rerio) | NM_130919 | [203] |
| VDR | Sea lamprey (Petromyzon marinus) | AY249863 | [20] |
| PXR | Human | AF061056, NM_003889 | [25, 127] |
| PXR | Rhesus monkey | AF454671 | [27] |
| PXR | Dog | AF454670 (partial) | [27] |
| PXR | Pig | AF454672 (partial) | [27] |
| PXR | Rabbit | AF188476, AF182217 | [27, 34] |
| PXR | Mouse | AF031814 | [24] |
| PXR | Rat | AF151377 | [39] |
| PXR (=CXR) | Chicken | AF276753 | [36] |
| PXR (=BXRα) | African clawed frog (Xenopus laevis) | BC041187 | [35, 87] |
| PXR (=BXRβ) | African clawed frog | AF305201 | [35, 87, 88] |
| PXR | Zebrafish | AF454673, AF502918 | [27] |
| PXR | Pufferfish (Fugu rubripes) | SINFRUT00000171584 (Ensembl) | [6] |
| CAR | Human | NM_005122 | [40] |
| CAR | Chimpanzee | ENSPTRT00000002884 (Ensembl) | Unpublished |
| CAR | Rhesus monkey | AY116212 | Unpublished |
| CAR | Dog | ENSCAFT00000020528 (Ensembl) | Unpublished |
| CAR | Mouse | NM_009803 | [41] |
| CAR | Rat | NM_022941 | [204] |
| CAR | Baikal seal (Phoca sibirica) | AB109553 | [205] |
| CAR | Northern fur seal (Callorhinus ursinus) | AB109554 | [205] |
Human and mouse PXRs were cloned and functionally expressed in 1998. In both species, PXR is highly expressed in liver and intestine. [23–25]. Exhaustive attempts have failed to find high-affinity (subnanomolar) PXR ligands; however, human PXR binds an impressive array of molecules with low (typically micromolar) affinity [23–27]. The highest affinity ligands for human PXR (e.g., hyperforin, the active component of the herbal antidepressant St. John’s wort) only have binding affinities in the tens of nanomolar range [28, 29]. The name pregnane X receptor derives from the activation of PXRs by pregnane (21-carbon or C21) steroids such as 5β-pregnan-3,20-dione [24, 25], although some estrane (C18) and androstane (C19) steroids also activate PXRs; the highest EC50 values of steroids for activating human PXR in reporter gene assays are low micromolar or barely submicromolar [23–25, 27, 30–34]. PXR has been cloned and functionally expressed from zebrafish, frog, chicken, and multiple mammalian species (Table 1) [23–27, 34–39].
CAR was first cloned in 1994 [40] with detailed functional analysis reported several years later [26, 41–44]. CAR differs from PXR and VDR in having high constitutive activity in the absence of ligand [42, 45–47]. CAR genes have so far been detected only in mammals (Table 1). The high constitutive activity of CAR means that some compounds act as ‘inverse agonists’ and decrease the level of constitutive activation while other compounds serve as agonists and further increase activation [41, 42, 48–50]. For the mouse CAR, for example, androstane steroids such as 5α-androstan-3α-ol (androstanol) or 5α-androst-16-en-3α-ol (androstenol) are efficacious inverse agonists [42].
Table 1 shows accession numbers for VDR, PXR, and CAR genes in all available vertebrate species. Based on current genetic data, multiple NR1I subfamily members have been found only in vertebrates. PXR has not yet been identified in jawless vertebrates. A single NR1I-like gene equally similar to VDR/PXR/CAR and a Drosophila NR is found in the draft genome of the urochordate Ciona intestinalis, an invertebrate reasonably closely related to vertebrates [51]. The functional properties of this invertebrate NR remain uncharacterized.
Overlapping transcriptional targets of VDR, PXR, and CAR
Other than the high-affinity endocrine effects mediated by calcitriol at VDRs, the NR1I subfamily members show considerable overlap in their transcriptional targets (Table 2), a topic that has been reviewed in detail elsewhere [52–58]. All three receptors are capable of inducing the expression of broad-specificity hepatic and intestinal phase I enzymes (e.g., CYP2C9 and 3A4) that play major roles in metabolizing xenobiotics and endogenous compounds. PXR and CAR also upregulate the expression of phase II conjugating enzymes as well as ‘phase III’ pumps such as P-glycoprotein (Table 2). In humans, xenobiotics such as rifampin or phenobarbital cause clinically important drug-drug or drug-hormone interactions by virtue of PXR or CAR activation. For example, the efficacious PXR activators rifampin and St. Johns wort increase clearance of the anti-rejection drug cyclosporine by inducing CYP3A4 and MDR1 expression, possibly leading to organ rejection in an allograft recipient [59]. Similarly, CAR and PXR activators increase metabolism of the estrogen component of combined oral contraceptives, potentially leading to unintended pregnancy [60–62].
Table 2.
Transcriptional Targets of NR1I Subfamily Members
| Human gene | NR1I member(s) upregulating expression in humans and/or rodents | Function | References |
|---|---|---|---|
| CYP2B6 | VDR, PXR, CAR | Metabolizing enzyme – endogenous function not well understood | [44, 206–208] |
| CYP2C9 | VDR, PXR, CAR | Metabolizing enzyme – endogenous function not well understood | [206, 209] |
| CYP3A4 | VDR, PXR, CAR | Broad specificity enzyme (steroid hormones, bile salts, xenobiotics) – major adult liver xenobiotic- metabolizing enzyme | [23, 31, 65, 128, 206, 208, 210–212] |
| CYP3A7 | PXR, CAR | Broad specificity enzyme (steroids, bile salts, xenobiotics) – major fetal liver xenobiotic- metabolizing enzyme | [213, 214] |
| UGT1A1 | PXR, CAR | Conjugating enzyme (bile acids, thyroxine, xenobiotics) | [188, 189, 208, 215, 216] |
| MDR1 (P-glycoprotein, ABCB1) | PXR, CAR | Broad specificity pump | [208, 212, 217, 218] |
| MRP2 (ABCC2) | PXR, CAR | Broad specificity pump | [208, 219] |
| CYP24 (25- Hydroxyvitamin 24-D3- hydroxylase) | PXR, VDR | Inactivation of 25- hydroxyvitamin D3 | [220, 221] |
| 5-aminolevulinic acid synthase-1 | PXR, CAR | Rate-limiting enzyme in heme synthesis | [222, 223] |
| Cytosolic sulfotransferase | CAR | Bile salt detoxification | [126] |
LIGAND SELECTIVITY OF NR1I SUBFAMILY MEMBERS
VDRs have relatively narrow ligand selectivity. VDRs bind calcitriol and related synthetic analogs with high affinity, and can distinguish between calcitriol and other vitamin D3 metabolites or precursors, [13, 14, 20, 22, 63, 64] and in general do not respond even to high concentrations of adrenocortical steroids, sex steroids, or cholesterol [13, 14]. Mouse and human VDRs are, however, activated by micromolar concentrations of lithocholic acid (3α-hydroxy-5β-cholan-24-oic acid; LCA) and its metabolites and synthetic derivatives [65], a function not shared by sea lamprey VDR [66]. As described below, activation by LCA is a shared property of mammalian PXRs and VDRs.
PXR has the broadest ligand specificity of the NR1I subfamily members, consistent with its flexible and large ligand-binding cavity [67–72], second only in size to peroxisome proliferator-activated receptor γ (NR1C3) (although NR1C3 has not yet been shown in binding studies to actually bind ligands as large as can bind to the human PXR) [73]. PXRs, particularly in mammals, are remarkably promiscuous with respect to ligand specificity [74, 75]. Human, rabbit, pig, and dog PXRs have especially broad specificity for activating compounds [27]. The ability of PXR to be activated by structurally diverse ligands parallels the broad substrate specificity of two important transcriptional targets of PXR: the cytochrome P450 (CYP) 3A subfamily (e.g., CYP3A4 and 3A7 in humans) [76–80] and the P-glycoprotein multidrug resistance pump (product of the ABCB1 gene) [81–84].
A variety of ligands are capable of activating human PXR, including prescription drugs (rifampin, nifedipine, indinavir), herbal drugs (St. Johns wort), steroids, environmental contaminants, endocrine disruptors, and bile salts [23–29, 34, 36, 37, 85, 86]. The structural basis of the ligand promiscuity of human PXR has been studied in several high-resolution crystal structures of human PXR, including separate structures of human PXR bound to three different ligands - SR12813 (experimental cholesterol-lowering drug) [67, 70], hyperforin (component of St. Johns wort) [68], and rifampin [72]. The human PXR LBD shares a number of structural features with other NRs, including the ligand-binding cavity in one hemisphere and an ‘α-helical sandwich’ of helices α1/α3, α4/α5/α8, and α7/α10 in the other hemisphere [67–70, 72]. However, the ligand-binding cavity of human PXR is large, smooth, and hydrophobic, which contrasts with endocrine NRs (including the VDR) that have compact ligand-binding cavities that closely resemble the shape of their specific ligands [69]. The human PXR ligand-binding cavity also shows considerable flexibility, expanding by 250 Ǻ3 to accommodate the ligand hyperforin [68]. There are a number of features of the human PXR LBD that are not found in other NRs and which contribute to its broad ligand specificity: a variable four-residue turn between helices α1 and α3, replacement of α6 by a large, flexible loop, and two additional β strands not observed in other NRs [67–70, 72].
There are considerable differences between species in terms of PXR activators. Human, dog, pig, rabbit, and chicken PXRs have very broad ligand specificity, accommodating large ligands such as rifampin while mouse PXR has a narrower ligand specificity [26, 27, 36]. Pregnenolone 16α-carbonitrile activates mouse but not human PXR, whereas hyperforin and rifampin activate human but not mouse PXR [23, 24, 26, 27, 31]. The PXRs from the African clawed frog Xenopus laevis deserve special mention in that these receptors completely lack the broad ligand specificity of other PXRs and have a tissue expression pattern different from other PXRs, being found not in drug-metabolizing organs like liver or intestine but in gonadal tissue and brain [27, 35, 87–89]. The frog PXRs are activated well only by benzoates, endogenous compounds with unique roles in frog development (hence the alternative term of benzoate X receptors, or BXRs, for the frog PXRs), and not by steroids or bile salts [27, 35, 66, 87]. The zebrafish PXR shares a number of steroid activators with mammalian PXRs but is only activated by a handful of xenobiotics [27, 66].
CAR also has the capacity to bind a range of ligands although not to the extent of PXR [26, 27]. Recently, three groups independently reported high-resolution crystallographic structures of human or mouse CARs bound to various ligands [45–47]. These structures provide insight into the high level of constitutive activity of CAR and to the ability of certain compounds, such as androstanol or androstenol in the mouse CAR [45], to act as inverse agonists.
Fig. (1) shows examples of endogenous compounds that activate VDR, PXR, and/or CAR and indicates ligands active at more than receptor. As can be seen, calcitriol is selective for VDRs, while mammalian VDRs and mammalian/chicken PXRs are activated by the secondary bile acid LCA [27, 65, 66, 86]. Androstane and pregnane steroids both affect PXRs and CARs at low micromolar concentrations with certain androstane steroids being efficacious inverse agonists of mouse CAR [23–25, 27, 31, 42]. The endogenous benzoate isolated from Xenopus laevis, 3-aminoethylbenzoate, has so far been found to be selective for BXRα, and inactive or weakly active at other PXRs, CARs, and VDRs [27, 35, 66, 87].
Fig. (1). Endogenous Ligands of VDR, PXR, and CAR.

Chemical structures of endogenous ligands for VDR, PXR, and CAR are shown, indicating compounds that overlap between receptors. 1,25-(OH)2-Vitamin D3 (calcitriol) is a selective high-affinity agonist for all VDRs and shows no activity at CARs or PXRs. Lithocholic acid, a potentially toxic evolutionarily ‘recent’ bile acid, activates mammalian VDRs and mammalian/chicken PXRs. VDRs and PXRs appear to have recently evolved to respond to LCA. Cyprinol sulfate, a 5α- C27 bile alcohol sulfate representative of the earliest bile salts to evolve in vertebrates, activates all PXRs except for the frog BXRs. 3-Aminoethylbenzoate, a compound isolated from Xenopus laevis and shown to have a role in Xenopus development, is fairly selective for Xenopus BXRα, especially in terms of the efficacy of its effect. Pregnane and androstane ligands overlap between CARs and PXRs with some steroids such as 5α-androstan-3α-ol showing strong inverse agonist actions at mouse CAR.
SEQUENCE VARIATION OF VDR, PXR, AND CAR BETWEEN ANIMAL SPECIES AND WITHIN HUMANS
Unusual sequence diversity of PXR and CAR genes across species
Most of the genes in the vertebrate NR superfamily are strongly conserved between vertebrate species. For example, amino acid sequence identities between human and mouse orthologous NR genes are typically greater than 95% in the DBD and greater than 85% in the LBD [1]. Not surprisingly, a study comparing human, mouse, and rat and another study comparing human, mouse, and chimpanzee revealed that genes in the NR superfamily are tightly conserved across vertebrate species and hence have likely been subjected to negative evolutionary selection [1, 90]. The only two clear exceptions are the LBDs of PXR and CAR [1, 66, 91].
Similar to the majority of the NR superfamily, VDR genes are highly conserved across species (Fig. (2)). Even the VDR from the sea lamprey, a jawless fish whose most recent common ancestor with mammals was over 450 million years ago [92], has 59% amino acid sequence identity to the human VDR [20]. The sequence conservation of VDRs is also demonstrated by the fact that only 4 of 22 ligand-contacting residues in VDRs show any amino acid sequence variation across all vertebrate species sequenced (Fig. (3A)). Only 1 of 22 of the ligand-contacting residues varies within mammals (Fig. (3A)).
Fig. (2). Sequence Alignment of the LBD of PXR, VDR, and CAR Genes Indicating Ligand-binding Residues.

The locations of the α-helices above the amino acid sequence are based on the structures determined from x-ray crystallography of human VDR [132] and human PXR [70]. Amino acid residues highlighted in bold type are residues show to directly interact with ligands in high-resolution, x-ray crystallographic structures of human VDR [132–134, 197], rat VDR [198], mouse CAR [45, 46], human CAR [47], and human PXR [67, 68, 70, 72].
Fig. (3). Contrasting Degrees of Sequence Conservation of Ligand-binding Residues in VDR, PXR, and CAR.
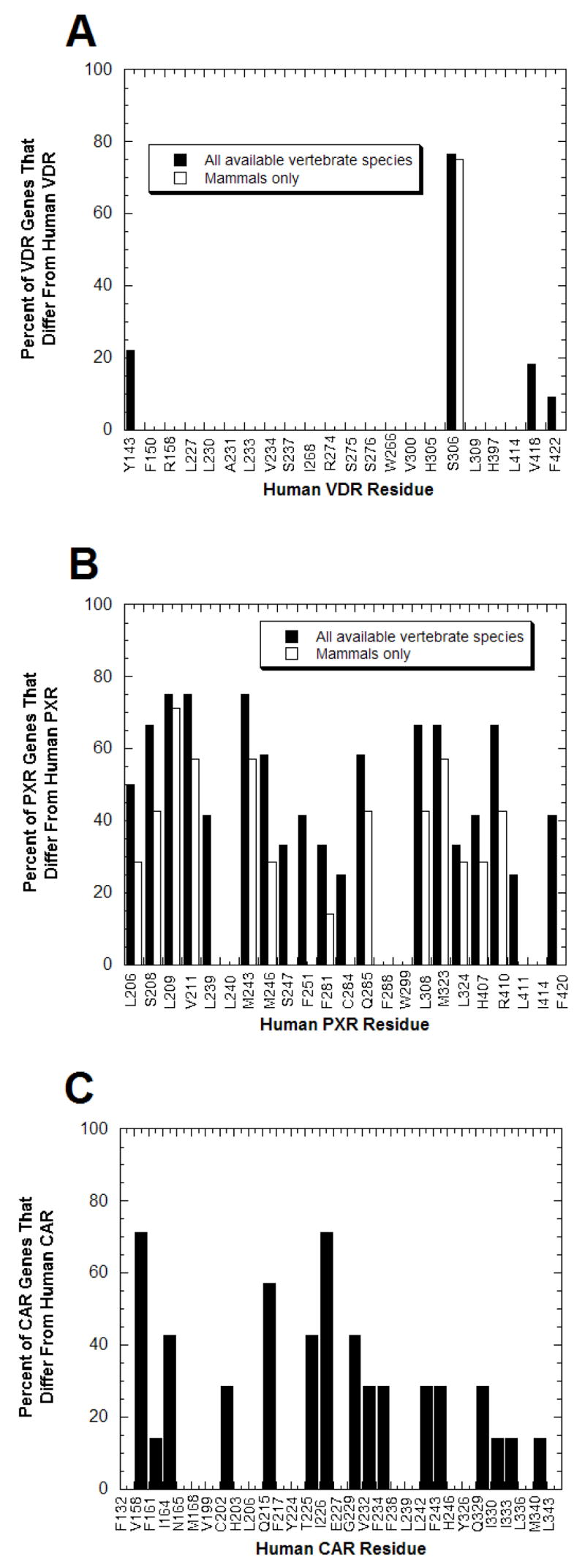
From the x-ray crystallographic structures of VDRs, PXRs, and CARs described in the legend to Fig. (2), the amino acid residues that directly interact with ligands are known. (A) Of the 22 amino acid residues shown to interact with ligands at human and rat VDRs, only 4 residues show any sequence variation across vertebrate species; the remaining 18 residues are completely conserved across all species. Due to partial sequence, data was missing for the four most C-terminal ligand-binding residues (corresponding to human VDR residues His397, Leu414, Val418, and F422) for crocodile, frog, fugu-β, lizard, snake, and turtle VDRs; data for the chimpanzee VDR was only available for the first two ligand-binding residues (corresponding to human VDR residues Tyr142 and Phe150). (B) PXRs contrast with VDRs in showing extensive amino acid sequence divergence at the positions corresponding to human PXR ligand-binding residues. Only 3 of 23 positions are conserved throughout the 13 vertebrate PXRs while for 9 of 23 residues, over half of the PXRs differ from the human PXR residue. (C) CARs also show significant sequence divergence at ligand-binding residues but not to the same magnitude as PXRs. The data is based on eight mammalian CAR sequences.
In contrast, a striking feature of PXR, and to a lesser degree CAR, is high cross-species sequence divergence in the LBD. The LBD of PXR shares amino acid identities of only 75% between human and rodent sequences and only 50% between human, teleost fish, and chicken sequences [1, 27]. The sequence identities of PXR and CAR across species are unusually low compared to the rest of vertebrates NRs, which tend to have comparable sequences identities between species 10–15% higher [1, 27]. PXR and CAR even show considerable sequence variation at amino acid positions corresponding to residues that interact directly with ligands in X-ray crystallographic structures of human PXR [67, 68, 70, 93], human CAR [47], and mouse CAR [45, 46] bound to various ligands (Fig. (3B,C)). There is even considerable variation of ligand-binding residues of PXR between mammalian species (Fig. (3B); compared closed versus open bars).
The cross-species variation in the LBDs of PXR and CAR is even more striking when DNA sequences are compared, in particular by analyzing the rate of nonsynonymous (changes amino acid sequence of a codon) and synonymous (does not change amino acid sequence) nucleotide substitution rates. The ratio of the rate of non-synonymous versus the rate of synonymous nucleotide variation (i.e., how many non-synonymous or synonymous changes have occurred in comparison to the total number of non-synonymous or synonymous changes possible; dN/dS or ω ratio) provides some indication into evolutionary selective forces acting on a given gene [94]. Synonymous substitutions are considered to be neutral, an assumption which is usually true, although there are exceptions [95]. For most gene comparisons, ω is less than one, often less than 0.1, reflective of negative or purifying selection to maintain a conserved amino acid sequence [96]. ω = 1 reflects neutral selection (a ratio that would be expected for a non-functional pseudogene) while ω > 1 suggests positive selection (non-synonymous substitutions that change amino acid sequence are actually favored over synonymous, ‘neutral’ substitutions). Large-scale comparisons of genes between species show that very few genes, or even gene domains, have ω values equal to or exceeding one [96]. In two-species comparisons within mammals (e.g., human vs. mouse or human vs. rat), PXR and CAR have ω ratios at least several-fold higher than the average for all other NR genes [1, 90].
More sophisticated multi-species phylogenetic analysis utilizing maximum likelihood methods [94, 97, 98] reveals that sub-populations of codons within PXR genes have ω ratios exceeding one [66, 91, 99]. Overall, the LBDs of PXR and CAR have sub-populations of codons with the highest ω ratios of any genes in the vertebrate NR superfamily [66, 91]. These results strongly suggest that natural selection has favored sequence diversity in the LBD of PXR and CAR, possibly to adapt to cross-species differences in important ligands. PXR and CAR may thus represent unusual examples of NR genes that have changed their ligand specificities across vertebrate species to adapt to cross-species differences in exogenous and/or endogenous toxic compounds [24, 27, 66, 100, 101].
The PXRs showing the strongest evidence for positive selection are actually the Xenopus laevis BXRs. Relative to other PXRs such as that from zebrafish and presumably to the ancestral PXR, the BXRs have lost broad specificity for ligands, gained high efficacy activation by endogenous benzoates, and altered tissue expression pattern to play a developmental role in the frog [35, 87, 89]. Phylogenetic analysis by maximum likelihood shows that 23 codons in BXRα and/or BXRβ show strong evidence for positive selection [66, 91]; all are in the LBD and 9 of 23 are orthologous to or adjacent to residues in human PXR shown to directly interact with ligand by x-ray crystallography [67, 68, 70, 72]. No other PXRs show such striking features consistent with positive selection although overall phylogenetic analyses of PXR genes across species strongly suggest that the LBD of PXRs has changed across species to evolutionary advantage. The same may be true for CAR genes but the current phylogenetic data is not as strong as that for PXR.
Tight conservation of VDR, PXR, and CAR within humans
The marked diversity of the PXR and CAR LBDs across vertebrates contrasts with detailed DNA resequencing studies of the human PXR and CAR genes showing non-synonymous mutations in the LBDs of these genes are rare, although variation in non-coding regions or due to splice variants may have clinical importance [102]. Resequencing of 100 individuals from several different ethnic groups for the PXR gene [103] and 70 individuals from three different ethnic groups for the CAR gene [99] showed very low nucleotide diversity (lower than the genome-wide averages for human genes) and no non-synonymous substitutions in the LBD of either gene. Sequencing of 253 Japanese subjects revealed only a single non-synonymous substitution in the CAR LBD [104]. The amino acid residue identified (valine-133) is located next to a ligand-binding residue of human CAR [47] and mouse CAR [46]. Sequencing of 205 Japanese subjects found non-synonymous PXR substitutions as single alleles in separate individuals [105]. These PXR mutations caused modest reductions in a transactivation assay using a mammalian cell line [106]. Two separate substitutions in the PXR LBD were discovered in a re-sequencing study of 74 Africans and 418 Caucasians [33]. The VDR gene also has a very low rate of non-synonymous nucleotide variation in the LBD although some polymorphisms elsewhere in the gene have functional importance [107]. Variation in the coding regions of VDR, PXR, or CAR do not account for well-described inter-individual differences in metabolism, such as variation in baseline activity of CYP3A4 in liver or intestine [102].
Overall, NR1I subfamily members show little variation in the LBD between human individuals. This particularly contrasts with the marked cross-species sequence variation of PXR and CAR described above. In addition, the nucleotide divergence between the human and chimpanzee PXR and CAR genes are lower than the average for other genes in the human genome [90, 99, 108]. This suggests that important ligands for PXR and CAR, at least in terms of influencing reproductive fitness, do not vary between humans, and perhaps not even between humans and other primates, but do vary between different animals. The next sections will look at variation of ligand specificity across species for VDR, CAR, and PXR and its possible evolutionary and physiologic significance.
ACTIVATION OF VDR, PXR, AND CAR BY ENDOGENOUS AND EXOGENOUS COMPOUNDS AND EVOLUTIONARY SIGNIFICANCE
Ligand specificity and the evolution of PXR and CAR
The broad ligand specificity of PXR and, to a lesser degree CAR, has suggested that a major function of these two receptors is to protect animals against toxic levels of exogenous and endogenous compounds [24, 109–114]. From an evolutionary standpoint, these toxic compounds may be exogenous (likely of dietary origin) or endogenous (hormones, bile salts). An important goal of comparative evolutionary studies of PXR and CAR is to explain why these receptors, as described above, show very little sequence variation within humans, and even between humans and other primates, but such striking cross-species sequence variation in the LBD and even at ligand-binding residues across mammalian and non-mammalian species. This suggests that key ligands for PXR and CAR vary across species either due to differences in diet or physiology. In contrast, for VDRs, the paucity of cross-species sequence variation in the LBD is consistent with little or no differences in key VDR ligands between animal species.
Bile salts
Bile salts such as cholic acid (3α,7α,12α-trihydoxy-5β-cholan-24-oic acid) are end-products of cholesterol metabolism and also function as surfactants for lipophilic compounds in the gut [115, 116]. Bile salts are synthesized in the liver and stored in the gall bladder (although some animals lack this organ). In general, most bile salts are not particularly toxic, and these compounds can circulate at micromolar concentrations in the plasma without much deleterious effects [116]. An exception is LCA, a mono-hydroxylated ‘secondary’ bile acid formed by the action of bacterial 7-dehydroxlases on primary bile acids such as chenodeoxycholic acid (3α,7α-dihydroxy-5β-cholan-24-oic acid; CDCA). LCA is poorly water-soluble and not reabsorbed well by the gut. At higher concentrations, LCA forms DNA adducts and inhibits DNA repair enzymes [117–119]. High colonic levels of LCA are implicated as a factor in colorectal cancer [120], and LCA promotes colon cancer in animal models [121].
Elegant mouse models have illustrated the importance of PXR- and CAR-mediated pathways in LCA toxicity. Both PXR and CAR knockout mice are more susceptible to exogenously administered LCA [86, 101, 122–125], with PXR/CAR ‘double-knockout’ mice being especially sensitive [122, 123]. Conversely, administration of PXR and CAR activators, or mice genetically engineered for higher expression of CAR or PXR, reduce the toxic sequelae of exogenous LCA exposure [101, 122, 123, 126]. LCA is a low potency PXR activator, with maximal effects occurring only at concentrations above 100 μM [27, 86, 100, 101, 127]. LCA does not directly affect CAR activity [27], although, as mentioned above, activating CAR with other ligands protects against exogenous LCA toxicity. In studies with PXR knockout mice exposed to high doses of LCA, it was noted that increased expression of CYP3A occurred even in mice totally deficient in PXR expression [101]. Later it was found that LCA and some of its metabolites also activate the VDR [65]. This finding provided physiologic importance to previous demonstrations that VDR activation upregulated CYP3A expression, particularly in the gut [128, 129].
LCA activation of VDR has been demonstrated so far only in human and mouse VDRs, and not in the most phylogenetically basal VDR from the sea lamprey [65, 66]. Human and mouse VDRs are only activated by LCA and its metabolites (e.g., 3-ketolithocholic acid) or synthetic derivatives (e.g., lithocholic acid acetate) and not by the majority of primary bile acids such as cholic acid, chenodeoxycholic acid, or the muricholic acids (found in rodents) [65, 66, 130, 131]. The narrow specificity of human and mouse VDRs for LCA and its metabolites contrasts with the broader specificity of human and other mammalian PXRs for primary and secondary bile acids [27, 85, 86, 100, 101, 127].
LCA is a by-product of evolutionarily more recent bile salt synthetic pathways typical of birds and mammals. The greater toxicity of LCA compared to other bile salts may have been the evolutionary driving force for mammalian (and perhaps avian) PXRs and VDRs to alter their ligand specificity to be activated by this compound [116]. The sensitivity of human and mouse VDRs to LCA (and its derivatives) is somewhat remarkable given the insensitivity of VDRs to steroid hormones and essentially all other bile salts. Molecular modeling and site-directed mutagenesis studies of human VDR, based on x-ray crystallographic data of human VDR bound to calcitriol and other ligands [132–134], show that LCA and calcitriol occupy overlapping binding sites on human VDR [130, 131, 135]. This suggests that slight alteration of the calcitriol binding pocket in VDR occurred during evolution to accommodate low-affinity binding of LCA, a feature presumably absent in ancestral animals whose bile salt pathways could not lead to LCA formation. In a mammalian cell culture system studying VDR function, LCA and calcitriol showed differences with regard to heterodimer association and comodulator activation; these results indicate perhaps that VDR is a bifunctional regulator, with high-affinity ‘endocrine’ effects mediated efficiently by calcitriol and low-affinity, detoxification effects (e.g., upregulation of CYP3A4 in the colon) mediated by LCA [135].
Although a majority of animal models exploring bile acid detoxification by VDR, PXR, and CAR have focused on LCA, other bile acids can play important roles in cholestasis. In severe cholestatic liver injury, serum bile acid concentrations reach high micromolar concentrations known to activate PXR [136]. This was demonstrated clearly in a mouse bile duct ligation model, in which hepatic CYP3A11 (ortholog of human CYP3A4) expression was upregulated even though serum levels of LCA were not increased; however, serum levels of 6β-hydroxylated bile acids were markedly increased [137]. Given that 6β-hydroxylated bile acids such as β-muricholic (3α,6β,7β-trihydroxy-5β-cholan-24-oic acid) and murideoxycholic acid (3α,6β-dihydroxy-5β-cholan-24-oic acid; also known as murocholic acid) are efficacious activators of mouse PXR but not mouse VDR [66], this suggests that hepatic CYP3A11 upregulation in the bile duct ligation model is PXR-mediated. PXR activation by high circulating levels of bile acids could be a protective response to cholestasis of various etiologies. Even if high circulating levels of some bile salts does not directly result in toxicity, the presence of elevated bile acids signifies likely impairment of hepatobiliary excretion of xenobiotics and other endogenous compounds. PXR activation would thus be an attempted adaptive response to increase metabolism and elimination of toxic compounds by alternative pathways.
The importance of PXR in bile salt metabolism and elimination is also illustrated by the rare disease cerebrotendinous xanthomatosis (CTX), an inborn error of metabolism caused by deficiency of CYP27A1. In humans, the enzyme deficiency of CTX leads to accumulation of C27 bile alcohols (which retain the entire carbon skeleton of cholesterol) and the pathological symptoms of xanthomas, gallstones, and neurological dysfunction [138]. Interestingly, knockout of the Cyp27a gene in mice did not phenocopy human CTX due to a dramatic increase of CYP3A activity seen in Cyp27a−/− mice but not observed in human CTX patients [139, 140]. The upregulation of CYP3A expression allowed the Cyp27a−/− mice to bypass the enzyme deficiency and detoxify the bile acid precursor 5β-cholestan-3α,7α,12α-triol. Two research groups later showed that 5β-cholestan-3α,7α,12α-triol is an efficacious activator of mouse, but not human, PXR [85, 127]. Because 5β-cholestan-3α,7α,12α-triol does not activate human PXR well (it is probably a very weak partial agonist at human PXR), individuals with CTX are unable to prevent pathological accumulation of 5β-cholestan-3α,7α,12α-triol and other bile acid precursors.
In contrast to the data in mammals described above, the first major study to compare multiple non-mammalian and mammalian PXRs found that the zebrafish PXR was not activated by a variety of bile acids and synthetic derivatives [27]. However, biliary bile salts vary significantly across vertebrate species, and the bile acids found in human, mice, and most other mammals are not present in all vertebrates [141–143]. Whereas most mammals and birds convert 27-carbon cholesterol predominantly to C24 bile acids such as cholic acid and chenodeoxycholic acid, conjugated to either glycine or taurine, jawless fish (hagfish, lampreys), cartilaginous fish (e.g., sharks, skates), and some teleost (bony) fish such as zebrafish synthesize C27 bile alcohols conjugated with sulfate.
In general, the phylogenically oldest fish (i.e., those that have the most distant common ancestor with mammals) utilize C27 bile alcohol pathways, with these bile alcohols accounting for nearly all biliary lipids [144, 145]. Relative to mammalian bile acids, C27 bile alcohols are often hydroxylated more extensively and retain the entire side-chain of cholesterol. In addition, many fish synthesize bile salts with an α rather than β configuration of the hydrogen at the 5 position of the steroid ring [142–144]. The zebrafish does not produce any C24 bile acids and instead synthesizes 5α-cyprinol (5α-cholestan-3α,7α,12α,26,27-pentol) sulfate [146, 147], a bile alcohol sulfate very similar to that found in the earliest vertebrates, the jawless fish [148, 149]. The bile salt synthetic pathway leading to 5α-C27 bile alcohol sulfates is a simpler pathway than that needed to produce C24 bile acids such as cholic acid (avoiding for example the need to cleave the cholesterol side-chain) and likely represents the first bile salt synthetic pathway to evolve in vertebrates [150]. Interestingly, some amphibians and even a few mammals (e.g., sea manatee, pony) retain the evolutionarily ‘early’ pathway leading to C27 bile alcohol sulfates as their dominant method of producing biliary bile salts [145].
In a functional assay, zebrafish PXR was activated efficaciously by its bile alcohol sulfate (cyprinol sulfate), scymnol (5β-cholestan-3α,7α,12α,24,26,27-hexol) sulfate (from the Spotted eagle ray, a cartilaginous fish), and essentially by no other bile salts [66]. Further, human, mouse, rat, rabbit, and chicken PXRs were all activated by cyprinol sulfate and scymnol sulfate [66]. Activation by C27 bile alcohol sulfates thus appears to be a ‘basal’ property of PXRs and has been retained as a vestigial function in mammalian and chicken PXRs, even though these animals, including humans, produce only minute quantities of C27 bile alcohols (except in rare diseases involving the bile acid biosynthetic pathway) [141, 142, 151, 152]. The ability to be activated by C24 bile acids is likely a more recent evolutionary innovation for PXRs and VDRs.
Overall, the variation of bile salts across species parallels the sequence variation of the PXR LBD. Bile salts vary little between primates, with only subtle differences in percentages of bile acid composition in biliary secretions, but do show differences between humans and other mammals. For example, α- and β-muricholic acids are the main primary bile acids in rodents while cholic and chenodeoxycholic acids are the dominant bile acids in humans and other primates [142, 143]. Even within mammals, there is evidence that PXRs have adapted to variations in biliary bile salts [66]. Thus, biliary bile salts are plausible endogenous ligands whose variation across species has influenced the ligand specificity of PXRs. This hypothesis can be strengthened by more extensive testing in additional species, particularly cartilaginous and jawless fish, and in reptiles.
The overlapping ability of VDR, PXR, and CAR to alter bile acid metabolism and elimination suggests that activators of these receptors may find therapeutic effect in treating cholestasis and diseases such as primary biliary cirrhosis where abnormally high levels of bile acids accumulate [153]. Indeed, the CAR activator phenobarbital and the PXR activator rifampin have shown some therapeutic benefit in the treatment of primary biliary cirrhosis [154–156]. Animal models demonstrate benefit of both PXR and CAR agonists in the treatment of cholestatic liver injury [157, 158]. It remains to be seen if selective VDR, PXR, and/or CAR agonists can be developed to detoxify bile acids and minimize other unwanted effects.
Steroid hormones
Whereas VDRs are basically insensitive to a wide variety of steroid hormones, PXRs and CARs both share the property of being activated or repressed by micromolar concentrations of androstane and pregnane steroids. This property is conserved across all PXRs, except for the divergent Xenopus laevis BXRs. For example, both 5β-pregnan-3,20-dione and 5α-androstan-3α-ol activate human, rhesus monkey, dog, pig, mouse, rat, rabbit, chicken, and zebrafish PXRs [27]. While it is tempting to view such conservation as significant, physiologic relevance of steroid effects on PXR and/or CAR has been difficult to prove. The concentrations of steroid hormones that individually affect PXR and CAR are much higher than concentrations typically found in human serum or plasma, even in pregnancy or fetal development. What has not been examined in detail is the ability of combinations of steroid hormones, for instance at levels found in pregnancy or fetal development, to activate PXRs or CARs.
A recently published well-designed clinical study clearly shows that CYP3A activity, as measured by N-demethylation of dextromethorphan, is increased approximately 35% throughput all trimesters of pregnancy [159], confirming previous more limited investigations of cortisol [160], nifedipine [161], and nelfinavir/indinavir [162] metabolism during pregnancy, suggesting that hormonal changes may influence CYP3A expression. However, hormonal changes during the menstrual cycle have generally not been shown to affect CYP3A expression. Despite a few reports of menstrual cycle differences in CYP3A activity or gene expression [163, 164], a number of investigations have revealed no menstrual cycle differences in the metabolism of CYP3A substrates midazolam [165, 166], dextromethorphan [167], or alfentanil [168], or any significant variation between pre- and postmenopausal women in metabolism of cortisol [169]. The increase in CYP3A during pregnancy may thus be mainly due to fetal or placental contribution [159, 170], although this may not explain the significant rise in CYP3A activity in the first trimester of pregnancy.
Whether hormonal factors during pregnancy increase CYP3A activity via PXR- or CAR-mediated pathways warrants more careful investigation. One possibility would be to expose human hepatocytes or cells that recombinantly express PXR or CAR to maternal serum or to combinations of hormones found in pregnancy. The elevation of CYP3A activity in pregnancy serves the possible function of protecting the developing fetus from harmful compounds and is a potential evolutionarily important adaptation. It will be interesting to see if this finding is seen in other animals and, if so, whether PXR and/or CAR mediate the effect.
The physiologic roles of the pregnane and androstane steroids most active at PXR and CAR are not well understood although some progress has been made in this area. A recent report provides evidence that PXR activation by 5β-dihydroprogesterone mediates chronic uterine relaxation during pregnancy via regulation of inducible nitric oxide synthase (iNOS) expression [171]. A response element for PXR/RXR had previously been demonstrated in the iNOS gene [172]. Several of the androstane steroids that strongly inhibit mouse CAR or activate mammalian PXRs have pheromone activities. These include 5α-androst-16-en-3α-ol and 5α-androst-16-en-3-one, musk-scented compounds with known pheromone activities in boars but unclear effects in humans [173–175]. The structure-activity relationships for activation of mouse CAR by androstanes [42] does not match with pheromonal activities of these compounds [174], suggesting that CAR does not mediate the pheromonal effects. The role of androstane and pregnane steroids such as 5β-pregnan-3,20-dione, 5α-androstan-3α-ol, or 5α-androst-16-en-3α-ol is basically unknown in non-mammalian species. Overall, the physiologic and evolutionary relevance of pregnane and androstane steroids as agonists or inverse agonists of PXR and CAR remains an open question.
Exogenous ligands
The impressive ability of PXR, and to a lesser extent, CAR to be activated by xenobiotics in humans suggests that a possible evolutionary function of these receptors is to detect toxic exogenous compounds, acquired through diet or other exposure to plants or other animals. Such a role may be analogous to the aryl hydrocarbon receptor, a key regulator of the CYP1A genes, which has recently been shown to be activated by dietary compounds in cow’s milk [176]. Somewhat surprisingly, evidence that dietary ligands activate PXR and CAR has been slow to accumulate, although vitamin E [177–179] and carotenoids [180] are now documented PXR activators. The identification of dietary ligands for PXR and CAR will be aided by cloning and functional expression of these genes from more species, particularly focusing on species that show diversity of diet (e.g., omnivores, carnivores, herbivores) and evolutionary history.
NR1I SUBFAMILY MEMBERS IN NON-MAMMALIAN SPECIES
In contrast to the wealth of data on mammalian NR1I subfamily members, there has been much less functional characterization of non-mammalian receptors. Based on current data, only mammals have three distinct NR1I members (i.e., VDR, PXR, and CAR), although some fish have two VDR genes and Xenopus laevis frogs have two PXR genes. CAR genes have only been detected in mammals (Table 1), although there is some uncertainty in classifying the chicken PXR (also known as the chicken X receptor or CXR), as this receptor has about equal sequence and functional similarity to both PXRs and CARs [27, 36, 181]. CAR genes have not been detected in teleost fish, amphibians, or in chickens, despite the completion or partial completion of genome sequencing projects for the pufferfish Fugu rubripes [6, 182], the tiger pufferish Tetraodon nigroviridis [3], the frog Xenopus tropicalis, and the chicken [183].
In agreement with the genome sequencing of chicken, which so far reveals only a single PXR/CAR gene, multiple experimental approaches, including extensive screening for additional PXR/CAR orthologs and RNA interference of CXR expression (to detect the presence of an additional xenobiotic-responsive receptor), have failed to provide evidence that chickens possess an additional NR1I subfamily member beyond PXR and VDR [181]. The CAR gene likely arose following a duplication of an ancestral PXR gene, followed by diversification [181]. The evolutionary benefits to mammals of having both PXR and CAR genes are currently unknown.
The role of VDR is regulating expression of hepatic and intestinal metabolizing enzymes in non-mammalian species has not been explored. Similar to many other genes that are duplicated in teleost fish as compared to mammalian genes [3–5], two VDR genes are found in some teleost fish, such as pufferfish [6] and Paralichthys olivaceus (Japanese flounder) [184], but not zebrafish (Table 1). Interestingly, the two VDR genes found in pufferfish and Japanese flounder have different tissue expression patterns. Pufferfish VDRβ is expressed only in gut, while VDRα shows fairly ubiquitous tissue expression (typical of mammalian VDRs), including brain, gill, heart, liver, and ovary [6]. Japanese flounder VDRβ is not expressed in liver or muscle while VDRα shows widespread tissue expression, with especially high amounts in intestine, heart, testis, and gill [184]. The single VDR so far cloned from the sea lamprey Petromyzon marinus was not detected in intestine but was cloned from skin and mouth [20]. The Xenopus laevis VDR gene is expressed in many adult tissues but is also strongly expressed during Xenopus development, peaking at the metamorphosis stage [64]. For all the non-mammalian VDRs described above, little functional characterization has been done except in some cases to show that the recombinantly expressed gene product binds calcitriol with high affinity and can activate VDR target genes [20, 64].
The dominant role of the Xenopus laevis BXRs in regulating early frog development [35, 87, 89], and of the detection of VDR in early frog embyos [64] and of PXR in larval zebrafish [185], suggests that PXR and VDR may generally be important developmental regulators in non-mammalian vertebrates. Developmental roles of PXR and CAR in mammals have not been described. PXR knockout, CAR knockout, and PXR/CAR double-knockout mice are phenotypically normal unless challenged with potentially toxic compounds such as LCA or xenobiotics [86, 101, 123, 186–190]. It remains to be seen if PXR and CAR mediate subtle functions during mammalian development.
In contrast to the extensive body of research on regulation of hepatic and intestinal metabolism/elimination (e.g., CYP3A expression) in mammals by NR1I subfamily members, this area has been much less studied outside mammals and birds. This differs considerably from the aryl hydrocarbon receptors (key regulators of CYP1A expression), for which fish models have provided considerable insight into mammalian receptor function, pharmacology, and toxicology [191]. Similar to mammals, some drugs and endogenous compounds can induce CYP3A gene expression in reptiles, amphibians, and fish. Chemically-induced upregulation of CYP3A expression has been demonstrated in microsomes from alligator liver by phenobarbital and 3-methylcholanthrene [192], in microsomes from a toad epithelial cell line by dexamethasone and corticosterone [193], in Atlantic cod by alkylphenols [194], in rainbow trout and killifish by ketoconazole [195], in adult zebrafish liver by pregnenolone 16α-carbonitrile (but not clotrimazole or nifedipine) [185], and in larval zebrafish foregut by rifampin and dexamethasone [196]. The molecular mechanism of CYP3A induction in the species mentioned above has not been determined. PXR is a logical candidate; however, the compounds shown to increase CYP3A expression in zebrafish adult liver (pregnenolone 16α-carbonitrile) [185] or larval foregut (rifampin, dexamethasone) [196] did not activate zebrafish PXR in an in vitro reporter assay [27], whereas clotrimazole and nifedepine, which did activate zebrafish PXR in vitro [27], did not induce CYP3A in adult zebrafish liver [185]. This raises the possibility that other NRs (e.g., glucocorticoid receptors) or other receptors regulate hepatic and intestinal metabolism and elimination. Overall, much work remains to be done with non-mammalian models of PXR and VDR regulation of xenobiotic and endogenous compound metabolism.
Fig. (4) shows a proposed phylogeny of the NR1I subfamily, taking into account functional data and tissue expression patterns. An unanswered question in the evolution of the NR1I subfamily is the functional properties of the closest common ancestor to PXR and VDR. Will this receptor have narrow ligand specificity and mediate predominantly endocrine functions like VDR or have broader ligand specificity and a major role in regulating metabolism and elimination of toxic compounds? Insights into this may be revealed if PXR can be characterized from a jawless fish such as hagfish or lamprey. Sequencing of the genome of the chordate Amphioxus, one of the closest invertebrate relatives to vertebrates, may also be helpful in this regard. Very little is known of the physiology of early fish and chordate invertebrates and of the role of NRs in these animals.
Fig. (4). Proposed Phylogeny of the NR1I Subfamily Showing Functional Characteristics.
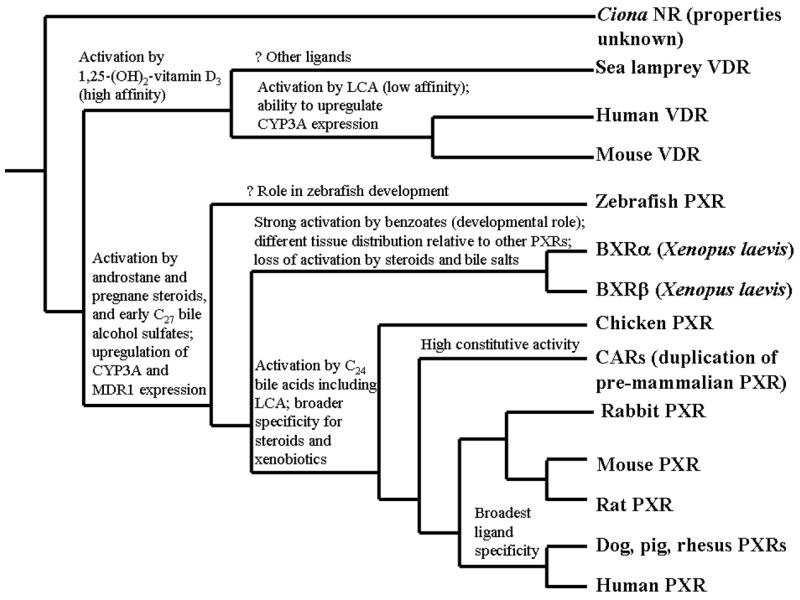
The phylogenetic tree is derived from known phylogenetic relationships between the animal species combined with pharmacological properties and tissue expression patterns. Characteristics included are activation by androstane steroids, pregnane steroids, C27 bile alcohol sulfates (representative of the earliest bile salts to evolve in vertebrates; current examples include cyprinol sulfate and scymnol sulfate), C24 bile acids (such as cholic acid or lithocholic acid, LCA), and benzoates (e.g., 3-aminoethylbenzoate in Xenopus laevis); ability to increase (upregulate) expression of CYP3A and MDR1; high constitutive (basal) activity; and tissue expression patterns. The possible developmental role of zebrafish PXR is highlighted by its strong expression in early life stages of the zebrafish [199].
CONCLUSION
PXR, VDR, and CAR perform overlapping functions in regulating metabolism and elimination of toxic endogenous and exogenous molecules. These three receptors also have an interesting evolutionary history which is just beginning to be untangled. Future work will focus on better understanding of how the unusually high sequence cross-species diversity of the CAR and PXR LBDs relates to variation in diet and physiology across animal species and whether non-mammalian model organisms such as zebrafish can provide additional insights into the function of human and mammalian NR1I subfamily members.
Acknowledgments
We thank Drs. Erin Schuetz, Lee Hagey, Anna Di Rienzo, and Kazuto Yasuda for their contribution to the original studies described in this review and for their helpful discussions. MD Krasowski is supported by a Clinical-Scientist Development Award from the National Institutes of Health (K08-GM074238-01A1). The authors also thank Dr. Holly Ingraham for helpful comments on the structural studies of the NR1I subfamily receptors.
References
- 1.Zhang Z, Burch PE, Cooney AJ, Lanz RB, Pereira FA, Wu J, Gibbs RA, Weinstock G, Wheeler DA. Genome Res. 2004;14(4):580–590. doi: 10.1101/gr.2160004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson-Rechavi M, Carpentier AS, Duffraisse M, Laudet V. Trends Genet. 2001;17(10):554–556. doi: 10.1016/s0168-9525(01)02417-9. [DOI] [PubMed] [Google Scholar]
- 3.Jaillon O, Aury JM, Brunet F, Petit JL, Strange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, Nicaud S, Jaffe D, Fisher SE, Lutfalla G, Dossat C, Segurens B, Dasilva C, Salanoubat M, Levy M, Boudet N, Castellano S, Anthouard V, Jubin C, Castelli C, Katinka M, Vacherie B, Biémont C, Skalli Z, Cattolico L, Poulain J, de Berardinis V, Cruard C, Duprat S, Brottier P, Coutanceau JP, Gouzy J, Parra G, Guigó R, Zody MC, Mesirov J, Lindblad-Toh K, Birren B, Nusbaum C, Kahn D, Robinson-Rechavi M, Laudet V, Schachter V, Quétier F, Saurin W, Scarpelli C, Wincker P, Lander ES, Weissenbach J, Crollius HR. Nature. 2004;431(7011):946–956. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- 4.Robinson-Rechavi M, Boussau B, Laudet V. Mol Biol Evol. 2004;21(3):580–586. doi: 10.1093/molbev/msh046. [DOI] [PubMed] [Google Scholar]
- 5.Escriva H, Manzon L, Youson J, Laudet V. Mol Biol Evol. 2002;19(9):1440–1450. doi: 10.1093/oxfordjournals.molbev.a004207. [DOI] [PubMed] [Google Scholar]
- 6.Maglich JM, Caravella JA, Lambert MH, Willson TM, Moore JT, Ramamurthy L. Nuc Acids Res. 2003;31(14):4051–4058. doi: 10.1093/nar/gkg444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Committee NRN. Cell. 1999;97(2):161–163. [Google Scholar]
- 8.Bertrand S, Brunet F, Escriva H, Parmentier G, Laudet V, Robinson-Rechavi M. Mol Biol Evol. 2004;21(10):1923–1937. doi: 10.1093/molbev/msh200. [DOI] [PubMed] [Google Scholar]
- 9.Mangelsdorf DJ, Evans RM. Cell. 1995;83(6):841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 10.Zhang XK, Hoffmann B, Tran PB, Graupner G, Pfahl M. Nature. 1992;355(6359):441–446. doi: 10.1038/355441a0. [DOI] [PubMed] [Google Scholar]
- 11.McDonnell DP, Mangelsdorf DJ, Pike JW, Haussler MR, O’Malley BW. Science. 1987;235(4793):1214–1217. doi: 10.1126/science.3029866. [DOI] [PubMed] [Google Scholar]
- 12.Burmester JK, Wiese RJ, Maeda N, DeLuca HF. Proc Natl Acad Sci USA. 1988;85(24):9499–9502. doi: 10.1073/pnas.85.24.9499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burmester JK, Maeda N, DeLuca HF. Proc Natl Acad Sci USA. 1988;85 (24):1005–1009. doi: 10.1073/pnas.85.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker AR, McDonnell DP, Hughes M, Crisp TM, Mangelsdorf DJ, Haussler MR, Pike JW, Shine J, O’Malley BW. Proc Natl Acad Sci USA. 1988;85(10):3294–3298. doi: 10.1073/pnas.85.10.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christakos S, Dhawan P, Liu Y, Peng X, Porta A. J Cell Biochem. 2003;88(4):695–705. doi: 10.1002/jcb.10423. [DOI] [PubMed] [Google Scholar]
- 16.Dusso AS, Brown AJ, Slatopolsky E. Am J Physiol Renal Physiol. 2005;289(1):F8–F28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 17.Holick MF. Recent Results Cancer Res. 2003;164(1):3–28. doi: 10.1007/978-3-642-55580-0_1. [DOI] [PubMed] [Google Scholar]
- 18.Nagpal S, Na S, Rathnachalam R. Endocrine Rev. 2005;26(5):662–687. doi: 10.1210/er.2004-0002. [DOI] [PubMed] [Google Scholar]
- 19.White JH. J Steroid Biochem Mol Biol. 2004;89–90(1–5):239–244. doi: 10.1016/j.jsbmb.2004.03.074. [DOI] [PubMed] [Google Scholar]
- 20.Whitfield GK, Dang HTL, Schluter SF, Bernstein RM, Bunag T, Manzon LA, Hsieh G, Dominguez CE, Youson JH, Haussler MR, Marchalonis JJ. Endocrinology. 2003;144(6):2704–2716. doi: 10.1210/en.2002-221101. [DOI] [PubMed] [Google Scholar]
- 21.MacDonald PN, Haussler CA, Terpening CM, Galligan MA, Reeder MC, Whitfield GK, Haussler MR. J Biol Chem. 1991;266(28):18808–18813. [PubMed] [Google Scholar]
- 22.Ross TK, Prahl JM, DeLuca HF. Proc Natl Acad Sci USA. 1991;88(15):6555–6559. doi: 10.1073/pnas.88.15.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blumberg B, Sabbagh W, Juguilon H, Bolado J, van Meter CM, Ong ES, Evans RM. Genes Dev. 1998;12(20):3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterstrom RH, Perlmann T, Lehmann JM. Cell. 1998;92(1):73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 25.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. J Clin Invest. 1998;102(5):1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moore LB, Parks DJ, Jones SA, Bledsoe RK, Consler TG, Stimmel JB, Goodwin B, Liddle C, Blanchard SG, Willson TM, Collins JL, Kliewer SA. J Biol Chem. 2000;275(20):15122–15127. doi: 10.1074/jbc.M001215200. [DOI] [PubMed] [Google Scholar]
- 27.Moore LB, Maglich JM, McKee DD, Wisely B, Willson TM, Kliewer SA, Lambert MH, Moore JT. Mol Endocrinol. 2002;16(5):977–986. doi: 10.1210/mend.16.5.0828. [DOI] [PubMed] [Google Scholar]
- 28.Wentworth JM, Agostini M, Love J, Schwabe JW, Chatterjee VKK. J Endocrinol. 2000;166(3):R11–R16. doi: 10.1677/joe.0.166r011. [DOI] [PubMed] [Google Scholar]
- 29.Moore LB, Goodwin B, Jones SA, Wisely GB, Serabjit-Singh CJ, Willson TM, Collins JL, Kliewer SA. Proc Natl Acad Sci USA. 2000;97(13):7500–7502. doi: 10.1073/pnas.130155097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ripp SL, Fitzpatrick JL, Peters JM, Prough RA. Drug Metab Dispos. 2002;30(5):570–575. doi: 10.1124/dmd.30.5.570. [DOI] [PubMed] [Google Scholar]
- 31.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstram A. Proc Natl Acad Sci USA. 1998;95(21):12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones SA, Moore LB, Shenk JL, Wisely GB, Hamilton GA, McKee DD, Tomkinson NCO, LeCluyse EL, Lambert MH, Willson TM, Kliewer SA, Moore JT. Mol Endocrinol. 2000;14(1):27–39. doi: 10.1210/mend.14.1.0409. [DOI] [PubMed] [Google Scholar]
- 33.Hustert E, Zibat A, Presecan-Siedel E, Eiselt R, Mueller R, Fub C, Brehm I, Brinkmann U, Eichelbaum M, Wojnowski L, Burk O. Drug Metab Dispos. 2001;29(11):1454–1459. [PubMed] [Google Scholar]
- 34.Savas U, Wester MR, Griffin KJ, Johnson EF. Drug Metab Dispos. 2000;28(5):529–537. [PubMed] [Google Scholar]
- 35.Blumberg B, Kang H, Bolado J, Chen H, Craig AG, Moreno TA, Umesano K, Perlmann T, De Robertis EM, Evans RM. Genes Dev. 1998;12(9):1269–1277. doi: 10.1101/gad.12.9.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Handschin C, Podvinec M, Meyer UA. Proc Natl Acad Sci USA. 2000;97(20):10769–10774. doi: 10.1073/pnas.97.20.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Handschin C, Podvinec M, Amherd R, Looser R, Ourlin JC, Meyer UA. J Biol Chem. 2002;277(33):29561–29567. doi: 10.1074/jbc.M202739200. [DOI] [PubMed] [Google Scholar]
- 38.Hartley DP, Dai X, He YD, Carlini EJ, Wang B, Huskey SW, Ulrich RG, Rushmore TH, Evers R, Evans DC. Mol Pharmacol. 2004;65(5):1159–1171. doi: 10.1124/mol.65.5.1159. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H, LeCulyse E, Liu L, Hu M, Matoney L, Zhu W, Yan B. Arch Biochem Biophys. 1999;368(1):14–22. doi: 10.1006/abbi.1999.1307. [DOI] [PubMed] [Google Scholar]
- 40.Baes M, Gulick T, Choi HS, Martinolli MG, Simha D, Moore DD. Mol Cell Biol. 1994;14(3):1544–1552. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi HS, Chung M, Tzameli I, Simha D, Lee YK, Seol W, Moore DD. J Biol Chem. 1997;272(38):23565–23571. doi: 10.1074/jbc.272.38.23565. [DOI] [PubMed] [Google Scholar]
- 42.Forman BM, Tzameli I, Choi HS, Chen J, Simha D, Seol W, Evans RM, Moore DD. Nature. 1998;395(6702):612–615. doi: 10.1038/26996. [DOI] [PubMed] [Google Scholar]
- 43.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. Mol Cell Biol. 1998;18(10):5652–5658. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. J Biol Chem. 1999;274(10):6043–6046. doi: 10.1074/jbc.274.10.6043. [DOI] [PubMed] [Google Scholar]
- 45.Shan L, Vincent J, Brunzelle JS, Dussault I, Lin M, Ianculescu I, Sherman MA, Forman BM, Fernandez EJ. Mol Cell. 2004;16(6):907–917. doi: 10.1016/j.molcel.2004.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suino K, Peng L, Reynolds R, Li Y, Cha JY, Repa JJ, Kliewer SA, Xu HE. Mol Cell. 2004;16(6):893–905. doi: 10.1016/j.molcel.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 47.Xu RX, Lambert MH, Wisely BB, Warren EN, Weinert EE, Waitt GM, Williams JD, Collins JL, Moore LB, Willson TM, Moore JT. Mol Cell. 2004;16(6):919–928. doi: 10.1016/j.molcel.2004.11.042. [DOI] [PubMed] [Google Scholar]
- 48.Huang W, Zhang J, Wei P, Schrader WT, Moore DD. Mol Endocrinol. 2004;18(10):2402–2408. doi: 10.1210/me.2004-0046. [DOI] [PubMed] [Google Scholar]
- 49.Frank C, Molnár F, Matilainen M, Lempiäinen H, Carlberg C. J Biol Chem. 2004;279(32):33558–33566. doi: 10.1074/jbc.M403946200. [DOI] [PubMed] [Google Scholar]
- 50.Maglich JM, Parks DJ, Moore LB, Collins JL, Goodwin B, Billin AN, Stoltz CA, Kliewer SA, Lambert MH, Willson TM, Moore JT. J Biol Chem. 2003;278(19):17277–17283. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- 51.Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, Harafuji N, Hastings KEM, Ho I, Hotta K, Huang W, Kawashima T, Lemaire P, Martinez D, Meinertzhagen IA, Necula S, Nonaka M, Putnam N, Rash S, Saiga H, Satake M, Terry A, Yamada L, Wang HG, Awazu S, Azumi K, Boore J, Branno M, Chin-bow S, DeSantis R, Doyle S, Francina P, Keys DN, Haga S, Hayashi H, Hino K, Imai KS, Kano S, Kobayashi K, Kobayashi M, Lee BI, Makabe KW, Manohar C, Matassi G, Medina M, Mochizuki Y, Mount S, Morishita T, Miura S, Nakayama A, Nishizaka S, Nomoto H, Ohta F, Oishi K, Rigoutsos I, Sano M, Sasaki A, Sasakura Y, Shoguchi E, Shin-i T, Spagnuolo A, Stainier D, Suzuki MM, Tassy O, Takatori N, Tokuoka M, Yagi K, Yoshizaki F, Wada S, Zhang C, Hyatt PD, Larimer F, Detter C, Doggett N, Galvina T, Hawkins T, Richardson P, Lucas S, Kohara Y, Levine M, Satoh N, Rokhsar DS. Science. 2002;298(5601):2157–2167. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- 52.Pascussi JM, Gerbal-Chaloin S, Drocourt L, Assénat E, Larrey D, Pichard-Garcia L, Vilarem MJ, Maurel P. Xenobiotica. 2004;34(7):633–664. doi: 10.1080/00498250412331285454. [DOI] [PubMed] [Google Scholar]
- 53.Schuetz EG. Curr Drug Metab. 2001;2(2):139–147. doi: 10.2174/1389200013338595. [DOI] [PubMed] [Google Scholar]
- 54.Sueyoshi T, Negishi M. Ann Rev Pharmacol Toxicol. 2001;41(1):123–143. doi: 10.1146/annurev.pharmtox.41.1.123. [DOI] [PubMed] [Google Scholar]
- 55.Mackenzie PI, Gregory PA, Gardner DA, Lewinsky RH, Jorgensen BR, Nishiyama T, Xie W, Radominska-Pandya A. Curr Drug Metab. 2003;4(3):249–257. doi: 10.2174/1389200033489442. [DOI] [PubMed] [Google Scholar]
- 56.Klaasen CD, Slitt AL. Curr Drug Metab. 2005;6(4):309–328. doi: 10.2174/1389200054633826. [DOI] [PubMed] [Google Scholar]
- 57.Runge-Morris M, Kocarek TA. Curr Drug Metab. 2005;6(4):299–307. doi: 10.2174/1389200054633871. [DOI] [PubMed] [Google Scholar]
- 58.Zhou J, Zhang J, Xie W. Curr Drug Metab. 2005;6(4):289–298. doi: 10.2174/1389200054633853. [DOI] [PubMed] [Google Scholar]
- 59.Van Buren D, Wideman CA, Ried M, Gibbons S, Van Buren CT, Jarowenko M, Flechner SM, Frazier OH, Cooley DA, Kahan BD. Transplant Proc. 1984;16(6):1642–1645. [PubMed] [Google Scholar]
- 60.Back DJ, Bates M, Bowden A, Breckenridge AM, Hall MJ, Jones H, MacIver M, Orme M, Perucca E, Richens A, Rowe PH, Smith E. Contraception. 1980;22(5):495–503. doi: 10.1016/0010-7824(80)90102-x. [DOI] [PubMed] [Google Scholar]
- 61.Schwarz UI, Büschel B, Kirch W. Br J Clin Pharmacol. 2003;55(1):112–113. doi: 10.1046/j.1365-2125.2003.t01-1-01716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skolnick JL, Stoler BS, Katz DB, Anderston WH. JAMA. 1976;236(12):1382. [PubMed] [Google Scholar]
- 63.Elaroussi MA, Prahl JM, DeLuca HF. Proc Natl Acad Sci USA. 1994;91(24):11596–11600. doi: 10.1073/pnas.91.24.11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li YC, Bergwitz C, Juppner H, Demay MB. Endocrinology. 1997;138(6):2347–2353. doi: 10.1210/endo.138.6.5210. [DOI] [PubMed] [Google Scholar]
- 65.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Science. 2002;296(5571):1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 66.Krasowski MD, Yasuda K, Hagey LR, Schuetz EG. Mol Endocrinol. 2005;19(7):1720–1739. doi: 10.1210/me.2004-0427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watkins RE, Davis-Searles PR, Lambert MH, Redinbo MR. J Mol Biol. 2003;331(4):815–828. doi: 10.1016/s0022-2836(03)00795-2. [DOI] [PubMed] [Google Scholar]
- 68.Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR. Biochemistry. 2003;42(6):1430–1438. doi: 10.1021/bi0268753. [DOI] [PubMed] [Google Scholar]
- 69.Watkins RE, Noble SM, Redinbo MR. Curr Opin Drug Discov Dev. 2002;5(1):150–158. [PubMed] [Google Scholar]
- 70.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. Science. 2001;292(5525):2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 71.Ingraham HA, Redinbo MR. Curr Opin StructBiol. 2005 in press. [Google Scholar]
- 72.Chrencik JE, Orans J, Moore LB, Xue Y, Peng L, Collins JL, Wisely GB, Lambert MH, Kliewer SA, Redinbo MR. Mol Endocrinol. 2005;19(5):1125–1134. doi: 10.1210/me.2004-0346. [DOI] [PubMed] [Google Scholar]
- 73.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, Rosenfeld MG, Willson TM, Glass CK, Milburn MV. Nature. 1998;395(6698):137–143. doi: 10.1038/25931. [DOI] [PubMed] [Google Scholar]
- 74.Orans J, Teotico DG, Redinbo MR. Mol Endocrinol. 2005 doi: 10.1210/me.2005-0156. in press. [DOI] [PubMed] [Google Scholar]
- 75.Carnahan VE, Redinbo MR. Curr Drug Metab. 2005;6(4):357–367. doi: 10.2174/1389200054633844. [DOI] [PubMed] [Google Scholar]
- 76.Ekins S, Bravi G, Wikel JH, Wrighton SA. J Pharmacol Exp Ther. 1999;291(1):424–433. [PubMed] [Google Scholar]
- 77.Wrighton SA, Schuetz EG, Thummel KE, Shen DD, Korzekwa KR, Watkins PB. Drug Metab Rev. 2000;32(3–4):339–361. doi: 10.1081/dmr-100102338. [DOI] [PubMed] [Google Scholar]
- 78.Quattrochi LC, Guzelian PS. Drug Metab Dispos. 2001;29(5):615–622. [PubMed] [Google Scholar]
- 79.Luo G, Guenthner T, Gan LS, Humphreys WG. Curr Drug Metab. 2004;5(6):483–505. doi: 10.2174/1389200043335397. [DOI] [PubMed] [Google Scholar]
- 80.Li AP, Kaminski DL, Rasmussen A. Toxicology. 1995;104(1–3):1–8. doi: 10.1016/0300-483x(95)03155-9. [DOI] [PubMed] [Google Scholar]
- 81.Ekins S, Kim RB, Leake BF, Dantzig AH, Schuetz EG, Lan LB, Yasuda K, Shepard RL, Winter MA, Schuetz JD, Wikel JH, Wrighton SA. Mol Pharmacol. 2002;61(5):974–981. doi: 10.1124/mol.61.5.974. [DOI] [PubMed] [Google Scholar]
- 82.Higgins CF. Curr Opin Cell Biol. 1993;5(4):684–687. doi: 10.1016/0955-0674(93)90140-l. [DOI] [PubMed] [Google Scholar]
- 83.Wacher VJ, Wu CY, Benet LZ. Mol Carcinogenesis. 1995;13(3):129–134. doi: 10.1002/mc.2940130302. [DOI] [PubMed] [Google Scholar]
- 84.Yu DK. J Clin Pharmacol. 1999;39(12):1203–1211. doi: 10.1177/00912709922012006. [DOI] [PubMed] [Google Scholar]
- 85.Goodwin B, Gauthier KC, Umetani M, Watson MA, Lochansky MI, Collins JL, Leitersdorf E, Mangelsdorf DJ, Kliewer SA, Repa JJ. Proc Natl Acad Sci USA. 2003;100(1):223–228. doi: 10.1073/pnas.0237082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. Proc Natl Acad Sci USA. 2001;98(6):3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grün F, Venkatesan RN, Tabb MM, Zhou C, Cao J, Hemmati D, Blumberg B. J Biol Chem. 2002;277(46):43691–43697. doi: 10.1074/jbc.M206553200. [DOI] [PubMed] [Google Scholar]
- 88.Nishikawa J, Saito K, Sasaki M, Tomigahara Y, Nishihara T. Biochem Biophys Res Comm. 2000;277(1):209–215. doi: 10.1006/bbrc.2000.3649. [DOI] [PubMed] [Google Scholar]
- 89.Heath LA, Jones EA, Old RW. Int J Dev Biol. 2000;44(1):141–144. [PubMed] [Google Scholar]
- 90.Clark AG, Glanowski S, Nielsen R, Thomas PD, Kejariwal A, Todd MA, Tanenbaum DM, Civello D, Lu F, Murphy B, Ferriera S, Wang G, Zheng X, White TJ, Sninsky JJ, Adams MD, Cargill M. Science. 2003;302(5652):1960–1963. doi: 10.1126/science.1088821. [DOI] [PubMed] [Google Scholar]
- 91.Krasowski MD, Yasuda K, Hagey LR, Schuetz EG. Nuclear Receptor. 2005;3(1):2. doi: 10.1186/1478-1336-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Forey P, Janvier P. Nature. 1993;361(6408):129–134. [Google Scholar]
- 93.Chen TC, Persons KS, Lu Z, Mathieu JS, Holick MF. J Nutr Biochem. 2000;11(5):267–272. doi: 10.1016/s0955-2863(00)00077-2. [DOI] [PubMed] [Google Scholar]
- 94.Yang Z, Bielawski JP. Trends Ecol Evol. 2000;15(12):496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pagani F, Raponi M, Baralle FE. Proc Natl Acad Sci USA. 2005;102(18):6368–6372. doi: 10.1073/pnas.0502288102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Endo T, Ikeo K, Gojobori T. Mol Biol Evol. 1996;13(5):685–690. doi: 10.1093/oxfordjournals.molbev.a025629. [DOI] [PubMed] [Google Scholar]
- 97.Yang Z, Nielsen R, Goldman N, Pedersen AK. Genetics. 2000;155(1):431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nielsen R, Yang Z. Genetics. 1998;148(3):929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thompson EE, Kuttab-Boulos H, Krasowski MD, Di Rienzo A. Hum Genomics. 2005;2(3):168–178. doi: 10.1186/1479-7364-2-3-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schuetz EG, Strom S, Yasuda K, Lecureur V, Assem M, Brimer C, Lamba J, Kim RB, Ramachandran V, Komoroski BJ, Venkataramanan R, Cai H, Sinal CJ, Gonzalez FJ, Schuetz JD. J Biol Chem. 2001;276(42):39411–39418. doi: 10.1074/jbc.M106340200. [DOI] [PubMed] [Google Scholar]
- 101.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. Proc Natl Acad Sci USA. 2001;98(6):3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lamba J, Lamba V, Schuetz E. Curr Drug Metab. 2005;6(4):369–383. doi: 10.2174/1389200054633880. [DOI] [PubMed] [Google Scholar]
- 103.Zhang J, Kuehl P, Green ED, Touchman JW, Watkins PB, Daly A, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Wrighton SA, Hancock M, Kim RB, Strom S, Thummel K, Russell CG, Hudson JR, Schuetz EG, Boguski MS. Pharmacogenetics. 2001;11(7):555–572. doi: 10.1097/00008571-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 104.Ikeda S, Kurose K, Ozawa S, Sai K, Hasegawa R, Komamura K, Ueno K, Kamakura S, Kitakaze M, Tomoike H, Nakajima T, Matsumoto K, Saito H, Goto Y, Kimura H, Katoh M, Sugai K, Minami N, Shirao K, Tamura T, Yamamoto N, Minami H, Ohtsu A, Yoshida T, Saijo N, Saito Y, Sawada J. Drug Metab Pharmacokin. 2003;18(6):413–418. doi: 10.2133/dmpk.18.413. [DOI] [PubMed] [Google Scholar]
- 105.Koyano S, Kurose K, Ozawa S, Saeki M, Nakajima Y, Hasegawa R, Komamura K, Ueno K, Kamakura S, Nakajima T, Saito H, Kimura H, Goto Y, Saitoh O, Katoh M, Ohnuma T, Kawai M, Sugai K, Ohtsuki T, Suzuki C, Minami N, Saito Y, Sawada J. Drug Metab Pharmacokin. 2002;17(6):561–565. doi: 10.2133/dmpk.17.561. [DOI] [PubMed] [Google Scholar]
- 106.Koyano S, Kurose K, Saito Y, Ozawa S, Hasegawa R, Komamura K, Ueno K, Kamakura S, Kitakaze M, Nakajima T, Matsumoto K, Akasawa A, Saito H, Sawada JI. Drug Metab Dispos. 2004;32(1):149–154. doi: 10.1124/dmd.32.1.149. [DOI] [PubMed] [Google Scholar]
- 107.Uitterlinden AG, Fang Y, van Meurs JBJ, Pols HAP, van Leeuwen JPTM. Gene. 2004;338(2):143–156. doi: 10.1016/j.gene.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 108.Ebersberger I, Metzler D, Schwarz C, Pääbo S. Am J Hum Genet. 2002;70(6):1490–1497. doi: 10.1086/340787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kliewer SA. J Nutr. 2003;133(7 Suppl):2444S–2447S. doi: 10.1093/jn/133.7.2444S. [DOI] [PubMed] [Google Scholar]
- 110.Kliewer SA, Goodwin B, Willson TM. Endocrine Rev. 2002;23(5):687–702. doi: 10.1210/er.2001-0038. [DOI] [PubMed] [Google Scholar]
- 111.Dussault I, Forman BM. Crit Rev Eukaryotic Gene Express. 2002;12(1):53–64. doi: 10.1615/critreveukaryotgeneexpr.v12.i1.30. [DOI] [PubMed] [Google Scholar]
- 112.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. Nature. 2000;407(6806):920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 113.Xie W, Barwick JL, Simon CM, Pierce AM, Safe S, Blumberg B, Guzelian PS, Evans RM. Genes Dev. 2000;14(23):3014–3023. doi: 10.1101/gad.846800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sonoda J, Rosenfeld JM, Xu L, Evans RM, Xie W. Curr Drug Metab. 2003;4(1):59–72. doi: 10.2174/1389200033336739. [DOI] [PubMed] [Google Scholar]
- 115.Hofmann AF. Arch Intern Med. 1999;159(22):2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 116.Hofmann AF. Drug Metab Rev. 2004;36(3–4):703–722. doi: 10.1081/dmr-200033475. [DOI] [PubMed] [Google Scholar]
- 117.Ogawa A, Murate T, Suzuki M, Nimura Y, Yoshida S. Jap J Cancer Res. 1998;89(11):1154–1159. doi: 10.1111/j.1349-7006.1998.tb00510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nagengast FM, Grubben MJ, van Munster IP. Eur J Cancer. 1995;31A(7–8):1067–1070. doi: 10.1016/0959-8049(95)00216-6. [DOI] [PubMed] [Google Scholar]
- 119.Hamada K, Umemoto A, Kajikawa A, Seraj MJ, Monden Y. Carcinogenesis. 1994;15(9):1911–1915. doi: 10.1093/carcin/15.9.1911. [DOI] [PubMed] [Google Scholar]
- 120.Owen RW, Dodo M, Thompson MH, Hill MJ. Nutr Cancer. 1987;9(2–3):73–80. doi: 10.1080/01635588709513914. [DOI] [PubMed] [Google Scholar]
- 121.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. J Nat Cancer Inst. 1974;53(4):1093–1097. doi: 10.1093/jnci/53.4.1093. [DOI] [PubMed] [Google Scholar]
- 122.Uppal H, Toma D, Saini SPS, Ren S, Jones TJ, Xie W. Hepatology. 2005;41(1):168–176. doi: 10.1002/hep.20512. [DOI] [PubMed] [Google Scholar]
- 123.Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. J Biol Chem. 2004;279(47):49517–49522. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 124.Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM. Proc Natl Acad Sci USA. 2002;99(21):13801–13806. doi: 10.1073/pnas.212494599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Saini SPS, Mu Y, Gong H, Toma D, Uppal H, Ren S, Li S, Poloyac SM, Xie W. Hepatology. 2005;41(3):497–505. doi: 10.1002/hep.20570. [DOI] [PubMed] [Google Scholar]
- 126.Saini SPS, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren S, Moore DD, Evans RM, Xie W. Mol Pharmacol. 2004;65(2):292–300. doi: 10.1124/mol.65.2.292. [DOI] [PubMed] [Google Scholar]
- 127.Dussault I, Yoo HD, Lin M, Fan M, Batta AK, Salen G, Erickson SK, Forman BM. Proc Natl Acad Sci USA. 2003;100(3):833–838. doi: 10.1073/pnas.0336235100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thummel KE, Brimer C, Yasuda K, Thottassery J, Senn T, Lin Y, Ishizuka H, Kharasch E, Schuetz J, Schuetz E. Mol Pharmacol. 2001;60(6):1399–1406. doi: 10.1124/mol.60.6.1399. [DOI] [PubMed] [Google Scholar]
- 129.Schmiedlin-Ren P, Thummel KE, Fisher JM, Paine MF, Watkins PB. Drug Metab Dispos. 2001;29(11):1446–1453. [PubMed] [Google Scholar]
- 130.Adachi S, Shulman AI, Yamamoto K, Shimomura I, Yamada S, Mangelsdorf DJ, Makishima M. Mol Endocrinol. 2004;18(1):43–52. doi: 10.1210/me.2003-0244. [DOI] [PubMed] [Google Scholar]
- 131.Adachi R, Honma Y, Masuno H, Karana K, Shimomura I, Yamada S, Makishima M. J Lipid Res. 2005;46(1):46–57. doi: 10.1194/jlr.M400294-JLR200. [DOI] [PubMed] [Google Scholar]
- 132.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. Mol Cell. 2000;5(1):173–179. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 133.Tocchini-Valentini G, Rochel N, Wurtz JM, Mitschler A, Moras D. Proc Natl Acad Sci USA. 2001;98(10):5491–5496. doi: 10.1073/pnas.091018698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tocchini-Valentini G, Rochel N, Wurtz JM, Moras D. J Med Chem. 2004;47(8):1956–1961. doi: 10.1021/jm0310582. [DOI] [PubMed] [Google Scholar]
- 135.Jurutka PW, Thompson PD, Whitfield GK, Eichhorst KR, Hall N, Dominguez CE, Hsieh JC, Haussler CA, Haussler MR. J Cell Biochem. 2005;94(5):917–943. doi: 10.1002/jcb.20359. [DOI] [PubMed] [Google Scholar]
- 136.Fischer S, Beuers U, Spengler U, Zwiebel FM, Koebe HG. Clin Chim Acta. 1996;251(2):173–186. doi: 10.1016/0009-8981(96)06305-x. [DOI] [PubMed] [Google Scholar]
- 137.Stedman C, Robertson G, Coulter S, Liddle C. J Biol Chem. 2004;279(12):11336–11343. doi: 10.1074/jbc.M310258200. [DOI] [PubMed] [Google Scholar]
- 138.Cali JJ, Hsieh CL, Francke U, Russell DW. J Biol Chem. 1991;266(12):7779–7783. [PMC free article] [PubMed] [Google Scholar]
- 139.Honda A, Salen G, Matsuzaki Y, Batta AK, Xu G, Leitersdorf E, Tint GS, Erickson SK, Tanaka N, Shefer S. J Biol Chem. 2001;276(37):34579–34585. doi: 10.1074/jbc.M103025200. [DOI] [PubMed] [Google Scholar]
- 140.Honda A, Salen G, Matsuzaki Y, Batta AK, Xu G, Leitersdorf E, Tint GS, Erickson SK, Tanaka N, Shefer S. J Lipid Res. 2001;42(2):291–300. [PubMed] [Google Scholar]
- 141.Hoshita T. Bile alcohols and primitive bile acids. In: Danielsson H, Sjovall J, editors. Sterols and bile acids. Amsterdam: Elsevier Science Publishers; 1985. pp. 279–302. [Google Scholar]
- 142.Hagey LR. Bile acid biodiversity in vertebrates: chemistry and evolutionary implications. Ann Arbor: UMI; 1992. [Google Scholar]
- 143.Hofmann AF, Schteingart CD, Hagey LR. Species differences in bile acid metabolism. In: Paumgartner G, Beuers U, editors. International Falk Symposium: Bile acids in liver diseases. Dordrecht: Kluwer Academic Publishers; 1995. pp. 3–30. [Google Scholar]
- 144.Une M, Hoshita T. Hiroshima J Med Sci. 1994;43(2):37–67. [PubMed] [Google Scholar]
- 145.Moschetta A, Xu F, Hagey LR, van Berge Henegouwen GP, van Erpecum KJ, Brouwers JF, Cohen JC, Bierman M, Hobbs HH, Steinbach JH, Hofmann AF. J Lipid Res. 2005;46(10):2221–2232. doi: 10.1194/jlr.M500178-JLR200. [DOI] [PubMed] [Google Scholar]
- 146.Goto T, Holzinger F, Hagey LR, Cerrè C, Ton-Nu HT, Schteingart CD, Steinbach JH, Shneider BL, Hofmann AF. J Lipid Res. 2003;44(9):1643–1651. doi: 10.1194/jlr.M300155-JLR200. [DOI] [PubMed] [Google Scholar]
- 147.Anderson IG, Briggs T, Haslewood GAD. Biophys J. 1964;90(2):303–308. doi: 10.1042/bj0900303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Haslewood GAD. Biophys J. 1966;100(1):233–237. [Google Scholar]
- 149.Haslewood GAD, Tökés L. Biophys J. 1969;114(2):179–184. doi: 10.1042/bj1140179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Haslewood GAD. J Lipid Res. 1967;8(6):535–550. [PubMed] [Google Scholar]
- 151.Nakagawa M, Une M, Takenaka S, Tazawa Y, Nozaki S, Imanaka T, Kuramoto T. Clin Chim Acta. 2001;314(1–2):101–106. doi: 10.1016/s0009-8981(01)00636-2. [DOI] [PubMed] [Google Scholar]
- 152.Yousef IM, Perwalz S, Lamireau T, Tuchweber B. Med Sci Monit. 2003;9(3):MT21–MT31. [PubMed] [Google Scholar]
- 153.Karpen SJ. Hepatology. 2005;42(2):266–269. doi: 10.1002/hep.20833. [DOI] [PubMed] [Google Scholar]
- 154.Cancado ELR, Leitao RMC, Carrilho FJ, Laudanna AA. Am J Gastroenterol. 1998;93(9):1510–1517. doi: 10.1111/j.1572-0241.1998.00472.x. [DOI] [PubMed] [Google Scholar]
- 155.Bachs L, Pares A, Elena M, Piera C, Rodes J. Lancet. 1989;1(8638):574–576. doi: 10.1016/s0140-6736(89)91608-5. [DOI] [PubMed] [Google Scholar]
- 156.Bachs L, Pares A, Elena M, Piera C, Rodes J. Gastroenterology. 1992;102(6):2077–2080. doi: 10.1016/0016-5085(92)90335-v. [DOI] [PubMed] [Google Scholar]
- 157.Stedman CAM, Liddle C, Coulter SA, Sonoda J, Alvarez JGA, Moore DD, Evans RM, Downes M. Proc Natl Acad Sci USA. 2005;102(6):2063–2068. doi: 10.1073/pnas.0409794102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, Zatloukal K, Trauner M. Hepatology. 2005;42(2):420–430. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- 159.Tracy TS, Venkataramanan R, Glover DD, Caritis SN. Am J Obstet Gynecol. 2005;192(2):633–639. doi: 10.1016/j.ajog.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 160.Homma M, Beckerman K, Hayashi S, Jayewardene AL, Oka K, Gambertoglio JG, Aweeka FT. J Pharmaceut Biomed Anal. 2000;23(4):629–635. doi: 10.1016/s0731-7085(00)00334-4. [DOI] [PubMed] [Google Scholar]
- 161.Barton JR, Prevost RR, Wilson DA, Whybrew WD, Sibai BM. Am J Obstet Gynecol. 1991;165(4 Pt 1):951–954. doi: 10.1016/0002-9378(91)90446-x. [DOI] [PubMed] [Google Scholar]
- 162.Kosel BW, Beckerman K, Hayashi S, Homma M, Aweeka FT. AIDS. 2003;17(8):1195–1199. doi: 10.1097/00002030-200305230-00011. [DOI] [PubMed] [Google Scholar]
- 163.Zhu B, Lin ZQ, Chen GL, Chen XP, Ou-Yang DS, Wang LS, Huang SL, Tan ZR, Zhou HH. Br J Clin Pharmacol. 2003;55(3):264–269. doi: 10.1046/j.1365-2125.2003.01728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sarkar MA, Vadlamuri V, Ghosh S, Glover DD. Drug Metab Dispos. 2003;31(1):1–6. doi: 10.1124/dmd.31.1.1. [DOI] [PubMed] [Google Scholar]
- 165.Kashuba AD, Bertino JS, Rocci ML, Kulawy RW, Beck DJ, Nafziger AN. Clin Pharmacol Ther. 1998;64(3):269–277. doi: 10.1016/S0009-9236(98)90175-8. [DOI] [PubMed] [Google Scholar]
- 166.Kharasch ED, Mautz D, Senn T, Leutz G, Cox K. J Clin Pharmacol. 1999;39(3):275–280. [PubMed] [Google Scholar]
- 167.Kashuba AD, Nafziger AN, Kearus HL, Leeder JS, Shirey CS, Gotschall R, Gaedigk A, Bertino JS. Pharmacogenetics. 1998;8(5):403–410. doi: 10.1097/00008571-199810000-00005. [DOI] [PubMed] [Google Scholar]
- 168.Kharasch ED, Russell M, Garton K, Lentz G, Bowdle TA, Cox K. Anesthesiology. 1997;87(1):26–35. doi: 10.1097/00000542-199707000-00005. [DOI] [PubMed] [Google Scholar]
- 169.Burstein AH, Reiss WG, Kantor E, Anderson GD. Pharmacotherapy. 1998;18(6):1271–1276. [PubMed] [Google Scholar]
- 170.Jacqz-Aigrain E, Funck-Brentano C, Cresteil T. Pharmacogenetics. 1993;3(4):197–204. doi: 10.1097/00008571-199308000-00004. [DOI] [PubMed] [Google Scholar]
- 171.Mitchell BF, Mitchell JM, Chowdhury J, Tougas M, Engelen SME, Senff N, Heijnen I, Moore JT, Goodwin B, Wong SH, Davidge ST. Am J Obstet Gynecol. 2005;192(4):1304–1312. doi: 10.1016/j.ajog.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 172.Toell A, Kröncke KD, Kleinert H, Carlberg C. J Cell Biochem. 2002;85(1):72–82. [PubMed] [Google Scholar]
- 173.Singer AG. J Steroid Biochem Mol Biol. 1991;39(4B):627–632. doi: 10.1016/0960-0760(91)90261-3. [DOI] [PubMed] [Google Scholar]
- 174.Weusten JJ, Legemat G, van der Wouw MP, Smals AG, Kloppenburg PW, Benraad T. J Steroid Biochem. 1989;32(5):689–694. doi: 10.1016/0022-4731(89)90513-x. [DOI] [PubMed] [Google Scholar]
- 175.Claus R, Alsing W. J Endocrinol. 1976;68(3):483–484. doi: 10.1677/joe.0.0680483. [DOI] [PubMed] [Google Scholar]
- 176.Xu H, Rajesan R, Harper P, Kim RB, Lonnerdal B, Yang M, Uematsu S, Hutson J, Watson-Macdonnell J, Ito S. Br J Pharmacol. 2005;146(2):296–305. doi: 10.1038/sj.bjp.0706319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Landes N, Pfluger P, Kluth D, Birringer M, Ruhl R, Bol GF, Glatt H, Brigelius-Flohe R. Biochem Pharmacol. 2003;65(2):269–273. doi: 10.1016/s0006-2952(02)01520-4. [DOI] [PubMed] [Google Scholar]
- 178.Brigelius-Flohé R. J Plant Physiol. 2005;162(7):797–802. doi: 10.1016/j.jplph.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 179.Traber MG. Arch Biochem Biophys. 2004;423(1):6–11. doi: 10.1016/j.abb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 180.Rühl R, Sczech R, Landes N, Pfluger P, Kluth D, Schweigert FJ. Eur J Nutrit. 2004;43(6):336–343. doi: 10.1007/s00394-004-0475-1. [DOI] [PubMed] [Google Scholar]
- 181.Handschin C, Blaettler S, Roth A, Looser R, Oscarson M, Kaufmann MR, Podvinec M, Gnerre C, Meyer UA. Nuclear Receptor. 2004;2(1):7. doi: 10.1186/1478-1336-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Aparicio S, Chapman J, Stupk E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, Gelpke MD, Roach J, Oh T, Ho IY, Wong M, Detter C, Verhoef F, Predki P, Tay A, Lucas S, Richardson P, Smith SF, Clark MS, Edwards YJ, Doggett NAZ, Tavtigian SV, Pruss D, Barnstead M, Evans C, Baden H, Powell J, Glusman G, Rowen L, Hood L, Tan YH, Elgar G, Hawkins T, Venkatesh B, Rokhsar D, Brenner S. Science. 2002;297(5585):1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- 183.Hillier LW, Miller W, Birney E, Warren W, Hardison RC, Ponting CP, Bork P, Burt DW, Groenen MA, Delany ME, Dodgson JB, Chinwalla AT, Cliften PF, Clifton SW, Delehaunty KD, Fronick C, Fulton RS, Graves TA, Kremitzk C, Layman D, Magrini V, McPherson JD, Miner TL, Minx P, Nash WE, Nhan MN, Nelson JO, Oddy LG, Pohl CS, Randall-Maher J, Smith SM, Wallis JW, Yang SP, Romanov MN, Rondelli CM, Paton B, Smith J, Morrice D, Daniels L, Tempest HG, Robertson L, Masabanda JS, Griffin DK, Vignal A, Fillon V, Jacobbson L, Kerje S, Andersson L, Crooijmans RP, Aerts J, van der Poel JJ, Ellegren H, Caldwell RB, Hubbard SJ, Grafham DV, Kierzek AM, McLaren SR, Overton IM, Arakawa H, Beattie KJ, Bezzubov Y, Boardman PE, Bonfield JK, Croning MD, Davies RM, Francis MD, Humphray SJ, Scott CE, Taylor RG, Tickle C, Brown WR, Rogers J, Buerstedde JM, Wilson SA, Stubbs L, Ovcharenko I, Gordon L, Lucas S, Miller MM, Inoko H, Shiina T, Kaufman J, Salomonsen J, Skjoedt K, Wong GK, Wang J, Liu B, Wang J, Yu J, Yang H, Nefedov M, Koriabine M, Dejong PJ, Goodstadt L, Webber C, Dickens NJ, Letunic I, Suyama M, Torrents D, von Mering C, Zdobnov EM, Makova K, Nekrutenko A, Elnitski L, Eswara P, King DC, Yang S, Tyekucheva S, Radakrishnan A, Harris RS, Chiaromonte F, Taylor J, He J, Rijnkels M, Griffiths-Jones S, Ureta-Vidal A, Hoffman MM, Severin J, Searle SM, Law AS, Speed D, Waddington D, Cheng Z, Tuzun E, Eichler E, Bao Z, Flicek P, Shteynberg DD, Brent MR, Bye JM, Huckle EJ, Chatterji S, Dewey C, Pachter L, Kouranov A, Mourelatos Z, Hatzigeorgiou AG, Paterson AH, Ivarie R, Brandstrom M, Axelsson E, Backstrom N, Berlin S, Webster MT, Pourquie O, Reymond A, Ucla C, Antonarakis SE, Long M, Emerson JJ, Betran E, Dupanloup I, Kaessmann H, Hinrichs AS, Bejerano G, Furey TS, Harte RA, Raney B, Siepel A, Kent WJ, Haussler D, Eyras E, Castelo R, Abril JF, Castellano S, Camara F, Parra G, Guigo R, Bourque G, Tesler G, Pevzner PA, Smit A, Fulton LA, Mardis ER, Wilson RK. Nature. 2004;432(7018):695–716. [Google Scholar]
- 184.Suzuki T, Suzuki N, Srivastava AS, Kurokawa T. Biochem Biophys Res Comm. 2000;270(1):40–45. doi: 10.1006/bbrc.2000.2378. [DOI] [PubMed] [Google Scholar]
- 185.Bresolin T, de Freitas Rebelo M, Bainy ACD. Comp Biochem Physiol Part C. 2005;140(3–4):403–407. doi: 10.1016/j.cca.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 186.Dai G, Wan YJ. Curr Drug Metab. 2005;6(4):341–355. doi: 10.2174/1389200054633862. [DOI] [PubMed] [Google Scholar]
- 187.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM. Nature. 2000;406(6794):435–439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 188.Xie W, Yeuh MF, Radominska-Pandya A, Saini SPS, Negishi Y, Bottroff BS, Cabrera GY, Tukey RH, Evans RM. Proc Natl Acad Sci USA. 2003;100(7):4150–4155. doi: 10.1073/pnas.0438010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, Moore DD. Proc Natl Acad Sci USA. 2003;100(7):4156–4161. doi: 10.1073/pnas.0630614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Assem M, Schuetz EG, Leggas M, Sun D, Yasuda K, Reid G, Zelcer N, Adachi M, Strom S, Evans RM, Moore DD, Borst P, Schuetz JD. J Biol Chem. 2004;279(21):22250–22257. doi: 10.1074/jbc.M314111200. [DOI] [PubMed] [Google Scholar]
- 191.Hahn ME. Chemico-Biol Inteact. 2002;141(1–2):131–160. doi: 10.1016/s0009-2797(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 192.Ertl RP, Bandiera SM, Buhler DR, Stegeman JJ, Winston GW. Toxicol Appl Pharmacol. 1999;157(3):157–165. doi: 10.1006/taap.1999.8669. [DOI] [PubMed] [Google Scholar]
- 193.Schuetz EG, Schuetz JD, Grogan WM, Naray-Fejes-Toth A, Fejes-Toth G, Raucy J, Guzelian P, Gionela K, Watlington CO. Arch Biochem Biophys. 1992;294(1):206–214. doi: 10.1016/0003-9861(92)90159-t. [DOI] [PubMed] [Google Scholar]
- 194.Hasselberg L, Meier S, Svardal A, Hegelund T, Celander MC. Aquat Toxciol. 2004;67(4):303–313. doi: 10.1016/j.aquatox.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 195.Hegelund T, Ottosson K, Rådinger M, Tomberg P, Celander MC. Environ Toxicol Chem. 2004;23(5):1326–1334. doi: 10.1897/03-155. [DOI] [PubMed] [Google Scholar]
- 196.Tseng HP, Hseu TH, Buhler DR, Wang WD, Hu CH. Toxicol Appl Pharmacol. 2005;205(3):247–258. doi: 10.1016/j.taap.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 197.Mizwicki MT, Keidel D, Bula CM, Bishop JE, Zanello LP, Wurtz JM, Moras D, Norman AW. Proc Natl Acad Sci USA. 2004;101(35):12876–12881. doi: 10.1073/pnas.0403606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Vanhooke JL, Benning MM, Bauer CB, Pike JW, DeLuca HF. Biochemistry. 2004;43(14):4101–4110. doi: 10.1021/bi036056y. [DOI] [PubMed] [Google Scholar]
- 199.Bainy ACD, Stegeman JJ. Marine Environ Res. 2004;58(1):133–134. [Google Scholar]
- 200.Kamei Y, Kawada T, Fukuwatari T, Ono T, Kato S, Sugimoto E. Gene. 1995;152(2):281–282. doi: 10.1016/0378-1119(94)00735-b. [DOI] [PubMed] [Google Scholar]
- 201.Lu Z, Hanson K, DeLuca HF. Arch Biochem Biophys. 1997;339(1):99–106. doi: 10.1006/abbi.1996.9864. [DOI] [PubMed] [Google Scholar]
- 202.Hughes S, Zelus D, Mouchiroud D. Mol Biol Evol. 1999;16(11):1521–1527. doi: 10.1093/oxfordjournals.molbev.a026064. [DOI] [PubMed] [Google Scholar]
- 203.Ciesielski F, Rochel N, Mitschler A, Kouzmenko A, Moras D. J Steroid Biochem Mol Biol. 2004;89–90(1–5):55–59. doi: 10.1016/j.jsbmb.2004.03.109. [DOI] [PubMed] [Google Scholar]
- 204.Yoshinari K, Sueyoshi T, Moore R, Negishi M. Mol Pharmacol. 2001;59(2):278–284. doi: 10.1124/mol.59.2.278. [DOI] [PubMed] [Google Scholar]
- 205.Sakai H, Iwata H, Kim EY, Tanabe S, Baba N. Marine Environ Res. 2004;58(2–5):107–111. doi: 10.1016/j.marenvres.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 206.Drocourt L, Ourlin JC, Pascussi JM, Maurel P, Vilarem MJ. J Biol Chem. 2002;277(28):25125–25132. doi: 10.1074/jbc.M201323200. [DOI] [PubMed] [Google Scholar]
- 207.Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA. Mol Pharmacol. 2001;60(3):427–431. [PubMed] [Google Scholar]
- 208.Maglich JM, Stoltz CM, Goodwin B, Hawkins-brown D, Moore JT, Kliewer SA. Mol Pharmacol. 2002;62(3):638–646. doi: 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- 209.Gerbal-Chaloin S, Pascussi JM, Pichard-Garcia L, Daujat M, Waechter F, Fabre JM, Carrère N, Maurel P. Drug Metab Dispos. 2001;29(3):242–251. [PubMed] [Google Scholar]
- 210.Goodwin B, Hodgson E, D’Costa DJ, Robertson GR, Liddle C. Mol Pharmacol. 2002;62(2):359–365. doi: 10.1124/mol.62.2.359. [DOI] [PubMed] [Google Scholar]
- 211.Rosenfeld JM, Vargas R, Xie W, Evans RM. Mol Endocrinol. 2003;17(7):1268–1282. doi: 10.1210/me.2002-0421. [DOI] [PubMed] [Google Scholar]
- 212.Synold TW, Dussault I, Forman BM. Nature Med. 2001;7(5):584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 213.Bertilsson G, Berkenstam A, Blomquist P. Biochem Biophys Res Comm. 2001;280(1):139–144. doi: 10.1006/bbrc.2000.4066. [DOI] [PubMed] [Google Scholar]
- 214.Pascussi JM, Jounaidi Y, Drocourt L, Domergue J, Balabaud C, Maurel P, Vilarem MJ. Biochem Biophys Res Comm. 1999;260(2):377–381. doi: 10.1006/bbrc.1999.0745. [DOI] [PubMed] [Google Scholar]
- 215.Sugatani J, Kojima H, Ueda A, Kaziaki S, Yoshinari K, Gong QH, Owens IS, Negishi M, Sueyoshi T. Hepatology. 2001;33(5):1232–1238. doi: 10.1053/jhep.2001.24172. [DOI] [PubMed] [Google Scholar]
- 216.Frank C, Gonzalez MM, Oinenon C, Dunlop TW, Carlberg C. J Biol Chem. 2003;278(44):43299–43310. doi: 10.1074/jbc.M305186200. [DOI] [PubMed] [Google Scholar]
- 217.Burk O, Arnold KA, Geick A, Tegude H, Eichelbaum M. Biol Chem. 2005;386(6):503–513. doi: 10.1515/BC.2005.060. [DOI] [PubMed] [Google Scholar]
- 218.Geick A, Eichelbaum M, Burk O. J Biol Chem. 2001;276(18):14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 219.Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, Tontonoz P, Kliewer S, Willson TM, Edwards PA. J Biol Chem. 2002;277(4):2908–2915. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- 220.Ohyama Y, Ozono K, Uchida M, Shinki T, Kato S, Suda T, Yamamoto O, Noshiro M, Kato Y. J Biol Chem. 1994;269(14):10545–10550. [PubMed] [Google Scholar]
- 221.Pascussi JM, Robert A, Nguyen M, Walrant-Debray O, Garabedian M, Martin P, Pineau T, Saric J, Navarro F, Maurel P, Vilarem MJ. J Clin Invest. 2005;115(1):177–186. doi: 10.1172/JCI21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Fraser DJ, Zumsteg A, Meyer UA. J Biol Chem. 2003;278(41):39392–39401. doi: 10.1074/jbc.M306148200. [DOI] [PubMed] [Google Scholar]
- 223.Podvinec M, Handschin C, Looser R, Meyer UA. Proc Natl Acad Sci USA. 2004;101(24):9127–9132. doi: 10.1073/pnas.0401845101. [DOI] [PMC free article] [PubMed] [Google Scholar]