

Abstract
Hanatoxin inhibits voltage-gated K+ channels by modifying the energetics of activation. We studied the molecular determinants and physical location of the Hanatoxin receptors on the drk1 voltage-gated K+ channel. First, we made multiple substitutions at three previously identified positions in the COOH terminus of S3 to examine whether these residues interact intimately with the toxin. We also examined a region encompassing S1–S3 using alanine-scanning mutagenesis to identify additional determinants of the toxin receptors. Finally, guided by the structure of the KcsA K+ channel, we explored whether the toxin interacts with the peripheral extracellular surface of the pore domain in the drk1 K+ channel. Our results argue for an intimate interaction between the toxin and the COOH terminus of S3 and suggest that the Hanatoxin receptors are confined within the voltage-sensing domains of the channel, at least 20–25 Å away from the central pore axis.
Keywords: gating modifier toxin, scanning mutagenesis, voltage-dependent gating, protein–protein interaction
INTRODUCTION
Voltage-gated K+ channels are tetramers with each subunit containing six transmembrane segments, termed S1–S6. As illustrated in Fig. 1, we can think of these channels as constructed from two types of domains: a central pore domain formed by S5–S6 and four surrounding voltage-sensing domains, each comprised of S1–S4. The structure of the pore domain is likely to be similar to the crystal structure of the KcsA K+ channel, a bacterial channel that is homologous to S5–S6 in voltage-gated K+ channels (Schrempf et al. 1995; Doyle et al. 1998; MacKinnon et al. 1998; Heginbotham et al. 1999). Thus, both S5 and S6 in voltage-gated K+ channels are membrane-spanning α helices with the linker between them forming a short pore helix and the selectivity filter. A growing body of evidence suggests that the voltage-sensing domains are constructed from S1 through S4 (Papazian et al. 1991, Papazian et al. 1995; Liman et al. 1991; Perozo et al. 1994; Planells-Cases et al. 1995; Aggarwal and MacKinnon 1996; Larsson et al. 1996; Mannuzzu et al. 1996; Seoh et al. 1996; Yang et al. 1996; Yusaf et al. 1996; Tiwari-Woodruff et al. 1997; Cha and Bezanilla 1997; Smith-Maxwell et al. 1998a,Smith-Maxwell et al. 1998b; Li-Smerin and Swartz 1998; Ledwell and Aldrich 1999; Monks et al. 1999; Hong and Miller 2000; Li-Smerin et al. 2000a). Compared with the pore domain, much less is known about the structure of this important region of voltage-gated K+ channels. As illustrated in Fig. 1 A, the first four transmembrane segments (S1, S2, S3, and S4) are thought to be membrane-spanning α helices (Monks et al. 1999; Hong and Miller 2000; Li-Smerin et al. 2000a). There is also evidence for additional helices in the extracellular linkers between S1 and S2 and between S3 and S4 (Li-Smerin et al. 2000a).
Figure 1.
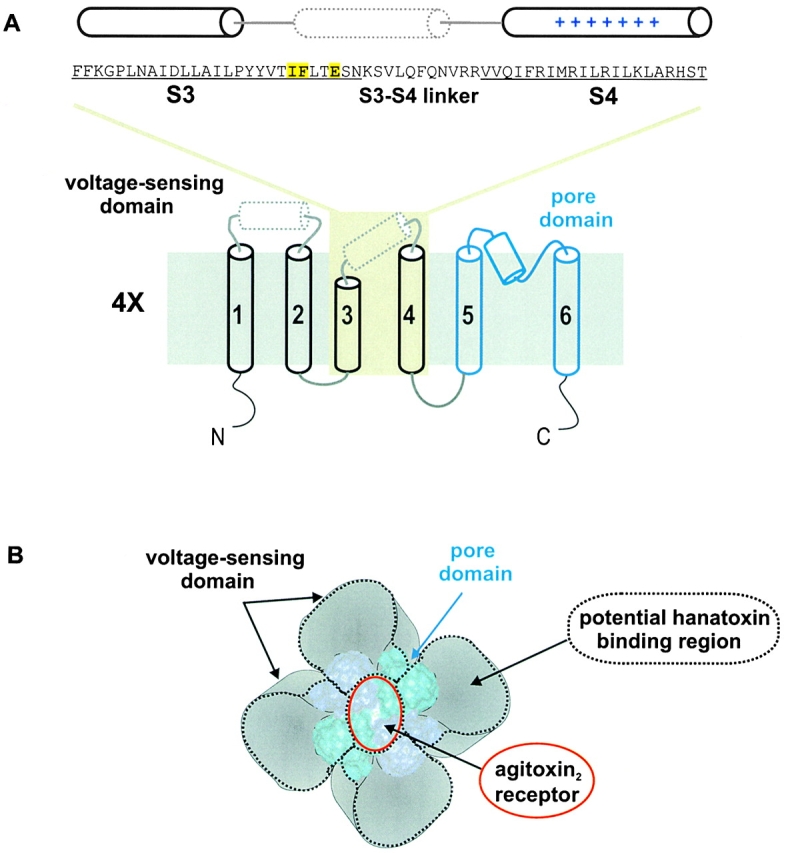
Topology and architecture of voltage-gated K+ channels. (A) Sequence of the drk1 K+ channel for the region starting at the NH2 terminus of S3 and ending at the COOH-terminal of S4 and the putative topology of a single α subunit. Yellow highlighting indicates the three positions (I273, F274, and E277) where point mutations have been found to alter the binding affinity of Hanatoxin (Swartz and MacKinnon 1997b). The putative demarcations of the S3 and S4 transmembrane segments, indicated by underlining, are based on Kyte-Doolittle hydrophobicity analysis. The cylinders above the sequence indicated the approximate positions of three α helices. The solid cylinders (for S3 and S4) are based on α-helical periodicity detected using alanine- and tryptophan-scanning mutagenesis (Hong and Miller 2000; Li-Smerin et al. 2000a). The dotted cylinder positioned near the COOH terminus of S3 and the S3–S4 linker was proposed based on helical periodicity detected with alanine-scanning mutagenesis (Li-Smerin et al. 2000a). The topology of a single K+ channel subunit shown (bottom) illustrates the first four transmembrane segments (S1–S4, in black) comprising the voltage-sensing domain and the last two transmembrane segments (S5 to S6, in blue) the pore domain. The dotted cylinder in the S1–S2 linker is based on helical periodicity detected with alanine-scanning mutagenesis (Li-Smerin et al. 2000a). The top is extracellular and the bottom is intracellular. (B) Architecture of voltage-gated K+ channels containing a central pore domain and four surrounding voltage-sensing domains drawn according to Li-Smerin et al. 2000b. The pore domain is represented by the crystal structure of KcsA (Doyle et al. 1998), a simple K+ channel homologous to the S5–S6 region of voltage-gated K+ channels. Adjacent pore domain subunits in the tetramer are illustrated using different shades of blue. The red ring over the ion conduction pore indicates the footprint of Agitoxin2 (∼20 × 30 Å), a pore-blocking toxin (MacKinnon et al. 1998). Each surrounding voltage-sensing domain is formed by S1–S4 transmembrane helices. The four areas surrounded by black dotted lines are regions that potentially interact with Hanatoxin, based on cohabitation of Hanatoxin and Agitoxin2. The overall dimensions of the channel are drawn using estimates of 80 × 80 Å from electron microscopy (Li et al. 1994).
A large number of protein toxins isolated from venomous animals interact with voltage-gated ion channels and either physically block ion conduction or modify voltage-dependent gating. The pore-blocking toxins interact with the outer vestibule of the channel, as illustrated by the receptor for Agitoxin2 in Fig. 1 B (MacKinnon and Miller 1988, MacKinnon and Miller 1989; Miller 1988, Miller 1995; Hurst et al. 1991; Stocker et al. 1991; Park and Miller 1992a,Park and Miller 1992b; Goldstein and Miller 1993; Garcia et al. 1994; Goldstein et al. 1994; Lu and MacKinnon 1997; Imredy et al. 1998; Jin and Lu 1998). In fact, these toxins were initially used to identify the pore-forming region of the channel and subsequently to probe the three-dimensional structure of this region (MacKinnon and Miller 1988; Hurst et al. 1991; Stampe et al. 1994; Stocker and Miller 1994; Goldstein et al. 1994; Hidalgo and MacKinnon 1995; Ranganathan et al. 1996; Naranjo and Miller 1996; MacKinnon et al. 1998). Importantly, many of the structural constraints obtained using pore-blocking toxins have been born out by the crystal structure of the KcsA K+ channel, recently solved by x-ray diffraction (Doyle et al. 1998; MacKinnon et al. 1998). In contrast to the pore-blocking toxins, the location and structural determinants of the receptors for gating modifier toxins are poorly understood, but presumably these toxins interact with regions of the channel involved in gating. The precedent set by the pore-blocking toxins suggests that gating modifier toxins should be very useful probes for studying these gating structures.
Hanatoxin is a protein toxin from spider venom that inhibits the drk1 voltage-gated K+ channel by binding to receptors on the extracellular face of the channel and modifying the energetics of gating (Swartz and MacKinnon 1995, Swartz and MacKinnon 1997a). This mechanism was suggested by the observation that toxin-bound channels can open, but require large depolarizing voltages to do so. Unlike the pore-blocking toxins that bind with 1:1 stoichiometry, four Hanatoxin molecules can bind to the external surface of a single K+ channel (Swartz and MacKinnon 1997a). An initial search for residues forming the Hanatoxin receptors identified three positions in the COOH-terminal part of S3 (I273, F274, and E277), where point mutations decrease toxin binding affinity (Fig. 1 A, yellow highlighted area) (Swartz and MacKinnon 1997b). A significant constraint on the location of the Hanatoxin receptors came from the observation that Agitoxin2 (Garcia et al. 1994), a pore-blocking toxin, can co-occupy the K+ channel along with Hanatoxin (Fig. 1) (Swartz and MacKinnon 1997b). The cohabitation of pore-blocking and gating-modifier toxins argues for peripheral locations of the Hanatoxin receptors, probably >15 Å from the central pore axis. The peripheral location of the Hanatoxin receptors fits nicely with the 4:1 stoichiometry between toxin and channel since these channels are homotetramers. However, the actual position of the Hanatoxin receptors remains poorly defined because the pore-blocking toxin receptor constitutes a relatively small fraction of the large (∼80 × 80 Å) extracellular surface of the channel (Li et al. 1994). Thus, Hanatoxin might interact with the channel anywhere within the black dotted lines in Fig. 1 B.
In this paper, we studied the structural components and location of the Hanatoxin receptors on the drk1 K+ channel. First, we examined whether the three previously identified residues in the COOH-terminal end of S3 interact with the toxin by looking for patterns arising from multiple substitutions at each position. Second, we examined the possible contribution of additional residues in forming the Hanatoxin receptors by Ala-scanning a previously unexplored region between the NH2-terminal side of S1 and the COOH-terminal end of S3. Finally, we explored the possibility that Hanatoxin interacts with residues on the peripheral surface of the pore domain. Our results are consistent with an intimate interaction between Hanatoxin and channel residues in the COOH-terminal part of S3. In addition, our results suggest that the toxin does not interact with the extracellular surface of the pore domain, but rather exclusively interacts with the voltage-sensing domains of the channel.
MATERIALS AND METHODS
Mutagenesis and Channel Expression
The cDNA encoding the wild-type drk1 K+ channel (Frech et al. 1989) used in this study contained several unique restriction sites that were previously introduced using PCR (Swartz and MacKinnon 1997b). To construct point mutations of the channel, mutant fragments were first generated by PCR and then ligated into appropriately digested vectors. Mutations were confirmed by dideoxy (Sanger et al. 1977) or automated DNA sequencing. cDNAs encoding wild-type and mutant channels were linearized with NotI and transcribed with T7 RNA polymerase.
Oocytes from Xenopus laevis frogs were removed surgically and incubated with agitation for 1–1.5 h in a solution containing (mM): 82.5 NaCl, 2.5 KCl, 1 MgCl2, 5 HEPES, and 2 mg/ml collagenase (Worthington Biochemical Corp.), pH 7.6 with NaOH. Defolliculated oocytes were injected with cRNA and incubated at 17°C in a solution containing (mM): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, 50 μg/ml gentamicin (GIBCO BRL), pH 7.6 with NaOH for 1–5 d before recording.
Examination of the Toxin-Channel Interaction
Two-electrode voltage-clamp recording techniques were employed to study the toxin-channel interaction using an OC-725C oocyte clamp (Warner Instruments). Oocytes were studied in a 160-μl recording chamber that was perfused with a solution containing (mM): 50 RbCl, 50 NaCl, 1 MgCl2, 0.3 CaCl2, and 5 HEPES, pH 7.6 with NaOH. Data were filtered at 2 kHz (eight-pole Bessel) and digitized at 10 kHz. Microelectrode resistances were between 0.2 and 1.2 MΩ when filled with 3 M KCl. All experiments were performed at room temperature (∼22°C).
The equilibrium dissociation constant for Hanatoxin binding to closed or resting channels was determined using very negative holding voltages (−120 to −80 mV) where no steady state inactivation could be detected. The fraction of unbound channels was estimated using depolarizations that were too weak to open toxin bound channels and too short to perturb the equilibrium for toxin binding to resting channels, as previously described (Swartz and MacKinnon 1997a,Swartz and MacKinnon 1997b). After toxin-bound channels are opened by strong depolarization, they close more rapidly than unbound channels after repolarization (Swartz and MacKinnon 1997a). We therefore examined the kinetics of deactivation using tail currents carried by 50 mM Rb+ (to slow deactivation) to confirm that toxin-bound channels did not contribute to the currents measured with weak depolarizations. For all channels, we recorded tail current voltage-activation relations in the absence and presence of different concentrations of toxin. After addition of the toxin to the recording chamber, the equilibration between toxin and channel was monitored using weak depolarizations elicited at 5–20-s intervals. Recovery from toxin inhibition after removing the toxin from the recording chamber was routinely monitored but not examined quantitatively because of complexities arising from the 4:1 stoichiometry between toxin and channel (Swartz and MacKinnon 1997a,Swartz and MacKinnon 1997b). The ratio of tail current (I/I0) recorded in the presence (I) and absence (I0) of toxin was calculated for various strength depolarizations, typically −50 to +60 mV. The value of I/I0 measured in the plateau phase at negative voltages (In/In 0), where toxin-bound channels do not open, was taken as equal to the fraction of unbound channels (see Fig. 2). The equilibrium dissociation constant (K d) for toxin binding was calculated according to:
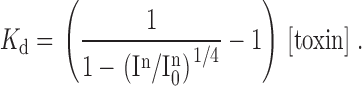 |
Figure 2.

Inhibition of the drk1 K+ channel by Hanatoxin. (A) Families of current records obtained from two-electrode voltage-clamp recording of oocytes expressing the wild-type (left) and two mutant drk1 K+ channels (F274R, middle; E277K, right), both in the absence and presence of 1 μM toxin. Currents were elicited by voltage steps beginning at −50 mV and incrementing in 5-mV steps from a holding voltage of −80 mV. 15 traces are shown for wild-type and F274R families (−50 to +20 mV), while 17 records are shown for the E277K family (−50 to +30 mV). Tail currents carried by Rb+ were elicited by repolarization to −50 mV. All traces are shown without subtraction of leak or capacitive currents. (B) Tail current voltage-activation relations for wild-type (left), F274R (middle), and E277K (right). Tail current amplitudes were measured 2–3 ms after repolarization in the absence (○) or presence (•) of 1 μM Hanatoxin. Same oocytes as in A. (C) Fraction of uninhibited tail current plotted against test voltage at different toxin concentrations for wild-type, F274R, and E277K. I is the tail current measured in the presence of toxin and I0 is the tail current measured in the absence of toxin. Data are shown as mean ± SEM for three to six cells at each toxin concentration.
This equation assumes four independent and equivalent binding sites on the drk1 K+ channel for Hanatoxin. For mutant channels with altered gating properties, all voltage protocols were adjusted appropriately so that the plateau phase in the I/I0–voltage relation was well defined. The fraction of uninhibited current (I/I0) was initially measured for all mutant channels using between 100 and 250 nM Hanatoxin. The concentration dependence for toxin inhibition was further examined using two or three toxin concentrations for all mutants displaying >10-fold changes in toxin K d in the initial screen.
Hanatoxin was purified from tarantula venom as previously described (Swartz and MacKinnon 1995). The native toxin preparation is an approximately equal mixture of two isoforms that differ only at position 13, where Hanatoxin1 contains serine and Hanatoxin2 contains alanine. The two isoforms most likely have similar binding affinities because the concentration dependence for inhibition of the channel by the native mixture and synthetic Hanatoxin1 are similar (Takahashi et al. 2000).
RESULTS
Hanatoxin inhibits the drk1 voltage-gated K+ channel by shifting activation to more depolarized voltages (Swartz and MacKinnon 1997a). We set out to localize the Hanatoxin receptors on the drk1 voltage-gated K+ channel by examining the effects of point mutations on toxin binding affinity. Wild-type and mutant channels were expressed in Xenopus oocytes and the interaction between toxin and channel were examined electrophysiologically. We determined the equilibrium dissociation constant for toxin binding to mutant and wild-type channels by studying the concentration dependence for toxin occupancy of the channel as previously described (Swartz and MacKinnon 1997a) (see materials and methods). In this approach, the fraction of unbound channels is estimated by measuring the fraction of uninhibited current at negative voltages where toxin bound channels do not open.
Effects of Multiple Substitutions in the COOH-terminal Part of S3
Mutations to Ala at three positions in the COOH-terminal part of S3 in the drk1 K+ channel were previously reported to decrease Hanatoxin binding affinity by ∼10–25-fold (Swartz and MacKinnon 1997b). In the wild-type channel, two of these residues are hydrophobic (I273 and F274) and one is acidic (E277). To explore possible interactions of these three channel residues with the toxin, we made multiple substitutions (one at a time) at each of these positions and determined the equilibrium dissociation constant for Hanatoxin binding to each mutant channel. Our approach was to look for patterns between changes in the toxin K d and the chemical nature of the substituted amino acids.
The measurement of toxin binding affinity is illustrated in Fig. 2 for the wild-type and two exemplary mutant channels (F274R and E277K). Voltage-activation relations were obtained in the absence and presence of various toxin concentrations using tail-current protocols (Fig. 2A and Fig. B). The fraction of uninhibited tail current was then measured for different strength depolarizations, as shown in Fig. 2 C. This fraction of uninhibited current in the plateau phase at negative voltages (In/In 0), which approximates the fraction of unbound channels, was used to calculate the K d for toxin binding to the K+ channel (see materials and methods) (Swartz and MacKinnon 1997a). In the absence of toxin, the voltage-activation relations for F274R and E277K displayed only small rightward shifts relative to the wild-type channel. In contrast, for both mutants, even very high toxin concentrations produced much less inhibition at negative voltages when compared with the wild-type channel, suggesting that the binding affinity of the toxin for these mutants is greatly diminished.
Fig. 3 shows the results for multiple substitutions at F274, where mutation to Ala was previously reported to reduce Hanatoxin binding affinity by ∼25-fold. The 14 substitutions made at F274 include 8 hydrophobic residues (Cys, Ala, Met, Pro, Tyr, Trp, Val, Ile), 2 basic residues (Lys, Arg), 2 acidic residues (Glu, Asp), Ser, and Gly. Fig. 3 A shows the dependence of In/In 0 on toxin concentration for the wild-type and two mutant channels, F274G and F274R. For the wild-type channel, the data were well described by a model assuming four equivalent and independent binding sites for Hanatoxin on the drk1 K+ channel with a K d of 103 nM for toxin binding to each site (see materials and methods). The two mutants, F274G and F274R, greatly reduced the toxin binding affinity with K d values of 4.4 and 61.6 μM, respectively. The normalized Hanatoxin K d values for all 14 substitutions at position 274 are summarized in Fig. 3 B. One prominent trend in the data is that substitutions with hydrophobic amino acids cause the smallest perturbation in Hanatoxin binding energy. The range of ΔΔG values [ΔΔG = −RT ln (K d wt/K d mut)] for hydrophobic residues (Ile, Val, Trp, Tyr, Pro, Met, Ala, Cys) is 1.2–1.8 kcal mol−1, while the range of ΔΔG values for nonhydrophobic residues (Gly, Asp, Ser, Glu, Arg, Lys) is 2.4–3.7 kcal mol−1. Although the hydrophobic residues caused the smallest perturbations, even these perturbations were quite significant, implying a very close and specific interaction with the toxin. These results are consistent with an intimate hydrophobic interaction between F274 and the toxin. It is also interesting that at position 274, substitutions with either Lys or Arg produce the largest perturbations, with ΔΔG values of 3.7 and 3.6 kcal mol−1, respectively. One possibility is that F274 is positioned close to a basic toxin residue in the channel-toxin complex (see discussion).
Figure 3.
Effects of multiple substitutions at position 274 on Hanatoxin binding affinity. (A) Concentration dependence for inhibition of the wild-type and two mutant drk1 K+ channels by Hanatoxin. In/In 0 is the value of I/I0 measured in the plateau phase at negative voltages (e.g., Fig. 2 C). Symbols are experimental data for the mean ± SEM of three to five cells. Solid lines correspond to In/In 0 = (1 − P)4, where P = [toxin]/([toxin] + K d), with K d values of 103 nM, 4.4 μM, and 61.6 μM for WT, F274G, and F274R, respectively. The equation assumes four equivalent and independent toxin binding sites per channel. (B) Normalized K d values for 14 substitutions at the position 274. Mean ± SEM (n = 3–15) for each mutant channel. The corresponding ΔΔG values are (kcal mol−1): 3.7 K, 3.6 R, 2.8 E, 2.6 S, 2.6 D, 2.4 G, 1.8 C, 1.6 A, 1.5 M, 1.5 P, 1.5 Y, 1.4 W, 1.4 V, and 1.2 I.
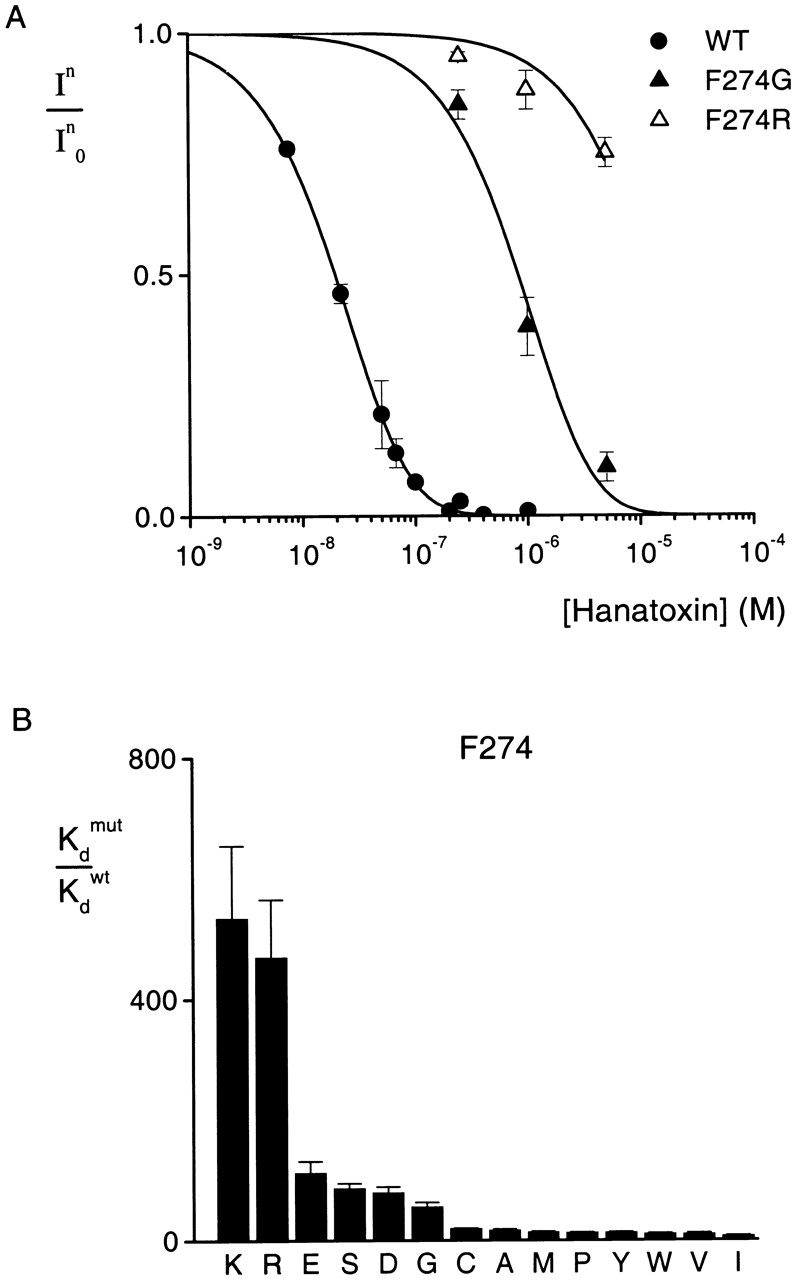
We next examined the effects of multiple substitutions at E277. Fig. 4 shows the concentration dependence of In/In 0 for two mutant channels, E277Y and E277K. Compared with the wild-type channels (same data as for Fig. 3), E277Y and E277K displayed greatly reduced binding affinities for Hanatoxin, with K d values of 2.5 and 17.3 μM, respectively. The normalized Hanatoxin K d values for all eight substitutions at position 277 are summarized in Fig. 4 B. An interesting correlation between the magnitude of the effects and the substituted amino acids emerges. Substitutions with basic residues have the largest effects (ΔΔG = 2.8 kcal mol−1), while those with neutral residues have more moderate effects (ΔΔG from 1.3 to 2.2 kcal mol−1). Mutation of E277 to Asp produced no discernible change in toxin binding affinity (ΔΔG = 0.04 kcal mol−1). The correlation between the charge of the side chain at 277 and toxin binding energy is to a first approximation consistent with a through-space electrostatic interaction between this position on the channel and a basic residue on the toxin. However, two observations argue for a more intimate interaction. First, substitutions with various neutral amino acids produce significantly different perturbations, with ΔΔG values ranging from 1.3 to 2.2 kcal mol−1. Second, the large values of ΔΔG for all mutations (1.3–2.8 kcal mol−1), with the exception of Asp, are more consistent with intimate or short-range interactions. We speculate that E277 interacts with a basic residue on Hanatoxin through formation of a salt bridge. In this case, the apparent tranquility observed upon substitution of Asp for Glu would imply some degree of conformational flexibility in either the toxin or the channel.
Figure 4.
Effects of multiple substitutions at position 277 on Hanatoxin binding affinity. (A) Concentration dependence for inhibition of the wild-type and two mutant drk1 K+ channels by Hanatoxin. In/In 0 is the value of I/I0 measured in the plateau phase at negative voltages (e.g., Fig. 2 C). Symbols are experimental data for the mean ± SEM of three to six cells. Solid lines correspond to In/In 0 = (1 − P)4, where P = [toxin]/([toxin] + K d), with K d values of 2.5 and 17.3 μM for E277Y and E277K, respectively. Data for the wild-type channel are the same as in Fig. 3. (B) Normalized K d values of eight substitutions at position 277. Mean ± SEM (n = 3–13) for each mutant channel. The corresponding ΔΔG values are (kcal mol−1): 2.8 R, 2.8 K, 2.2 W, 2.0 Y, 1.6 G, 1.5 Q, 1.3 N, and 0.04 D.
The results of 13 substitutions at position 273 are shown in Fig. 5. The mutation to Tyr produces the largest change in toxin binding affinity (ΔΔG = 2.8 kcal mol−1). Substitutions with Thr, Arg, Asp, Ala, Gly, and Cys cause moderate changes (ΔΔG = 1.5–1.7 kcal mol−1) in toxin binding affinity, while substitutions with Val, Met, Trp, Phe, His, and Leu produce only minor changes (ΔΔG ≤ 0.5 kcal mol−1). These results are more complex than observed at either F274 or E277. The observation that most hydrophobic substitutions (Val, Met, Trp, Phe, and Leu) produced little change in toxin binding affinity with ΔΔG < 0.3 kcal mol−1 seems to hint at a hydrophobic interaction between position 273 and the toxin. However, the pattern is not simple because mutation to Tyr produces the largest perturbations (ΔΔG = 2.8 kcal mol−1) and very significant perturbations (1.6 and 1.7 kcal mol−1) were seen for mutation to Ala and Cys, two smaller hydrophobic residues. A partial explanation might be that I273 is involved in a hydrophobic interaction with the toxin and that there is a requirement for both a large and hydrophobic side-chain at position 273.
Figure 5.
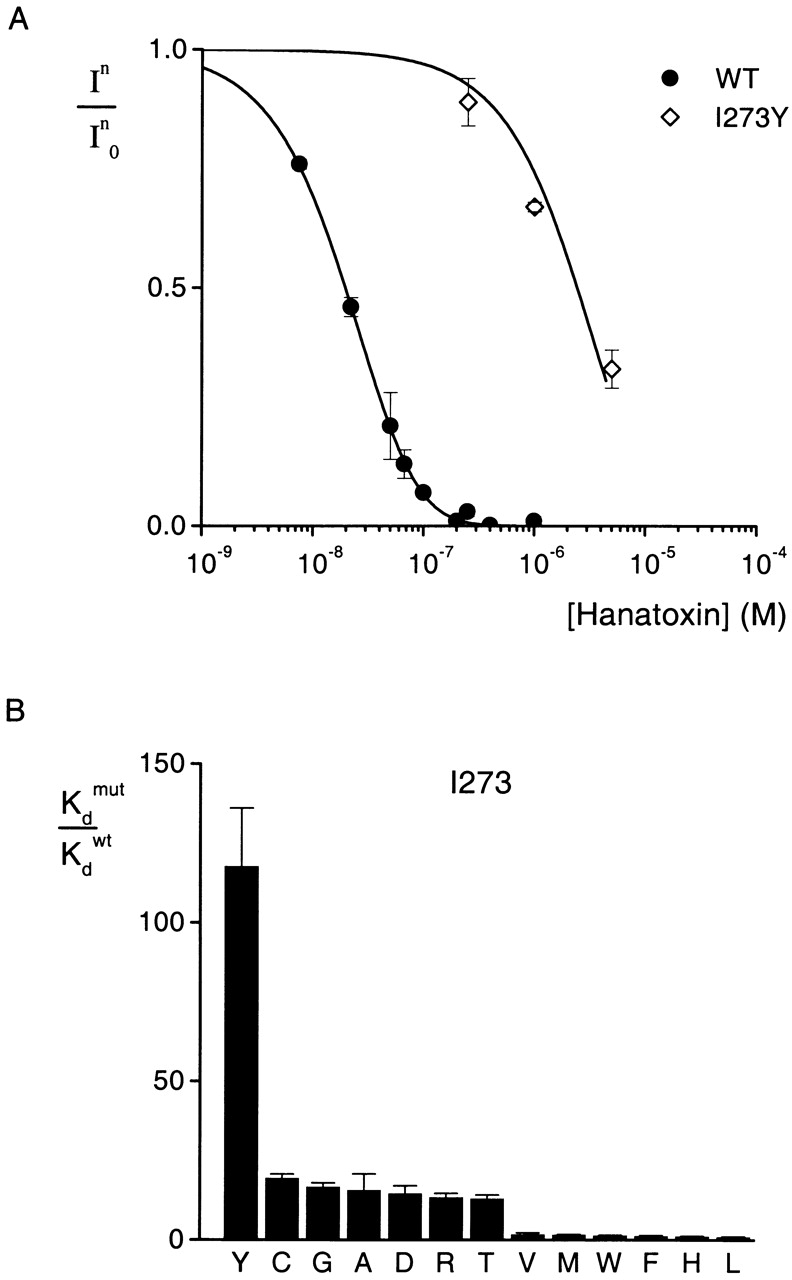
Effects of multiple substitutions at position 273 on Hanatoxin binding affinity. (A) Concentration dependence for inhibition of the wild-type and a mutant drk1 K+ channel by Hanatoxin. In/In 0 is the value of I/I0 measured in the plateau phase at negative voltages (e.g., Fig. 2 C). Symbols are experimental data for the mean ± SEM of three to four cells. Solid lines correspond to In/In 0 = (1 − P)4, where P = [toxin]/([toxin] + K d), with a K d value of 12.9 μM for I273Y. Data for the wild-type channel are the same as presented in Fig. 3. (B) Normalized K d values of 13 substitutions at I273. Mean ± SEM (n = 3–10) for each mutant channel. The corresponding ΔΔG values are (kcal mol−1): 2.8 Y, 1.7 C, 1.6 G, 1.6 A, 1.6 D, 1.5 R, 1.5 T, 0.3 V, 0.3 M, 0.2 W, 0.1 F, 0.05 H, 0.03 L.
Alanine-scanning Mutagenesis from the NH2-terminal part of S1 through the COOH-terminal Edge of S3
The above results are consistent with intimate interactions between three residues in the COOH-terminal end of S3 and Hanatoxin. While the surface area at the toxin–channel interface is unknown, the dimensions of Hanatoxin (∼20 × 25 Å) argue that many channel residues, perhaps 10 or more, are located at the toxin–channel interface (Takahashi et al. 2000). We therefore continued to search for residues contributing to the Hanatoxin receptors by Ala-scanning from the NH2-terminal side of S1 through the COOH-terminal edge of S3 (started at K185 and ended at Y269). Fig. 6 shows the normalized Hanatoxin K d values for the 85 mutants made in this region (S1–S3), together with the previous results for an Ala-scan of the S3–S4 linker and the S4 segment (Swartz and MacKinnon 1997b), shown for comparison. A Kyte-Doolittle hydrophobicity index profile for this region is shown to mark the approximate position of each transmembrane segment (S1–S4). In contrast to the previously observed large perturbations in the COOH-terminal part of S3 (and small changes and in S4), the mutations in S1 through most of S3 caused no more than threefold changes in the toxin K d. These results suggest that residues from the NH2-terminal end of S1 to the NH2-terminal part of S3 do not appear to be critical components of the Hanatoxin receptors.
Figure 6.

Alanine-scanning mutagenesis of S1 to S3. Bar graph plot of normalized Hanatoxin K d values for mutations spanning from K185 in S1 to T311 in S4. Data for Y270 in the COOH terminal of S3 through T311 in S4 are presented from a previous study (Swartz and MacKinnon 1997b) for comparison. Letters and numbers indicate the wild-type residues and their positions, respectively. All residues were mutated to Ala, except for the native Ala residues, which were mutated to Tyr. K d values are mean ± SEM with n = 3 or 4 for each mutant obtained using between 100 and 250 nM Hanatoxin. The solid line superimposed on the bar graph is a 17-residue window analysis of the Kyte-Doolittle hydrophobicity index (Kyte and Doolittle 1982).
Does Hanatoxin Interact with Residues on the Peripheral Surface of the Pore Domain?
From previous experiments, we know that Agitoxin2, a well-studied pore-blocking toxin, and Hanatoxin can simultaneously occupy the drk1 K+ channel (Swartz and MacKinnon 1997b). Agitoxin2 belongs to a family of closely related toxins that bind over the central pore axis and have dimensions of ∼20 × 30 Å (MacKinnon and Miller 1988; Miller 1988; Park and Miller 1992a; Hidalgo and MacKinnon 1995; Krezel et al. 1995; Ranganathan et al. 1996; MacKinnon et al. 1998). Thus, co-occupancy of the two toxins indicates that the Hanatoxin receptors must be located in peripheral regions on the extracellular surface of the channel, probably at least 15 Å away from the central pore axis (Fig. 7 B). However, from the structure of the KcsA K+ channel (Doyle et al. 1998), we can infer that a large fraction of the extracellular surface of the pore domain lies outside the footprint of Agitoxin2 (MacKinnon et al. 1998) and therefore represents a potential interaction surface for Hanatoxin. To test for a possible interaction between Hanatoxin and the peripheral surface of the pore domain in the drk1 K+ channel, we aligned the sequences for the transmembrane part of KcsA (TM1 through TM2) and the pore domain of the drk1 K+ channel (S5 through S6) (Fig. 7 A). The alignment shows that >50% of the residues in this region are conserved between the two types of K+ channels. From the alignment, we identified 18 residues on the pore domain of drk1 that correspond to residues located on the perimeter of the extracellular surface of KcsA (Fig. 7 A, asterisks, and B, gray side chains). Most residues were mutated to Ala and the binding affinity of Hanatoxin was determined. As apparent from the normalized Hanatoxin K d values in Fig. 7 C, the largest change in Hanatoxin binding affinity was slightly less than twofold. These results argue against an intimate interaction between Hanatoxin and the extracellular surface of the pore domain, and thereby suggest that the toxin binding sites are confined within the voltage-sensing domains of the channel.
Figure 7.

Effects of the mutations in the peripheral surface of the pore domain. (A) Sequence alignment for the pore domain of the drk1 K+ channel and transmembrane part of KcsA. Numbers above and below the sequences indicate the positions of the residues for each channel. Yellow highlighting marks conserved residues between the two K+ channels. Cylinders indicate the demarcations of the two transmembrane α helices (TM1 and TM2 for KcsA and S5 and S6 for the drk1 K+ channel) and the pore helix. *18 positions in the drk1 K+ channel where mutations were made and the effects on Hanatoxin binding affinity were determined. All residues were mutated to Ala except A348L, W365F, and L384F. (B) Location of KcsA residues (shown in gray) that correspond to those mutated in the drk1 K+ channel. Gray residues correspond to those marked by asterisks in A. The crystal structure of the tetrameric KcsA channel shown for a top view (left) or a side view (right). Numbers correspond to residues in the drk1 K+ channel. Yellow dotted rings show two orientations of the Agitoxin2 footprint. (C) Normalized K d values for 18 mutations in the pore domain of the drk1 K+ channel. Data are mean ± SEM with n = 3 or 4 for each mutant channel. Dashed line marks a normalized K d value of 1.
DISCUSSION
Previous studies support the idea that Hanatoxin interacts with four receptors on the voltage-gated K+ channel and inhibits the channel by modifying the energetics of gating (Swartz and MacKinnon 1997a,Swartz and MacKinnon 1997b). The objective of the present study was to explore two questions. First, what is the molecular composition of the Hanatoxin receptors? Second, where are these receptors located in the structure of the voltage-gated K+ channel?
In the first part of this paper, we examined the effects of multiple substitutions at three positions in the COOH-terminal end of S3, where mutations to Ala were previously found to alter toxin binding affinity. Our results for both I273 and F274 are consistent with hydrophobic interactions between these positions and hydrophobic residues on the toxin. The Glu residue at position 277 also seems to interact intimately with the toxin, perhaps by way of a salt bridge with a basic residue on the toxin. The solution structure of Hanatoxin was recently solved by NMR spectroscopy (Takahashi et al. 2000). The toxin is composed of two β strands, one turn of a 310 helix, with four chain reversals. While the surface of the toxin that interacts with the K+ channel remains to be experimentally identified, a channel interacting face has been proposed based on a structural comparison of different gating modifier toxins (Takahashi et al. 2000). Two types of gating modifier toxins that slow inactivation of Na+ channels are the α-scorpion toxins and the sea anemone toxins. Hanatoxin and these Na+ channel toxins each have a face that contains a large hydrophobic patch surrounded by basic and acidic residues. This structural feature is illustrated in the surface rendering of Hanatoxin in Fig. 8 A. Our results would be consistent with F274, and possibly I273, interacting with the hydrophobic cluster on the toxin. E277 might interact with one of the many basic residues (through formation of a salt bridge) that surround the hydrophobic patch. While our results with F274 are consistent with this residue participating in a hydrophobic interaction, they also show that the most dramatic perturbations result from mutations to either Lys or Arg. This might be an indication that in the toxin–channel complex, F274 is located close to a basic toxin residue, a quite feasible scenario for the face of Hanatoxin shown in Fig. 8 A. It is interesting that the face of Hanatoxin proposed to interact with the channel resembles other well-studied protein–protein interfaces. For example, the interface between growth hormone and its receptor shows a patch of hydrophobic residues, where tight hydrophobic contacts exist, surrounded by polar residues that participate in hydrogen bonds and salt bridges (Clackson and Wells 1995; Bogan and Thorn 1998; Clackson et al. 1998).
Figure 8.
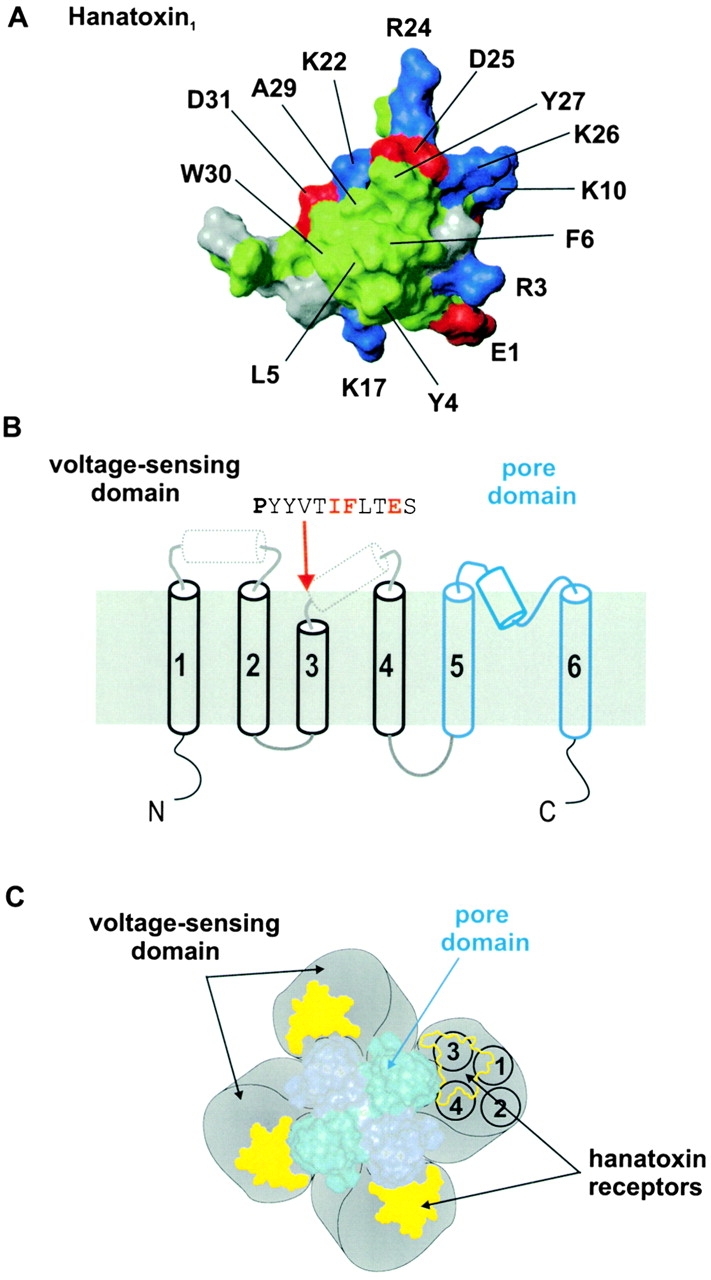
Molecular determinants and localization of the Hanatoxin receptors on the drk1 K + channel. (A) Surface rendering for one face of Hanatoxin1. The structure is a solution structure solved by NMR spectroscopy (Takahashi et al. 2000). The cluster of hydrophobic residues (green) surrounded by basic (blue) and acid (red) residues represents a surface feature common to Hanatoxin1, CsEV (an α-scorpion toxin) and ATXIII (a sea anemone toxin). (B) Putative topology and secondary structure of voltage-gated K+ channel α subunits. The first four transmembrane segments (S1–S4, in black) comprise the voltage-sensing domain and the last two transmembrane segments (S5–S6, in blue) in the pore domain. See Fig. 1 legend for details of secondary structure. The top is extracellular and the bottom is intracellular. A short stretch of sequence is shown for the COOH-terminal part of S3, beginning with the P268 (bold), a residue that is conserved in voltage-gated K+ channels. Red lettering indicates the positions of the three residues (I273, F274, and E277) that interact intimately with Hanatoxin. (C) Architecture of a voltage-gated K+ channel containing a central pore domain and four surrounding voltage-sensing domains drawn according to Li-Smerin et al. 2000b. Black circles shown for one voltage-sensing domain illustrate the four transmembrane (S1–S4) helices contained within, positioned according to Li-Smerin et al. 2000a. Yellow patches illustrate the Hanatoxin receptors confined within the voltage-sensing domains.
The dimensions of Hanatoxin (∼20 × 25 Å) strongly suggest that many channel residues, certainly more than three, are located at the interaction surface between toxin and channel. To search for additional components of the toxin receptors, we first systematically mutated the region spanning from the NH2 terminus of S1 to the COOH terminus of S3. All mutants in this region displayed binding affinities for Hanatoxin that are comparable to the wild-type channel (Fig. 6). The other region that we examined for potential interactions with Hanatoxin was the extracellular surface of the pore domain. From the known structure of the KcsA K+ channel, we can infer that the extracellular surface of the pore domain in voltage-gated K+ channels has sufficient area to accommodate Agitoxin2, a pore-blocking toxin, and at least portions of four Hanatoxin molecules. When we mutated 18 residues (representing 72 positions in the homotetramer) on the peripheral surface of the pore domain, we found no significant alterations in Hanatoxin binding affinity (Fig. 7). These results argue that Hanatoxin does not interact with the extracellular surface of the pore domain and therefore most likely interacts exclusively with the voltage-sensing domains. Since the extracellular surface of the KcsA K+ channel has dimensions of ∼45 × 45 Å (Doyle et al. 1996), we conclude that the Hanatoxin receptors must be located at least 20–25 Å away from the central pore axis in voltage-gated K+ channels, as illustrated by the Hanatoxin footprints in Fig. 8 C.
It is remarkable that mutations of only three residues in the entire voltage-sensing domain (S1–S4) of the drk1 K+ channel have large effects on Hanatoxin binding affinity. One possibility is that Ala-scanning mutagenesis may have missed residues that contribute either weakly or moderately to the interaction with toxin because some substitutions to Ala represent rather conservative changes. Another very interesting possibility is that the toxin also interacts with the NH2-terminal part of S4, where a previous Ala-scan identified several positions where mutations have small effects (three- to fivefold) on Hanatoxin binding affinity (Swartz and MacKinnon 1997b). We did not examine this region in the present study because the previously reported effects are rather small and therefore difficult to study. However, the large number of “negative” mutations observed elsewhere in the channel strengthens the possibility that there is an important interaction between Hanatoxin and the NH2-terminal part of S4. While we probably have not found all the residues that lie at the interface with Hanatoxin, we may have identified the residues (I273, F274, E277) that dominate the energetics of the toxin–channel interaction. This notion is consistent with other examples of protein–protein interactions, where only a few residues can account for most of the binding energy even though many more residues lie at the interface. Two notable examples are the barnase:barnstar and growth hormone:receptor complexes. In both instances, when all residues present at the protein–protein interfaces are mutated to Ala, only ∼25% of them are found to cause substantial perturbations in binding energy (Schreiber and Fersht 1995; Clackson and Wells 1995; Bogan and Thorn 1998; Clackson et al. 1998; Conte et al. 1999).
Several recent studies examining the secondary structure of the first four transmembrane segments (S1–S4) in voltage-gated K+ channels suggest that all four segments are membrane-spanning α helices (Monks et al. 1999; Hong and Miller 2000; Li-Smerin et al. 2000a). The identified interaction of Hanatoxin with the COOH terminus of S3, together with the absence of an interaction with the pore domain, suggests that the toxin interacts with the extracellular surface of this bundle of four helices. However, S1, S2, and the linker between them do not appear to interact strongly with the toxin, suggesting that the Hanatoxin receptors are located over S3 and perhaps S4. The S3 segment is interesting in that only the intracellular or NH2-terminal two-thirds, up to a conserved proline (P268 in drk1), exhibits α-helical character (Hong and Miller 2000; Li-Smerin et al. 2000a). While there is no evidence for α-helical structure for the COOH-terminal part of S3, there is evidence for α-helical structure in the nearby linker between S3 and S4 (Li-Smerin et al. 2000a). One possibility is that S3 is composed of two helices separated by a short nonhelical region near P268, as illustrated in Fig. 8 B. In this case, the three residues implicated in forming the Hanatoxin receptors (I273, F274, and E277) would be located either within or near the beginning of the second helix. The spacing of these three residues is compatible with helical structure in this region, with one side of the helix comprising the external surface where intimate interactions with the toxin occur.
It is interesting that gating modifier toxins, perhaps as a general rule, interact with the equivalent region of voltage-gated K+, Na+, and Ca2+ channels. The present results are consistent with an intimate interaction between Hanatoxin and E277 in the drk1 K+ channel. Mutations at E1613 in the fourth repeat of the brain IIA Na+ channel alter the binding affinity of α-scorpion toxin and sea anemone toxin, two types of toxins that slow inactivation (Rogers et al. 1996). Also, mutation of E1658 in repeat four of the α1A voltage-gated Ca2+ channel disrupts the binding of ω-Aga-IVA (Winterfield and Swartz, unpublished observations), a gating modifier toxin for the Ca2+ channel (McDonough et al. 1997). In sequence alignments, both E1613 in the Na+ channel and E1658 in the Ca2+ channel are at positions equivalent to E277 in the drk1 K+ channel. This connection between different gating modifier toxins is paralleled by several reported examples where gating modifier toxins interact rather promiscuously with different types of voltage-gated ion channels. Grammotoxin, a gating modifier of the α1A voltage-gated Ca2+ channel, also inhibits the drk1 K+ channel and the binding affinity of grammotoxin to the K+ channel is altered by mutations at I273, F274, or E277 (Li-Smerin and Swartz 1998). In addition, Hanatoxin can bind to and inhibit the α1A Ca2+ channel. Another example is kurtoxin, an α-scorpion toxin that slows inactivation of the brain IIA Na+ channel and also inhibits T-type voltage-gated Ca2+ channels by shifting activation to more depolarized voltages (Chuang et al. 1998). Taken together, these results suggest that gating modifier toxins interact with a common region of the voltage-sensing domain in different voltage-gated ion channels and that this region probably adopts a well conserved three-dimensional structure.
Acknowledgments
We thank Dave Hackos, Zhe Lu, and Rod MacKinnon for helpful discussions, Rosalind Chuang for making some of the mutants, and J. Nagle and D. Kauffman in the National Institute of Neurological Disorders and Stroke DNA Sequencing Facility.
References
- Aggarwal S.K., MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- Bogan A.A., Thorn K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Cha A., Bezanilla F. Characterizing voltage-dependent conformational changes in the Shaker K+ channel with fluorescence. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Chuang R.S., Jaffe H., Cribbs L., Perez-Reyes E., Swartz K.J. Inhibition of T-type voltage-gated calcium channels by a new scorpion toxin. Nat. Neurosci. 1998;1:668–674. doi: 10.1038/3669. [DOI] [PubMed] [Google Scholar]
- Clackson T., Ultsch M.H., Wells J.A., de Vos A.M. Structural and functional analysis of the 1:1 growth hormone:receptor complex reveals the molecular basis for receptor affinity. J. Mol. Biol. 1998;277:1111–1128. doi: 10.1006/jmbi.1998.1669. [DOI] [PubMed] [Google Scholar]
- Clackson T., Wells J.A. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- Conte L.L., Chothia C., Janin J. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., MacKinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Doyle D.A., Lee A., Lewis J., Kim E., Sheng M., MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domainmolecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- Frech G.C., VanDongen A.M., Schuster G., Brown A.M., Joho R.H. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature. 1989;340:642–645. doi: 10.1038/340642a0. [DOI] [PubMed] [Google Scholar]
- Garcia M.L., Garcia-Calvo M., Hidalgo P., Lee A., MacKinnon R. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 1994;33:6834–6839. doi: 10.1021/bi00188a012. [DOI] [PubMed] [Google Scholar]
- Goldstein S.A., Miller C. Mechanism of charybdotoxin block of a voltage-gated K+ channel. Biophys. J. 1993;65:1613–1619. doi: 10.1016/S0006-3495(93)81200-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein S.A., Pheasant D.J., Miller C. The charybdotoxin receptor of a Shaker K+ channelpeptide and channel residues mediating molecular recognition. Neuron. 1994;12:1377–1388. doi: 10.1016/0896-6273(94)90452-9. [DOI] [PubMed] [Google Scholar]
- Heginbotham L., LeMasurier M., Kolmakova-Partensky L., Miller C. Single Streptomyces lividans K+ channels. Functional asymmetries and sidedness of proton activation. J. Gen. Physiol. 1999;114:551–560. doi: 10.1085/jgp.114.4.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo P., MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- Hong K.H., Miller C. The lipid–protein interface of a Shaker K+ channel. J. Gen. Physiol. 2000;115:51–58. doi: 10.1085/jgp.115.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst R.S., Busch A.E., Kavanaugh M.P., Osborne P.B., North R.A., Adelman J.P. Identification of amino acid residues involved in dendrotoxin block of rat voltage-dependent potassium channels. Mol. Pharmacol. 1991;40:572–576. [PubMed] [Google Scholar]
- Imredy J.P., Chen C., MacKinnon R. A snake toxin inhibitor of inward rectifier potassium channel ROMK1. Biochemistry. 1998;37:14867–14874. doi: 10.1021/bi980929k. [DOI] [PubMed] [Google Scholar]
- Jin W., Lu Z. A novel high-affinity inhibitor for inward-rectifier K+ channels. Biochemistry. 1998;37:13291–13299. doi: 10.1021/bi981178p. [DOI] [PubMed] [Google Scholar]
- Krezel A.M., Kasibhatla C., Hidalgo P., MacKinnon R., Wagner G. Solution structure of the potassium channel inhibitor agitoxin 2caliper for probing channel geometry. Prot. Sci. 1995;4:1478–1489. doi: 10.1002/pro.5560040805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Larsson H.P., Baker O.S., Dhillon D.S., Isacoff E.Y. Transmembrane movement of the Shaker K+ channel S4. Neuron. 1996;16:387–397. doi: 10.1016/s0896-6273(00)80056-2. [DOI] [PubMed] [Google Scholar]
- Ledwell J.L., Aldrich R.W. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J. Gen. Physiol. 1999;113:389–414. doi: 10.1085/jgp.113.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Unwin N., Stauffer K.A., Jan Y.N., Jan L.Y. Images of purified Shaker potassium channels. Curr. Biol. 1994;4:110–115. doi: 10.1016/s0960-9822(94)00026-6. [DOI] [PubMed] [Google Scholar]
- Liman E.R., Hess P., Weaver F., Koren G. Voltage-sensing residues in the S4 region of a mammalian K+ channel. Nature. 1991;353:752–756. doi: 10.1038/353752a0. [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y., Hackos D.H., Swartz K.J. Alpha-helical structural elements within the voltage-sensing domains of a K+ channel J. Gen. Physiol. 115 2000. 33 49a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin Y., Hackos D.H., Swartz K.J. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel Neuron. 25 2000. 411 423b [DOI] [PubMed] [Google Scholar]
- Li-Smerin Y., Swartz K.J. Gating modifier toxins reveal a conserved structural motif in voltage-gated Ca2+ and K+ channels. Proc Natl Acad Sci USA. 1998;95:8585–8589. doi: 10.1073/pnas.95.15.8585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z., MacKinnon R. Purification, characterization, and synthesis of an inward-rectifier K+ channel inhibitor from scorpion venom. Biochemistry. 1997;36:6936–6940. doi: 10.1021/bi9702849. [DOI] [PubMed] [Google Scholar]
- MacKinnon R., Cohen S.L., Kuo A., Lee A., Chait B.T. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 1998;280:106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- MacKinnon R., Miller C. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J. Gen. Physiol. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R., Miller C. Mutant potassium channels with altered binding of charybdotoxin, a pore-blocking peptide inhibitor. Science. 1989;245:1382–1385. doi: 10.1126/science.2476850. [DOI] [PubMed] [Google Scholar]
- Mannuzzu L.M., Moronne M.M., Isacoff E.Y. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- McDonough S.I., Mintz I.M., Bean B.P. Alteration of P-type calcium channel gating by the spider toxin ω-Aga–IVA. Biophys. J. 1997;72:2117–2128. doi: 10.1016/S0006-3495(97)78854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. Competition for block of a Ca2+-activated K+ channel by charybdotoxin and tetraethylammonium. Neuron. 1988;1:1003–1006. doi: 10.1016/0896-6273(88)90157-2. [DOI] [PubMed] [Google Scholar]
- Miller C. The charybdotoxin family of K+ channel-blocking peptides. Neuron. 1995;15:5–10. doi: 10.1016/0896-6273(95)90057-8. [DOI] [PubMed] [Google Scholar]
- Monks S.A., Needleman D.J., Miller C. Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 1999;113:415–423. doi: 10.1085/jgp.113.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranjo D., Miller C. A strongly interacting pair of residues on the contact surface of charybdotoxin and a Shaker K+ channel. Neuron. 1996;16:123–130. doi: 10.1016/s0896-6273(00)80029-x. [DOI] [PubMed] [Google Scholar]
- Papazian D.M., Shao X.M., Seoh S.A., Mock A.F., Huang Y., Wainstock D.H. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- Papazian D.M., Timpe L.C., Jan Y.N., Jan L.Y. Alteration of voltage-dependence of Shaker potassium channel by mutations in the S4 sequence. Nature. 1991;349:305–310. doi: 10.1038/349305a0. [DOI] [PubMed] [Google Scholar]
- Park C.S., Miller C. Interaction of charybdotoxin with permeant ions inside the pore of a K+ channel Neuron. 9 1992. 307 313a [DOI] [PubMed] [Google Scholar]
- Park C.S., Miller C. Mapping function to structure in a channel-blocking peptideelectrostatic mutants of charybdotoxin Biochemistry. 31 1992. 7749 7755b [DOI] [PubMed] [Google Scholar]
- Perozo E., Santacruz-Toloza L., Stefani E., Bezanilla F., Papazian D.M. S4 mutations alter gating currents of Shaker K+ channels. Biophys. J. 1994;66:345–354. doi: 10.1016/s0006-3495(94)80783-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planells-Cases R., Ferrer-Montiel A.V., Patten C.D., Montal M. Mutation of conserved negatively charged residues in the S2 and S3 transmembrane segments of a mammalian K+ channel selectively modulates channel gating. Proc. Natl. Acad. Sci. USA. 1995;92:9422–9426. doi: 10.1073/pnas.92.20.9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan R., Lewis J.H., MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- Rogers J.C., Qu Y., Tanada T.N., Scheuer T., Catterall W.A. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3–S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G., Fersht A.R. Energetics of protein–protein interactionsanalysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Schrempf H., Schmidt O., Kummerlen R., Hinnah S., Muller D., Betzler M., Steinkamp T., Wagner R. A prokaryotic potassium ion channel with two predicted transmembrane segments from Streptomyces lividans . EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:5170–5178. doi: 10.1002/j.1460-2075.1995.tb00201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoh S.A., Sigg D., Papazian D.M., Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. Role of the S4 in cooperativity of voltage-dependent potassium channel activation J. Gen. Physiol. 111 1998. 399 420a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation J. Gen. Physiol. 111 1998. 421 439b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampe P., Kolmakova-Partensky L., Miller C. Intimations of K+ channel structure from a complete functional map of the molecular surface of charybdotoxin. Biochemistry. 1994;33:443–450. doi: 10.1021/bi00168a008. [DOI] [PubMed] [Google Scholar]
- Stocker M., Miller C. Electrostatic distance geometry in a K+ channel vestibule. Proc. Natl. Acad. Sci. USA. 1994;91:9509–9513. doi: 10.1073/pnas.91.20.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M., Pongs O., Hoth M., Heinemann S.H., Stühmer W., Schroter K.H., Ruppersberg J.P. Swapping of functional domains in voltage-gated K+ channels. Proc. R. Soc. Lond. B Biol. Sci. 1991;245:101–107. doi: 10.1098/rspb.1991.0094. [DOI] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites Neuron. 18 1997. 665 673a [DOI] [PubMed] [Google Scholar]
- Swartz K.J., MacKinnon R. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels Neuron. 18 1997. 675 682b [DOI] [PubMed] [Google Scholar]
- Takahashi H., Kim J.I., Min H.J., Sato K., Swartz K.J., Shimada I. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K+ channelscommon surface features of gating modifier toxins. J. Mol. Biol. 2000;297:771–780. doi: 10.1006/jmbi.2000.3609. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff S.K., Schulteis C.T., Mock A.F., Papazian D.M. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 1997;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., George A.L., Jr., Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Yusaf S.P., Wray D., Sivaprasadarao A. Measurement of the movement of the S4 segment during the activation of a voltage-gated potassium channel. Pflügers Arch. 1996;433:91–97. doi: 10.1007/s004240050253. [DOI] [PubMed] [Google Scholar]