

Abstract
Alterations in tissue-specific gene expression greatly affect cell function. Transcription factors (TFs) interact with cis-acting binding sites in noncoding enhancer promoter regions. Transposable elements (TEs) are abundant and similarly represented among mammalian genomes. TEs are important in gene regulation, but their function is not well understood. We have characterized a TE containing functional TF-binding sites for the carcinogen-activated dioxin receptor xenobiotic responsive element (XRE) and the epithelial–mesenchymal transition regulator Slug (Slug site). A Mus promoter database was scanned for XREs to predict coregulation with other TFs. We identified an overrepresented (1,398 genes) B1 retrotransposon containing XRE and Slug sites within 35 bp of each other (designated as B1-X35S). This B1-X35S retrotransposon differed from classic B1s by the presence of the Slug site and by its differential nucleotide conservation outside the X35S region. Phylogenetically, B1-X35S appeared recently in evolution, close to the B1-B subfamily. Comparative gene expression in 61 mouse tissues revealed that B1-X35S-containing genes had lower median expression levels than those with canonical B1 TEs, suggesting a repressive role for X35S. Indeed, X35S was functional and able to bind aryl hydrocarbon (dioxin) receptor (AhR) and Slug and, importantly, to repress cis-reporter genes. Moreover, AhR and Slug were recruited to X35S in vivo and repressed the endogenous expression of X35S-containing genes. Our results demonstrate the existence of a widely present B1 subfamily in the mouse. Because AhR and Slug are relevant in tumor development and differentiation, X35S may represent a genome-wide regulatory mechanism and a tool to modulate gene expression.
The critical mechanisms regulating gene transcription have been defined (1, 2). However, the spatial, temporal, and developmental regulation of gene expression is not well understood for most genes. The aim of many studies was to determine how gene expression directs the phenotype of a given organism and how genomes evolve. Transcriptional control of gene expression requires protein–DNA interactions at the proximal promoter. However, in most cases, transcription rates also depend on the interaction of transcription factors (TFs) with a complex combination of binding sites that assemble together to form promoter enhancers (1, 3–6).
In an attempt to clarify the role of TFs in cell physiology and development, several protocols have been developed to map protein–DNA interactions (7). Complex genomic searches of sequence information contained in databases such as Ensembl (8) allow prediction of new protein–protein interactions (9), functional protein domains (10), and active TF-binding sites (3). Most eukaryotic cells accumulate transposable elements (TEs) that eventually represent 45–50% of their genomes (11). Despite their parasitic nature, several reports indicate that TEs regulate gene expression (12–15). Furthermore, because TEs are able to recruit silencing machinery, TEs may be epigenetic regulators of gene expression (15). Thus, a relevant question is whether TEs carrying combinations of TF-binding sites represent a genome-wide mechanism to regulate gene expression.
Aryl hydrocarbon (dioxin) receptor (AhR) is a ligand-activated TF of the basic helix–loop–helix (bHLH) family of transcriptional regulators that has a relevant role in xenobiotic metabolism (16, 17). Ligand-bound AhR translocates from the cytosol to the nucleus, heterodimerizes with the AhR nuclear translocator (ARNT), and binds to xenobiotic responsive elements (XREs) located in the promoter of target genes (17). Recent evidence implicates AhR in cell proliferation and differentiation, organ homeostasis, cell adhesion, cell migration, and cancer (18–20). These functions presumably require AhR to be integrated in regulatory pathways that control gene expression in coordination with other transcription factors.
In this study, we provide experimental support for this hypothesis by identifying and characterizing a new subfamily of B1 retrotransposons [B1-X35S (XRE-35 bp-Slug)] that contain functional AhR- and Slug-binding sites. The repression of gene transcription attributable to the binding of AhR and Slug to B1-X35S in vitro and in vivo may be physiologically important in the modulation of gene expression.
Results and Discussion
We used a Perl script to identify mouse promoters in which the carcinogen-activated AhR-binding site, XRE (5′-GCGTG-3′), colocalized with other known TF consensus elements (Fig. 1A). From this screening, the site for the zinc finger Slug (5′-CAGGTG-3′) was found to colocalize with XRE in 23,459 sequences of mouse proximal upstream genomic regions. Interestingly, in 1,673 sites in 1,398 genes, the XRE and Slug sites were separated from each other by a distance of 35 bp [Fig. 1B and supporting information (SI) Table 1]. The arrangement of XRE and Slug sites in this X35S element was significantly overrepresented compared with both random and theoretical distributions (Fig. 1B, red and blue lines, respectively). The consensus X35S element and its flanking sequence shared high sequence homology with mouse short interspersed DNA element (SINE)-B1 retrotransposons of the B1-A, -B, -C, and -D subfamilies (Fig. 1C). B1 is a rodent 136-bp retrotransposable SINE highly abundant in the Mus genome. B1s, as primate Alu elements, were originated from an ancient 7SL RNA gene (21). Importantly, X35S was unique among B1s because of three specific nucleotide modifications, (i) a C56T transition generating the Slug site, (ii) an A8G transition, and (iii) a T115 deletion.
Fig. 1.

Identification and characterization of the murine B1-X35S retrotransposon. (A) Schematic workflow of the bioinformatics analysis in which random sites were used to filter TF-binding sites. The mouse promoter database consists of mouse proximal upstream genomic regions. (B) Analysis of the relative location of the XRE and Slug sites. The XRE and Slug sites are separated by 35 bp in >1,500 mouse proximal upstream genomic regions. Any two random sites would distribute relative to each other as indicated by the red line. Theoretical distribution of 5- and 6-mer sites in a random genome would produce the result shown by the dashed blue line. (C) Sequence alignment (Clustal) of B1-X35S and the most recent subfamilies of B1 retrotransposons B1-A, -B, -C, and -D (according to ref. 21). Conserved nucleotides are highlighted in black boxes. The position of X35S is indicated at the top. XRE and Slug-binding sites are also shown. (D) Phylogenetic tree for the alignment shown in C. The tree was generated with Phylip (Joe Felsenstein, University of Washington, Seattle) by using the neighbor-joining algorithm. BS numbers correspond to the bootstrapping values (n = 10,000 replicates) for the two nearest branches with respect to the B1-X35S subfamily. Sequence identity between B1-X35S and B1-B subfamilies is also shown.
Based on these differences, we propose that X35S is a previously uncharacterized subfamily of B1 retroposons hereafter named B1-X35S. Sequence analysis by the neighbor-joining algorithm produced a phylogenetic tree (Fig. 1D) in which B1-X35S was located closer to recently evolved B1s (B1-B and B1-A) (22), with the highest homology to B1-B (97.8). Thus, B1-X35S may have arisen from a master B1 copy rather than from the combination of point mutations preexisting in other B1 subfamilies.
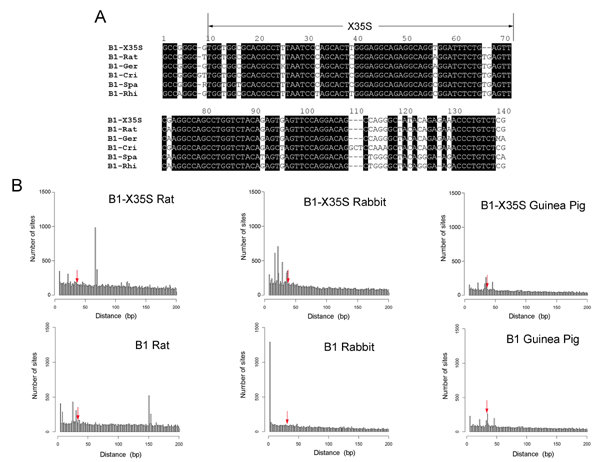
Because B1 retrotransposons are also present in other rodent species, we aligned mouse B1-X35S with B1s from the genera Rattus, Gerbillinae, Cricetidae, Spalacidae, and Rhizomyidae (SI Fig. 5). None of the consensus B1 sequences in these genera contained the X35S element (SI Fig. 5A). The screen for the presence of XRE and Slug sites in rat, rabbit, and guinea pig proximal upstream genomic region databases revealed an absence of X35S elements (SI Fig. 5B Upper). Furthermore, the canonical consensus B1 sequence was also absent in upstream genomic regions of these rodent species (SI Fig. 5B Lower). The Genome Browser chain alignment of two representative genes with B1-X35S in their upstream regulatory regions in mouse (i.e., Dad1 and Tbc1d1, see below) further confirms its absence in the rat (SI Fig. 6). Thus, the deficiency of X35S elements in the rat, rabbit, and guinea pig genomes can be explained by the fact that B1 retrotransposons are physically absent from their upstream genomic regions.
Because B1 SINE elements are stably integrated and represent useful phylogenetic markers (22), the B1-X35S may be an additional tool to analyze the origin and distribution of the genus Mus. Sequencing of additional mouse genomes should allow a more complete mapping of B1-derived retrotransposons in the mouse.
We further analyzed the genomic properties of B1-X35S within the B1 family. By using the Perl script, we scanned mouse proximal upstream genomic regions for the coexistence of XRE and the nucleotide sequence substituting for the Slug-binding site in the consensus B1 (CAGGCG). More than 2,000 sites contained XRE and CAGGCG sequences within 35 bp of each other (Fig. 2A), indicating that, as for X35S, the consensus B1 element (X35B) was also overrepresented in the mouse genome. A clue to the function of these elements came from the observation that both X35S and X35B tend to accumulate in upstream promoter regions, rather than in random positions in the mouse genome (Fig. 2B). This preferential distribution in the vicinity of gene-rich regions is not unique to these elements but is a feature common to SINE elements (23). When analyzed within upstream promoter regions, the incidence of X35S from +1 to −5,000 decreased close to the transcription start site of genes (SI Fig.7). Therefore, B1-X35S may regulate enhanced, rather than basal, gene expression. Considering the potential silencing effect of TEs (15), the location of B1-X35S at a distance from the transcription start site could minimize detrimental effects of B1-X35S on basal transcription.
Fig. 2.

Comparison between B1-X35S and consensus B1. (A) Scan of the mouse proximal upstream genomic database for XRE and the nucleotide sequence that substitutes the Slug binding site in consensus B1 (CAGGCG). A set of >2,000 sites contained XRE and CAGGCG within 35 bp of each other. Theoretical distribution of 5-mer and 6-mer sites in a random genome would produce the result shown by the dashed blue line. (B) Differential location of B1-X35S and B1 in promoter or other regions in the mouse genome. Scanning was performed as indicated in Materials and Methods by using 23,459 random regions of 5 kbp in length. (C) Conservation analysis of B1-X35S (red) and B1 (blue) retrotransposons. Each value represents the mean conservation obtained for a window of 20 nucleotides. A set of 500 sequences was used for each B1 subfamily. The X35S region is indicated. (D) The nucleotide position-specific weight matrix of 1,673 X35S sites. Inside the X35S, nucleotide homology exceeds 90%, whereas outside of the X35S region (black vertical lines), the sequence conservation decreases. The consensus sequence is shown at the bottom, with the XRE and Slug sites boxed. (E) Comparative gene expression analysis for genes lacking the XRE and the Slug-binding site (random, black), containing B1-X35S (red), or B1 (blue). Each set included 800 genes whose expression was analyzed in a panel of 61 mouse tissues from the Gene Expression Atlas. The data are shown as the median for each tissue. The horizontal dashed lines represent the mean of all of the tissues. The data were normalized by the robust multichip average method. Differences in the medians were statistically significant, as assessed by paired t test (random vs. B1-X35S, random vs. B1, and B1-X35S vs. B1; P < 2.2 × 10−16). The Mann–Whitney statistical analysis for the mean B1-X35S vs. B1 gene expression was significant for the mouse tissues indicated (P < 0.05).
B1-X35S and canonical B1 differed in their sequence conservation (Fig. 2C). A subset of 500 B1-containing genes was analyzed for nucleotide conservation with respect to the consensus sequences of B1-A, -B, -C, and -D retrotransposons. We found that conservation did not markedly change along the B1 sequence (Fig. 2C, blue). B1-X35S genes, on the contrary, had a maximum degree of conservation within the X35S element that decreased at the flanking regions (Fig. 2C, red). In addition, overall sequence conservation in B1-X35S retroposons was lower than in B1, except for the X35S element, which remained at similar levels. Alignment of B1-X35S in the 1,398 genes revealed >90% conservation within the X35S element, with only five nucleotides being <80% conserved (Fig. 2D). Thus, evolutionary pressure may be acting to conserve the X35S sequence in B1-X35S retroposons.
To determine the functionality of the X35S element, we first analyzed a panel of 61 tissues from the mouse gene expression atlas (24). We examined the median expression levels of three sets of genes (800 genes per set): genes lacking B1 (random), genes containing B1, and genes containing B1-X35S retroposons (Fig. 2E). We found that the B1-X35S-containing genes had lower median expression levels than the B1-containing genes in every tissue analyzed. This difference was statistically significant (P < 0.05) in at least 11 tissues (Fig. 2E, compare blue and red lines, and SI Tables 2 and 3). In addition, both B1-X35S- and B1-containing genes had higher median expression levels than those in the random set (black line). These genomic data provide support for the functionality of X35S in vivo, but more detailed studies should be performed on specific genes in particular tissues.
We next determined whether AhR and Slug proteins interacted in the same protein complexes under endogenous cellular conditions. The close localization of the XRE and Slug sites in X35S could allow functional interaction between the two binding proteins, and they may have a combined effect on X35S activity. As shown in Fig. 3A, the anti-Slug antibody coimmunoprecipitated AhR and Slug in nuclear fractions of mouse hepatoma Hepa 1 cells (lane 2). Because both proteins are transcriptional regulators (16, 25), this finding suggests that AhR and Slug may act together in regulating the X35S element.
Fig. 3.

Functional analyses of X35S in vitro. (A) Nuclear extracts from basal Hepa 1 cells were immunoprecipitated with the anti-Slug antibody, and the AhR protein was detected by Western blotting. The total extract was used as positive control. The efficiency of the immunoprecipitation was determined by analyzing the protein remaining in the supernatants. (B) The DNA-binding affinity for AhR and HA–Slug on X35S. WT X35S was end-labeled with biotin and coupled to streptavidin paramagnetic particles. Nuclear extracts from untreated (DMSO) or TCDD-treated (10 nM for 90 min) Hepa 1 cells were incubated with X35S. AhR and HA–Slug binding was determined by Western immunoblotting by using antibodies specific for each protein. Where indicated, Hepa 1 cells were transfected with HA–Slug, AhR, or both expression vectors. The binding of the AhR dimerization partner ARNT was also determined under the same experimental conditions. (C) The DNA-binding affinity of AhR and HA–Slug to the mutated XRE (Xmut35S) or Slug site (X35Smut). Experimental conditions were as indicated above. (D) Luciferase reporter assays were used to analyze X35S activity in vitro. Hepa 1 cells were left untreated (DMSO) or treated with 10 nM TCDD for 24 h. Where indicated, cultures were transfected with HA–Slug, AhR, or both. Firefly luciferase activity was normalized by renilla luciferase (relative luciferase activity). The data shown are means ± SE from three experiments in triplicate. Differences among experimental conditions are significant at P = 0.001 (**).
We then used DNA-binding affinity assays to analyze whether nuclear AhR and Slug bound the X35S element in vitro (Fig. 3B). The high affinity ligand dioxin [2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)] was used to induce nuclear translocation and transcriptional activation of AhR (17). AhR had a weak basal interaction with X35S (Fig. 3B, lane 2) that increased after receptor activation by TCDD (Fig. 3B, lane 3). AhR transfection, by itself, did not significantly alter basal binding (Fig. 3B, lane 6), whereas TCDD treatment of AhR-transfected cells maximally enhanced receptor binding (Fig. 3B, lane 7). HA–Slug transfection did not affect basal binding of AhR to X35S (Fig. 3B, lane 4), and its cotransfection with AhR in TCDD-treated cells did not show an additive effect (Fig. 3B, lane 9). This suggests that the binding of AhR to XRE is independent of the binding of Slug to the Slug site. ARNT, the dimerization partner of AhR (16–18), markedly increased its binding to X35S after receptor activation by TCDD (Fig. 3B, lanes 3, 5, 7, and 9), suggesting that the regulation of XRE in X35S required AhR/ARNT dimerization.
Slug had very low expression levels in Hepa 1 cells (data not shown) and no detectable interaction with X35S under basal or TCDD-treated conditions (lanes 2 and 3). Transfection of HA–Slug increased HA–Slug binding to X35S, although it was unaffected by TCDD (lanes 4 and 5). Interestingly, the binding of HA–Slug to X35S increased after cotransfection with activated AhR (lane 9). This suggests that, in vitro, AhR binding to XRE may have a positive effect on the interaction of Slug with the Slug site.
The XRE mutation (Xmut35S), but not the Slug site mutation (X35Smut), decreased both basal and TCDD-induced AhR binding to X35S (Fig. 3C Upper; compare lanes 1–3 and 2–4). Likewise, Slug binding was inhibited by the Slug site mutation but not by the XRE mutation (Fig. 3C Lower; compare lanes 2–4). However, some residual binding of AhR to Xmut35S (Fig. 3C Upper, lane 2) and of HA–Slug to X35Smut (Fig. 3C Lower, lane 4) remained. This residual binding may be attributable to incomplete efficacy of the mutations introduced. Nevertheless, because both proteins coimmunoprecipitated, AhR bound to Slug (and vice versa) could be detected in the DNA-binding assay as a result of Slug binding to the Slug site, despite mutation of XRE. Our findings suggest that (i) XRE and Slug sites are functional in X35S, (ii) AhR and Slug can bind independently to their consensus binding sites in X35S, (iii) AhR may positively influence the binding of Slug to the Slug site in vitro, (iv) mutation of the XRE or the Slug site only affects the binding of its specific binding protein, and (v) AhR and Slug can interact in a protein complex on X35S.
Because both AhR and Slug bind X35S, we analyzed their influence on X35S transcriptional activity in luciferase reporter assays (Fig. 3D). X35S had high basal activity (Fig. 3D, lane 2) that was repressed by Slug transfection (Fig. 3D, lane 4). AhR transfection also decreased X35S activity, although to a lesser extent (Fig. 3D, lane 6). Cotransfection of AhR and HA–Slug did not show an additive effect (Fig. 3D, lane 8), probably because of the strong repression produced by HA–Slug alone (Fig. 3D, lane 4). TCDD only slightly increased AhR-dependent repression and had no effect on HA–Slug-induced repression (Fig. 3D, lanes 5 and 7). X35S mutated in the XRE and the Slug site (Xmut35Smut) did not respond to AhR transfection (Fig. 3D, compare lanes 12 and 6) and was significantly less responsive to HA–Slug (Fig. 3D, compare lanes 11 and 4). Altogether, these data indicated that the interactions of AhR and Slug with their respective binding sites conferred repressor activity to B1-X35S, with Slug being a stronger repressor than AhR.
We next determined whether AhR and Slug repressed the transcription of B1-X35S-containing genes in vivo. We selected mouse genes not known to be regulated by AhR or Slug and with the B1-X35S located at various distances from the transcription start site. These genes included Lpp (cell adhesion-related lipoma preferred partner) (26), obesity-risk gene Tbc1d1 (27), and Dad1 (defender against cell death 1) (28). Real-time PCR showed that HA–Slug transfection decreased Lpp, Tbc1d1, and Dad1 mRNA levels (Fig. 4A). TCDD-activated AhR also repressed the expression of these target genes, with the greatest effect on Dad1. Although AhR and Slug cotransfection in the presence of TCDD repressed gene expression in cells, this additive effect was not evident in vivo. TCDD alone (empty vector) increased Dad1 mRNA levels, likely through an unrelated mechanism because AhR and Slug repressed its transcription.
Fig. 4.

Transcriptional repression of X35S-contaning genes by AhR and Slug. (A) Cells were transfected with HA–Slug, AhR, or both, and Lpp, Tbc1d1, and Dad1 mRNA expression was determined by real-time RT-PCR. Where indicated, cells were treated with 10 nM TCDD for 24 h (gray bars). Gene expression was normalized to Gapdh. The data are shown as the differences in amplification cycles between basal cells (transfected with empty pCDNA vector, TCDD-untreated) and those under each experimental condition. A difference in expression corresponding to 0.5 cycle is indicated by the dotted line. The boxes represent the position of X35S in the promoter of each gene analyzed. The data shown are means ± SE from three experiments performed in triplicate. Differences among experimental conditions are significant at P < 0.01 (**) or P = 0.05 (*). (B) ChIP analyses of AhR and HA–Slug binding to X35S in the promoter of Lpp, Tbc1d1, and Dad1 genes. ChIP experiments were performed by using AhR- and Slug-specific antibodies in cotransfected cultures. AhR-transfected cells were treated with 10 nM TCDD for 90 min. Positive controls were performed by amplifying input extracts. Negative controls were performed in the absence of antibody. The experiment was repeated three times with similar results. E/V, empty vector; S, Slug-binding site; X, XRE.
ChIP was used to detect the binding of AhR and Slug to X35S in the promoter of Lpp, Tbc1d1, and Dad1 (Fig. 4B) in vivo. TCDD-activated AhR and HA–Slug were recruited to X35S in the promoters of all three genes. The apparent higher binding of Slug to X35S in vivo may reflect a stronger repressor activity, although it could be also attributable to differences in protein affinity to DNA and/or antibody efficiency in the immunoprecipitates.
X35S has AhR- and Slug-dependent repressive activity for cis-acting promoters in vitro and in vivo. This provides a mechanism that integrates two different signaling pathways in the control of gene expression. Current experiments are underway to characterize whether or not AhR and Slug cooperate in X35S regulation. Although the general scheme for AhR- and Slug-dependent X35S repression appears to apply to the genes analyzed here, their different response to AhR and Slug suggests that additional factors may contribute to this pathway. Potential candidates include epigenetic mechanisms, because eukaryotic cells tend to inhibit TE activity by the recruitment of silencing machinery (15).
In summary, we report the identification and characterization of, to our knowledge, a previously uncharacterized B1-derived SINE retroposon, designated B1-X35S, in the Mus genus. Activation of B1-X35S can repress the expression of physiologically relevant genes by recruiting the transcription factors AhR and Slug. The identification of functional X35S elements in the mouse genome supports the importance of this sequence in transcriptional regulation, genomic stability, and evolution (14). AhR and Slug are well known transcription factors relevant in carcinogenesis and epithelial–mesenchymal transition. The mechanism of transcriptional regulation described here could allow the identification of novel cellular processes requiring coordinated activity of AhR and Slug. Furthermore, B1-X35S represents a model to study the mechanisms through which TFs integrate signals from different pathways.
Materials and Methods
Reagents, Antibodies, Plasmids, and Cell Lines.
DMSO, TCDD, and anti-actin (A2066) were from Sigma–Aldrich. Alpha-MEM was from BioWest. FBS was from BioWhittaker and was heat-inactivated before use. OPTI-MEM and Lipofectamine Plus were from Invitrogen. Protein A/G plus agarose, anti-Slug (sc-10437), and anti-ARNT (sc-8076) were from Santa Cruz Biotechnology. Anti-AhR (MA1-514) and secondary antibodies were obtained from Affinity Bioreagents. pcDNA3-AhR and pcDNA3-HA–Slug were used to express AhR and HA–Slug, respectively. Mouse Hepa 1 cells were purchased from Interlab Cell Line Collection (ICLC ATL98016) and used between passages 2 and 5. Cells were cultured at 37°C in a 5% CO2 atmosphere in alpha-MEM containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin.
In Silico Genomic Analysis for TF-Binding Sites.
A mouse promoter database was built with the first 5 kbp of upstream sequence from 23,459 mouse genes. A Perl script found genomic regions in which the XRE site (5′-GCGTG-3′) was localized within 200 bp of other TF-binding sites. Further analyses of these genomic regions were performed by using the R statistical language (Department of Statistics and Mathematics, Vienna University). The theoretical distribution of the probable colocalization of these sites was also simulated as number of total nucleotides/(probability XRE × probability TF-binding site). An unsupervised random approach was used to ensure statistical significance of the data. For this, the same Perl script was run with 200 random pairs of 5- and n-mer sequences (n = length of the second TF-binding site). The relative orientation of the XRE/Slug site dimers for the 35-bp data was determined (>95% with same structure but with no favored orientation toward +1 (either XRE reverse/Slug site forward or vice versa). We found 1,673 sites in 1,398 mouse genes with a 35-bp distance between XRE and Slug site and with the same structure (X35S region) (see SI Table 1). To generate DNA position weight matrices for B1-X35S (for Mus and other rodents) and B1 (mouse) retrotransposon sequences were aligned with Clustal software (29).
Nuclear Extracts, Immunoprecipitation, and Western Blotting.
Hepa 1 cells were washed and collected in ice-cold PBS. Nuclear extracts were obtained as described previously (30) and immunoprecipitated by using 700 μg of nuclear protein and 2.5 μg of anti-Slug antibody (31). After SDS/PAGE, gels were transferred to nitrocellulose membranes by electroblotting and blocked for 2 h at room temperature in TBS-T [50 mM Tris·HCl (pH 7.5), 150 mM NaCl, 0.2% Tween 20] containing 7% nonfat milk. Blots were sequentially incubated with the primary and secondary antibodies, washed in TBS-T, and visualized by using the SuperSignal luminol substrate (Pierce) and BioMax Light films (Kodak).
DNA-Binding Affinity.
The interaction of AhR and Slug with X35S in vitro was studied by DNA-binding affinity assays, essentially as described previously (32). Nuclear extracts were used for these assays. Forward and reverse oligonucleotides containing the X35S sequence (1 μg/μL) were mixed in 320 μl of buffer [10 mM Tris·HCl (pH 8), 50 mM NaCl, 10 mM MgCl2, 1 mM DTT] and annealed by overnight cooling from 95°C to room temperature. Aliquots of 100 μl of Streptavidin MagneSphere Paramagnetic Particles (Amersham Pharmacia) were washed twice with 1 ml of DA I buffer [10 mM Tris·HCl (pH 7.5), 1 mM EDTA, 2 M NaCl]. Beads were incubated for 30 min at room temperature in 70 μl of DA I buffer plus 30 μl of annealing buffer and sequentially washed with 1 ml of DA I buffer and 1 ml of DA II buffer [10 mM Tris·HCl (pH 7.5), 20 mM Hepes, pH 7.5, 5% glycerol, 170 mM NaCl, 0.5 mM EDTA, 0.5 mM MgCl2, 0.5% Triton X-100, and 0.5 mM DTT]. Nuclear extracts were mixed with 5 μg of poly(dI–dC) and added to the washed beads in 200 μl of DA II buffer. The mixture was gently rotated at 4°C for 2 h. Then the beads were washed five times with 1 ml of DA III buffer [16 mM Hepes (pH 7.6), 100 mM NaCl, 0.4 mM MgCl2, 1% glycerol]. Samples were denatured by heating for 5 min at 95°C in 5 μl of Laemmli buffer. Samples were analyzed by SDS/PAGE and Western blotting as described above. Oligonucleotide sequences were as follows: WT X35S forward, 5′-CGCGTTGGTGGGYRCACGCCTTTAAATCCCAGC A C TYGGGAGGCAGAGGCAGGTGGATTTCTGAGTTC-3′; WT X35S reverse, 5′-TCG A GAACTCAGAAATCCACCTGCCTCTGCCTCCCRAGTGCTGGGATTAAAAG GCGTGYRCCCACCAA-3′; mutant XRE (Xmut35S) forward, 5′-CGCGTTGGTGGGYRCTATCCTTTAAATCCCAGCACTYGGGAGGC A G AGGCAGGTG G A TTTCTGAGTTC-3′; Xmut35S reverse, 5′-TCGAGAACTCAGAAATCCACCTGCCTCTGCCTCCCRAGTGCTGGGATTAAAAGGATAGYRCCCACCAA-3′; mutant Slug (X35Smut) forward, 5′-CGCGTTGGTGGGYRCACGCCTTTAAATC C C AGCACTYGGGAGGCAGAGGCAGGCAGATTTCTGAGTTC-3′; X35Smut reverse, 5′AACTCAGAAATCTGCCTGCCTCTGCCTCCCRAGTGCTGGGATTAAAAG GC GTGYRCCCACCAA-3′; XRE and Slug double mutant (Xmut35Smut) forward, 5′-CGCGTTGGTGGGYRCTATCCTTTAAATCCCAGCACTYGGGAGGC A G A G G CAGGCAGATTTCTGAGTTC-3′; and Xmut35Smut reverse, 5′-TCGAGAACTCAGAAATCTGCCTGCCTCTGCCTCCCRAGTGCTGGGATTAAAAG GATAG Y R C C CACCAA-3′. XRE and Slug sequences are underlined and mutations are in boldface. “R” stands for adenine and guanine and “Y” for thymidine and cytosine. Reverse oligonucleotides were biotinylated at their 5′ ends for coupling to Streptavidin MagneSphere Paramagnetic Particles.
Luciferase Reporter Gene Assays.
The double-stranded oligonucleotides listed above were cloned into the pGL2 basic vector. Luciferase assays were done by using the dual-glow luciferase kit, according to the manufacturer's instructions. The pRLTK vector was used as normalization control. Firefly and renilla luciferase activities were determined in a Microtiter Plate Luminometer MLX (Dynex Technologies).
Real-Time PCR.
Total RNA was isolated by using the RNeasy kit. Retrotranscription, cDNA purification, and real-time PCR were performed as described previously (33). The primers used were as follows: Dad1 forward, 5′-GACTTCCTCTTTGCCAGCAC-3′; Dad1 reverse, 5′-GAAGAAGCCATGTGCAACAA-3′; Tbc1d1 forward, 5′-AACACCCTGAGTCATTTCCC-3′; Tbc1d1 reverse, 5′-CGCATGAGTTTCCTTTGAGA-3′; Lpp forward, 5′-GGTCCATCCTCTGGACAAAT-3′; Lpp reverse, 5′-TACCAACACAGGTTGGAGGA-3′; Gapdh forward, 5′-TGAAGCAGGCATCTGAGGG-3′; and Gapdh reverse, 5′-CGAAGGTGCAAGACTCGGAG-3′. Gene expression was normalized to Gapdh levels.
ChIP.
ChIP was performed as described previously (34). In brief, protein–DNA complexes were immunoprecipitated overnight at 4°C by using 4 μg of Slug or AhR antibodies. After eluting the DNA from the immunoprecipitates, PCR amplification was performed in a 25-μl reaction mixture by using oligonucleotides amplifying the X35S element in the promoter of each gene. The primer sequences were as follows: Dad1 forward, 5′-CTGGCCTCCAACTCAGGAAT-3′; Dad1 reverse, 5′-GGGAGAGTAGAGGGGGCATA-3′; Tbc1d1 forward, 5′-CAGAAATCCACCTGCCTCTG-3′; Tbc1d1 reverse, 5′-CACCTGCATACACTGGCATG-3′; Lpp forward, 5′-TGGCTGTCCTGGAACTCACT-3′; Lpp reverse, 5′-TGCAGTGGGACAAAATGATG-3′. The input fraction was amplified from serial dilutions of total DNA. Negative controls were performed in the absence of antibody. The amplified DNA was separated on 2.5% agarose gels and visualized by staining with ethidium bromide.
Statistical Analyses.
Data are shown as means ± SEM. Statistical comparison between treatments was performed by using Prism 4.0 software (GraphPad). One-way ANOVA, followed by Dunn's post test, was applied. Other specific statistics are indicated in the corresponding figure legends.
Supplementary Material
ACKNOWLEDGMENTS.
We thank Dr. Amparo Cano (Instituto de Investigaciones Biomedicas “Alberto Sols,” Madrid, Spain) for providing the HA–Slug expression vector and Dr. Jaime Correa-Bordes for suggestions and helpful comments. This work was supported by Spanish Ministry of Education and Sciences Grant SAF2005-00130 (to P.M.F.-S). A.C.R. was supported by a fellowship from the Spanish Ministry of Education and Sciences. D.A.B. and J.M.C.-G. were fellows from the Junta de Extremadura.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0708366105/DC1.
References
- 1.Levine M, Tjian R. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 2.Adams MD. Curr Opin Genet Dev. 2005;15:628–633. doi: 10.1016/j.gde.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 4.Murakami K, Kojima T, Sakaki Y. BMC Genomics. 2004;5:16. doi: 10.1186/1471-2164-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman BP, Nibu Y, Pfeiffer BD, Tomancak P, Celniker SE, Levine M, Rubin GM, Eisen MB. Proc Natl Acad Sci USA. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson DS, Mortazavi A, Myers RM, Wold B. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- 8.Hubbard TJ, Aken BL, Beal K, Ballester B, Caccamo M, Chen Y, Clarke L, Coates G, Cunningham F, Cutts T, et al. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M. Science. 2003;302:449–453. doi: 10.1126/science.1087361. [DOI] [PubMed] [Google Scholar]
- 10.Fomenko DE, Xing W, Adair BM, Thomas DJ, Gladyshev VN. Science. 2007;315:387–389. doi: 10.1126/science.1133114. [DOI] [PubMed] [Google Scholar]
- 11.Kidwell MG, Lisch DR. Evolution Int J Org Evolution. 2001;55:1–24. doi: 10.1111/j.0014-3820.2001.tb01268.x. [DOI] [PubMed] [Google Scholar]
- 12.Britten RJ. Proc Natl Acad Sci USA. 1996;93:9374–9377. doi: 10.1073/pnas.93.18.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, et al. Nature. 2004;430:471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 14.Kazazian HH., Jr Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 15.Slotkin RK, Martienssen R. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 16.Nebert DW, Dalton TP. Nat Rev Cancer. 2006;6:947–960. doi: 10.1038/nrc2015. [DOI] [PubMed] [Google Scholar]
- 17.Whitlock JP., Jr Annu Rev Pharmacol Toxicol. 1999;39:103–125. doi: 10.1146/annurev.pharmtox.39.1.103. [DOI] [PubMed] [Google Scholar]
- 18.Barouki R, Coumoul X, Fernandez-Salguero PM. FEBS Lett. 2007;581:3608–3615. doi: 10.1016/j.febslet.2007.03.046. [DOI] [PubMed] [Google Scholar]
- 19.Furness SG, Lees MJ, Whitelaw ML. FEBS Lett. 2007;581:3616–3625. doi: 10.1016/j.febslet.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 20.McMillan BJ, Bradfield CA. Mol Pharmacol. 2007;72:487–498. doi: 10.1124/mol.107.037259. [DOI] [PubMed] [Google Scholar]
- 21.Kriegs JO, Churakov G, Jurka J, Brosius J, Schmitz J. Trends Genet. 2007;23:158–161. doi: 10.1016/j.tig.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Kass DH, Raynor ME, Williams TM. J Mol Evol. 2000;51:256–264. doi: 10.1007/s002390010087. [DOI] [PubMed] [Google Scholar]
- 23.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 24.Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, et al. Proc Natl Acad Sci USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peinado H, Olmeda D, Cano A. Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 26.Petit MM, Meulemans SM, Van de Ven WJ. J Biol Chem. 2003;278:2157–2168. doi: 10.1074/jbc.M206106200. [DOI] [PubMed] [Google Scholar]
- 27.Stone S, Abkevich V, Russell DL, Riley R, Timms K, Tran T, Trem D, Frank D, Jammulapati S, Neff CD, et al. Hum Mol Genet. 2006;15:2709–2720. doi: 10.1093/hmg/ddl204. [DOI] [PubMed] [Google Scholar]
- 28.Nakashima T, Sekiguchi T, Kuraoka A, Fukushima K, Shibata Y, Komiyama S, Nishimoto T. Mol Cell Biol. 1993;13:6367–6374. doi: 10.1128/mcb.13.10.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santiago-Josefat B, Pozo-Guisado E, Mulero-Navarro S, Fernandez-Salguero PM. Mol Cell Biol. 2001;21:1700–1709. doi: 10.1128/MCB.21.5.1700-1709.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benitez DA, Pozo-Guisado E, Clementi M, Castellon E, Fernandez-Salguero PM. Br J Cancer. 2007;96:1595–1604. doi: 10.1038/sj.bjc.6603755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang LK, Chung JY, Hong YR, Ichimura T, Nakao M, Liu ST. Nucleic Acids Res. 2005;33:6528–6539. doi: 10.1093/nar/gki956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomez-Duran A, Mulero-Navarro S, Chang X, Fernandez-Salguero PM. J Cell Biochem. 2006;97:380–392. doi: 10.1002/jcb.20637. [DOI] [PubMed] [Google Scholar]
- 34.Mulero-Navarro S, Carvajal-Gonzalez JM, Herranz M, Ballestar E, Fraga MF, Ropero S, Esteller M, Fernandez-Salguero PM. Carcinogenesis. 2006;27:1099–1104. doi: 10.1093/carcin/bgi344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}