

Abstract
Epidermodysplasia verruciformis (EV) is a genodermatosis associated with skin cancers that results from a selective susceptibility to related human papillomaviruses (EV HPV). Invalidating mutations in either of two genes (EVER1 and EVER2) with unknown functions cause most EV cases. We report that EVER1 and EVER2 proteins form a complex and interact with the zinc transporter 1 (ZnT-1), as shown by yeast two-hybrid screening, GST pull-down, and immunoprecipitation experiments. In keratinocytes, EVER and ZnT-1 proteins do not influence intracellular zinc concentration, but do affect intracellular zinc distribution. EVER2 was found to inhibit free zinc influx to nucleoli. Keratinocytes with a mutated EVER2 grew faster than wild-type keratinocytes. In transiently and stably transfected HaCaT cells, EVER and ZnT-1 down-regulated transcription factors stimulated by zinc (MTF-1) or cytokines (c-Jun and Elk), as detected with luciferase assays. To get some insight into the control of EV HPV infection, we searched for interaction between EVER and ZnT-1 and oncoproteins of cutaneous (HPV5) and genital (HPV16) genotypes. HPV16 E5 protein binds to EVER and ZnT-1 and blocks their negative regulation. The lack of a functional E5 protein encoded by EV HPV genome may account for host restriction of these viruses.
Genetic predisposition to infection with infectious agents such as oncogenic human papillomaviruses (HPVs) is still poorly substantiated. Some of these viruses (HPV 16 and 18) induce anogenital carcinomas, in particular carcinomas of the uterine cervix (1–3). Others (HPV5 and HPV8) are associated with skin carcinomas developing in patients suffering from epidermodysplasia verruciformis (EV) (4, 5). This rare autosomal recessive dermatosis (OMIM 226400) is associated with an abnormal susceptibility to a specific group of related HPV genotypes (EV HPVs). EV patients develop disseminated, persistent, flat wart-like or macular skin lesions early in childhood. Patients infected with EV HPV type 5 or 8 are at high risk of developing intraepithelial or invasive nonmelanoma skin cancers in their early adult life. EV carcinomas harbor a high copy number of HPV genomes, which are maintained as episomes. It is worth stressing that trace amounts of EV HPV DNA are detected by nested PCR approaches in a high proportion of normal skin or hair follicles of healthy subjects and renal transplant recipients, implying asymptomatic infections (6, 7). We have shown that antibodies to HPV5 are generated in epidermal repair processes observed in psoriasis and bullous diseases or in burn patients, pointing to a possible reservoir for EV HPV (8, 9). In addition, a putative role of these viruses in skin carcinogenesis in the general population is suspected (10, 11). Thus, EV represents an attractive model to analyze host genetics factors in the outcome of EV HPV infection.
We have demonstrated that EV is caused by homozygous mutations in either EVER1 or EVER2 gene, which are also known as TMC6 and TMC8, respectively (12, 13). These two novel adjacent genes are located on 17q25.3 and encode integral membrane proteins that have been located in the ER. EVER1 and EVER2 proteins belong to the family of transmembrane channel-like (TMC) proteins (14). The EVER proteins are highly conserved in mouse, fish, Drosophila melanogaster, and Caenorhabditis elegans, which indicates important functions that have yet to be determined. As suggested for TMC1 protein, it can be speculated that EVER1 and EVER2 proteins underlie ion channel or signal transduction activities.
Our aim was to determine the functions of EVER proteins in normal cells and to get some insight in the control of EV HPV infection by these proteins. The first objective was to identify the cellular proteins interacting with EVER proteins using the yeast two-hybrid approach (15). Cellular partners with known function could bring clues to EVER function and cellular signaling pathways affected by EVER mutation in patients suffering from EV. We report that EVER1 and EVER2 form a complex and interact with the zinc transporter ZnT-1 protein. We found that EVER2 influences intracellular localization of free zinc and down-regulates the activity of transcription factors induced by zinc or EGF. This inhibition is alleviated by HPV16 E5 protein, which was found to interact with EVER1, EVER2, and ZnT1. The lack of E5 open reading frame in the EV HPV genome (16) may account for the host restriction of these HPVs in the general population.
RESULTS AND DISCUSSION
EVER proteins interact with the zinc transporter ZnT-1
To elucidate the physiological role of EVER proteins, we first performed a yeast two-hybrid screening to find out their cellular partners. Because of transmembrane domains into EVER proteins (13, 14), we used a region conserved among the TMC proteins (TMC domain) as bait to screen a cDNA library obtained from human HaCaT keratinocytes (17). We found that EVER1 and EVER2 interacted with the zinc transporter 1 (ZnT-1), which has previously been described in hamster cells as a plasma membrane zinc transporter responsible for zinc efflux and resistance to zinc-mediated toxicity (18). Up to 10 different zinc transporter proteins have been identified, and it is suggested that they play an important role in zinc homeostasis (19). Most ZnT proteins have been found associated with endosomes, Golgi, or ER. ZnT-1 has a vesicular localization, but is also at the plasma membrane (20).
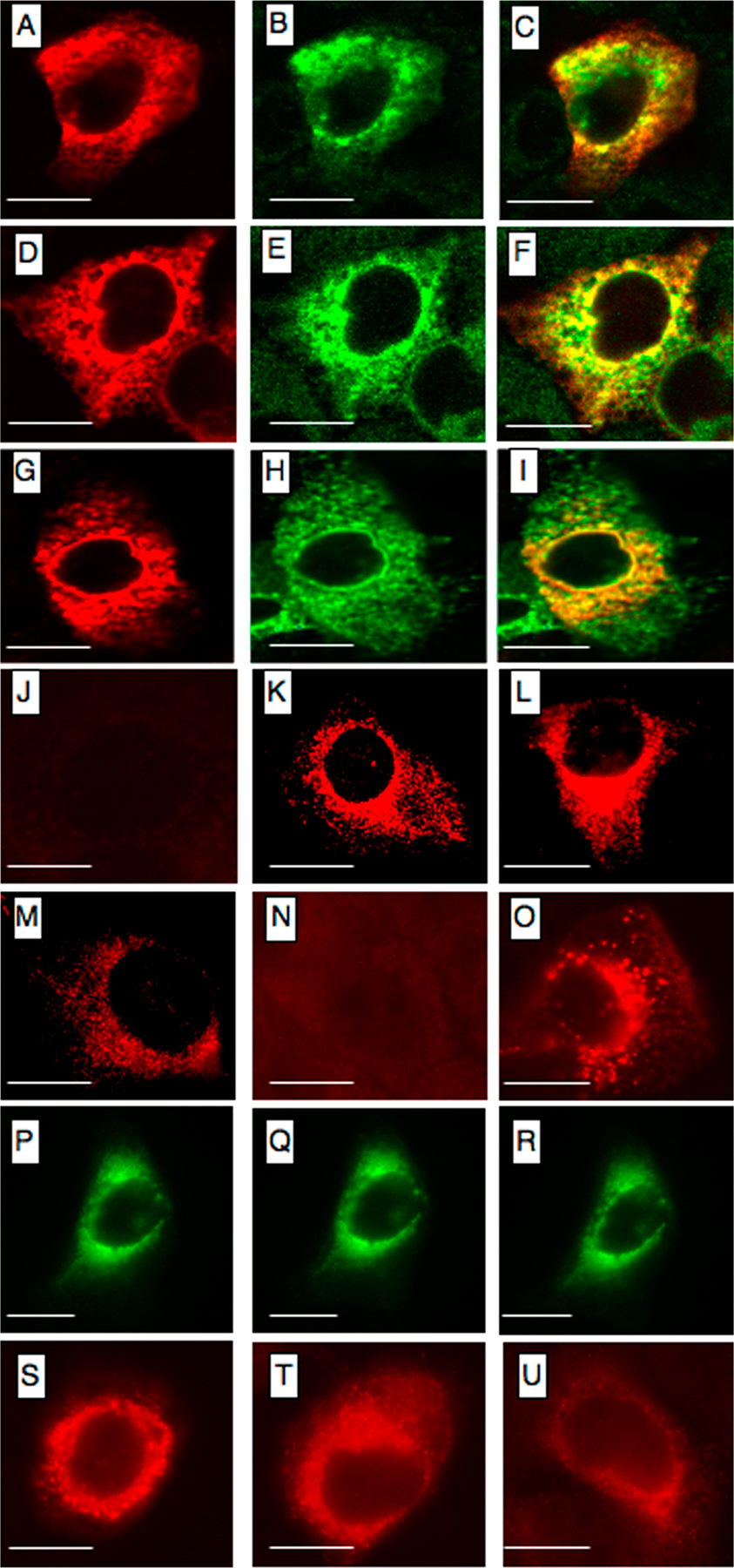
GST pull-down experiments indicated that full-length EVER1 and EVER2 associate with ZnT-1 (Fig. 1 A). As illustrated for EVER2, only the TMC domain of EVER reacted with ZnT-1, whereas no interaction was observed with the 3′ COOH region. In addition, coimmunoprecipitation experiments confirmed that EVER and ZnT-1 proteins form a complex (Fig. 1 B). Interestingly, in contrast to the previous study from hamster cells (18), ZnT-1 was found to be located not in the plasma membrane, but in the cytoplasm in human keratinocytes (Fig. 1 C). We confirmed ZnT-1 localization with a set of constructs with different tags in transiently or stably transfected cells, and also in living cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071311/DC1). ZnT-1 and EVERs were located mainly in ER, as they partially colocalized with calnexin, which is a marker of ER, but also in the nuclear membrane and Golgi apparatus (Fig. 1 C and Fig. S1). Similar location was observed in other keratinocyte cell lines (Caski, HeLa, and SKV). This is in agreement with a recent article reporting that, in mouse cells, ZnT-1 can be located in the cytoplasm (20). These results suggest the existence of an EVER1–EVER2–ZnT-1 complex in the ER.
Figure 1.

EVER proteins form a complex with ZnT-1. (A) BHK21 cells were transfected with plasmids encoding FLAG-tagged ZnT-1 and GST-tagged EVER proteins or truncated GST-EVER2 corresponding to the TMC or C-terminal region. Crude lysates (CL) or pulled down (PD) proteins were immunoblotted (IB) with the indicated antibodies. (B) BHK21 cells were transfected with plasmids encoding FLAG-EVER proteins and GFP-ZnT-1 or GFP alone. Crude lysates (CL) were immunoprecipitated by anti-GFP antibody (IP) and immunoblotted (IB) with indicated antibodies. Lines indicate where the original gel was spliced. (C) HaCaT cells transiently coexpressing FLAG-EVER proteins and GFP-ZnT-1 were incubated with mouse monoclonal anti-FLAG antibodies and, subsequently, with anti–mouse CY-3–conjugated secondary antibodies. Colocalization of EVERs (red) and ZnT-1 (green) is presented on merged images (yellow). Bars, 5 μm.
EVER and ZnT-1 regulate intracellular distribution of free zinc
Zinc is a trace element that is essential for a large variety of metalloenzymes and transcription factors. Because of toxicity for cells, free zinc activates several protective mechanisms, such as induction of zinc transporters (ZnT-1 and -2) and metal binding proteins like the metallothioneins MT-1 and -2 (19, 21). As a consequence, free zinc in cells is extremely low and tightly controlled, and even small fluctuations in free zinc level can lead to significant changes in signal transduction or activity of transcription factors. We asked whether in keratinocytes ZnT-1 as well as EVERs can serve as zinc effluxers, inhibiting intracellular zinc retention. To this end, HaCaT cells were transiently transfected with EVER or ZnT-1. We found that incubation of keratinocytes with high concentrations of zinc leads to increase in total cellular zinc content of cells (Fig. 2 A) and that ZnT-1 and EVER neither influence zinc accumulation in these cells nor confer resistance to zinc (Fig. 2 B). Similar results were obtained with HaCaT cells stably transfected with EVER and ZnT-1 (unpublished data). In addition, no difference in zinc content and toxicity was detected for keratinocyte cell lines that we isolated from a Polish EV patient with a homozygous EVER2 mutation (T150fsX3; unpublished data) and a healthy subject (Fig. 2, C and D). These data suggest that in human keratinocytes, ZnT-1 and EVERs do not serve as zinc effluxers and they do not inhibit total zinc accumulation, although their possible role in redistribution of zinc inside keratinocytes cannot be excluded.
Figure 2.

EVERs and ZnT-1 do not influence intracellular zinc accumulation and cell resistance to zinc. HaCaT cells (A and B) were transiently transfected with plasmid encoding ZnT-1, EVER1, EVER2, or control empty plasmid. Keratinocytes (C and D) were isolated from a patient suffering from EV (EVER2−/−) and from a healthy subject (EVER2+ /+). The cells were incubated for 24 h in a standard culture medium (5 μM basal zinc) or in the medium supplemented with indicated concentrations of zinc. The total intracellular zinc content was determined by flame absorption spectrometry (A and C) (29) and the cell number was determined by measurement of DNA (B) or protein (D) content in cell lysates. All the experiments were performed in triplicate. Error bars represent ± the SD.
We hypothesized that the ZnT-1–EVER complex could be involved in transport of free zinc inside keratinocytes, leading to changes in its local concentration. We used zinquin as a specific fluorescent indicator that has the potential to detect free zinc in different cellular compartments, including the nucleus (22, 23). We showed that free zinc accumulates in the nucleus, mainly in nucleoli (Fig. 3 A). Importantly, the concentration of free zinc in nucleoli, measured as a mean fluorescence, was significantly higher in the keratinocytes with a mutated EVER2 gene (EVER2−/− cells), as compared with the cells with a wild-type gene (EVER2+/+ cells; Fig. 3 B). No difference in the number of nucleoli was observed between EVER2−/− and EVER2+/+ keratinocytes (unpublished data). This suggests that in human keratinocytes, EVER2 regulates free zinc distribution and modulates its influx to nucleoli, which are a place for an extensive synthesis of ribosomal RNA and could also be involved in sensing cellular stresses (24). Although the exact mechanism and significance of this phenomenon remain to be determined, it seems probable that changes in nuclear and nucleolar zinc concentration could strongly influence function of the cell. Importantly, we observed that EVER2−/− keratinocytes grew faster than EVER2+/+ keratinocytes (Fig. 3 C).
Figure 3.

EVER2 influences intracellular distribution of free zinc and cell growth. (A) Keratinocytes with wild-type (EVER2+ /+) or mutated (EVER2−/−) gene were loaded with free zinc-specific indicator (Zinquin), incubated for 15 min in 140 μM zinc, and analyzed by phase-contrast microscopy (left) or fluorescence (right). A strong fluorescence is visible in nucleoli in both cell lines (arrows). Bars, 10 μm. (B) The fluorescence intensity in nucleoli was measured in 90 cells and presented as a percentage of fluorescence of EVER2+/+ cells. (C) EVER2−/− and EVER2+/+ keratinocytes were grown in triplicate in standard culture medium, and cell numbers were determined at different times as indicated. P < 0.01, as determined by Students' t test. Error bars represent ± the SD.
EVER and ZnT-1 inhibit transcription factor activities induced by zinc and cytokines
Zinc has recently been considered as a novel intracellular second messenger (25). In addition, it is well known that zinc ions activate the synthesis of ZnT-1 and metallothioneins through the metal-regulatory transcription factor MTF-1 (19, 21). Thus, we studied whether ZnT-1 and EVERs can modulate activity of MTF-1. In transiently or stably transfected HaCaT cells, full-length EVER and ZnT-1 down-regulated zinc-stimulated activity of MTF-1, as measured by transactivation of a construct with a luciferase gene under the control of a minimal responsive promoter for MTF-1 (pMT1/luc; Fig. 4, A and C). In contrast, an increase in luciferase activity was detected in cells expressing mutated EVER1 or EVER2 proteins (Fig. 4 B) or with a mutation in the EVER2 gene (Fig. 4 D). It is worth stressing that EVER1 and EVER2 mutant proteins that do not contain the conserved TMC domain were found to lose their interaction with ZnT-1. Treatment of HaCaT cells constitutively expressing ZnT-1 with two different ZnT-1–specific siRNAs increased the MTF-1/luc activity by four- to fivefold, whereas no effect was observed with the negative control siRNA (Fig. 4 E). All these data indicate that ZnT-1 and EVERs are negative regulators of MTF-1 activity. This might be because of the EVER–ZnT-1–dependent changes in the distribution of free zinc, although interference of the EVER–ZnT-1 complex with activation of MTF-1 (e.g., phosphorylation) cannot be excluded.
Figure 4.

EVERs and ZnT-1 down-regulate transcription factor activity. (A–D) EVER and ZnT-1 inhibit zinc-induced transcription factor MTF-1. HaCaT cells were transiently (A and B) or stably (C) transfected with control plasmid (pCINeo) or plasmids expressing full-length ZnT-1, EVER1, and EVER2 proteins, mutant EVER1* (R94X) (13) and EVER2* (T150fsX3) proteins or TMC and 3′COOH regions of EVER1 and EVER2. HaCaT cells, as well as keratinocytes with a wild-type (+/+) or mutant (−/−) EVER2 gene (D), were transfected with a luciferase reporter plasmid (pMT1/luc) under the control of transcription factor MTF-1 and grown for 24 h at different ZnSO4 concentrations (A, C, and D) or at 120 μM ZnSO4 (B). (E) HaCaT cells stably expressing the control pCiNeo plasmid (mock) or ZnT-1 were transfected with pMT1/luc and Renilla luciferase plasmids in the presence of control negative siRNA or two different siRNAs specific for ZnT-1 (siRNA#1 and siRNA#2). After 24 h, cells were harvested for luciferase activities. The results were scored as the ratio of firefly luciferase activity normalized to Renilla luciferase activity. (F–J) EVER and ZnT-1 inhibit transcription factors. HaCaT cells expressing the indicated plasmids were transfected with a mixture of plasmids encoding the transactivation domain of c-Jun (F and G) and Elk-1 (H) fused to the Gal4 DNA binding domain and a reporter luciferase plasmid containing binding sites for Gal4 (Gal4/luc). HaCaT cells (I) and keratinocytes with a wild-type (+/+) or mutant (−/−) EVER2 gene (J) were also transfected with a luciferase reporter gene under the control of AP-1 transcription factors (pAP-1/luc). The cells were grown without fetal calf serum and supplemented with 50 ng/ml EGF (F and H–J) or 10 ng/ml TGFα (G). Luciferase activities and cell number were determined as described in the Materials and methods. Values ± the SD obtained with c-Jun, Elk, and AP-1 expression plasmids in the presence of control pCiNeo plasmid were taken as 100%. *, P < 0.05; #, P < 0.01.
Thus, we focused on the influence of EVER and ZnT-1 proteins on specific signal transduction pathways leading to activation of transcription factors. Indeed, exposure of cells to zinc have been found to induce stimulation of the c-Jun N-terminal kinase (JNK) signaling pathway through phosphoinositide 3-kinase (PI3K) leading to synthesis of transcription factor like c-Jun, which belongs to the AP-1 family, and Elk-1 (26). To evaluate the phosphorylation-dependent activation of transcription factors, we generated a chimeric protein containing the transactivation domain of c-Jun or Elk-1 fused to the Gal4 DNA binding domain (Gal4-BD). This synthetic transcription factor is able to recognize Gal4 binding sites upstream of a minimal promoter that drives expression of the firefly luciferase reporter gene (unpublished data). In human keratinocytes stimulated with EGF, EVER inhibited activity of c-Jun transactivation domain (Fig. 4 F). Similar effects were observed after stimulation with TGF-α (Fig. 4 G), TGF-β, or anisomycin (not depicted). Regions of EVER proteins corresponding to the conserved TMC domain (14) were as effective as full-length proteins, whereas the 3′COOH region, as well as mutated EVER1 and EVER2 proteins, displayed no significant effect (Fig. 4 F). Interestingly, the influence of EVER and ZnT-1 is not limited to c-Jun only, but seems to be more general because similar effects were exerted on the transactivation domain of Elk-1 (Fig. 4 H). Although the underlying mechanism is not clear, it is likely that EVER-induced changes in free zinc concentration could play a role.
It must be stressed that all the experiments were performed using artificial synthetic transcription factors. To investigate the influence of ZnT-1 and EVERs on the natural transcription factor present in human keratinocytes, we studied transactivation of a luciferase reporter plasmid with a minimal responsive promoter for AP-1 transcription factors (pAP-1/luc). ZnT-1 and EVER inhibited luciferase expression (Fig. 4 I), whereas a significantly increased luciferase activity was observed in EVER2−/− keratinocytes (Fig. 4 J).
These data indicate that ZnT-1 and EVER are negative regulators of AP-1. Because AP-1 is a key transcription factor for HPV (2), a mutation in either EVER gene should facilitate the transcription of the viral genome, particularly the expression of E6 and E7 genes.
HPV16 E5 protein inhibits EVER and ZnT-1 activities
Although EV HPV are responsible for asymptomatic infections that are widespread in the general population (6, 7), they induce lesions only in EV patients where high amount of virions are produced. It must be stressed that EV patients have an abnormal susceptibility to EV HPV, but are not prone to infection with other cutaneous or genital HPV, such as the oncogenic genital HPV16 and HPV18 (4, 5). It can be speculated that EVER and ZnT-1 proteins are involved in the control of EV HPV expression. This prompted us to use a coimmunoprecipitation assay to search for a possible interaction between EVER1, EVER2, or ZnT-1 and early proteins specific for cutaneous and genital HPVs. This study was further justified by the recent study showing an interaction between HPV16 E5 and ZnT-1 in a two-hybrid assay (27). Importantly, the EV HPV genome lacks an E5 open reading frame (16).
No interaction was detected between EVER or ZnT-1 proteins and E6 or E7 of HPV5, HPV9, a nononcogenic EV HPV, and HPV16 (unpublished data). In contrast, it was found that the E5 protein of HPV16 (16E5) binds to EVER1 and EVER2 and, as expected with ZnT-1, also binds with an EVER1–EVER2 complex (Fig. 5 A). In addition, EVER and ZnT-1 colocalized with E5 in transiently transfected HaCaT cells (Fig. 5 B). Moreover, in keratinocytes transfected with ZnT-1 or EVER, 16E5 prevented ZnT-1/EVER-mediated inhibition of MTF-1 transcriptional activity (Fig. 5 C). Similarly, 16E5 blocked EVER2-mediated down-regulation of c-Jun transactivation domain activity in HaCaT cells with constitutive expression of EVER2 (Fig. 5 D). More importantly, 16 E5 increased luciferase activity by a factor of 1.6 also in EVER2+/+ keratinocytes (Fig. 5 E). Interestingly, no effect of 16 E5 protein was observed in EVER2−/− keratinocytes, which further suggests EVER complex as an important target for 16 E5 in keratinocytes. On the other hand, EVER2 deficiency seems to compensate the lack of functional 16 E5 protein, as shown with truncated 16 E5 corresponding to the N- or C-terminal half of the protein (Fig. 5 E). This led to MTF-1 transcriptional activity comparable to one induced by 16 E5 in wild-type keratinocytes. All these data indicate that 16 E5 is able to interact with endogenous EVER and ZnT-1 and to counteract their down-regulation.
Figure 5.

HPV16 E5 protein binds to EVER1, EVER2, and ZnT-1 and interferes with their activity. (A) Clear lysates (CL) from HaCaT cells expressing FLAG-EVER and/or FLAG-ZnT-1, together with GFP-16 E5 or GFP alone, were immunoprecipitated (IP) with anti-GFP antibody and subsequently immunoblotted (IB) with the indicated antibodies. Immunoprecipitation experiments with ZnT-1 and HPV16 E5 gave a band of unknown origin with a migration pattern similar to that of EVER2. (B) HaCaT cells were transiently transfected with plasmids encoding FLAG-EVERs or ZnT-1 and GFP-16 E5 and stained with anti-FLAG monoclonal antibodies, followed by incubation with CY-3–conjugated secondary antibodies. Colocalization of EVER/ZnT-1 (red) and 16 E5 (green) is presented on merged images (yellow). Bars, 10 μm. (C) HaCaT cells were transiently transfected with pMT1/luc and plasmids expressing EVER or ZnT-1 or control empty plasmid in the absence (E5-) or presence (E5 + ) of a plasmid encoding 16 E5. Cells were cultured for 24 h in medium with 140 μM zinc, and luciferase activity was measured. (D) HaCaT cells stably expressing control pCiNeo plasmid or EVER2 were starvated and transfected with a plasmid encoding 16 E5 (or control plasmid) and with a mixture of plasmids expressing the luciferase reporter gene (pGal4/luc) and chimeric Gal4/c-Jun protein. Cells were incubated for 24 h in a culture medium supplemented with 50 ng/ml EGF, and luciferase activity was determined. (E) Keratinocytes with a wild-type (+/+) or mutant (−/−) EVER2 gene were transiently transfected with pMT1/luc, together with the control empty plasmid (control) or plasmids expressing 16 E5 or truncated 16 E5 corresponding to the N-terminal (M1 to I51) and C-terminal (I51 to T83) parts of the viral protein. Cells were cultured for 24 h in medium with 40 μM zinc, and luciferase activity was measured. *, P < 0.05; #, P < 0.01.
These findings have some important implications, not only for EV HPV but also for genital HPV. It can be assumed that interaction between the E5 protein of genital HPV and EVER–ZnT-1 complex might facilitate the high level of free zinc and AP-1 activity needed for expression of viral genome (2). Interestingly, the E8 protein of the cottontail rabbit papillomavirus that shares structural similarities with HPV16 E5 protein was also found to interact with ZnT-1 (27). Furthermore, The E8–ZnT-1 interaction was found to be required for AP-1 activation. The lack of an E5 or E8 open reading frame in the genome of EV HPV (17) may explain the blocking of replication of these viruses in the general population. Mutation in either of the EVER genes may alleviate host restriction and favor viral replication. These data suggest that inhibition of EVER–ZnT-1 complex by E5 protein (genital HPV) or mutation (EV HPV) is crucial for the papillomavirus life cycle.
However, the importance of the EVER–ZnT-1 complex is probably not limited to the control of virus life cycle in keratinocytes. It has been reported that most of EV patients have an impaired cell-mediated immunity and, as was hypothesized, that EV could represent a primary deficiency of extrinsic immunity against the β papillomaviruses (28). Thus, it can be assumed that in addition to inhibition of EV HPV expression, regulation of zinc homeostasis by EVER and ZnT-1 proteins play a role in immune response, through the secretion of antiviral effectors such as cytokines, chemokines, or growth factors. Indeed, our preliminary data indicate that the proinflammatory cytokine IL-6 is down-regulated in EVER2−/− cells (unpublished data).
MATERIALS AND METHODS
Cell lines and culture conditions.
Cell lines were cultured at 37°C with 5% CO2 in MEM (human HaCaT keratinocyte line) or Dulbecco's minimum essential medium (hamster BHK21 cell line) supplemented with 10% fetal bovine serum, 2 mmol/liter glutamine, 1% penicillin-streptomycin-gentamicin antibiotics, and 0.5% fungizone. Keratinocytes were obtained from hair follicles (29) from a healthy subject and from a Polish EV patient. The samples were taken according to the approval of the Local Ethical Committee at the Medical University of Warsaw. The informed consent was obtained from EV family individuals.
Yeast two-hybrid analysis.
DNA fragments corresponding to the TMC region of EVER1 (aa L514 to W673) and EVER2 (aa E362 to S530) were obtained by PCR amplification. Fragments were cloned into plasmid pGBKT7 containing the GAL4 DNA-binding domain and used as bait to screen a human keratinocyte (HaCaT) cDNA library for proteins that were capable of interacting with EVER regions. Yeast transformation and two-hybrid screening assays were done as previously described (17).
Plasmids.
Full-length EVER1, EVER2, and ZnT-1 and DNA fragments corresponding to the conserved TMC and C-terminal regions of EVER1 (L514 to W673 and Q674 to A805) and EVER2 (E362 to S530 and A622 to L725) were obtained by PCR amplification. Fragments were cloned and tagged with FLAG epitope or fused to GST or GFP protein using Gateway recombination technology (Invitrogen) as described by Mendoza et al. (17).
Immunoprecipitation and GST-pull down assays.
BHK21 cells were plated in P6 culture plates and grown to 50–80% confluence. Cells were transfected by the PEI method with a combination of recombinant plasmids, and cell extracts were prepared for immunoprecipitation and GST pulldown as previously described (17).
Immunofluorescence microscopy.
HaCaT cells plated on 10-mm glass coverslips (200,000 cells) were transfected with different recombinant plasmids using the PEI transfection methods (17). 24 h later, the transfected cells were washed thrice with cold PBS, fixed in 3% paraformaldehyde, and permeabilized with 0.1% Triton X-100. Cells were incubated with primary mouse monoclonal or rabbit polyclonal antibodies for 1 h at 37°C. After washings, cells were incubated for 60 min with CY3-conjugated goat anti–mouse antibodies or FITC-conjugated anti–rabbit antibodies. Images were obtained using a TCS4D confocal microscope (Leica) (13). As negative controls, replicate samples were incubated with protein-blocking solution instead of primary antibodies.
For the detection of free zinc by fluorescence (22), the keratinocytes were grown to subconfluence on 30-mm plastic/glass dishes (MatTek Corporation), washed twice with incubation buffer (10 mM Hepes, 120 mM NaCl, 5.4 mM KCl, 5 mM glucose, 1 mM CaCl2, 1 mM MgCl2, 1 mM NaH2PO4, and 3 g/liter BSA, pH 7.35), and loaded with 25 μM Zinquin-E (Ex/Em 368nm/490nm; Qbiogene) in incubation buffer for 30 min at 37°C. The cells were washed twice with incubation buffer without BSA, and ZnSO4 (140 μM) was added for 15 min. The cells were investigated by fluorescence microscopy before and after incubation with zinc and photographs were taken at 200-fold magnification on a microscope (Axioplan 2; Carl Zeiss, Inc.) using a 365/520-nm filter.
Atomic absorption.
For determination of the total cellular zinc content, 3 × 106 cells were washed with PBS twice and collected in 1 ml of PBS. 50 μl were taken to determine the protein content of the samples using the Bio-Rad protein assay (Bio-Rad Laboratories). The remaining cell suspension was centrifuged, and the pellet was treated with 100 μl 33% H2O2 and 100 μl 65% HNO3 at 60°C for 1 h, followed by an overnight incubation at 85°C. Samples were dissolved in 0.5 ml 0.2% HNO3 and the zinc concentration was determined by flame atomic absorption (22), using an atomic absorption spectrometer (model 2380; PerkinElmer).
Luciferase assay.
HaCaT cells were plated in P24 culture plates, grown to 50–80% confluence, and transfected by the PEI method (18). In each cotransfection experiment, total DNA (0.5 μg) was adjusted with control pCiNeo DNA. To estimate transfection efficiency, the cells were also transfected with the TK promoter-driven Renilla-luciferase plasmid (0.1 μg). After 24 h, cells were washed with phosphate-buffered saline and overlaid with 100 μl of passive lysis buffer (Promega) for 15 min at room temperature. The extracts were centrifuged 1 min at 14,000 g, and firefly and Renilla luciferase activities were measured by using the Dual-Glo luciferase kit assay (Promega) and a Lumat LB 9507 luminometer (Berthold Technologies). All experiments were performed 6–12 times to ensure reproducibility. Because expression of EVER and ZnT-1 proteins were found to down-regulate both Firefly and Renilla luciferase activities, data were normalized to cell number as determined by DNA content by fluorescence of bis benzamidine H33258 (30) or protein content using Bio–Rad protein assay. The statistical significance of data were calculated by Student's t test. For all analyses, P < 0.05 was considered significant.
Small interfering RNA (siRNA)–mediated ZnT-1 silencing.
HaCaT cells constitutively expressing ZnT-1 were cotransfected with siRNA (20 pmol) oligonucleotides, pMT1/luc plasmid, and Renilla luciferase plasmid using Lipofectamine 2000 reagent (Invitrogen) according to the instructions of the manufacturer. Two different predesigned siRNAs specific for ZnT-1 (siRNA ID#117632 and siRNA ID#117633), as well as a negative control siRNA, were purchased from Ambion. Luciferase activities were tested 24 h after transfection. Experiments were performed in quadruplicate to ensure reproducibility.
Online supplemental material.
Fig. S1 shows the localization of EVER and ZnT-1 proteins by immunofluorescence using a confocal microscope (model TCS4D; Leica). The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20071311/DC1.
Supplemental Material
Acknowledgments
We thank E. Perret for technical expertise in confocal microscopy. We would particularly like to thank Professors S. Jabonska and S. Majewski (Department of Dermatology, The Medical University of Warsaw), and the EV patients whose support and cooperation were essential for collection of material. We would like to thank Dr. Y. Mechulam (Ecole Polytechnique, Gif-sur-Yvette, France) for his assistance with flame absorption spectrometry.
This work was supported in part by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM 2004 00 2308) and the Ligue Nationale contre le Cancer (Contrat n° R05/75-129 and Contrat n° RS07/75-75). M. Lazarczyk was supported by a fellowship from the Foundation for Polish Science.
The authors have no conflicting financial interests.
References
- 1.Orth, G. 1999. Papillomaviruses - human (Papovaviridae): General Features. In Encyclopedia of Virology. Second edition. R.G. Webster and A. Granhoff, editors. Academic Press Ltd., London. 1105–1114.
- 2.Howley, P.M., and D.R. Lowy. 2001. Papillomaviruses and their replication. Fields Virology. Vol. 2. Lippincott Williams and Wilkins, Philadelphia. 2197–2230.
- 3.zur Hausen, H. 2002. Papillomavirus and cancer: from basic studies to clinical application. Nat. Rev. Cancer. 2:342–350. [DOI] [PubMed] [Google Scholar]
- 4.Orth, G. 1987. Epidermodysplasia verruciformis. In The Papovaviridae: The Papillomaviruses. N.P. Salzman and P.M. Howley, editors. Vol. 2. Plenum Press New York. 199-243.
- 5.Majewski, S., S. Jablonska, and G. Orth. 1997. Epidermodysplasia verruciformis. Immunological and nonimmunological surveillance mechanisms: role in tumor progression. Clin. Dermatol. 15:321–334. [DOI] [PubMed] [Google Scholar]
- 6.Antonsson, A., O. Forslund, H. Ekberg, G. Sterner, and B.G. Hansson. 2000. The ubiquity and impressive skin papillomaviruses suggest a commensalic nature of these viruses. J. Virol. 74:11636–11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Struijk, L., J.N. Bouwes Bavinck, P. Wanningen, E. van der Meijden, R.G. Westendorp, I.J. Ter Schegget, and M.C. Feltkamp. 2003. Presence of human papillomavirus DNA in plucked eyebrow hairs is associated with a history of cutaneous squamous cell carcioma. J. Invest. Dermatol. 121:1531–1535. [DOI] [PubMed] [Google Scholar]
- 8.Favre, M., G. Orth, S. Majewski, S. Baloul, A. Pura, and S. Jablonska. 1998. Psoriasis: a possible reservoir for human papillomavirus type 5, the virus associated with skin carcinomas of epidermodysplasia verruciformis. J. Invest. Dermatol. 110:311–317. [DOI] [PubMed] [Google Scholar]
- 9.Favre, M., S. Majewski, B. Noszczyk, F. Maienfisch, A. Pura, G. Orth, and S. Jablonska. 2000. Antibodies to human papillomavirus type 5 are generated in epidermal repair processes. J. Invest. Dermatol. 114:403–407. [DOI] [PubMed] [Google Scholar]
- 10.Pfister, H. 2003. Chapter 8: human papillomavirus and skin cancer. J. Natl. Cancer Inst. 31:52–56. [DOI] [PubMed] [Google Scholar]
- 11.Orth, G. 2005. Human papillomaviruses associated with epidermodysplasia verruciformis in non-melanoma skin cancers: guilty or innocent? J. Invest. Dermatol. 125:xii–xiii. [DOI] [PubMed] [Google Scholar]
- 12.Ramoz, N., A. Taieb, L.-A. Rueda, L.-S. Montoya, B. Bouadjar, M. Favre, and G. Orth. 2000. Evidence for a nonallelic heterogeneity of epidermodysplasia verruciformis with two susceptibility loci mapped to chromosome regions 2p21-p24 and 17q25. J. Invest. Dermatol. 114:1148–1153. [DOI] [PubMed] [Google Scholar]
- 13.Ramoz, N., L.-A. Rueda, B. Bouadjar, L.-S. Montoya, G. Orth, and M. Favre. 2002. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat. Genet. 32:579–581. [DOI] [PubMed] [Google Scholar]
- 14.Keresztes, G., H. Mutai, and S. Heller. 2003. TMC and EVER genes belong to a larger novel family, the TMC gene family encoding transmembrane proteins. BMC Genomics. 4:24–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fields, S., and O. Song. 1989. A novel genetic system to detect protein-protein interactions. Nature. 340:245–246. [DOI] [PubMed] [Google Scholar]
- 16.Bravo, I.G., and A. Alonso. 2004. Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. J. Virol. 78:13613–13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mendoza, J.A., Y. Jacob, P. Cassonnet, and M. Favre. 2006. Human papillomavirus type 5 E6 oncoprotein represses the transforming growth factor β signaling pathway by binding to SMAD3. J. Virol. 80:12420–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmiter, R.D., and S.D. Findley. 1995. Cloning and functional characterization of a mammalian zinc transporter that confers resistance to zinc. EMBO J. 14:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cousins, R.J., J.P. Liuzzi, and L.A. Lichten. 2006. Mammalian zinc transport, trafficking, and signals. J. Biol. Chem. 281:24085–24089. [DOI] [PubMed] [Google Scholar]
- 20.Liuzzi, J.P., J.A. Bobo, L.A. Lichten, D.A. Samuelson, and R.J. Cousins. 2004. Responsive transporter genes within the murine intestinal-pancreatic axis form a basis of zinc homeostasis. Proc. Natl. Acad. Sci. USA. 101:14355–14360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bi, Y., R.D. Palmiter, K.M. Wood, and Q. Ma. 2004. Induction of metallothionein I by phenolic antioxidants requires metal-activated transcription factor 1 (MTF-1) and zinc. Biochem. J. 380:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berendji, D., V. Kolb-Bachofen, K.L. Meyer, O. Grapenthin, H. Weber, V. Wahn, and K.-D. Krönke. 1997. Nitric oxide mediates intracytoplasmic and intranuclear zinc release. FEBS Lett. 405:37–41. [DOI] [PubMed] [Google Scholar]
- 23.Haase, H., and D. Beyersmann. 2002. Intracellular zinc distribution and transport in C6 rat glioma cells. Biochem. Biophys. Res. Commun. 296:923–928. [DOI] [PubMed] [Google Scholar]
- 24.Lam, Y.W., L. Trinkle-Mulcahy, and A.I. Lamond. 2005. The nucleolus. J. Cell Sci. 118:1335–1337. [DOI] [PubMed] [Google Scholar]
- 25.Yamasaki, S., K. Sakata-Sogawa, A. Hasegawa, T. Suzuki, K. Kabu, E. Sato, T. Kurosaki, S. Yamashita, M. Tokunaga, K. Nishida, and T. Hirano. 2007. Zinc is a novel intracellular second messenger. J. Cell Biol. 177:637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eom, S.J., E.Y. Kim, J.E. Lee, H.J. Kang, J. Shim, S.U. Kim, B.J. Gwag, and E.J. Choi. 2001. Zn(2+) induces stimulation of the c-Jun N-terminal kinase signaling pathway through phosphoinositide 3-Kinase. Mol. Pharmacol. 59:981–986. [DOI] [PubMed] [Google Scholar]
- 27.Nonnenmacher, M., J. Salmon, Y. Jacob, G. Orth, and F. Breitburd. 2006. Cottontail rabbit papillomavirus E8 protein is essential for wart formation and provides new insights into viral pathogenesis. J. Virol. 80:4890–4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orth, G. 2006. Genetics of epidermodysplasia verruciformis: insights into host defense against papillomaviruses. Semin. Immunol. 18:362–374. [DOI] [PubMed] [Google Scholar]
- 29.Hoeller, D., B. Huppertz, T.C. Roos, P. Poblete Gutiérrez, H.F. Merk, J. Frank, and F.K. Jugert. 2001. An improved and rapid method to construct skin equivalents from hair follicles and fibroblasts. Exp. Dermatol. 10:264–271. [DOI] [PubMed] [Google Scholar]
- 30.Palmiter, R.D. 2004. Protection against zinc toxicity by metallothionein and zinc transporter 1. Proc. Natl. Acad. Sci. USA. 101:4918–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}