

Abstract
A new series of androgen receptor targeted agents (ARTA) was prepared and tested in androgen-dependent and -independent prostate cancer cell lines. These agents were bicalutamide analogs with isothiocyanato substituted B-rings. Also, the linker sulfone of R-bicalutamide was maintained or replaced with several alternative linkages including ether, amine, N-methylamine, thioether, and methylene (in this case the product was a racemic mixture) functional groups at the X-position. To expand the structure-activity relationship (SAR) of these arylisothiocyanato AR ligands, B-ring halogenated arylisothiocyanato ligands were also prepared and tested. The arylisothiocyanato AR ligands showed strong binding affinities to AR ranging from 0.6 to 54 nM. Among them, thioether and ether linkages demonstrated high binding affinities (0.6 and 4.6 nM, respectively) and selective cell growth inhibition (approximately 3- to 6-fold) for LNCaP, an androgen-dependent prostate cancer cell line, when compared to the androgen independent prostate cell lines (DU145, PC-3, and PPC-1) and a bladder cell line (TSU-Pr1). However, the ligands were inactive (IC50>100 mM) in a normal monkey kidney cell line (CV-1) that was used as the control for non-specific toxicity.
Keywords: Prostate cancer (CaP), Selective androgen receptor modulators (SARMs), Isothiocyanate, Bicalutamide, Androgen receptor target agents (ARTA), Non-steroidal, Irreversible, Androgen receptor (AR)
1. Introduction
Prostate cancer (CaP) is a major health hazard for men living in United States1 and developed countries. Approximately 90% of prostate tumors are androgen-dependent.2 Androgens, primarily testosterone (T) and dihydrotestosterone (DHT), bind to the androgen receptor (AR) and support the development and maintenance of normal prostate tissue but more importantly can have a pathologic role in prostate cancer patients by promoting the growth of the tumor.3 Antiandrogen therapy to block these pathological effects of endogenous androgens has been the golden standard for unrespectable prostate cancer (disease spread beyond prostate). However, despite the high initial response rate to hormonal therapy, almost all patients relapse and become hormone refractory.4 Our effort is directed at finding potent non-steroidal AR inhibitors that are effective even in hormone refractory prostate cancer.5 Until now, the blockage of the pharmacological effects of androgens by non-steroidal AR ligands was achieved through the use of nilutamide, flutamide, and bicalutamide in advanced CaP patients.6–8
All current AR antagonists are reversible ligands that interact with the AR via non-covalent bonds such as hydrophobic, electrostatic, and hydrogen bond interactions. Unlike a reversible ligand, an irreversible ligand can bind to the receptor via a covalent bond that permanently attaches the ligand to the receptor. Our laboratory has a long interest in the discovery of novel anticancer drugs that act via the AR. Recently, we reported that an isothiocyanate substituent at the para-position of the B-ring of thioether and sulfonyl-linked derivatives of bicalutamide bound tightly and may form a covalent bond to the AR.9 In this study, we expand our previous efforts10,11 regarding non-steroidal AR ligands to include bicalutamide analogs with the potential to bind irreversibly to the AR. In pursuit of this objective, chiral arylisothiocyanato derivatives were synthesized and tested in the prostate cancer cell lines LNCaP, DU145, PC-3, PPC-1, and a bladder cancer cell line (TSU-Pr1) as well as a normal monkey kidney cell line (CV-1). In this report, our previous ligands S-23 and R-35 (Table 1)10,11 were tested on CaP cell lines as selective androgen receptor modulators (SARMs) and diversified at the X-position and halogenated on the B-ring. This work was carried out to find suitable ligands, which are selective AR-dependent CaP inhibitors with strong AR binding affinity and no cytotoxicity on normal cell lines, and to address the key issues in enhancing AR binding affinities of bicalutamide derivatives and their use in prostate cancer therapy. First, we focused on which linker was most suitable for AR potential prostate cancer therapy. With this objective, we prepared and tested several analogs with ether, methylene, thioether, sulfone, amino, and methylamino groups as outlined in general structure S/R-1 (Fig. 1). The synthetic methods for these linker variants are shown in Scheme 1–Scheme 5. Our second goal was the determination of the pharmacological effects of halogenation at the 2- or 3-position of the 4-isothiocyanato substituted B-ring derivatives as outlined in general structure S-2 (Fig. 1). Kim et al.12 reported the para B-ring substituent is a major structural determinant of in vivo disposition and activity of non-steroidal AR agonists (a.k.a. selective androgen receptor modulators or SARMs). SARMs are a new set of non-steroidal AR ligands with potential therapeutic activity similar to testosterone. They may be used orally with androgenic and anabolic activity and have fewer side effects than testosterone. Chen et al.13 reported that halogenation at the 2- or 3-position of the B-ring increased their binding affinities to AR. To further probe the SAR of 2- or 3-halogenated SARMs, we tested the AR binding affinity and cancer cell growth inhibition of the 2- or 3-halogenated isothiocyanato ligands. Our last goal was the characterization of SAR A-ring para-position regarding AR affinity and cell growth inhibition. We compared the NO2 or CN group on the A-ring of our analogs. The in vitro prostate cancer directed cell growth inhibitory properties of the chiral and racemic AR ligands in Figure 1 were characterized. The AR binding affinity and the concentration of the ligand that inhibited cell growth by 50% (IC50) were determined by radioligand competitive binding assay and sulforhodamine B (SRB) assay, respectively. The AR binding affinities and cell growth inhibitory effect of these compounds (S and/or R)-23 ~ (S and/or R)-37 with diverse substituents on the aromatic A and B rings are reported.
Table 1.
Structures and pharmacological activities of arylisothiocyanato SARMs against prostate cancer cell lines (LNCaP, DU145, PC-3, and PPC-1), bladder cancer cell line (TSU-Pr1), and normal monkey kidney cell line (CV-1) (IC50 in µM)
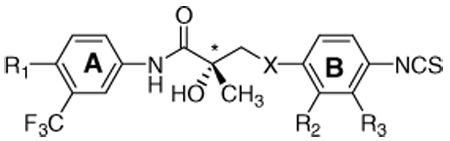 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| R1 | X | R2 | R3 | IC50(µM) |
Ki (nM) on hAR | ||||||
| LNCaP | DU145 | PC-3 | PPC-1 | TSU-Pr1 | CV-1 | ||||||
| DHT | 0.43 ± 0.01 | ||||||||||
| R-bicalutamidea | 45.9 ± 8.8 | 74.7 ± 12.2 | 71.6 ± 6.7 | 39.3 ± 1.8 | 8.3 ± 1.6 | 38.1± 9.2 | 11.0 ± 2.0 | ||||
| S-23 | NO2 | O | H | H | 17.4 ± 4.9 | 74.7 ± 5.9 | 59.1 ± 3.3 | 74.7 ± 5.9 | 57.1 ± 2.3 | >100 | 4.6 ± 0.310 |
| R-23b | NO2 | O | H | H | 24.9 ± 1.7 | 28.7 ± 1.7 | 15.7 ± 1.4 | 22.3 ± 0.1 | 28.7 ± 1.7 | 44.3 ± 1.1 | 36.2 ± 2.8 |
| S-24 | CN | O | H | H | 17.8 ± 12.8 | 76.8 ± 8.2 | 69.5 ± 4.8 | 43.2 ± 14.4 | 54.8 ± 3.1 | N.D. | 8.1 ± 0.3 |
| S-25 | NO2 | O | H | F | 13.9 ± 2.9 | 50.8 ± 6.0 | 47.2 ± 2.9 | 34.3 ± 5.1 | 45.3 ± 6.1 | N.D. | 12.6 ± 1.1 |
| S-26 | CN | O | H | F | 66.3 ± 14.9 | 76.0 ± 6.9 | 62.1 ± 12.2 | 56.9 ± 5.1 | 54.8 ± 3.9 | N.D. | 6.3 ± 1.0 |
| S-27 | NO2 | O | H | Cl | 30.6 ± 11.0 | 72.1 ± 3.3 | 41.2 ± 8.2 | 43.5 ± 7.4 | 39.8 ± 7.3 | N.D. | 11.9 ± 2.7 |
| S-28 | CN | O | H | Cl | 52.0 ± 15.3 | 55.0 ± 6.1 | 48.6 ± 2.5 | 53.0 ± 10.7 | 51.1 ± 6.7 | N.D. | 8.6 ± 0.9 |
| S-29 | NO2 | O | F | H | 23.8 ± 2.0 | 47.9 ± 3.9 | 39.5 ± 1.4 | 31.8 ± 1.3 | 49.0 ± 1.0 | N.D. | 34.6 ± 6.5 |
| S-30 | NO2 | O | Cl | H | 20.3 ± 1.4 | 29.4 ± 2.9 | 26.8 ± 1.6 | 22.2 ± 1.2 | 26.9 ± 1.1 | N.D. | 31.1 ± 1.8 |
| 31c | NO2 | CH2 | H | H | 20.6 ± 3.8 | 23.4 ± 1.3 | 21.1 ± 2.7 | 20.7 ± 0.9 | 23.8 ± 3.0 | N.D. | 31.8 ± 1.8 |
| S-32 | NO2 | NH | H | H | 21.5 ± 0.7 | 29.4 ± 2.9 | 32.4 ± 2.1 | 26.5 ± 1.3 | 38.3 ± 1.0 | N.D. | 21.4 ± 7.0 |
| S-33 | NO2 | NCH3 | H | H | 13.8 ± 0.4 | N.D. | 27.8 ± 1.8 | 21.9 ± 1.2 | 14.7 ± 0.3 | N.D. | 41.2 ± 5.9 |
| R-34 | NO2 | S | H | H | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 54.8 ± 3.0 |
| R-35 | CN | S | H | H | 10.6 ± 7.1 | 88.3 ± 8.2 | 91.1 ± 6.3 | 86.9 ± 4.1 | 65.6 ± 4.2 | >100 | 0.6 ± 0.111 |
| S-35b | CN | S | H | H | 64.0 ± 5.9 | >100 | >100 | >100 | >100 | >100 | 430 ± 13 |
| R-36 | NO2 | SO2 | H | H | 29.4 ± 1.6 | 59.3 ± 1.9 | 58.9 ± 4.1 | 43.9 ± 3.1 | 43.3 ± 3.7 | N.D. | 14.4 ± 4.4 |
| R-37 | CN | SO2 | H | H | 42.7 ± 5.3 | 81.5 ± 28.7 | 70.1 ± 7.6 | 69.4 ± 1.8 | 59.2 ± 6.7 | >100 | 41.0 ± 2.011 |
N.D., not determined.
Reversible ligand; tested on above cell lines and AR binding affinity determined.
Opposite enantiomeric (inactive) isomers synthesized using the chiral auxiliary starting from l-proline (vs d-proline for the other compounds via the same synthetic procedure as S-23 and R-35).
Racemic mixture.
Figure 1.
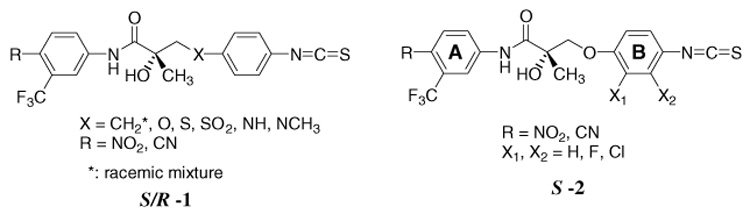
General structures of putative irreversible SARMs.
Scheme 1.
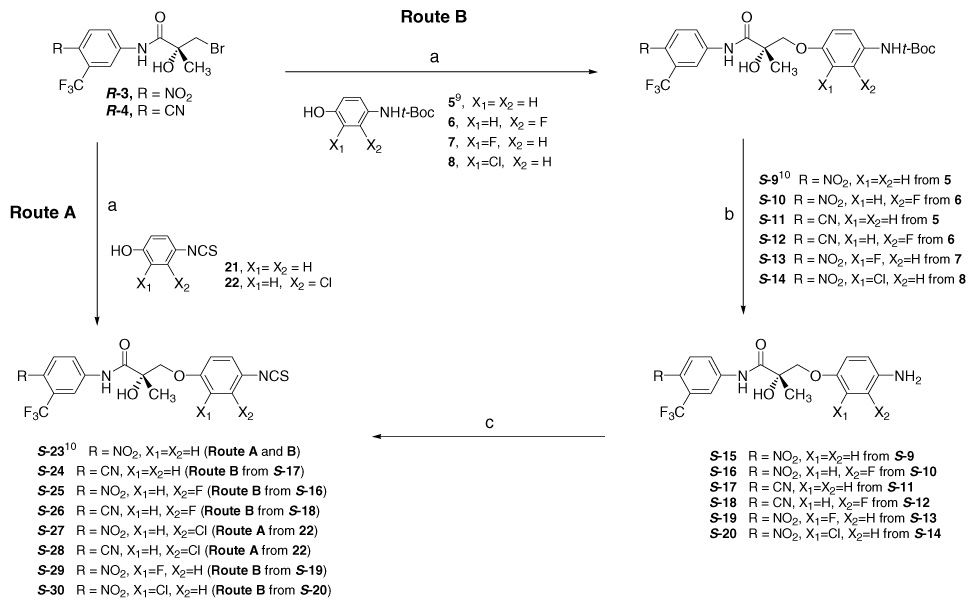
Reagents and conditions: (a) i—K2CO3, acetone, reflux, ii—substituted phenols (5, 6, 7, 8, 21 or 22), K2CO3, methylethylketone, reflux; (b) EtOH, AcCl, 0 °C; (c) CSCl2, aq NaHCO3, CHCl3.
Scheme 5.
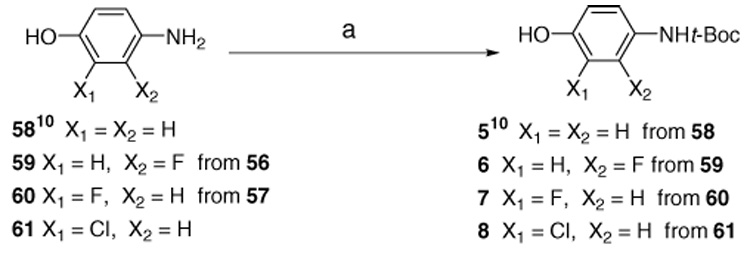
Reagents and conditions: (a) Et3N, di-tert-butyl dicarbonate, THF.
2. Chemistry
A total of 17 arylisothiocyanato derivatives (S and/or R)-23 to (S and/or R)-37 (Table 1) with several linkages (X = O, CH2, NH, NCH3, S, and SO2) were prepared as discussed in Scheme 1–Scheme 4, out of which 14 novel SARMs were newly synthesized. The target arylisothiocyanato derivatives were mainly synthesized by two known methods (Leclerc14 and Marhefka10). Scheme 1 describes the synthesis of ether-linked analogs. The protected arylamines (S-9 to S-14) were converted to free arylamines (S-15 to S-20) by deprotection under acidic conditions using AcCl and absolute EtOH.15 The resulting free amines (S-15 to S-20) were then converted to arylisothiocyanato derivatives (S-23 to S-26, S-29, and S-30) by reaction with thiophosgene in heterogeneous (chloroform/water) co-solvents under basic condition.14 Arylisothiocyanato S-23 was obtained by a straightforward procedure adopted by Marhefka et al.10 through the epoxide intermediate, by using 4-isothiocyanatophenol (21). With 3-chloro-4-isothiocyanatophenol (22), compounds S-27 and S-28 were prepared by a similar procedure from the amidobromide R-3 and R-4. The propionamide derivatives R-3 and R-4 were prepared from R-3-bromo-2-hydroxy-2-methylpropionic acid and 4-nitro or 4-cyano-3-trifluoromethylaniline as reported.11
Scheme 4.
Reagents and conditions: (a) NaH, THF, 0 °C; (b) CSCl2, aq NaHCO3, CHCl3; (c) 3-chloroperoxy-benzoic acid, CH2Cl2.
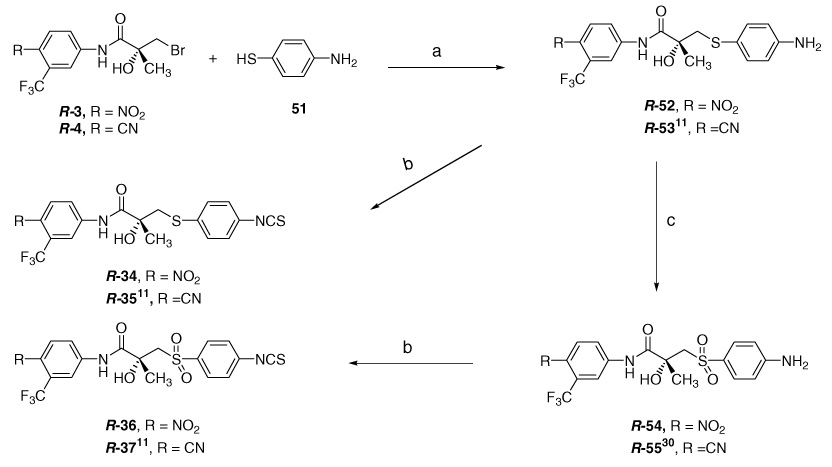
Derivatives having CH2, NH, N-CH3, S, and SO2 linkages (31, S-32, S-33, and R-34 ~ R-37) were prepared from corresponding precursors (42, S-49, S-50, and R-52 ~ R-55) as shown in Scheme 2–Scheme 4. The synthetic route outlined in Scheme 2 provided the optimized method for the methylene linkage (−CH2−) 31, which is a racemic mixture.
Scheme 2.
Reagents and conditions: (a) i—Me3SiCN, ZnI2 (cat.), CH2Cl2, ii—HCl, AcOH, reflux; (b) H2, Pd/C, MeOH, 30 psi; (c) i—SOCl2, THF, DMF (cat.), ii—4-nitro-3-(trifluoromethyl)aniline (41), Et3N; (d) CSCl2, aq NaHCO3, CHCl3.
Nitrophenyl butanone 38 was prepared by refluxing 1-bromomethyl-4-nitrobenzene and pentane-2,4-dione with K2CO3 in ethanolic medium.17 Trimethylsilyl cyanide (Me3SiCN) with catalytic zinc iodide (ZnI2) provided the cyanohydrin intermediates,18 which on subsequent hydrolysis under acidic conditions afforded the racemic butyric acid 39 in 91% yield. The nitro group of compound 3916 was converted to corresponding amine 40 by catalytic reduction. The aniline precursor 42 was prepared by coupling of hydroxybutyric acid 40 with 4-nitro-3-tri.uoromethylaniline (41). In order to prevent acid chloride of 40 from undergoing self condensation, the nitroaniline 41 was rapidly added to the solution under ice-cold conditions, followed by triethylamine (Et3N) to gain desired anilino product 42 in reasonable yield of 68%.
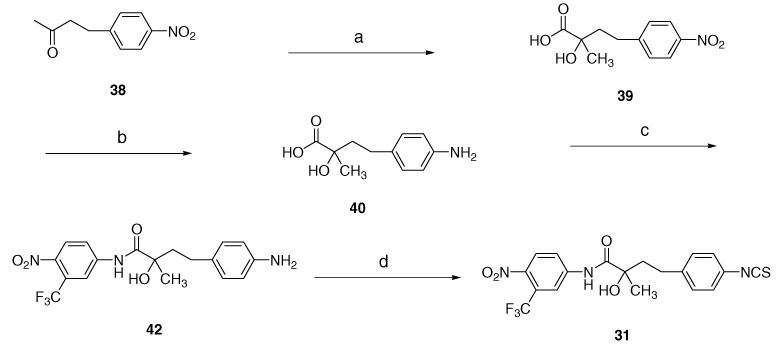
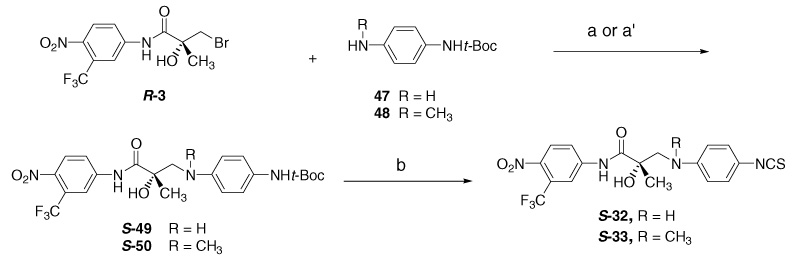
The S-32 and S-33 were synthesized from N-protected S-49 and S-50, respectively (Scheme 3) by deprotection under acidic condition to produce the corresponding aniline precursors (not isolated), and then to target amino-linked S-32, S-33 in 80% and 67% overall yields. The coupling of R-bromoanilide R-3 with N-protected diamine 47/48 was carried out under the following two basic conditions. The first method employed sodium hydride (NaH) in THF for N-methyl di-amino compound 48 to undergo coupling with R-bromoanilide R-3 in 78% yield. However, the protected diamino derivative 47 under sodium hydride condition failed to yield the desired product S-49. A solution of 47 and R-3 in THF, on re.uxing with Et3N, afforded amino-linked precursor S-49 in 63% yield. The amino-protected diamine 4719 was prepared from 4-nitroaniline (43) through amino-protection in presence of Et3N to yield (4-nitrophenyl)-carbamic acid tert-butyl ester (44),20 which was reduced catalytically in EtOAc at 30 psi to give the amino-protected diamine 47. The same reduction of nitro group of methyl-(4-nitrophenyl)amine (45) gave N-methylbenzene-1,4-diamine (46)21 quantitatively, and this was used for the preparation of compound 48 by adding di-tert-butyl dicarbonate (Boc anhydride).
Scheme 3.
Reagents and conditions: (a) Et3N, THF, reflux, for S-49 from 47. (a′) NaH, THF, 0 °C, for S-50 from 48; (b) i—EtOH, AcCl, 0 °C, ii—CSCl2, aq NaHCO3, CHCl3.
The S- and SO2-linked ligands (R-34 to R-37) were prepared from corresponding amines (R-52 to R-55) by Leclerc’s method14 as shown in Scheme 4. The thioamine R-52 and R-53, precursors for the R-36 and R-37 respectively, were oxidized to produce sulfone R-54 and R-55 following a literature procedure.11,17 The R-bromoanilide R-3 and R-4 were synthesized from d-proline as the chiral auxiliary.11 We also tested the other isomeric arylisothiocyanato compounds, R-23 and S-35, synthesized from l-proline in order to optimize the ligand for inhibition of prostate cancer cell lines.
The selective amino-protection for aminophenol (5–8), which used the synthesis of ether linkage (X = O) S-9 to S-14, is depicted in Scheme 5. The reduction of the nitrophenol22 was carried out using Pd/C with H2, followed by amino-protection with Boc anhydride, to obtain the desired products 5, 6, 7, and 8.
3. Results and discussion
A novel series of arylisothiocyanato 1,3-disubstituted-2-hydroxy-2-methylpropionamide AR ligands were prepared (Table 1) with several variations of the linkers (O, CH2, NH, N-CH3, S, and SO2; S/R–23, S-24, 31, S-32, S-33, R-34, S/R-35, R-36, and R-37) and modifications in position and degree of halogenation (2-, or 3-positions with F/Cl substitution on B-ring; S-25 to S-30). Irreversibly binding ligands have been proven to be very useful pharmacological tools in a number of systems.23 The reaction of isothiocyanato compounds may provide a unique mechanism for suppressing cell proliferation of prostate cancer cell growth. In this study, the isothiocyanate groups are capable of reacting with nucleophilic side chains that contain thiol (cysteine), hydroxyl (threonine or serine), or amino (lysine) functionalities to form a covalent bond with the AR and thus irreversibly block its pathological role in prostate cancer.5,24
The SARMs were tested for AR binding affinities as well as cell growth inhibition on prostate cancer cell lines (LNCaP, DU145, PC-3, and PPC-1), bladder cancer cell line (TSU-Pr1), and a normal renal cell line (CV-1), which was included as the control for non-specific cell growth inhibition (Table 1). We tested the pharmacological activities of the isothiocyanato AR ligands against prostate/bladder cancer cell lines and normal cell line (IC50 in µM) as shown in Table 1. Generally, the isomers that are analogs to R-bicalutamide (R- for X = S and SO2, S- for X = O) have higher AR binding affinity as described in our previous report.11 The cell growth inhibition results for the ether-linked S-23 showed an IC50 value on LNCaP of S-23 (18.8 µM) which is 1.5 times higher than R-23 (28.8 µM). In this study, the in vitro cell proliferation data also indicated that S-23 was more selective in inhibiting cell growth by 3- to 4-fold in the AR-dependent cell line LNCaP compared to the AR-independent prostate/bladder cancer cell lines (DU145, PC-3, PPC-1, and TSU-Pr1). This suggests that the S-isomer of ether-linked isothiocyanates generally demonstrated higher binding affinity and better selectivity for the AR-positive prostate tumors. The A-ring substitution of the isothiocyanato ligands also has an effect on the AR binding affinity. The presence of nitro group in para-position of S-23 demonstrated slightly stronger AR binding affinity than the corresponding cyano analog as in case of S-24. The methylene (CH2) linked 31 was a racemic mixture that inhibited the proliferation of various prostate cancer cell lines (i.e. had no selectivity for AR-dependent versus independent cell growth inhibition) despite intermediate levels of the AR binding affinity (Ki). Amino-linked ligands (S-32 and S-33) also showed mild inhibitions of CaP cell lines (13.8 ~ 32.4 µM) with intermediate Ki values (21.4 nM for S-32, 41.2 nM for S-33). The N-methyl ligand S-33 showed more activity against LNCaP (13.8 µM) and TSU-Pr1 (14.7 µM) cell lines but the binding affinity to AR was less than S-32 (21.5 µM for LNCaP, 38.3 µM for TSU-Pr1). We saw a trend of S-33 being more active in cancer cell lines but binding less to AR than S-32. Thus, N-methyl ligand S-32 lowered AR binding but not anticancer activity. The thioether-linked, nitro group containing R-34, ad a mild binding affinity, but was not tested in the CaP cell lines. However, the cyano A-ring variant, R-35, had selective androgen-dependent (10.6 µM on LNCaP) inhibition and no significant cell growth inhibition on the normal cell line (>100 µM on CV-1). Compound R-35 also had the strongest AR binding affinity (0.6 nM) as previously reported.11 The thioether-linked enantiomeric isomer, S-35, bound very weakly to AR (Ki = 430 nM), showed sixfold less activity against the androgen-dependent prostate cancer cell line (LNCaP), and no cell growth inhibition on normal cells (>100 µM, CV-1). However, ether-linked enantiomeric isomer, R-23 (28.8 µM for LNCaP), had similar LNCaP inhibition as its isomer S-23 (18.8 µM) despite much weaker AR binding affinity (141 vs 4.6 nM). The sulfone ligands R-36 and R-37 retained the selectivity for AR-dependent cell growth inhibition in LNCaP cells. R-36 showed selectivity for AR-dependent (29.4 µM for LNCaP), greater than AR-independent prostate carcinomas (43.9 µM for PPC-1). And, R-37 showed 42.7 µM for AR-dependent LNCaP cell line, greater than 69.3 µM for the AR-independent PPC-1 cell line. Comparison of the irreversibly binding isothiocyanate analog R-37 with R-bicalutamide revealed that there was no significant cell growth inhibition on normal cell lines (CV-1; >100 µM for R-37 vs 38.1 µM for R-bicalutamide), despite weak inhibition on bladder cancer cell lines (TSU-Pr1; 59.2 µM for R-37 vs 8.3 µM for R-bicalutamide) and its weak binding affinity to the AR (Ki; 41.0 nM for R-37 vs 11.0 nM for R-bicalutamide). Earlier studies disclosed that dihalogenation of the B-ring increased the binding affinity of 4-halogenated compounds on AR.13 The 2- or 3-halogenated 4-isothiocyanato (NCS) compounds S-25 to S-30 were synthesized. Unfortunately the addition of halogens on the B-ring did not consistently enhance activity. The 3- uoro B-ring S-25 and the 3-chloro B-ring S-27 had approximate 2- to 3-fold lower binding affinities (12.6 and 11.9 nM, respectively) than the non-halogenated variant S-23. As a result, only relatively modest to no LNCaP-selective cell growth inhibition was observed (13.9 and 30.6 µM on LNCaP, respectively, vs greater than or equal to 34.3 and 39.8 µM on other CaP cell lines). The cyano A-ring variants S-26 and S-28, and the 2-halogenated, NO2 group-containing S-29 and S-30 compounds had no selectivity on AR-dependent line (LNCaP) in spite of stronger Ki values in some cases (6.3 and 8.6 nM for S-26 and S-28, respectively). In conclusion, ether-linked S-23 and thioether-linked R-35 arylisothiocyanato linkage ligands are effective on androgen-dependent prostate cancer cells (LNCaP) and have no effect on androgen-independent cell lines (DU145, PC-3, PPC-1, and TSU-Pr1). Also, S-23 and R-35 demonstrated high binding affinities to hAR (4.6 and 0.6 nM, respectively). The current effort was an attempt to improve our binding affinity and selectivity in AR-dependent prostate cancer cell growth inhibition by the synthesis and testing of 2- and 3-halogenated ether linkages and the variants at X-position. We found in this study that CaP activities of SARMs are dependent on their linkers and substituents on B-ring. Unfortunately, the 2-, or 3- halogenation on the 4-isothiocyanato B-ring derivatives reported here did not produce an improved CaP cell growth inhibition profile. Replacing the NO2 group with CN on the A-ring of isothiocyanato SARMs produced several compounds that had significant AR binding affinity and/or favorable (AR-dependent) CaP cell growth inhibition profiles. Despite our SAR exploration around S-23 and R-35, these compounds still remain the best compounds to date for irreversible AR-targeted therapies for hormone-resistant prostate cancer as exhibited by their strong selectivity for CaP inhibition. We do not see a good correlation between AR binding and CaP inhibition (e.g. S-23 vs R-23). In order to provide a rationale for the observations, we will continue to investigate the requirements for cell growth inhibition in prostate cancer cell lines with compounds structurally related to S-23 and R-35, and we plan to examine selected compounds for in vivo activity in prostate cancer animal models.
4. Experimental
4.1. Biological tests
4.1.1. Cell culture.
Four prostate cancer cell lines (LNCaP, DU145, PC-3, and PPC-1), a bladder cancer cell line (TSU-Pr1), and a normal monkey kidney cell line (CV-1) were obtained from ATCC. Prostate cancer cells and bladder cancer cell were grown in RPMI-1640 medium and CV-1 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 2 mM l-glutamine supplemented with 10% fetal bovine serum (FBS) and maintained in a 5% CO2/95% air humidified atmosphere at 37 °C, respectively.
4.1.2. Assay for cell growth inhibition (sulforhodamine B assay)
Viable cells were quantified using the sulforhodamine B(SRB) assay. Cells were seeded for one day prior to the addition of different drugs at a range of concentrations (0–100 µM). After 96 h incubation, cells were fixed by the addition of 50 µL of 50% cold trichloroacetic acid and incubated at 4 °C for 1 h. The plate was washed five times with tap water and allowed to air-dry. The cells were then stained with 0.4% SRB dissolved in 1% acetic acid for 20 min at room temperature. Unbound SRB was washed away with five washes of 1% acetic acid. The plate was allowed to air-dry, bound SRB stain representing surviving cells was dissolved in 200 µL of Tris base (10 mM). The optical density was determined at 540 nm on a microplate reader (Dynex Technologies, Chantilly, VA). Mean values were obtained from at least five wells per treatment condition. Plots of percent inhibition of cell growth versus drug concentration were constructed, and the concentration that inhibited cell growth by 50% relative to the untreated control (IC50) was determined by non-linear least squares regression using WinNonlin software (version 4.1).
4.1.3. Assay for androgen receptor binding affinity
The AR binding affinity of tested compounds was determined by a radioligand competitive binding assay with cytosolic AR prepared from rat ventral prostates. The AR preparation was incubated with 1 nMof [³H]mibolerone (MIB), 1 µM triamcinolone acetonide at 4 °C for 18 h, in the presence of increasing concentrations of the test compound (10 different concentrations ranging from 10−2 to 104 nM) or in the absence of the compound. After incubation, the protein-bound radioactivity was separated from free radioactivity by hydroxyapatite (HAP) precipitation. The bound radioactivity was then extracted from HAP by ethanol, and counted in a Beckman LS6000 liquid scintillation counter (Beckman Instruments Inc., Palo Alto, CA). The specific binding of [³H]MIB at each concentration of the compound of interest (B) was obtained after subtracting the non-specific binding of [³H]MIB, and expressed as the percentage of the specific binding in the absence of the compound of interest (B0). The concentration of compound that reduced the specific binding of [³H]MIB by 50% (IC50) was determined by computer-fitting the data to the following equation using Win-Nonlin (Pharsight Corporation, Mountain View, CA): B = B0 × [1 − C/(IC50 + C)], where C is the concentration of the compound of interest. The apparent equilibrium binding constant (Ki) of the compound of interest was calculated by Ki = Kd × IC50/(Kd + L),where Kd is the equilibrium dissociation constant of [³H]MIB (Kd = 0.19 nM; determined in preliminary experiments) and L is the concentration of [³H]MIB used in the experiment (L = 1 nM).
4.2. Synthesis and physical data for SARMs
Melting points were determined using a Thomas-Hoover capillary apparatus and are uncorrected. The NMR spectra were recorded on Brucker ARX 300 (300 MHz for ¹H NMR and 75 Hz for 13CNMR) or Varian Inova FT spectrometers (500 MHz for ¹H NMR and 125 MHz for 13C NMR). The chemical shifts are reported in ppm downfield relative to tetramethylsilane. Spectral data were consistent with assigned structures. Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA, and found values were within 0.4% of the theoretical values. Analytical thin-layer chromatography was carried out on pre-coated silica gel (Merck Kieselgel 60 F254 layer thickness 0.25 mm). Flash column chromatography was carried out using Silica gel (230–400 mesh, Merck).
4.3. General procedure for ether-linked compounds S-23 to S-30
The target isothiocyanate compounds S-23 to S-30 were prepared by two synthetic methods (Route A and/or B) as outlined below in Scheme 1.
4.3.1. Route A
Compounds S-23, S-27, and S-28 were synthesized by coupling of chiral intermediate R-3 or R-4 with appropriate phenols (21 or 22). The synthesis of target compounds was achieved by a previously reported method.10 Briefly, the mixture of bromide R-3 or R-4 (0.81 mmol) and potassium carbonate (2.4 mmol) in acetone medium (30 mL) was heated to reflux for 30 min. After complete conversion of starting bromide R-3 or R-4 to corresponding epoxides as monitored by TLC, the solvent was evaporated under reduced pressure to give a yellowish residue. To the residue, suspended in methyl-ethylketone or isopropanol (30 mL), substituted phenol (0.81 mmol) and an additional amount of potassium carbonate (1.6 mmol) were added. The reaction mixture was refluxed for 2–4 h. The solvent in the mixture was removed under reduced pressure. The residue was dissolved in EtOAc (50 mL), washed with brine, aqueous ammonium chloride, and water. The ethyl acetate extract was dried by MgSO4, concentrated and purified by flash column chromatography using EtOAc/hexane as an eluent to give target compounds. The overall reaction was carried out in one-pot two-step process as shown in Scheme 1 and the yields ranged from 23% to 48%.
4.3.2. Route B
Compounds S-23 to S-26 and S-29 to S-30 were synthesized by coupling of chiral intermediate R-3 or R-4 with appropriate protected phenols (5, 6, 7 or 8) to get S-9 to S-14. Removal of the Boc group under acidic condition afforded free amine intermediates S-15 to S-20. Treating with thiophosgene under basic condition afforded the target compounds S-23 to S-26 and S-29 to S-30. Compounds S-9 to S-14 were prepared by synthetic method A by coupling of chiral intermediate R-3 or R-4 with protected phenols (5, 6, 7 or 8 in Scheme 5; yield 57 to 77% for ether linkages). Acetyl chloride (10 mL) was added dropwise to a solution of the protected amine (2 mmol) in EtOH (30 mL) under ice-cold conditions. The reaction mixture was raised to room temperature and stirred for 3 h. The completion of the reaction was determined by the disappearance of starting material (compounds S-9 to S-14), as monitored by TLC. Under reduced pressure, the solvent was removed to give crude white or brown foam of aniline S-15 to S-20. A solution of the amines in CHCl3 (30 mL) and NaHCO3 (841 mg, 10 mmol) in 30 mL H2O was cooled in an ice-bath and stirred vigorously. Thiophosgene (0.31 mL, 4 mmol) was slowly added to the solution and the mixture was stirred at room temperature for 5 h. The reaction mixture in CHCl3 was washed with water (50 mL 3X), dried with MgSO4 and purified using flash column chromatography using either CH2Cl2/hexane or EtOAc/hexane as an eluent to obtain S-23 to S-30, R-23 and S-35 as a yellowish solid. Yield 38–85%. The protected 4-amino phenol 5, 6, 7, and 8 were prepared from corresponding 58, 59,25 60,26 and 6127 by using Boc anhydride with Et3N in THF solvent (Scheme 5).28 Amino compound 59 was obtained from nitro phenol 56 by reduction with H2 on Pd/C on the Parr hydrogenation apparatus (mp 136.5–137.5 °C; lit.29 mp 138–139 °C).
4.4. (2-Fluoro-4-hydroxyphenyl)carbamic acid tert-butyl ester (6)
Yield 27% (from 59); brown oil; Rf 0.17 (EtOAc/hexane 1:5); ESI-MS; m/z: 226.0 [M–H]−; ¹H NMR (CDCl3) δ 7.54 (t, J = 8.1 Hz, 1H, OH), 7.01 (br s, 1H, NH), 6.59–6.46 (m, 3H, ArH), 1.55 (s, 9H, CH3); 13C NMR (CDCl3) δ 154.4 (d, JC–F = 155.1 Hz), 153.0 (d, JC–F = 10.9 Hz), 152.2, 123.3, 117.7 (d, JC–F = 11.5 Hz), 110.8 (d, JC–F = 3.2 Hz), 103.0 (d, JC–F = 22.4 Hz), 80.7, 27.8; Anal. Calcd for C11H14FNO3: C, 58.14; H, 6.21; N, 6.16. Found: C, 58.19; H, 6.26; N, 6.28.
4.5. (3-Fluoro-4-hydroxyphenyl)carbamic acid tert-butyl ester (7)
Yield 56% (from 60); white solid; Rf 0.43 (EtOAc/hexane 1:2); mp 122–123 °C; ESI-MS; m/z: 226.0 [M–H]−; ¹H NMR (CDCl3) δ 7.31 (d, JH–F = 12.0 Hz, 1H, ArH), 6.91–6.80 (m, 2H, ArH), 6.47 (br s, 1H, OH), 5.54 (br s, 1H, NH), 1.52 (s, 9H, CH2); 13C NMR: (CDCl3) δ 151.9, 150.4 (d, JC–F = 254.3 Hz), 139.0 (d, JC–F = 14.3 Hz), 130.9, 116.7, 114.9, 107.3 (d, JC–F = 23.8 Hz), 80.3, 27.8; Anal. Calcd for C11H14FNO3: C, 58.14; H, 6.21; N, 6.16. Found: C, 58.20; H, 6.15; N, 6.15.
4.6. (3-Chloro-4-hydroxyphenyl)carbamic acid tert-butyl ester (8)
Yield 65% (from 61); white solid; Rf 0.48 (EtOAc/hexane 1:2); mp 77–79 °C; ESI-MS; m/z: 241.9 [M–H]−; ¹H NMR (CDCl3) δ 7.55 (s, 1H, ArH), 7.03 (dd, J = 8.9, 2.4 Hz, 1H, ArH), 6.92 (d, J = 8.9 Hz, 1H, ArH), 6.38 (br s, 1H, NH), 5.44 (br s, 1H, OH), 1.51 (s, 9H, CH3); 13C NMR (CDCl3) δ 151.5, 147.8, 132.2, 120.3, 120.1, 119.8, 116.5, 81.1, 28.7; Anal. Calcd for C11H14ClNO3: C, 54.22; H, 5.79; N, 5.75. Found: C, 54.15; H, 5.74; N, 5.72.
4.7. R-{4-[2-Hydroxy-2-(4-nitro-3-trifluoromethylphenylcarbamoyl) propoxy]phenyl} carbamic acid tert-butyl ester (R-9)
Prepared by Route B; yield 75% (from S-3 with 5, S-3 is opposite isomer of R-3 synthesized from l-proline); yellowish solid; Rf 0.22 (EtOAc/hexane 1:2); ESI-MS; m/z: 498.0 [M–H]−; mp 145–147 °C; ¹H NMR (CDCl3) δ 9.20 (br s, 1H, NH), 8.10 (s, 1H, ArH), 8.01 (m, 2H, ArH), 7.27 (d, J = 8.6 Hz, 2H, ArH), 6.84 (d, J = 8.6 Hz, 2H, ArH), 6.40 (br s, 1H, NH), 4.42 (d, J = 9.0 Hz, 1H, CH2), 3.95 (d, J = 9.0 Hz, 1H, CH2), 3.54 (br s, 1H, OH), 1.58 (s, 3H, CH3), 1.51 (s, 9H, CH3); Anal. Calcd for C22H24F3N3O7: C, 52.91; H, 4.84; N, 8.41. Found: C, 52.77; H, 4.91; N, 8.42.
4.8. S-{2-Fluoro-4-[2-hydroxy-2-(4-nitro-3-trifluoromethylphenylcarbamoyl)propoxy] phenyl}carbamic acid tert-butyl ester (S-10)
Prepared by Route B; yield 59% (two steps from R-3 with 6); yellowish solid; Rf 0.22 (EtOAc/hexane 1:2); ESI-MS; m/z: 516.0 [M–H]−; mp 54–57 °C; ¹H NMR (CDCl3) δ 9.23 (br s, 1H, NH), 8.11 (s, 1H, ArH), 8.03 (m, 2H, ArH), 7.89 (br s, 1H, NH), 8.68 (m, 2H, ArH), 6.54 (m, 1H, ArH), 4.43 (d, J = 9.0 Hz, 1H, CH2), 3.96 (d, J = 9.0 Hz, 1H, CH2), 3.59 (br s, 1H, OH), 1.59 (s, 3H, CH3), 1.53 (s, 9H, CH3); 13C NMR (CDCl3) δ 172.1, 153.1 (d, JC–F = 10.2 Hz), 152.3 (d, JC–F = 242.8 Hz), 152.2, 142.7, 140.9, 126.5, 125.0 (q, JC–F = 34.0 Hz), 121.6, 121.3 (q, JC–F = 272.0 Hz), 121.2, 120.5 (d, JC–F = 10.7 Hz), 117.7 (q, JC–F = 5.9 Hz), 109.8 (d, JC–F = 3.3 Hz), 102.3 (d, JC–F = 23.0 Hz), 80.5, 75.3, 72.6, 27.6, 22.4; Anal. Calcd for C22H23F4N3O7: C, 51.07; H, 4.48; N, 8.12. Found: C, 51.27; H, 4.67; N, 8.17.
4.9. S-{4-[2-(4-Cyano-3-trifluoromethylphenylcarbamoyl)- 2-hydroxypropoxy]phenyl} carbamic acid tert-butyl ester (S-11)
Prepared by Route B; yield 57% (two steps from R-4 with 5); white solid; Rf 0.27 (EtOAc/hexane 2:3); mp: 156–158 °C; ESI-MS; m/z: 478.1 [M–H]−; ¹H NMR (CDCl3) δ 9.17 (br s, 1H, NH), 8.10 (s, 1H, ArH), 7.96 (d, J = 8.4 Hz, 1H, ArH), 7.80 (d, J = 8.4 Hz, 1H, ArH), 7.27 (d, J = 8.1 Hz, 2H, ArH), 6.84 (d, J = 8.1 Hz, 2H, ArH), 6.43 (br s, 1H, NH), 4.42 (d, J = 9.0 Hz, 1H, CH2), 3.95 (d, J = 9.0 Hz, 1H, CH2), 3.58 (br s, 1H, OH), 1.57 (s, 3H, CH3), 1.50 (s, 9H, CH3); Anal. (C23H24F3N3O5 · H2O) C, H, N. Anal. Calcd for C23H24F3N3O5: C, 57.62; H, 5.05; N, 8.76. Found: C, 57.41; H, 5.10; N, 8.66.
4.10. S-{4-[2-(4-Cyano-3-trifluoromethylphenylcarbamoyl)-2-hydroxypropoxy]-2-fluoro phenyl}carbamic acid tert-butyl ester (S-12)
Prepared by Route B; yield 65% (two steps from R-4 with 6); yellowish solid; Rf 0.12 (EtOAc/hexane 1:2); mp: 56–59 °C; ESI-MS; m/z: 496.2 [M–H]−; ¹H NMR (CDCl3) δ 9.20 (br s, 1H, NH), 8.13 (d, J = 2.1 Hz, 1H, ArH), 7.98 (dd, J = 8.7, 2.1 Hz, 1H, ArH), 7.89 (br s, 1H, NH), 7.82 (d, J = 8.7 Hz, 1H, ArH), 7.68 (m, 2H, ArH), 6.53 (m, 1H, ArH), 4.42 (d, J = 9.0 Hz, 1H, CH2), 3.96 (d, J = 9.0 Hz, 1H, CH2), 3.63 (br s, 1H, OH), 1.58 (s, 3H, CH3), 1.52 (s, 9H, CH3); 13C NMR (CDCl3) δ 172.1, 153.1 (d, JC–F = 10.1 Hz), 152.3 (d, JC–F = 242.6 Hz), 152.2, 140.9, 135.3, 133.7 (q, JC–F = 32.6 Hz), 121.6 (q, JC–F = 272.4 Hz), 121.3, 121.2, 120.5 (d, JC–F = 10.7 Hz), 116.8 (q, JC–F = 4.9 Hz), 114.9, 109.8 (d, JC–F = 3.2 Hz), 104.1 (d, JC–F = 2.0 Hz), 102.3 (d, JC–F = 23.0 Hz), 80.5, 75.2, 72.6, 27.7, 22.4; Anal. Calcd for C23H23F4N3O5: C, 55.53; H, 4.68; N, 8.45. Found: C, 55.26; H, 4.70; N, 8.29.
4.11. S-{3-Fluoro-4-[2-hydroxy-2-(4-nitro-3-trifluoromethylphenylcarbamoyl)propoxy]phenyl}carbamic acid tert-butyl ester (S-13)
Prepared by Route B; yield 31% (two steps from R-3 with 7); brown solid; Rf 0.28 (EtOAc/hexane 2:3); mp: 69–72 °C; ESI-MS; m/z: 516.1 [M–H]; ¹H NMR (CDCl3) δ 9.39 (br s, 1H, NH), 8.11 (s, 1H, ArH), 7.94 (m, 2H, ArH), 7.28 (br s, 1H, NH), 6.71–6.77 (m, 3H, ArH), 4.41 (d, J = 9.0 Hz, 1H, CH2), 4.23 (br s, 1H, OH), 3.97 (d, J = 9.0 Hz, 1H, CH2), 1.55 (s, 3H, CH3), 1.44 (s, 9H, CH3); 13C NMR: (CDCl3) δ 172.6, 152.3, 152.2 (d, JC–F = 242.8 Hz), 143.7, 141.4, 140.8 (d, JC–F = 11.2 Hz), 133.2 (d, JC–F = 11.2 Hz), 126.7, 124.9 (q, JC–F = 33.8 Hz), 121.8, 121.2 (q, JC–F = 272.0 Hz), 117.9 (q, JC–F = 5.8 Hz), 116.5 (d, JC–F = 1.8 Hz), 113.9, 107.4 (d, JC–F = 23.3 Hz), 75.0, 74.7, 65.2, 27.9, 22.2; Anal. Calcd for C22H23F4N3O7: C, 51.07; H, 4.48; N, 8.12. Found: C, 51.06; H, 4.63; N, 7.99.
4.11.1. S-{3-Chloro-4-[2-hydroxy-2-(4-nitro-3-trifluoromethylphenylcarbamoyl) propoxy]phenyl}carbamic acid tert-butyl ester (S-14)
Prepared by Route B; yield 44% (two steps from R-3 with 8); brown solid; Rf 0.28 (EtOAc/hexane 2:3); mp 71–74 °C; ESI-MS; m/z: 532.0 [M–H]−; ¹H NMR (CDCl3) δ 9.39 (br s, 1H, NH), 8.12 (s, 1H, ArH), 7.92 (m, 2H, ArH), 7.43 (br s, 1H, NH), 7.06 (m, 1H, ArH), 6.87 (s, 1H, ArH), 6.77 (d, J = 8.7 Hz, 1H, ArH), 4.34 (d, J = 9.0 Hz, 1H, CH2), 4.21 (br s, 1 H, OH), 3.96 (d, J = 9.0 Hz, 1H, CH2), 1.63 (s, 3H, CH3), 1.47 (s, 9H, CH3); 13C NMR (CDCl3) δ 173.3, 153.0, 149.2, 143.4, 141.8, 133.8, 127.2, 125.3 (q, JC–F = 34.5 Hz), 123.8, 122.5, 121.9 (q, JC–F = 241.9 Hz), 121.2, 118.7 (q, JC–F = 5.9 Hz), 118.6, 115.6, 75.7, 74.7, 66.0, 28.4, 23.0; Anal. Calcd for C22H23ClF3 N3O7: C, 49.88; H, 4.71; N, 7.27. Found: C, 49.96; H, 4.70; N, 7.30.
4.12. S-3-(4-Aminophenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethylphenyl) propionamide (S-15)
Prepared from S-9 by Route B; brown solid; Rf 0.29 (CHCl3/MeOH 20:1); mp 138–140 °C; ESI-MS; m/z: 398.0 [M–H]−; 1H NMR (CD3OD) δ 8.38 (d, J = 1.8 Hz, 1H, ArH), 8.17 (dd, J = 8.7, 1.8 Hz, 1H, ArH), 8.04 (d, J = 8.7 Hz, 1H, ArH), 7.11 (d, J = 9.0 Hz, 2H, ArH), 6,98 (d, J = 9.0 Hz, 2H, ArH), 4.32 (d, J = 9.6 Hz, 1H, CH2), 4.03 (d, J = 9.6 Hz, 1H, CH2), 1.52 (s, 3H, CH3); 13C NMR (CDCl3) δ 172.6, 150.3, 142.6, 141.1, 140.6, 126.5, 124.6 (q, JC–F = 33.9 Hz), 121.5, 121.1 (q, JC–F = 272.0 Hz), 117.7 (q, JC–F = 5.9 Hz), 115.9 (2C), 115.7 (2C), 75.3, 73.0, 22.4; Anal. Calcd for C17H16F3N3O5: C, 51.13; H, 4.04; N, 14.27. Found: C, 51.35; H, 4.06; N, 14.19.
4.13. S-3-(4-Aminophenoxy)-N-(4-cyano-3-trifluoromethylphenyl)-2-hydroxy-2-methylpropionamide (S-17)
Prepared from S-11 by Route B; brown solid; Rf 0.24 (EtOAc/hexane 2:1); mp 177–179 °C; ESI-MS; m/z: 378.1 [M–H]−; ¹H NMR (DMSO-d6) δ 10.54 (br s, 1H, NH), 8.57 (d, J = 1.8 Hz 1H, ArH), 8.32 (dd, J = 8.7, 1.8 Hz, 1H, ArH), 8.11 (d, J = 8.7 Hz, 1H, ArH), 6.63 (d, J = 9.0 Hz, 2H, ArH), 6.48 (d, J = 9.0 Hz, 2H, ArH), 6.17 (br s, 1H, OH), 4.61 (br s, 2H, NH2), 4.08 (d, J = 9.6 Hz, 1H, CH2), 3.85 (d, J = 9.6 Hz, 1H, CH2), 1.18 (s, 3H, CH3); 13C NMR (DMSO-d6) δ 174.7, 149.8, 143.2, 142.7, 136.2, 131.6 (q, JC–F = 33.8 Hz), 122.6, 122.4 (q, JC–F = 264.1 Hz), 117.3 (q, JC–F = 4.9 Hz), 115.8, 115.6 (2C), 114.8 (2C), 101.8, 75.0, 74.5, 22.9; Anal. Calcd for C18H16F3N3O3 0.2H2O: C, 56.46; H, 4.32; N, 10.97. Found: C, 56.35; H, 4.26; N, 11.09.
4.14. S-3-(4-Amino-3-fluorophenoxy)-N-(4-cyano-3-trifluoromethylphenyl)-2-hydroxy-2-methylpropionamide (S-18)
Prepared from S-12 by Route B; brown solid; Rf 0.33 (EtOAc/hexane 1:1); mp 179–181 °C; ESI-MS; m/z: 395.9 [M–H]−; ¹H NMR (DMSO-d6) δ 10.55 (br s, 1H, NH), 8.56 (d, J = 1.8 Hz, 1H, ArH), 8.30 (dd, J = 8.7, 1.8 Hz, 1H, ArH), 8.10 (d, J = 8.7 Hz, 1H, ArH), 6.68 (m, 2H, ArH), 6.50 (dd, J = 8.7, 2.1 Hz, 1H, ArH), 6.21 (br s, 1H, OH), 4.63 (br s, 2H, NH2), 4.10 (d, J = 9.6 Hz, 1 H, CH2), 3.86 (d, J = 9.6 Hz, 1H, CH2), 1.40 (s, 3H, CH3); 13C NMR (DMSO-d6) δ 174.6, 150.9 (d, JC–F = 180.9 Hz), 149.4 (d, JC–F = 64.5 Hz), 143.2, 136.2, 131.5 (q, JC–F = 31.4 Hz), 130.1, 122.6, 122.5 (q, JC–F = 271.7 Hz), 117.4, 116.6 (q, JC–F = 5.9 Hz), 115.8, 111.1, 103.0, 101.9, 74.9, 74.6, 22.9.
4.15. S-3-(4-Amino-2-fluorophenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl phenyl)propionamide (S-19)
Prepared from S-13 by Route B; brown solid; Rf 0.17 (EtOAc/hexane 1:1); mp 103–105 °C; ESI-MS; m/z: 416.1 [M–H]−; ¹H NMR (DMSO-d6) δ 10.71 (br s, 1H, NH), 10.25 (br s, 2H, NH2), 8.57 (s, 1H, ArH), 8.35 (d, J = 8.1 Hz, 1H, ArH), 8.19 (d, J = 8.1 Hz, 1H, ArH), 7.35–7.24 (m, 2H, ArH), 7.15 (m, 1H, ArH), 4.31 (d, J = 9.3 Hz, 1H, CH2), 4.13 (d, J = 9.3 Hz, 1H, CH2), 1.46 (s, 3H, CH3); 13C NMR (DMSO-d6) δ 174.3, 151.2 (d, JC–F = 244.9 Hz), 145.8 (d, JC–F = 10.3 Hz), 143.2, 141.7, 127.3, 125.5, 123.1, 122.2 (q, JC–F = 33.0 Hz), 122.1 (q, JC–F = 271.4 Hz), 119.3 (d, JC–F = 3.2 Hz), 118.3 (q, JC–F = 5.9 Hz), 116.3 (d, JC–F = 1.8 Hz), 111.4 (d, JC–F = 21.7 Hz), 75.0, 74.9, 22.8.
4.16. S-3-(4-Amino-2-chlorophenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl phenyl)propionamide (S-20)
Prepared from S-14 by Route B; yellowish solid; Rf 0.15 (EtOAc/hexane 1:1); mp 108–110 °C; ESI-MS; m/z: 432.1 [M–H]−; ¹H NMR (DMSO-d6) δ 10.71 (br s, 1H, NH), 10.27 (br s, 2H, NH2), 8.57 (s, 1H, ArH), 8.34 (d, J = 7.8 Hz, 1H, ArH), 8.18 (d, J = 7.8 Hz, 1H, ArH), 7.42 (s, 1H, ArH), 7.30 (br s, 2H, ArH), 4.25 (d, J = 9.0 Hz, 1H, CH2), 4.19 (d, J = 9.0 Hz, 1H, CH2), 1.48 (s, 3H, CH3); 13C NMR (DMSO-d6) δ 174.2, 153.2, 143.2, 141.6, 127.2, 125.8, 124.3, 123.2, 122.9, 122.2 (q, JC–F = 32.9 Hz), 122.0 (q, JC–F = 271.4 Hz), 121.8, 118.3 (q, JC–F = 5.8 Hz), 114.9, 75.1, 74.9, 22.6.
4.17. S-2-Hydroxy-3-(4-isothiocyanatophenoxy)-2-methyl-N-(4-nitro-3-trifluoromethyl phenyl)propionamide (S-23)10
Prepared by Route B; (from S-9 through S-15, S-9 synthesized starting from d-proline); ESI-MS; m/z: 439.8 [M–H]−; Anal. Calcd for C18H14F3N3O5S: C, 48.98; H, 3.20; N, 9.52. Found: C, 49.10; H, 3.31; N, 9.44.
4.18. R-2-Hydroxy-3-(4-isothiocyanatophenoxy)-2-methyl-N-(4-nitro-3-trifluoromethyl phenyl)propionamide (R-23)
Prepared by Route B (R-23 is opposite isomer of S-23); yield 81% (two steps from R-9 through R-15, R-9 synthesized starting from l-proline); yellowish solid; Rf 0.21 (EtOAc/hexane 3:2); ESI-MS; m/z: 439.8 [M–H]−; ¹H NMR (CDCl3) δ 9.14 (br s, 1H, NH), 8.04 (s, 1H, ArH), 7.92 (m, 2H, ArH), 7.08 (d, J = 8.4 Hz, 2H, ArH), 6,80 (d, J = 8.4 Hz, 2H, ArH), 4.40 (d, J = 9.0 Hz, 1H, CH2), 3.93 (d, J = 9.0 Hz, 1H, CH2), 3.39 (br s, 1H, OH), 1.53 (s, 3H, CH3); 13C NMR (DMSO-d6) δ 172.7, 156.8, 143.5, 141.7, 135.2, 127.2 (2C), 125.4 (q, JC–F = 34.1 Hz), 125.3, 122.4, 122.6 (q, JC–F = 272.0 Hz), 118.5 (q, JC–F = 5.8 Hz), 116.0 (2C), 76.0, 73.0, 23.1; Anal. Calcd for C18H14F3N3O5S: C, 48.98; H, 3.20; N, 9.52. Found: C, 48.71; H, 3.25; N, 9.57.
4.19. S-N-(4-Cyano-3-trifluoromethylphenyl)-2-hydroxy-3-(4-isothiocyanatophenoxy)-2-methyl-propionamide (S-24)
Prepared by Route B; yield 84% (two steps from S-11 through S-17); brown solid; Rf 0.46 (EtOAc/hexane 1:1); mp: 41–43 °C; ESI-MS; m/z: 420.1 [M–H]−; ¹H NMR (CDCl3) δ 9.14 (br s, 1H, NH), 8.13 (d, J = 1.8 Hz, 1H, ArH), 7.99 (dd, J = 8.4, 1.8 Hz, 1H, ArH), 7.84 (d, J = 8.4 Hz, 1H, ArH), 7.20 (d, J = 9.0 Hz, 2H, ArH), 6.91 (d, J = 9.0 Hz, 2H, ArH), 4.49 (d, J = 9.0 Hz, 1H, CH2), 4.02 (d, J = 9.0 Hz, 1H, CH2), 3.39 (br s, 1H, OH), 1.62 (s, 3H, CH3); 13C NMR (CDCl3) δ 171.9, 156.0, 140.8, 135.4, 134.2, 126.6, 126.3 (q, JC–F = 33.8 Hz), 124.7, 122.0 (q, JC–F = 270.2 Hz), 121.3, (2 C), 116.7 (q, JC–F = 4.9 Hz), 115.2 (2C), 114.8, 104.1, 75.3, 72.2, 22.4; Anal. Calcd for C19H14F3N3O3S: C, 54.15; H, 3.35; N, 9.97. Found: C, 54.07; H, 3.44; N, 9.61.
4.20. S-3-(3-Fluoro-4-isothiocyanatophenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)propion-amide (S-25)
Prepared by Route B; yield 85% (two steps from S-10 through S-16); yellowish solid; Rf 0.24 (EtOAc/hexane 2:3); mp 42–45 °C; ESI-MS; m/z: 458.0 [M–H]−; ¹H NMR (CDCl3) δ 9.24 (br s, 1H, NH), 8.15 (br s, 1H, ArH), 8.08–8.00 (m, 2H, ArH), 7.14 (t, J = 8.7 Hz, 1H, ArH), 6.78–6.68 (m, 2H, ArH), 4.50 (d, J = 9.0 Hz, 1H, CH2), 4.05 (d, J = 9.0 Hz, 1H, CH2), 3.44 (br s, 1H, OH), 1.64 (s, 3H, CH3); 13C NMR (CDCl3) δ 174.5, 156.0, 154.1 (q, JC–F = 144.9 Hz), 143.2, 136.2, 131.4 (q, JC–F = 126.9 Hz), 129.4, 122.6, 122.5, 122.4 (q, JC–F = 272.0 Hz), 117.3 (q, JC–F = 5.8 Hz), 115.8 (q, JC–F = 287.3 Hz), 103.3, 103.0, 102.4 (q, JC–F = 82.4 Hz), 74.8, 74.7, 22.9; Anal. Calcd for C18H13F4N3O5S: C, 47.06; H, 2.85; N, 9.15. Found: C, 46.98; H, 2.87; N, 9.10.
4.21. S-N-(4-Cyano-3-trifluoromethylphenyl)-3-(3-fluoro-4-isothiocyanatophenoxy)-2-hydroxy-2-methylpropionamide (S-26)
Prepared by Route B; yield 79% (two steps from S-12 through S-18); white solid; Rf 0.23 (EtOAc/hexane 2:3); mp 43–46 °C; ESI-MS; m/z: 438.1 [M–H]−; ¹H NMR (CDCl3) δ 9.17 (br s, 1H, NH), 8.14 (d, J = 1.2 Hz, 1H, ArH), 7.99 (dd, J = 8.4, 1.2 Hz, 1H, ArH), 7.82 (d, J = 8.4 Hz, 1H, ArH), 7.15 (t, J = 8.4 Hz, 1H, ArH), 6.76–6.66 (m, 2H, ArH), 4.47 (d, J = 9.0 Hz, 1H, CH2), 4.02 (d, J = 9.0 Hz, 1H, CH2), 3.49 (br s, 1H, OH), 1.62 (s, 3H, CH3); 13C NMR (CDCl3) δ 171.8, 158.4 (d, JC–F = 158.4 Hz), 156.9, 140.9, 139.9, 135.5, 135.3, 133.6 (q, JC–F = 32.7 Hz), 126.4, 121.4 (q, JC–F = 272.4 Hz), 121.3, 116.8 (q, JC–F = 4.9 Hz), 114.9, 110.3 (d, JC–F = 3.4 Hz), 104.1, 103.3 (d, JC–F = 22.2 Hz), 75.2, 72.6, 22.4; Anal. Calcd for C19H13F4N3O3S: C, 51.94; H, 2.98; N, 9.56. Found: C, 51.71; H, 3.13; N, 9.41.
4.22. S-3-(3-Chloro-4-isothiocyanatophenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)-propionamide (S-27)
Prepared by Route A; yield 38% (two steps from R-3 with 22); yellowish solid; Rf 0.25 (EtOAc/hexane 2:3); mp 44–46 °C; ESI-MS; m/z: 474.0 [M–H]−; ¹H NMR (CDCl3) δ 9.23 (br s, 1H, NH), 8.14 (m, 1H, ArH), 8.01 (m, 2H, ArH), 7.18 (d, J = 9.0 Hz, 1H, ArH), 6.99 (d, J = 2.7 Hz, 1H, ArH), 6.81 (dd, J = 9.0, 2.7 Hz, 1H, ArH), 4.48 (d, J = 9.0 Hz, 1H, CH2), 4.02 (d, J = 9.0 Hz, 1H, CH2), 3.47 (br s, 1H, OH), 1.62 (s, 3H, CH3); 13C NMR (CDCl3) δ 171.8, 156.1, 142.8, 140.9, 137.8, 132.2, 128.4, 126.8, 126.5, 124.8 (q, JC–F = 34.1 Hz), 121.6, 121.8 (q, JC–F = 272.0 Hz), 117.8 (q, JC–F = 5.9 Hz), 116.0, 113.6, 75.3, 72.5, 22.4; Anal. Calcd for C18H13ClF3N3O5S: C, 45.44; H, 2.75; N, 8.83. Found: C, 45.63; H, 2.92; N, 8.74.
4.23. S-3-(3-Chloro-4-isothiocyanatophenoxy)-N-(4-cyano-3-trifluoromethylphenyl)-2-hydroxy-2-methylpropionamide (S-28)
Prepared by Route A; yield 48% (two steps from R-4 with 22); white solid; Rf 0.24 (EtOAc/hexane 2:3); mp 50–53 °C; ESI-MS; m/z: 454.4 [M–H]−; ¹H NMR (CDCl3) δ 9.21 (br s, 1H, NH), 8.14 (d, J = 1.2 Hz, 1H, ArH), 7.98 (dd, J = 8.7, 1.2 Hz, 1H, ArH), 7.81 (d, J = 8.7 Hz, 1H, ArH), 7.16 (d, J = 9.0 Hz, 1H, ArH), 6.99 (d, J = 2.7 Hz, 1H, ArH), 6.80 (dd, J = 9.0, 2.7 Hz, 1H, ArH), 4.46 (d, J = 9.0 Hz, 1H, CH2), 4.02 (d, J = 9.0 Hz, 1H, CH2), 3.65 (br s, 1H, OH), 1.61 (s, 3H,CH3); 13CNMR (CDCl3) δ 171.9, 156.2, 140.9, 135.7, 135.3, 133.5 (q, JC–F = 32.7 Hz), 132.1, 126.8, 123.1, 121.6 (q, JC–F = 272.4 Hz), 121.3, 116.8 (q, JC–F = 4.9 Hz), 116.0, 114.9, 113.6, 104.1, 75.2, 72.6, 22.4; Anal. Calcd for C19H13ClF3N3O3S: C, 50.06; H, 2.87; N, 9.22. Found: C, 50.13; H, 3.04; N, 9.08.
4.24. S-3-(2-Fluoro-4-isothiocyanatophenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoro methylphenyl)propionamide (S-29)
Prepared by Route B; yield 73% (two steps from S-13 through S-19); yellowish solid; Rf 0.14 (EtOAc/hexane 1:2); mp 51–53 °C; ESI-MS; m/z: 457.9 [M–H]−; ¹H NMR (CDCl3) δ 9.19 (br s, 1H, NH), 8.12 (s, 1H, ArH), 8.01 (m, 2H, ArH), 6.99 (m, 3H, ArH), 4.51 (d, J = 9.0 Hz, 1H, CH2), 4.07 (d, J = 9.0 Hz, 1H, CH2), 3.60 (br s, 1 H, OH), 1.63 (s, 3H, CH3); 13C NMR: (CDCl3) δ 172.6, 152.5 (d, JC–F = 247.7 Hz), 145.3 (d, JC–F = 10.9 Hz), 143.6, 141.6, 137.1, 127.4, 126.3, 126.1, 125.5 (q, JC–F = 34.1 Hz), 122.4, 121.9 (q, JC–F = 272.0 Hz), 118.6 (d, JC–F = 5.9 Hz), 116.6, 114.5 (d, JC–F = 21.3 Hz), 75.9, 74.6, 23.1; Anal. Calcd for C18H13F4N3O5S: C, 47.95; H, 3.60; N, 8.06. Found: C, 48.08; H, 3.57; N, 8.09.
4.25. S-3-(2-Chloro-4-isothiocyanatophenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)-propionamide (S-30)
Prepared by Route B; yield 77% (two steps from S-14 through S-20); yellowish solid; Rf 0.14 (EtOAc/hexane 1:2); mp 57–59 °C; ESI-MS; m/z: 473.9 [M–H]−; ¹H NMR (CDCl3) δ 9.18 (br s, 1H, NH), 8.13 (s, 1H, ArH), 8.02 (m, 2H, ArH), 7.29 (d, J = 2.4 Hz, 1H ArH), 7.14 (dd, J = 8.7, 2.4 Hz, 1H ArH), 6.95 (d, J = 8.7 Hz, 1H ArH), 4.49 (d, J = 9.0 Hz, 1H, CH2), 4.09 (d, J = 9.0 Hz, 1H, CH2), 3.62 (br s, 1H, OH), 1.66 (s, 3H, CH3); 13C NMR: (CDCl3) δ 172.7, 152.5, 143.2, 141.6, 137.2, 127.7, 127.3, 126.3, 125.6, 125.4 (q, JC–F = 34.0 Hz), 124.1, 122.5, 121.9 (q, JC–F = 272.1 Hz), 118.7 (q, JC–F = 5.9 Hz), 115.0, 75.7, 74.3, 23.2; Anal. Calcd for C18H13ClF3N3O5S · 0. 2C4H8O2: C, 45.76; H, 2.98; N, 8.52. Found: C, 45.97; H, 3.02; N, 8.46.
4.26. Preparation for methylene-linked compound 31
4.26.1. 2-Hydroxy-2-methyl-4-(4-nitrophenyl)butyric acid (39)
To the solution of the nitro ketone 38 (710 mg, 3.7 mmol) in anhydrous CH2Cl2 (20 mL) were added dropwise trimethylsilyl cyanide (0.54 mL, 4.0 mmol) and catalytic amounts of zinc iodide (ZnI2) at ice-bath under argon atmosphere. The reaction mixture was stirred for 1 h at room temperature. The completion of the reaction was determined by the disappearance of starting material (compound 38), as monitored by TLC. After removing the solvent under reduced pressure, concd hydrochloric acid (5 mL) and acetic acid (5 mL) were added to the mixture and heated to reflux for 30 min. The solution was diluted with water (100 mL), extracted with EtOAc (2× 30 mL), and washed with satd NaHCO3 solution. The aqueous layer was acidified with 3 N hydrochloric acid, extracted with Et2O, concentrated under reduced pressure, and crystallized from CH2Cl2/hexane to obtain compound 39 (805 mg, 91% yield) as white solid; mp 110–111 °C; ESI-MS; m/z: 237.9 [M–H]−; ¹H NMR (DMSO-d6) δ 12.6 (br s, 1H, CO2H), 8.16 (d, J = 8.7 Hz, 2H ArH), 7.46 (d, J = 8.7 Hz, 2H ArH), 3.33 (br s, 1H, OH), 2.84 (m, 1H CH2), 2.64 (m, 1H CH2), 1.96 (m, 1H CH2), 1.86 (m, 1H CH2), 1.32 (s, 3H, CH3); 13C NMR: (DMSO-d6) δ 177.2, 150.5, 145.8, 129.5 (2C), 123.4 (2C), 73.0, 41.1, 29.7, 25.8; Anal. Calcd for C11H13NO5: C, 55.23; H, 5.48; N, 5.86. Found: C, 55.34; H, 5.52; N, 5.87.
4.26.2. 4-(4-Aminophenyl)-2-hydroxy-2-methylbutyric acid (40)
The solution of compound 39 (635 mg, 2.7 mmol) and Pd/C (64 mg) in MeOH (10 mL) was reduced with H2 gas on Parr apparatus under 30 psi pressure for 3 h. The mixture was filtered through Celite 521 and concentrated on reduced pressure, and then, crystallized from CH2Cl2/hexane to obtain compound 40 (quantitative yield) as white solid; mp 158–161 °C; ESI-MS; m/z: 208.4 [M–H]−; ¹H NMR (DMSO-d6) δ 6.79 (d, J = 8.1 Hz, 2H, ArH), 6.47 (d, J = 8.1 Hz, 2H, ArH), 3.33 (br s, 1H, OH), 2.55 (m, 1H CH2), 2.30 (m, 1H CH2), 1.83 (m, 1H CH2), 1.65 (m, 1H CH2), 1.28 (s, 3H, CH3); 13C NMR: (DMSO-d6) δ 177.5, 146.2, 129.1, 128.4 (2C), 114.1 (2C), 73.2, 42.6, 28.8, 25.9.
4.26.3. 4-(Aminophenyl)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethylphenyl) butyramide (42)
To the solution of compound 40 (230 mg, 1.1 mmol) in THF (10 mL) were added thionyl chloride in dropwise manner (0.14 mL, 1.8 mmol) and catalytic amounts of DMF at ice-water bath. The mixture was stirred for 1 h at room temperature. Nitroaniline 41 (189 mg, 0.9 mmol) was added, followed by dropwise addition of triethylamine (0.19 mL, 1.4 mmol), to the solution. The mixture was stirred for 3 h at room temperature, concentrated under reduced pressure, poured into EtOAc (20 mL), washed with satd NaHCO3, brine, dried with MgSO4, and purified with flash column chromatography using EtOAc/hexane (1:1) as an eluent to gain compound 42 (246 mg, yield 68%) as brown solid; Rf 0.25 (EtOAc/hexane 1:1); mp 52–55 °C; ESI-MS; m/z: 396.0 [M–H]−; ¹H NMR (CDCl3) δ 9.15 (br s, 1H, NH), 8.07 (s, 1H, ArH), 7.99 (m, 2H, ArH), 6.97 (d, J = 8.1 Hz, 2H, ArH), 6.60 (d, J = 8.1 Hz, 2H, ArH), 3.15 (br s, 3H, NH2 & OH), 2.69 (m, 1H, CH2), 2.58 (m, 1H, CH2), 2.32 (m, 1H, CH2), 1.92 (m, 1H, CH2), 1.56 (s, 3H, CH3); 13C NMR: (CDCl3) δ 174.6, 144.8, 143.2, 141.9, 130.8, 129.4, 127.2, 125.5 (q, JC–F = 34.1 Hz), 122.2, 122.0 (q, JC–F = 272.0 Hz), 118.4 (q, JC–F = 5.9 Hz), 115.7, 77.1, 42.0, 29.4, 27.3; Anal. Calcd for C18H18F3N3O4: C, 54.41; H, 4.57; N, 10.58. Found: C, 54.44; H, 5.52; N, 10.67.
4.26.4. 2-Hydroxy-4-(4-isothiocyanatophenyl)-2-methyl-N-(4-nitro-3-trifluoromethyl phenyl)butyramide (31)
Prepared by Route B (followed by Method c in Scheme 1); yellowish oil; Yield 81% (from 42); Rf 0.24 (EtOAc/hexane 1:2); ESI-MS; m/z: 438.1 [M–H]−; ¹H NMR (CDCl3) δ 9.27 (br s, 1H, NH), 8.14 (s, 1H, ArH), 7.98 (m, 2H, ArH), 7.14 (d, J = 8.4 Hz, 2H, ArH), 7.07 (d, J = 8.4 Hz, 2H, ArH), 2.90 (br s, 1H, OH), 2.81 (m, 1H CH2), 2.63 (m, 1H CH2), 2.33 (m, 1H, CH2), 1.95 (m, 1H, CH2), 1.62 (s, 3H, CH3); 13C NMR: (CDCl3) δ 174.5, 143.1, 141.9, 140,7, 135.1, 129.7 (2C), 129.2, 127.2, 125.9 (2C), 125.4 (q, JC–F = 33.9 Hz), 122.3, 121.9 (q, JC–F = 272.0 Hz), 118.4 (q, JC–F = 5.9 Hz), 76.6, 41.9, 29.8, 27.2; Anal. Calcd for C19H16F3N3O4S: C, 51.93; H, 3.67; N, 9.56. Found: C, 52.04; H, 3.52; N, 9.67.
4.27. Preparation for amine- and N-methylamine-linked compounds S-32 and S-33
The amino linkages of S-32 and S-33 were directly introduced by two synthetic methods (a and a′) from protected S-49 and S-50 through the deprotection without isolating corresponding aniline moieties.
4.27.1. S-{4-[2-Hydroxy-2-(4-nitro-3-trifluoromethylphenylcarbamoyl) propylamino]phenyl}carbamic acid tert-butyl ester (S-49)
Method (a) in Scheme 3: To a solution of 47 (300 mg, 1.44 mmol) and R-3 (490 mg, 1.44 mmol) in THF (30 mL) was added triethylamine (0.37 mL, 2.6 mmol). The mixture was refluxed overnight. After cooling, the mixture was concentrated under reduced pressure, poured into EtOAc (30 mL), washed with water and dried with MgSO4, concentrated under reduced pressure, and purified by flash column chromatography to gain compound S-49 (412 mg, 63%) as yellowish solid. mp 78–81 °C; ESI-MS; m/z: 497.2 [M–H]−; ¹H NMR (CDCl3) δ 9.26 (br s, 1H, NH), 8.04 (s, 1H, ArH), 7.90 (m, 2H, ArH), 7.07 (d, J = 7.5 Hz, 2H, ArH), 6.57 (d, J = 7.5 Hz, 2H, ArH), 6.43 (br s, 1H, NH), 4.13 (br s, 2H, OH & NH), 3.70 (d, J = 12.9 Hz, 1H, CH2), 3.07 (d, J = 12.9 Hz, 1H, CH2), 1.51 (s, 3H, CH3); 13C NMR: (CDCl3) δ 174.0, 153.5, 143.8, 142.3, 141.2, 128.7, 126.3 (2C), 124.6 (q, JC–F = 33.9 Hz), 121.7 (2C), 121.4, 121.3 (q, JC–F = 272.0 Hz), 117.8 (q, JC–F = 5.9 Hz), 113.8, 79.9, 75.4, 52.3, 27.2, 23.6; Anal. Calcd for C22H25F3N4O6: C, 53.01; H, 5.06; N, 11.24. Found: C, 52.92; H, 5.16; N, 11.29.
4.27.2. S-(4-{[2-Hydroxy-2-(4-nitro-3-trifluoromethylphenylcarbamoyl)propyl]methylamino}phenyl)carbamic acid tert-butyl ester (S-50)
Method (a′) in Scheme 3: To a suspension of sodium hydride (1.2 mmol) in THF (10 mL) was added protected aniline (48, 1.1 mmol) at ice-bath under argon and raised room temperature. After 30 min, a solution of R-3 (1.1 mmol) in THF (10 mL) was added to the suspension at ice-bath. The mixture was stirred overnight under argon. Water (10 mL) was added carefully, and the mixture was extracted with EtOAc (3× 20 mL). The combined organic extracts were dried with MgSO4, concentrated under reduced pressure and puri.ed with flash column chromatography to gain compound S-50 (390 mg, yield 78%) as brown solid; Rf 0.19 (EtOAc/hexane 1:2); mp 84–85 °C; ESI-MS; m/z: 511.0 [M–H]−; ¹H NMR (CDCl3) δ 9.36 (br s, 1H, NH), 8.07 (s, 1H, ArH), 7.97 (m, 2H, ArH), 7.19 (d, J = 8.6 Hz, 2H, ArH), 7.82 (d, J = 8.6 Hz, 2H, ArH), 6.48 (br s, 1H, NH), 4.13 (br s, 1H, OH), 3.85 (d, J = 14.6 Hz, 1 H, CH2), 3.33 (d, J = 14.6 Hz, 1H, CH2), 2.82 (s, 3H, NCH3), 1.52 (s, 3H, CH3), 1.50 (s, 9H, CH3); 13C NMR: (CDCl3) δ 174.5, 152.8, 146.4, 142.5, 141.2, 130.3, 126.4 (2C), 124.6, (q, JC–F = 33.9 Hz), 121.6, 121.2 (q, JC–F = 271.9 Hz), 120.1, 117.8 (q, JC–F = 5.9 Hz), 115.8 (2C), 79.8, 74.9, 62.0, 40.6, 27.8, 24.3; Anal. Calcd for C23H27F3N4O6: C, 53.90; H, 5.31; N, 10.93. Found: C, 53.78; H, 5.30; N, 10.67.
4.27.3. S-2-Hydroxy-3-(4-isothiocyanatophenylamino)-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)propionamide (S-32)
Prepared by S-49 (Route B in Scheme 1, followed by Method (c); yellowish solid; yield 80% (two steps from S-49); Rf 0.29 (EtOAc/hexane 2:3); mp 63–66 °C; ESI-MS; m/z: 439.2 [M–H]−; ¹H NMR (CDCl3) δ 9.20 (br s, 1H, NH), 8.13 (s, 1H, ArH), 8.00 (m, 2H, ArH), 7.17 (d, J = 8.4 Hz, 2H, ArH), 6.98 (d, J = 8.4 Hz, 2H, ArH), 4.47 (d, J = 9.0 Hz, 1H, CH2), 4.01 (d, J = 9.0 Hz, 1H, CH2), 3.42 (br s, 1H, OH), 1.62 (s, 3H, CH3); 13C NMR (CDCl3) δ 172.7, 156.8, 143.5, 141.6, 135.2, 127.3 (2C), 126.0 (q, JC–F = 34.0 Hz), 125.8, 122.3, 121.9 (q, JC–F = 272.0 Hz), 118.5 (q, JC–F = 5.9 Hz), 117.7, 116.0 (2C), 76.0, 73.0, 22.9; Anal. Calcd for C18H15F3N4O4S: C, 49.09; H, 3.43; N, 12.72. Found: C, 48.92; H, 3.26; N, 12.53.
4.27.4. S-2-Hydroxy-3-[(4-isothiocyanatophenyl)methylamino]-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)propionamide (S-33)
Prepared by S-50 (Route B in Scheme 1, followed by Method (c); brown oil; yield 67% (two steps from S-50); Rf 0.28 (EtOAc/hexane 2:3); ESI-MS; m/z: 453.1 [M–H]−; ¹H NMR (CDCl3) δ 9.27 (br s, 1H, NH), 8.09 (s, 1H, ArH), 7.98 (m, 2H, ArH), 7.08 (d, J = 8.4 Hz, 2H, ArH), 7.81 (d, J = 8.4 Hz, 2 H, ArH), 3.94 (d, J = 15.0 Hz, 1H, CH2), 3.57 (d, J = 15.0 Hz, 1H, CH2), 3.28 (br s, 1H, OH), 2.97 (s, 3H, NCH3), 1.60 (s, 3H, CH3); 13C NMR: (CDCl3) δ 173.6, 148.9, 142.7, 141.0, 132.7, 126.5, 126.2 (2C), 124.7 (q, JC–F = 34.2 Hz), 121.6, 121.2 (q, JC–F = 272.0 Hz), 120.4, 117.8 (q, JC–F = 5.9 Hz), 113.4 (2C), 76.9, 60.6, 39.7, 24.2; Anal. Calcd for C19H17F3N4O4S: C, 50.22; H, 3.77; N, 12.33. Found: C, 50.25; H, 3.85; N, 12.18.
4.28. Preparation for thioether- and sulfone-linked compounds, R-34 to R-37
Compounds R-34 to R-37 were prepared by coupling the propanamide derivatives (R-3, R-4) with 4-aminothiophenol (51) and subsequent conversion of the amine (R-52 to R-55) to isothiocyanate (R-34 to R-37) by a procedure similar to Leclerc’s method14 as shown in Scheme 4.
Synthesis of R-52 and R-53: To a suspension of sodium hydride (3 mmol) in THF (10 mL) was added 4-aminothiophenol (51, 2.7 mmol) at ice-bath under argon and later raised to room temperature. After 30 min, a solution of R-3 or R-4 (2.7 mmol) in THF (30 mL) was added to the suspension at ice-bath. The mixture was stirred under argon for 20 h. Water (30 mL) was added carefully, and the mixture was extracted with EtOAc (3× 30 mL). The combined organic extracts were dried with MgSO4, concentrated under reduced pressure, and purified with flash column chromatography using EtOAc/hexane (2:3) as an eluent to gain compound R-52 or R-53 as below.
Synthesis of R-54 and R-55: To a solution of R-52 or R-53 (1.8 mmol) in methylene chloride (20 mL) was added 3-chloroperoxybenzoic acid (935 mg, 5.4 mmol) at room temperature. The mixture was stirred for 2 h, and then washed with sodium sulfite solution and water, dried with MgSO4, concentrated under reduced pressure, and purified with flash column chromatography as an eluent of EtOAc/hexane (2:3) to gain compound R-54 or R-55 as below.
4.28.1. R-3-(4-Aminophenylsulfanyl)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)propionamide (R-52)
Yellowish oil; yield 85% (from R-3 with 51); Rf 0.26 (EtOAc/hexane 2:3); ESI-MS; m/z: 414.1 [M–H]−; ¹H NMR (CDCl3) δ 9.04 (br s, 1H, NH), 7.92 (m, 2H, ArH), 7.74 (m, 1H, ArH), 7.26 (d, J = 8.4 Hz, 2H, ArH), 6.60 (d, J = 8.4 Hz, 2H, ArH), 3.78 (d, J = 14.4 Hz, 1H, CH2), 3.68 (br s, 3H, NH2 & OH), 2.93 (d, J = 14.4 Hz, 1H, CH2), 1.51 (s, 3H, CH3); 13C NMR: (CDCl3) δ 172.9, 146.6, 146.4, 142.4, 141.0, 133.3 (2C), 126.2, 124.5 (q, JC–F = 33.8 Hz), 121.4, 121.3 (q, JC–F = 272.0 Hz), 117.6 (q, JC–F = 5.9 Hz), 115.2 (2C), 74.3, 45.8, 24.1; Anal. Calcd for C17H16F3N3O4S·0.1H2O: C, 48.94; H, 3.91; N, 10.07. Found: C, 49.02; H, 3.87; N, 10.17.
4.28.2. R-3-(4-Aminophenylsulfanyl)-N-(4-cyano-3-trifluoromethylphenyl)-2-hydroxy-2-methylpropionamide (R- 53)11
Prepared from R-4 and 51; white solid; ESI-MS; m/z: 394.1 [M–H]−; ¹H NMR (CDCl3) δ 9.01 (br s, 1H, NH), 7.93 (d, J = 1.7 Hz, 1H ArH), 7.69 (d, J = 8.4 Hz, 1H ArH), 7.62 (dd, J = 8.1, 1.7 Hz, 1H ArH), 7.17 (d, J = 8.4 Hz, 2H, ArH), 6.38 (d, J = 8.4 Hz, 2H ArH), 3.72 (br s, 3 H, NH2 & OH), 3.65 (d, J = 14.1 Hz, 1H, CH2), 2.92 (d, J = 14.1 Hz, 1H, CH2), 1.47 (s, 3H, CH3); 13C NMR: (CDCl3) δ 173.0, 146.4, 141.0, 135.1, 134.0, 133.8, 133.2 (q, JC–F = 32.8 Hz), 130.2, 125.1, 121.4 (q, JC–F = 275.4 Hz), 116.7 (q, JC–F = 5.0 Hz), 115.1, 103.8, 74.5, 45.9, 25.8; Anal. Calcd for C18H16F3N3O2S: C, 54.68; H, 4.08; N, 10.63. Found: C, 54.44; H, 4.02; N, 10.6.
4.28.3. R-3-(4-Aminobenzenesulfonyl)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl phenyl)propionamide (R-54)
Yellowish solid; yield 71% (from R-52); Rf 0.12 (EtOAc/hexane 2:3); mp 90–93 °C; ESI-MS; m/z: 445.9 [M–H]−; ¹H NMR (CDCl3) δ 9.30 (br s, 1H, NH), 8.03 (s, 1H, ArH), 7.92 (d, J = 8.7 Hz, 1H, ArH), 7.82 (d, J = 8.7 Hz, 1H, ArH), 7.56 (d, J = 8.4 Hz, 2H, ArH), 6.54 (d, J = 8.4 Hz, 2H, ArH), 5.35 (br s, 1H, OH), 4.34 (br s, 2H, NH2), 4.02 (d, J = 14.4 Hz, 1H, CH2), 3.43 (d, J = 14.4 Hz, 1H, CH2), 1.57 (s, 3H, CH3); 13C NMR: (CDCl3) δ 171.4, 151.7, 142.6, 141.0, 130.4, 129.7 (2C), 126.2, 124.3 (q, JC–F = 34.0 Hz), 121.8, 121.1 (q, JC–F = 272.0 Hz), 117.8 (q, JC–F = 5.9 Hz), 113.3 (2C), 73.7, 61.2, 22.4; Anal. Calcd for C17H16 F3N3O6S: C, 45.64; H, 3.60; N, 9.39. Found: C, 45.82; H, 3.71; N, 9.40.
4.28.4. R-3-(4-Aminophenylsulfonyl)-N-(4-cyano-3-trifluoromethylphenyl)-2-hydroxy-2-methylpropionamide (R-55).30
White solid; mp 157.5–159 °C; ESI-MS; m/z: 426.0 [M–H]−; ¹H NMR (DMSO-d6) δ 10.35 (br s, 1H, NH), 8.44 (d, J = 1.2 Hz, 1H, ArH), 8.21 (dd, J = 8.4, 1.2 Hz, 1H, ArH), 8.06 (d, J = 8.4 Hz, 1H, ArH), 7.43 (d, J = 8.7 Hz, 2H, ArH), 6.52 (d, J = 8.7 Hz, 2H, ArH), 6.29 (br s, 1 H, OH), 6.02 (br s, 2H, NH2), 3.76 (d, J = 14.4 Hz, 1H, CH2), 3.47 (d, J = 14.4 Hz, 1H, CH2), 1.39 (s, 3H, CH3); 13C NMR: (CDCl3) δ 173.8, 153.5, 143.2, 136.0, 131.3 (q, JC–F = 31.5 Hz), 129.8, 125.4, 122.8, 122.2 (q, JC–F = 272.1 Hz), 117.4 (q, JC–F = 5.0 Hz), 115.8, 112.3, 101.8, 73.2, 63.9, 27.1; Anal. Calcd for C18H16F3N3O4S: C, 50.58; H, 3.77; N, 9.83. Found: C, 50.40; H, 3.88; N, 9.63.
The thioether-linked isothiocyanate compounds (R-34 and R-35) and sulfone-linked compounds (R-36 and R-37) were prepared as shown in Scheme 4. The synthetic methods of the following compounds R-34, R-35, R-36, and R-37 were same as method c of Route B in Scheme 1.
4.28.5. R-2-Hydroxy-3-(4-isothiocyanatophenylsulfanyl)-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)propionamide (R-34)
Prepared from R-52; brown oil; yield 76%; Rf 0.36 (EtOAc/hexane 2:3); ESI-MS; m/z: 455.9 [M–H]−; ¹H NMR (CDCl3) δ 9.09 (br s, 1H, NH), 7.98 (br s, 2H, ArH), 7.86 (m, 1H, ArH), 7.37 (d, J = 7.8 Hz, 2H, ArH), 7.06 (d, J = 7.8 Hz, 2H, ArH), 3.74 (d, J = 13.8 Hz, 1H, CH2), 3.48 (br s, 1H, OH), 3.22 (d, J = 13.8 Hz, 1H, CH2), 1.59 (s, 3H, CH3); 13C NMR: (CDCl3) δ 172.3, 142.7, 140.8, 136.4, 133.0, 131.0, 129.9 (2C), 126.5, 125.9 (2C), 124.7 (q, JC–F = 34.0 Hz), 121.5, 121.2 (q, JC–F = 272.0 Hz), 117.7 (q, JC–F = 5.9 Hz), 75.1, 44.1, 25.6; Anal. Calcd for C18H14F3N4O2S2: C, 47.26; H, 3.08; N, 9.19. Found: C, 47.36; H, 3.03; N, 9.01.
4.28.6. R-N-(4-Cyano-3-trifluoromethylphenyl)-2-hydroxy-3-(4-isothiocyanatophenyl sulfanyl)-2-methylpropionamide (R-35).11
Prepared from R-53; white solid; ESI-MS; m/z: 336.1 [M–H]−; ¹H NMR (CDCl3) δ 9.05 (br s, 1H, NH), 7.97 (s, 1H, ArH), 7.79 (m, 2H, ArH), 7.36 (d, J = 7.5 Hz, 2H, ArH), 7.04 (d, J = 7.5 Hz, 2H, ArH), 3.73 (d, J = 13.8 Hz, 1H, CH2), 3.51 (br s, 1H, OH), 3.20 (d, J = 13.8 Hz, 1H, CH2), 1.58 (s, 3H, CH3). 13C NMR: (CDCl3) δ 173.2, 141.5, 137.1, 136.0, 134.1 (q, JC–F = 32.6 Hz), 133.8, 131.7, 130.5, 126.3, 122.3 (q, JC–F = 272.4 Hz), 122.0, 117.4 (q, JC–F = 4.8 Hz), 115.7, 104.7, 75.8, 44.8, 26.4; Anal. Calcd for C19H14F3N3O2S2: C, 52.17; H, 3.23; N, 9.61. Found: C, 52.09; H, 3.16; N, 9.53.
4.28.7. R-2-Hydroxy-3-(4-isothiocyanatobenzenesulfonyl)-2-methyl-N-(4-nitro-3-trifluoromethylphenyl)propionamide (R-36)
Prepared from R-54; yellowish oil; yield 81%; Rf 0.22 (EtOAc/hexane 1:1); ESI-MS; m/z: 487.9 [M–H]−; ¹H NMR (CDCl3) δ 9.29 (br s, 1H, NH), 8.03 (s, 1H, ArH), 7.94 (m, 4H, ArH), 7.32 (d, J = 8.1 Hz, 2H, ArH), 4.99 (br s, 1H, OH), 4.06 (d, J = 14.4 Hz, 1H, CH2), 3.56 (d, J = 14.4 Hz, 1H, CH2), 1.63 (s, 3H, CH3); 13C NMR: (CDCl3) δ 171.2, 142.8, 140.7, 140.0, 137.4, 136.6, 129.1 (2C), 126.4, 125.9 (2C), 124.6 (q, JC–F = 34.0 Hz), 121.7, 121.2 (q, JC–F = 272.0 Hz), 117.9 (q, JC–F = 5.9 Hz), 74.0, 61.5, 20.5; Anal. Calcd for C18H14F3N3O6S2: C, 44.17; H, 2.88; N, 8.59. Found: C, 43.99; H, 2.76; N, 8.53.
4.28.8. R-N-(4-Cyano-3-trifluoromethylphenyl)-2-hydroxy-3-(4-isothiocyanatophenyl sulfonyl)-2-methylpropionamide (R-37).11
Prepared from R-55; white solid; m/z: 467.7 [M–H]−; ¹H NMR (DMSO-d6) δ 10.33 (br s, 1H, NH), 8.41 (s, 1H, ArH), 8.19 (d, J = 8.4, 1H, ArH), 8.08 (d, J = 8.4 Hz, 1H, ArH), 7.90 (d, J = 8.4 Hz, 2H, ArH), 7.51 (d, J = 8.4 Hz, 2H, ArH), 6.46 (br s, 1H, OH), 3.98 (d, J = 14.7 Hz, 1H, CH2), 3.74 (d, J = 14.7 Hz, 1H, CH2), 1.41 (s, 3H, CH3); 13C NMR: (CDCl3) δ 173.5, 143.0, 139.0, 136.5, 136.1, 134.8, 131.4 (q, JC–F = 31.4 Hz), 130.1, 126.2, 122.7, 122.4 (q, JC–F = 272.0 Hz), 117.4 (q, JC–F = 5.1 Hz), 115.7, 102.0, 73.0, 63.3, 27.2; Anal. Calcd for C19H14F3N3O4S2: C, 48.61; H, 3.01; N, 8.95. Found: C, 48.71; H, 2.95; N, 8.79.
Acknowledgments
This work was supported by grants from National Institutes of Health (NIH-5R01DK065227-02 and -03) and GTx-Inc.
References and notes
- 1.Greenlee RT, Hill-Harmon MB, Murray T, Thun M. CA-Cancer J. Clin. 2001;51:15. doi: 10.3322/canjclin.51.1.15. [DOI] [PubMed] [Google Scholar]
- 2.Clement OO, Freeman CM, Hartmann RW, Handratta VD, Vasaitis TS, Brodie AMH, Njar VCO. J. Med. Chem. 2003;46:2345. doi: 10.1021/jm020576u. [DOI] [PubMed] [Google Scholar]
- 3.McConnell JD. Urol. Clin. North Am. 1991;18:1. [PubMed] [Google Scholar]
- 4.Carter BS, Carter HB, Isaacs JT. Prostate. 1990;16:187. doi: 10.1002/pros.2990160302. [DOI] [PubMed] [Google Scholar]
- 5.(a) Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Proc. Natl. Acad. Sci. U.S.A. 2005;102:6201. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Reddy VK, Sarkar A, Valasinas A, Marton LJ, Basu HS, Frydman BJ. Med. Chem. 2001;44:404. doi: 10.1021/jm000310s. [DOI] [PubMed] [Google Scholar]; (c) Valasinas A, Sarkar A, Reddy VK, Marton LJ, Basu HS, Frydman B. J. Med. Chem. 2001;44:390. doi: 10.1021/jm000309t. [DOI] [PubMed] [Google Scholar]; (d) Frydman B, Bhattacharya S, Sarkar A, Drandarov K, Chesnov S, Guggisberg A, Popaj K, Sergeyev S, Yurdakul A, Hesse M, Basu HS, Marton LJ. Med. Chem. 2004;47:1051. doi: 10.1021/jm030437s. [DOI] [PubMed] [Google Scholar]; (e) Le HT, Schaldach CM, Firestone GL, Bjeldanes LF. J. Biol. Chem. 2003;278:21136. doi: 10.1074/jbc.M300588200. [DOI] [PubMed] [Google Scholar]; (f) Ohtsu H, Xiao Z, Ishida J, Nagai M, Wang H-K, Itokawa H, Su C-Y, Shih C, Chiang T, Chang E, Lee Y, Tsai M-Y, Chang C, Lee K-H. J. Med. Chem. 2002;45:5037. doi: 10.1021/jm020200g. [DOI] [PubMed] [Google Scholar]; (g) Matias PM, Carrondo MA, Coelho R, Thomaz M, Zhao X-Y, Wegg A, Crusius K, Egner U, Donner P. J. Med. Chem. 2002;45:1439. doi: 10.1021/jm011072j. [DOI] [PubMed] [Google Scholar]; (h) Brandy SF, Pawluczyk JM, Lumma PK, Feng D-M, Wai JM, Jones R, DeFeoJones D, Wong BK, Miller-Stein C, Lin JH, Oliff A, Freidinger RM, Garsky VM. J. Med. Chem. 2002;45:4706. doi: 10.1021/jm020139f. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kassouf W, Tanguay S, Aprikian AG. J. Urol. 2003;169:1742. doi: 10.1097/01.ju.0000057795.97626.66. [DOI] [PubMed] [Google Scholar]; (b) Dukes M, Furr BJ, Hughes LR, Tucker H, Woodburn JR. Steroids. 2000;65:725. doi: 10.1016/s0039-128x(00)00179-3. [DOI] [PubMed] [Google Scholar]
- 7.Schroder FH, Whelan P, de Reijke TM, Kurth KH, Pavone-Macaluso M, Mattelaer J, van Velthoven RF, Debois M, Collette L. Eur. Urol. 2004;45:457. doi: 10.1016/j.eururo.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 8.(a) James KD, Ekwuribe NN. Synthesis. 2002;7:850. [Google Scholar]; (b) Schellhammer PF, Davis JW. Clin. Prostate Cancer. 2004;2:213. doi: 10.3816/cgc.2004.n.002. [DOI] [PubMed] [Google Scholar]
- 9.Mukherjee A, Kirkovsky LI, Kimura Y, Marvel MM, Miller DD, Dalton JT. Biochem. Pharmacol. 1999;58:1259. doi: 10.1016/s0006-2952(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 10.Marhefka CA, Gao W, Chung K, Kim J, He Y, Yin D, Bohl CE, Dalton JT, Miller DD. J. Med. Chem. 2004;47:993. doi: 10.1021/jm030336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kirkovsky L, Mukherjee A, Yin D, Dalton JT, Miller DD. J. Med. Chem. 2000;43:581. doi: 10.1021/jm990027x. [DOI] [PubMed] [Google Scholar]
- 12.Kim J, Wu D, Hwang DJ, Miller DD, Dalton JT. J. Pharmacol. Exp. Ther. 2005;315:230. doi: 10.1124/jpet.105.088344. [DOI] [PubMed] [Google Scholar]
- 13.Chen J, Hwang DJ, Chung K, Bohl CE, Miller DD, Dalton JT. Endocrinology. 2005;146:5444. doi: 10.1210/en.2005-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leclerc G, Amlaiky N, Decker N, Schartz J, Schneyder DD. Eur. J. Med. Chem. 1983;18:379. [Google Scholar]
- 15.Hwang DJ, Kim SN, Choi JH, Lee YS. Bioorg. Med. Chem. 2001;9:1429. doi: 10.1016/s0968-0896(01)00013-x. [DOI] [PubMed] [Google Scholar]
- 16.He Y, Yin D, Perera M, Kirkovsky L, Stourman N, Li W, Dalton JT, Miller DD. Eur. J. Med. Chem. 2002;37:619. doi: 10.1016/s0223-5234(02)01335-1. [DOI] [PubMed] [Google Scholar]
- 17.(a) Romesberg FE, Flanagan ME, Uno T, Schultz PG. J. Am. Chem. Soc. 1998;120:5160. [Google Scholar]; (b) Bartoli G, Bosco M, Dalpozzo R, Todesco PE. J. Org. Chem. 1986;51:3694. [Google Scholar]
- 18.(a) Tucker H, Crook JW, Chesterson GJ. J. Med. Chem. 1988;31:954. doi: 10.1021/jm00400a011. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Carroll GL, Truesdale LK. J. Org. Chem. 1974;39:914. [Google Scholar]; (c) Hoye TR, Kurth MJ. J. Org. Chem. 1979;44:3461. [Google Scholar]
- 19.Galzi JL, Mejean A, Ilien B, Mollereau C, Meunier JC, Goeldner M, Hirth C. J. Med. Chem. 1990;33:2456. doi: 10.1021/jm00171a020. [DOI] [PubMed] [Google Scholar]
- 20.Leardini R, Zanardi G. Synthesis. 1982;3:225. [Google Scholar]
- 21.(a) Kessler P, Chatrenet B, Goeldner M, Hirth C. Synthesis. 1990;11:1065. [Google Scholar]; (b) Lurdes M, Almieda S, Grehn L, Ragnarsson UJ. Chem. Soc. Perkin Trans. 1. 1988:1905. [Google Scholar]
- 22.(a) Dyatkin AB, Gong Y, Miskowski TA, Kimball ES, Prouty SM, Fisher MC, Santulli RJ, Schneider CR, Wallace NH, Hornby PJ, Diamond C, Kinney WA, Maryanoff BE, Diamiano BP, He W. Bioorg. Med. Chem. 2005;13:6693. doi: 10.1016/j.bmc.2005.07.022. [DOI] [PubMed] [Google Scholar]; (b) Moreau E, Fortin S, Desjardins M, Rousseau JL, Petitclerc E, C-Gaudreault R. Bioorg. Med. Chem. 2005;13:6703. doi: 10.1016/j.bmc.2005.07.048. [DOI] [PubMed] [Google Scholar]
- 23.(a) Zhang X, Neamati N, Lee YK, Orr A, Brown RD, Whitaker N, Pommier Y, Burke TR. Bioorg. Med. Chem. 2001;9:1649. doi: 10.1016/s0968-0896(01)00075-x. [DOI] [PubMed] [Google Scholar]; (b) Shaw E. Physiol. Rev. 1970;50:244. doi: 10.1152/physrev.1970.50.2.244. [DOI] [PubMed] [Google Scholar]
- 24.Marhefka CA, Moore BM, II, Bishop TC, Kirkovsky L, Mukherjee A, Dalton JT, Miller DD. J. Med. Chem. 2001;44:1729. doi: 10.1021/jm0005353. [DOI] [PubMed] [Google Scholar]
- 25.(a) Haga N, Endo Y, Kataoka K-I, Yamaguchi K, Shudo K. J. Am. Chem. Soc. 1992;114:9795. [Google Scholar]; (b) Hodgson NJ. Chem. Soc. 1941:645. [Google Scholar]
- 26.Okada H, Koyanagi T, Yamada N, Haga T. Chem. Pharm. Bull. 1991;39:2308. doi: 10.1248/cpb.39.2308. [DOI] [PubMed] [Google Scholar]
- 27.Christiansen WG. J. Am. Chem. Soc. 1923;45:2191. [Google Scholar]
- 28.Xue C-B, He X, Roderick J, Corbett RL, Duan JJ-W, Liu R-Q, Covington MB, Newton RC, Trzaskos JM, Magolda RL, Wexler RR, Decicco CP. Bioorg. Med. Chem. Lett. 2003;13:4293. doi: 10.1016/j.bmcl.2003.09.056. [DOI] [PubMed] [Google Scholar]
- 29.(a) Lipowska M, Patonay G, Strekowski L. Synth. Commun. 1993;23:3087. [Google Scholar]; (b) Hodgson N. J. Chem. Soc. 1941:645. [Google Scholar]
- 30.(a) Bohl CE, Chang C, Mohler ML, Chen J, Miller DD, Swaan PW, Dalton JT. J. Med. Chem. 2004;47:3765. doi: 10.1021/JM0499007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Mol. Pharmacol. 2003;63:211. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]