

Abstract
Lytic replication of Kaposi's sarcoma-associated herpesvirus (KSHV) promotes the progression of Kaposi's sarcoma (KS), a dominant malignancy in patients with AIDS. While 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-induced KSHV reactivation from latency is mediated by the protein kinase C δ and MEK/ERK mitogen-activated protein kinase (MAPK) pathways, we have recently shown that the MEK/ERK, JNK and p38 MAPK pathways modulate KSHV lytic replication during productive primary infection of human umbilical vein endothelial cells (Pan, H.Y., Xie, J.P., Ye, F.C., Gao, S.-J., 2006. J. Virol. 80, 5371-82). Here, we report that, besides the MEK/ERK pathway, the JNK and p38 MAPK pathways also mediate TPA-induced KSHV reactivation from latency. The MEK/ERK, JNK and p38 MAPK pathways were constitutively activated in latent KSHV-infected BCBL-1 cells. TPA treatment enhanced the levels of activated ERK and p38 but not those of activated JNK. Inhibitors of all three MAPK pathways reduced TPA-induced production of KSHV infectious virions in BCBL-1 cells in a dose-dependent fashion. The inhibitors blocked KSHV lytic replication at the early stage(s) of reactivation, and reduced the expression of viral lytic genes including RTA, a key immediate-early transactivator of viral lytic replication. Activation of MAPK pathways was necessary and sufficient for activating the promoter of RTA. Furthermore, we showed that the activation of RTA promoter by MAPK pathways was mediated by their downstream target AP-1. Together, these findings suggest that MAPK pathways might have general roles in regulating the life cycle of KSHV by mediating both viral infection and switch from viral latency to lytic replication.
Keywords: Kaposi's sarcoma, KSHV, reactivation, MAPK pathways, AP-1, RTA, TPA
Introduction
Infection by Kaposi's sarcoma-associated herpesvirus (KSHV), also known as human herpesvirus 8 (HHV8), is linked to the development of Kaposi's sarcoma (KS), a malignancy frequently seen in untreated AIDS patients (Greene et al., 2007). In the last two decades, KS has evolved as the most common cancer in some African regions, and could account for up to 30% of all types cancer in both HIV-infected and uninfected adult and pediatric patients (Orem et al., 2004). In these regions, KSHV seroprevalence reaches up to 70% in the general population (Greene et al., 2007). With the spread of the AIDS epidemics, KSHV and KS have emerged as an important health problem worldwide. Besides KS, KSHV is also associated with some other malignancies such as primary effusion lymphoma (PEL) and multicentric Castleman's disease (Greene et al., 2007).
Like other herpesviruses, the life cycle of KSHV consists of two phases: a latent phase during which the virus persists as episomes expressing several viral latent genes; and a lytic phase during which the viral genomes replicate in a linear form expressing most of the more than 80 viral lytic genes, and produces infectious virions (Dourmishev et al., 2003). In KS lesions, the majority of tumor cells are latently infected by KSHV, indicating an essential role for viral latent infection in tumor development (Dupin et al., 1999); however, a small number of KS tumor cells also undergo lytic infection (Staskus et al., 1997; Zhong et al., 1996). Results from recent laboratory and clinical studies have indicated that these lytic cells might promote KS tumors through several synergetic mechanisms including: the production of infectious virions for spreading to new target cells, the induction of angiogenic and inflammatory cytokines during virus spread and lytic replication, and the generation of KSHV-encoded angiogenic and inflammatory cytokines, and regulatory molecules (Greene et al., 2007). Thus, it is essential to understand the mechanism controlling KSHV lytic replication.
So far KSHV lytic replication has been examined primarily with cell lines isolated from lymphomas or peripheral blood mononuclear cells of PEL patients (Cesarman et al., 1995; Gao et al., 1996b; Renne et al., 1996). These cells are latently infected by KSHV but can be reactivated into lytic replication by chemical inducers such as 12-O-tetradecanoyl-phorbol-13-acetate (TPA). These studies have identified the protein of replication and transcriptional activator (RTA) encoded by ORF50 as a central viral protein that controls KSHV lytic replication (Lukac et al., 1998; Sun et al., 1998; Xu et al., 2005). The expression of RTA leads to the transactivation of cascades of viral lytic genes and the replication of viral genomes, which results in full viral lytic replication. Consequently, RTA is necessary and sufficient for activating KSHV from latency (Lukac et al., 1998; Sun et al., 1998; Xu et al., 2005).
Multiple factors control the expression of RTA and KSHV lytic replication. Both epigenetic modifications and cellular signaling pathways play important roles in regulating the expression of RTA. The RTA promoter is associated with histone deacetylases 1, 5, and 7 in latent PEL cells (Lu et al., 2003). Treatment with butyrate induces KSHV reactivation, which correlates with the rapid association of Ini1/Snf5, a component of the Swi/Snf family of the chromatin remodeling proteins. TPA treatment leads to the demethylation of RTA promoter and KSHV reactivation from latency (Chen et al., 2001). TPA induction of KSHV reactivation is also mediated by calcium-dependent calcineurin signaling and protein kinase C (PKC) δ pathway (Deutsch et al., 2004; Zoeteweij et al., 2001). More recently, it was shown that epinephrine and norepinephrine could efficiently reactivate KSHV from latency via β-adrenergic activation of the protein kinase A (PKA) signaling pathway (Chang et al., 2005). The CCAAT/enhancer-binding protein-α (C/EBPα) is induced during the early stages of KSHV reactivation, and overexpression of C/EBPα alone is sufficient to activate the RTA promoter in transient-cotransfection reporter gene assays (Wang et al., 2003). Hypoxia and inflammatory cytokines including interferon-γ and oncostatin M have been shown to induce KSHV reactivation but the underlying mechanisms remain undefined (Blackbourn et al., 2000; Chang et al., 2000; Davis et al., 2001; Mercader et al., 2000; Monini et al., 1999). While the primary target of the Notch signaling pathway RBP-Jκ (CSL) mediates RTA activation of KSHV lytic genes (Liang et al., 2002), overexpression of intracellular-activated Notch1 is sufficient to reactivate KSHV from latency in PEL cells (Lan et al., 2006). The role of NF-κB pathway in KSHV lytic replication remains controversial. In PEL cells, inhibition of NF-κB led to KSHV reactivation and the expression of lytic proteins (Brown et al., 2003); however, it was reported that NF-κB induction was required for the production of KSHV infectious virions in TPA-induced PEL cells (Sgarbanti et al., 2004).
We have recently shown that KSHV activates the MEK/ERK, JNK and p38 mitogen-activated protein kinase (MAPK) pathways during primary infection in a productive infection system of human umbilical vein endothelial cells (HUVEC), and these three pathways modulate not only the early viral entry events but also subsequent viral lytic replication (Xie et al., 2005; Pan et al., 2006). It is believed that KSHV lytic replication during productive primary infection and reactivation from latency share similar features. Both are involved with similar steps and viral lytic cycle genes, and as a common consequence, both generate infectious virions. However, the signaling pathways controlling KSHV lytic replication and their destinies in the two processes might differ. During primary infection, the viral genomes are newly delivered into the cells without being subjected to epigenetic modifications. At the same time, KSHV alters cellular pathways such as MAPK pathways, some of which could determine whether the virus goes into latency or lytic replication in a cell. In contrast, during KSHV reactivation, the latent genomes are epigenetically modified, and could respond differently to cellular signals or cues. In spite of the differences of the two systems, the findings from the productive primary infection system indicate that KSHV reactivation from latency might also be modulated by the MEK/ERK, JNK and p38 MAPK pathways (Pan et al., 2006). Indeed, two recent studies have shown that KSHV reactivation from latency is modulated by the MEK/ERK pathways (Cohen et al., 2006; Ford et al., 2006). In this report, we show that KSHV reactivation from latency is modulated not only by the MEK/ERK pathway but also by the JNK and p38 pathways. We further show that all three MAPK pathways mediate KSHV reactivation through AP-1, which binds to the promoter of RTA leading to its expression and activation of the KSHV lytic replication program.
Results
Activation of ERK, JNK and p38 MAPK pathways in BCBL-1 cells
We used BCBL-1 cells infected with a recombinant KSHV, BAC36, to define the roles of MAPK pathways in KSHV reactivation (Zhou et al., 2002). BAC36 contains a GFP marker, which facilitates the titration of infectious virions (Gao et al., 2003). We first determined the status of the ERK, JNK and p38 MAPK pathways in BCBL-1 cells. We detected robust constitutive activation of all three pathways in BCBL-1 cells (Fig. 1A). TPA treatment did not alter the expression levels of total ERK, JNK and p38. Similarly, the levels of activated JNK (pJNK) were not altered after TPA induction. In contrast, the level of activated ERK (pERK) started to increase as early as 0.08 h post-induction (hpi) and peaked at 12 hpi (Fig. 1A). At the peak of induction, the pERK levels in TPA-induced cells were 3.5-fold of that of mock-induced cells (Fig. 1B). Similar levels of pERK induction by TPA were previously reported (Ford et al., 2006). After 3 day post-induction (dpi), no difference of pERK was observed between uninduced- and TPA-induced cells (Fig. 1A and B). Activated p38 (pp38) was transiently and marginally induced by TPA at the early time points (Fig. 1A). The levels of pp38 in TPA-induced cells were 1.7- and 1.6-fold of that of mocked-induced cells at 0.08 and 0.17 hpi, respectively, but dropped to similar levels afterwards (Fig. 1C). Similar results were also observed in BCBL-1 cells without BAC36 (data not shown), indicating that the above observed results were not due to the presence of BAC36.
Fig. 1.

Constitutive activation of the ERK, JNK and p38 MAPK pathways in uninduced BCBL-1 cells, and transient enhanced activation of the ERK but not JNK and p38 pathways by TPA. (A) Western-blotting with specific antibodies to detect the phosphorylated forms of ERK (first set of panels), p38 (third set of panels) and JNK (sixth set of panels), and total ERK (second set of panels), p38 (fourth set of panels) and JNK (seventh set of panels) in cells induced with TPA for different lengths of time (0-144 h). An anti-β-tubulin antibody was used to normalize the sample loading (fifth and eighth panels). The ERK and p38 blots were run with one set of samples while the JNK blots were run with another set of samples. (B) Quantification of pERK levels in mock- and TPA-induced cells. (C) Quantification of pp38 levels in mock- and TPA-induced cells.
Inhibitors of MAPK pathways reduced the production of infectious virions during TPA-induced KSHV reactivation from latency
To determine the involvements of MAPK pathways in TPA-induced KSHV reactivation from latency, we employed specific pharmacological inhibitors to target the pathways: U0126 for MEKs, the immediate upstream kinases of the ERK pathway, JNK inhibitor II for the JNK pathway, and SB203580 for the p38 pathway. We induced the BCBL-1 cells with TPA for 5 day in the presence of these inhibitors at the recommended concentrations, i.e., 10 μM for the MEK inhibitor, and 50 μM for the JNK and p38 inhibitors. At the end of induction, culture supernatants were collected and titrated for the presence of infectious KSHV virions by infecting 293 cell cultures. The numbers of GFP-positive cells representing virus titers were counted at 2 day post-infection. Supernatants from uninduced cells with or without treatment with the inhibitors of MAPK pathways had no infectious virions. As expected, we observed high levels of infectious virions in the range of 4-5 × 105 GFP-positive cells/ml (GFU) in cells induced with TPA alone (Fig. 2A). Inhibitors of all three MAPK pathways significantly reduced virion production in TPA-induced cells as indicated by the reduced numbers of GFP-positive cells in the titration target 293 cultures (Fig. 2A). Quantification of the GFP-positive cell numbers showed that the MEK, JNK and p38 inhibitors reduced virion production by 90%, 98%, and 94%, respectively (Fig. 2B).
Fig. 2.

Inhibitors of MAPK pathways reduced the production of infectious virions in TPA-induced KSHV reactivation from latency in BCBL-1 cells. (A) Effects of inhibitors of MAPK pathways on TPA-induced virion production. Supernatants from cells treated with the inhibitors were titrated at 5 dpi for infectious virions by infecting 293 cells and recording GFP-positive cells at 2 days post-infection. Both JNK inhibitor II (JNK inhibitor) and SB203580 (p38 inhibitor) were used at 50 μM while U0126 (MEK inhibitor) was used at 10 μM. (B) Quantification of relative virus titers in supernatants from cells treated with inhibitors of MAPK pathways as described in (A). (C-D) The effects of inhibitors of MAPK pathways on the infectivity of KSHV inoculums. Virus inoculums were incubated with the inhibitors of MAPK pathways for 5 days at 37 °C before titration (C), or with freshly reconstituted inhibitors (D) immediate before titration. The same concentrations of the inhibitors described in (A) were used for the experiments. (E-G) Inhibitors of MEK (E), JNK (F) and p38 (G) pathways reduced the production of TPA-induced KSHV infectious virions in a dose-dependent manner. The experiments were independently carried out four times except those in (C and D) which were carried out twice, each with three repeats. Results presented were the averages with standard deviations from one experiment.
We had previously shown that addition of inhibitors of MAPK pathways at the early stages of KSHV primary infection (before 4 h post-infection) reduced virus infectivity (Pan et al., 2006). These observations raised the issue that the reduced virus titers in cells treated with the inhibitors of MAPK pathways observed in Fig. 2A and B could simply be due to the effects of the inhibitors on virus infectivity. Thus, we carried out further investigations to define the effects of the inhibitors on KSHV reactivation. In the primary infection study, the inhibitors were freshly reconstituted, and immediately used for the infectivity experiments (Pan et al., 2006). In the current study, we performed virus titration at 5 dpi. The half-life of the inhibitors under our current experimental conditions was unknown. Therefore, we determined the effects of the inhibitors on KSHV infectivity under the current experimental conditions. We added MEK, p38, and JNK inhibitors at their working concentrations of 10 μM, 50 μM, and 50 μM, respectively, to a virus preparation titrated to have a titer of 5X105 GFU, and incubated the virus preparations at 37 °C for five days. We then determined the infectivity of these treated virus preparations. As shown in Fig 2C, all the inhibitors no longer had any effects on KSHV infectivity. Similar results were obtained when the virus preparations were incubated at 4 °C for five days with the inhibitors (data not shown). As previously reported, addition of freshly reconstituted inhibitors to the virus inoculum significantly reduced KSHV infectivity (Fig. 2D). These results indicated that the inhibitors had lost their effectiveness after five days of incubation at either 37 °C or 4 °C. Thus, we concluded that the reduction of infectious virions resulting from treatment with the inhibitors of MAPK pathways during TPA-induced KSHV reactivation were genuinely due to the effects of the inhibitors on the early stages of virus reactivation rather than on the virus infectivity in the subsequent titration experiments.
To further confirm the specificity of the inhibitors on the production of infectious virions during KSHV reactivation, we conducted a dose-dependent experiment. In addition, we assayed the kinetics of virion production from 1 to 5 dpi. We induced BCBL-1 cells with TPA in the presence of the inhibitors at different concentrations: U0126 at 2.5, 5, 10, and 20 μM, and both JNK Inhibitor II and SB23580 at 12.5, 25, 50, and 100 μM. We collected the supernatants daily for up to 5 dpi, stored them at 4 °C, and titrated all of the supernatants at 5 dpi. As shown in Fig. 2E-G, the production of infectious virions increased as the time of TPA induction progressed from 1 dpi to 5 dpi in cultures without the inhibitors. As expected, inhibitors of all three MAPK pathways reduced virion production in a dose-dependent fashion. At high concentrations of the inhibitors (20 μM for U0126, 50 and 100 μM for JNK Inhibitor II, and 100 μM for SB23580), the production of infectious virions was totally abolished. These results clearly demonstrated dose-dependent effects of the inhibitors on virion production during TPA-induced KSHV reactivation from latency.
Inhibitors of MAPK pathways targeted the early stages of TPA-induced KSHV reactivation from latency
The above results indicated that the inhibitors of MAPK pathways were likely to reduce virion production by affecting the early stage(s) of KSHV reactivation as the inhibitors appeared to have a short half-life under the experimental condition (Fig. 2C). To further defined the stage(s) of KSHV reactivation that were modulated by the MAPK pathways, we treated the TPA-induced BCBL-1 cells with the inhibitors of MAPK pathways at different time points post-induction. While addition of the inhibitors at the onset of TPA induction (0 dpi) reduced more than 90% of virion production, addition of the inhibitors at 1 and 2 dpi reduced the virion production at much lower rates: 25% and 22% for the MEK inhibitor, 32% and 27% for the JNK inhibitor, and 15% and 8% for the p38 inhibitor, respectively (Fig. 3A). Addition of the inhibitors at 3 and 4 dpi had only marginal or no detectable effect. To further confirm that the inhibitors primarily exerted their effects at the early stage(s) of KSHV reaction, we added them at the onset of TPA-induction, washed them away at 2 dpi, and titrated the supernatants at 5 dpi. As shown in Fig. 3B, the inhibitors effectively reduced virion production, albeit they were washed away at 2 dpi. These results confirmed that the inhibitors primarily targeted the early stage(s) of KSHV reactivation.
Fig. 3.
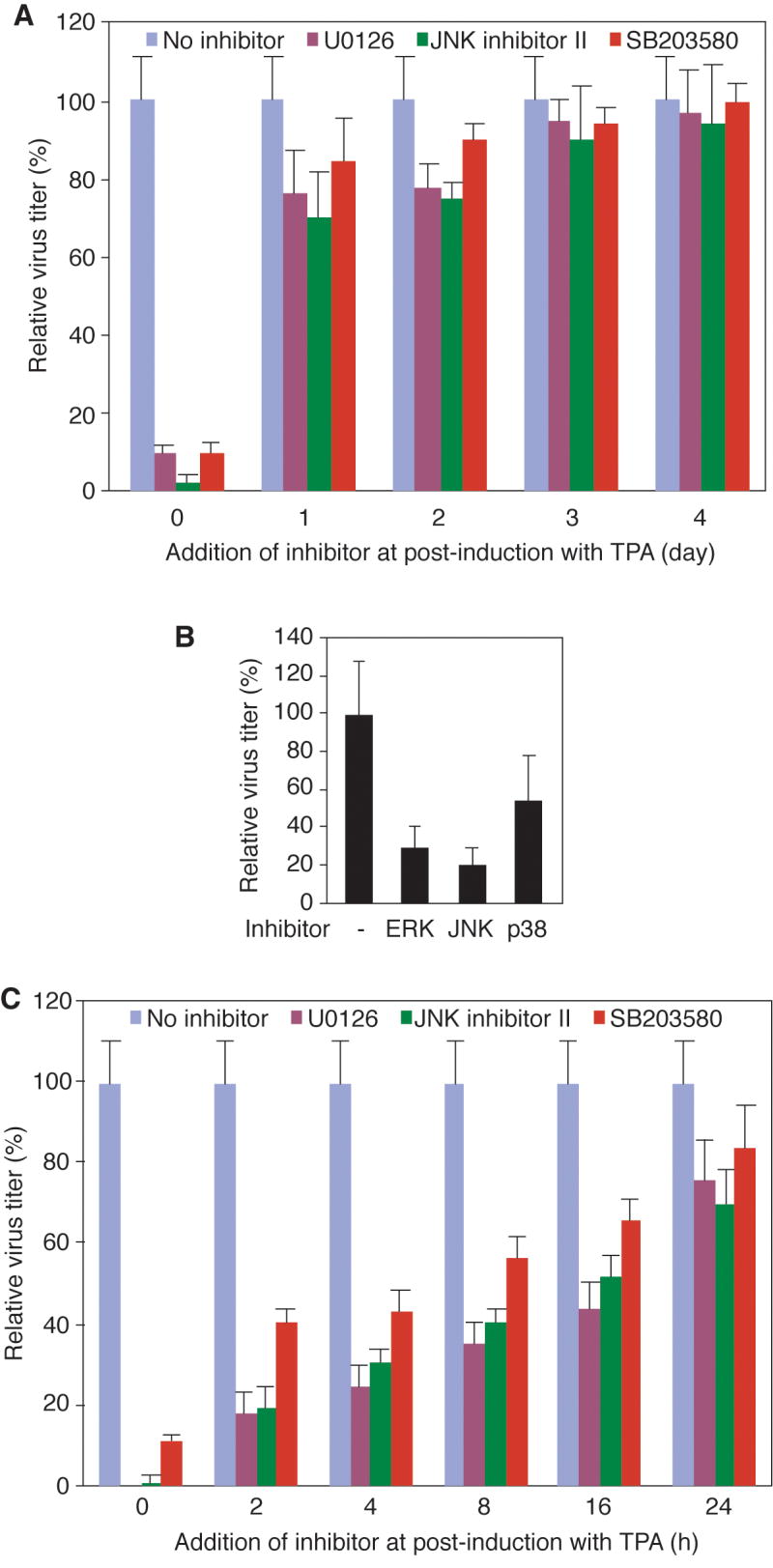
Inhibitors of MAPK pathways affected the early stages of TPA-induced KSHV reactivation from latency in BCBL-1 cells. (A) TPA-induced cells were treated with inhibitors of MEK, JNK, and p38 MAPK pathways at the onset of TPA induction or at 1, 2, 3, and 4 dpi, and supernatants were titrated for infectious virions at 5 dpi. (B) TPA-induced cells were treated with inhibitors of MAPK pathways at the onset of TPA induction. Both TPA and the inhibitors were washed away at 2 dpi and replaced with fresh medium without any inducer and inhibitors. The supernatants were collected and titrated for infectious virions at 5 dpi. (C) TPA-induced cells were treated with inhibitors of MEK, JNK, and p38 MAPK pathways at the onset of TPA induction or at 2, 4, 8, 16 and 24 hpi, and the supernatants were titrated for infectious virions at 5 dpi. All the experiments were independently carried out three times, each with three repeats. Results presented were the averages with standard deviations from one representative experiment.
To better define the stage(s) of KSHV reactivation modulated by the MAPK pathways, we added the inhibitors at different hpi within the first day of TPA induction (Fig. 3C). Again, addition of the inhibitors at the onset of TPA induction (0 hpi) reduced more than 90% of virion production. Addition of the inhibitors at the later time points (2-16 hpi) had reduced inhibitory effects on virion production. The later the inhibitors were added, the less inhibitory effects were observed. At 2 hpi, their inhibitory effects were reduced to 83%, 81% and 61% for the MEK, JNK and p38 inhibitors, respectively. At 16 hpi, the inhibitory effects were further reduced to 55%, 47% and 34%, respectively (Fig. 3C). Together, these results indicated that the MAPK pathways modulated the early stage(s) of KSHV reactivation.
Inhibitors of MAPK pathways reduced the expression of viral lytic genes during TPA-induced KSHV reactivation from latency
Efficient KSHV lytic replication depends on the expression of viral lytic genes. During TPA-induced KSHV reactivation, viral lytic genes are expressed in a cascade fashion with immediate-early (IE) genes such as RTA being expressed at as early as 6 hpi, followed by the early genes and late genes (Sun et al., 1999). Based on the above results, we hypothesized that inhibitors of MAPK pathways reduced the expression of viral lytic genes to affect KSHV reactivation. Thus, we examined the effects of the inhibitors on the expression of KSHV genes by reverse transcription quantitative real-time PCR (RT-qPCR). BCBL-1 cells induced with TPA with or without the inhibitors were collected at 48 hpi and analyzed for a set of KSHV genes representing different viral gene classes. These included a latent gene vFLIP (ORF-K13), an IE gene RTA, two early genes ORF59 and vIL-6 (ORF-K2), and a late gene ORF-K8.1. TPA treatment increased the expression of vFLIP transcripts by 3-fold and lytic transcripts by 7- to 13-fold (Fig. 4A). Inhibitors of MAPK pathways had marginal effects on the expression of vFLIP transcripts; however, they inhibited TPA-induction of all viral lytic genes analyzed. The expression of RTA was reduced 80%, 77% and 57% by the MEK, JNK and p38 inhibitors, respectively (Fig. 4A). The inhibitory effects of the inhibitors on the expression of KSHV lytic genes are in agreement with their inhibitory effects on the production of infectious virions.
Fig. 4.

Inhibitors of MAPK pathways suppressed the expression of viral lytic genes during TPA-induced KSHV reactivation from latency in BCBL-1 cells. (A) Inhibitors of MAPK pathways suppressed the expression of KSHV lytic transcripts but not latent transcripts during KSHV reactivation. Cells were induced with TPA in the presence of inhibitors of MAPK pathways, and analyzed for the expression of vFLIP, RTA, ORF59, vIL-6 and K8.1 transcripts at 48 hpi by RT-qPCR. (B) Inhibitors of MAPK pathways suppressed the protein expression of a KSHV lytic gene ORF59 during KSHV reactivation. Cells were treated as described in (A) and analyzed for the expression of ORF59 protein at 48 hpi by immunofluorescence antibody staining using a specific monoclonal antibody. (C) Quantification of the effects of inhibitors of MAPK pathways on the expression of ORF59 protein during KSHV reactivation as described in (B). (D) Inhibitors of MAPK pathways had no effect on the expression of KSHV latent protein LANA during KSHV reactivation. Cells were treated as described in (A) and analyzed for the expression of LANA protein at 48 hpi by immunofluorescence antibody staining using a specific monoclonal antibody. All the experiments were independently carried out three times, each with three repeats. Results presented in (A, C and D) were the averages with standard deviations from one representative experiment.
To confirm the effects of inhibiting MAPK pathways on the expression of KSHV lytic transcripts, we further analyzed the protein expression of ORF59. BCBL-1 cells induced with TPA with or without the inhibitors of MAPK pathways were collected at 48 hpi and analyzed for ORF59 protein expression by immunofluorescence antibody assay (IFA) using a specific monoclonal antibody (Fig. 4B). TPA treatment increased ORF59-positive cells from 2.5% to 16.5% (Fig. 4C); however, MEK, JNK and p38 inhibitors reduced ORF59-positive cells to 6.5%, 6.6% and 2.4%, respectively. In contrast, the percentage of cells expressing KSHV major latent nuclear antigen LANA (LNA) encoded by ORF73 was neither induced by TPA nor reduced by the inhibitors, and remained at close to 100% (Fig. 4D). Similar results were also observed when the cells were examined at 5 dpi (data not shown). Since LANA is the only KSHV protein that is necessary and sufficient for mediating the replication and persistence of KSHV episome in cells (Ballestas et al., 1999; Ye et al., 2004), these results indicated that the reduced levels of KSHV reactivation was not due to the loss of KSHV genomes.
Inhibitors of MAPK pathways reduced TPA activation of the RTA promoter
RTA is a central protein that controls KSHV reactivation. RTA directly or indirectly activates the expression of ORF59, vIL-6, and K8.1 (Deng et al., 2007). Since the expression of RTA transcripts was induced by TPA but reduced by the inhibitors of MAPK pathways, we further determined whether the RTA promoter was modulated by MAPK pathways. First, we examined the effect of TPA induction on the activity of the RTA promoter. Bjab cells were transfected with a full-length RTA promoter luciferase reporter construct, R-914. At 48 h post-transfection, the transfected cells were assayed for luciferase activities. Before collection, the cells were treated with TPA for different lengths of time. As shown in Fig. 5A, TPA treatment increased the activity of the RTA promoter reporter, which peaked at around 6 hpi. At the peak of induction, TPA treatment increased the RTA promoter reporter activity by as much as 4.8-fold. Based on these results, we induced the cells with TPA for 6 h in the subsequent experiments.
Fig. 5.

TPA-induced activation of the RTA promoter was mediated by multiple MAPK pathways. (A) Kinetics of TPA activation of the RTA promoter reporter. Bjab cells were transfected with a full-length RTA promoter reporter construct, and assayed for luciferase activity at 48 h post-transfection. Prior to harvest, cells were treated with 20 ng/ml of TPA for 0, 1, 2, 4, 6, 8, 12, and 15 h. (B) Inhibitors of MAPK pathways suppressed TPA activation of the RTA promoter reporter in a dose-dependent fashion. Bjab cells transfected with a RTA promoter reporter construct for 42 h were treated with 20 ng/ml of TPA for 6 h with or without inhibitors of JNK, MEK and p38 pathways, lysed, and assayed for luciferase activity. The inhibitors were used at the following concentrations: JNK inhibitor II (JNK inhibitor) at 12.5, 25, 50 and 100 μM; U0126 (MEK inhibitor) at 2.5, 5, 10, and 20 μM; and SB203580 (p38 inhibitor) at 12.5, 25, 50 and 100 μM. (C) Dominant negative constructs (DNs) of MAPK pathways inhibited TPA activation of the RTA promoter reporter in a dose-dependent fashion. Bjab cells transfected with a RTA promoter reporter construct together with a vector control (V) or different doses of DNs of JNK, MEK and p38 MAPK pathways for 42 h were treated with TPA for 6 h, lysed, and assayed for luciferase activity. (D) Overexpression of active forms (CAs) of ERK, JNK or p38 was sufficient to activate the RTA promoter, which was abolished by their respective DNs. Bjab cells transfected with a RTA promoter reporter construct together with a vector control or a CA of ERK, JNK or p38 with or without the presence of their respective DNs for 48 h were lysed and assayed for luciferase activity. The experiments were carried out three times, each with three repeats. Results presented were the averages with standard deviations from one experiment.
Next, we examined the effects of modulating MAPK pathways on the RTA promoter reporter activity. Again, Bjab cells were transfected with the RTA promoter reporter, R-914, and treated with TPA for 6 h with or without the inhibitors of MAPK pathways at different concentrations. As shown in Fig. 5B, all three inhibitors reduced TPA induction of RTA promoter reporter activities in a dose-dependent fashion. At the recommended working concentrations, the inhibitors of MEK, JNK and p38 pathways (10 μM for the MEK inhibitor, and 50 μM for the JNK and p38 inhibitors) reduced the reporter activities by 76%, 62% and 58%, respectively (Fig. 5B).
We then used dominant negative constructs (DNs) to inhibit the MAPK pathways and assay their roles in modulating the RTA promoter. Bjab cells were co-transfected with the RTA promoter reporter with different concentrations of the DNs, and assayed for luciferase activities at 48 h post-transfection. Before cell collection, the cells were treated with TPA for 6 h. As shown in Fig. 5C, DNs of all three MAPK pathways inhibited TPA induction of RTA promoter reporter activities in a dose-dependent fashion. At the highest doses, the ERK, JNK and p38 DNs inhibited the reporter activities by 82%, 84% and 82%, respectively (Fig. 5C). Together, these results indicated that the MAPK pathways were essential for TPA-induced activation of the RTA promoter.
We further determined whether activation of each of the MAPK pathways was sufficient to activate the RTA promoter. Bjab cells were co-transfected with the RTA promoter reporter with constitutively active constructs (CAs) of MAPK pathways, and assayed for luciferase activities at 48 h post-transfection. As shown in Fig. 5D, CAs of the ERK, JNK and p38 pathways activated the RTA promoter reporter by 15.2-, 11.0-and 9.8-fold, respectively. Co-transfection with their respective DNs abolished the activation of the RTA promote reporter by the CAs (Fig. 5D). These results indicated that the MAPK pathways were not only necessary but also sufficient for activating the RTA promoter.
MAPK pathways modulated TPA activation of the RTA promoter through AP-1
To map the region in the RTA promoter that mediated TPA induction, we carried out reporter assays with a set of RTA promoter serial deletion constructs. These deletion constructs were previously used to map the responsive region during KSHV productive primary infection in HUVEC (Pan et al., 2006). As shown in Fig. 6A, the region from -15 to -259 contributed the most to TPA activation of the RTA promoter. This region was also previously mapped to be responsive to the modulation of MAPK pathways during KSHV primary infection (Pan et al., 2006). Furthermore, an AP-1 site was identified in this region to be targeted by the MAPK pathways during KSHV primary infection (Pan et al., 2006). Based on our current and previous findings, it appeared that the AP-1 site also mediated the activation of the RTA promoter during TPA-induced KSHV reactivation. Thus, we used the reporter R-259 and its mutant construct R-259mut with the AP-1 site mutated to define the role of AP-1 in TPA-induced activation of the RTA promoter (Fig. 6B). While TPA induced a 4.3-fold increase of the wild-type R-259 reporter activity, mutation of the AP-1 site totally abolished TPA induction of the mutant R-259mut reporter activity (Fig. 6C). As expected, DNs of the MAPK pathways reduced TPA activation of the R-259 reporter activities (Fig. 6C). Specifically, DNs of the ERK, JNK and p38 pathways reduced 100%, 70% and 59% of the reporter activities, respectively. Similar results were obtained when inhibitors of the MAPK pathways were used though the inhibitory effects were slightly lower (Fig. 6D). The MEK, JNK and p38 inhibitors reduced 61%, 59% and 49% of the R-259 reporter activities, respectively. Similar to the full-length RTA reporter construct, activation of the MAPK pathways with CAs also increased the activities of the R-259 reporter but not the mutant R-259mut reporter (Fig. 6E). CAs of the ERK, JNK and p38 pathways activated the R-259 reporter by 4.6-, 3-, and 2-fold, respectively, which was inhibited by co-transfection with their respective DNs (Fig. 6E).
Fig. 6.

Identification of the dominant cis-element in RTA promoter activated by TPA. (A) Deletion analysis of the RTA promoter to identify the region responsive to TPA induction. Bjab cells transfected with different RTA promoter deletion reporter constructs for 42 h were treated with 20 ng/ml of TPA for 6 h, lysed, and assayed for luciferase activity. (B) Illustration of a wild type reporter (R-259) in the RTA promoter region (-15 to -259) responsive to TPA induction. R-259mut was a corresponding mutant reporter with the AP-1 site ablated. (C) Dominant negative constructs (DNs) of MAPK pathways inhibited TPA activation of the R-259 reporter while the mutant R-259mut reporter was not responsive to TPA treatment. Bjab cells transfected with R-259 or R-259mut reporter constructs together with a vector control (V) or DNs of the JNK, MEK and p38 MAPK pathways for 42 h were treated with TPA for 6 h, lysed and assayed for luciferase activity. (D) Inhibitors of MAPK pathways suppressed TPA activation of the R-259 reporter. Bjab cells transfected with R-259 or R-259mut reporter constructs for 42 h were treated with 20 ng/ml of TPA for 6 h with or without inhibitors of JNK, MEK and p38 pathways, lysed and assayed for luciferase activity. Both JNK inhibitor II (JNK inhibitor) and SB203580 (p38 inhibitor) were used at 50 μM while U0126 (MEK inhibitor) was used at 10 μM. (E) Overexpression of an active form (CA) of ERK, JNK or p38 was sufficient to activate the R-259 reporter but not the mutant R-259mut reporter. Bjab cells transfected with R-259 or R-259mut reporter constructs together with a vector control or a CA of ERK, JNK or p38 with or without their respective DNs for 48 h were lysed and assayed for luciferase activity. (F) DN of c-Fos or c-Jun inhibited TPA activation of the R-259 reporter. Bjab cells transfected with R-259 or R-259mut reporter constructs together with a vector control (V) or DN of c-Fos, c-Jun, or both for 42 h were treated with TPA for 6 h, lysed and assayed for luciferase activity.
Since mutation of the AP-1 site affected modulation of the RTA promoter by the MAPK pathways, of which AP-1 is a downstream target, we directly examined the role of AP-1 in the regulation of TPA-induced activation of the RTA promoter. DNs of either c-Fos or c-Jun reduced TPA activation of the R-259 reporter by 100% and 65%, respectively (Fig. 6F). In consistency with these results, we have previously shown that overexpression of CA of either c-Fos or c-Jun alone is sufficient to activate the RTA promoter, which is inhibited by co-transfection with their respective DNs (Pan et al., 2006). We further conducted electrophoretic mobility shift assay (EMSA) to examine the direct binding of the AP-1 protein complex to the RTA promoter. As shown in Fig. 7, wild-type probe from the putative AP-1 site in the RTA promoter formed a DNA-protein complex when nuclear extract of uninduced BCBL-1 cells was used. The intensity of this complex was significantly increased upon TPA induction. Competition with the unlabeled wild-type cold probe but not the mutant probe abolished the DNA-protein complex. IgG of an antibody to c-Fos or c-Jun but not that a control antibody supershifted the DNA-protein complex, indicating that it was indeed an AP-1 complex. In contrast, the mutant probe did not form any AP-1 DNA-protein complex with nuclear extracts from either uninduced or TPA-induced BCBL-1 cells (Fig. 7). Together, these results clearly demonstrated that MAPK pathways mediated the activation of the RTA promoter through AP-1.
Fig. 7.

Increased binding of AP-1 complexes to a putative AP-1 cis-element in the RTA promoter during TPA-induced KSHV reactivation from latency in BCBL-1 cells. Nuclear extracts from uninduced cells (lanes 3-4) or cells induced with 20 ng/ml of TPA for 6 h (lanes 5-16) were subjected to electrophoretic mobility shift assay using a 32P-labeled probe of the putative AP-1 cis-element from the RTA promoter (W) or its mutant with the putative AP-1 site mutated (M). The labeled probes alone were shown in lanes 1 and 2, respectively. In the competition assay, 100X excess amounts of unlabeled probes were added to the reactions (lanes 7-10). Addition of 1 μg of an antibody to c-Fos (lanes 11-12), or c-Jun (lanes 13-14), respectively, supershifted the AP-1-probe complex, while addition of 1 μg of IgG of a control antibody did not (lanes 15-16).
Discussion
The mechanisms controlling KSHV lytic replication are complex. Whether KSHV undergoes latent or lytic replication might depend on multiple factors including: the status of cellular signaling pathways, cell cycle, extracellular factors, cell types, stages of viral infection, and the expression of viral proteins. In this report, we have shown that the MEK/ERK, JNK and p38 MAPK pathways regulate TPA-induced KSHV reactivation from latency. These results are consistent with those of KSHV productive primary infection (Pan et al., 2006). Although KSHV lytic replication during productive primary infection and reactivation from latency have similar as well as distinct features, our previous and current studies have shown that both processes are regulated by all three MAPK pathways. These two studies have identified AP-1 as the downstream target of the three MAPK pathways that mediates the expression of RTA, the key transactivator of KSHV lytic replication, during viral primary infection and reactivation from latency (Pan et al., 2006). AP-1 also mediates TPA-induced expression of several other KSHV lytic genes such as MTA (ORF57) and RAP (ORF-K8) as well as the origins of lytic replication (oriLyts) (Byun et al., 2002; AuCoin et al., 2004; Wang et al., 2004). Together, these findings suggest that the MAPK pathways might have general roles in regulating different stages/phases of KSHV life cycle.
Several KSHV proteins and the binding of KSHV glycoproteins to cellular receptors mediate the activation of AP-1 and MAPK pathways. Two KSHV gene products, LANA and vFLIP, directly activate AP-1 (An et al., 2002; An et al., 2003; An et al., 2004). vFLIP and five other KSHV gene products: vGPCR (ORF74), vPK (ORF36), LAMP (ORF-K15), kaposin B (ORF-K12) and ORF49, activate MAPK pathways (Akula et al., 2002; An et al., 2003; Bais et al., 1998; Brinkmann et al., 2003; Hamza et al., 2004; McCormick and Ganem, 2005; Xie et al., 2005; Gonzalez et al., 2006). Some of these viral genes such as vGPCR, vPK and ORF49 are viral lytic genes, and thus their expression could lead to a positive feedback in viral lytic replication through the activation of MAPK pathways; however, other viral genes such as LANA and vFLIP are latent genes, and thus could activate the AP-1 and MAPK pathways during KSHV latency. In fact, we detected constitutive activation of all three MAPK pathways in latent KSHV-infected BCBL-1 cells (Fig. 1). Constitutive activation of both the JNK and p38 pathways in KSHV-infected latent cells has not been reported before but has been shown for the MEK/ERK pathway (Cohen et al., 2006; Ford et al., 2006). It is unclear why the majority of the cells remain latent since forcing activation of the MAPK pathways or AP-1 is sufficient to activate the expression of RTA (Fig. 5D and 6E) (Pan et al., 2006). There are several possible explanations. 1) There could be thresholds of activated levels of these pathways that are required for the activation of RTA and viral lytic replication. 2) The activation of the downstream targets by MAPK pathways could vary during viral latency and reactivation, some of which could have favorable while others could have repressive effects on viral lytic replication. For example, the AP-1 complexes could be formed by different components including c-Fos, FosB, c-Jun, JunB, JunD, Fra-1 and Fra-2, some of which have positive while others have repressive effects on gene expression (Shaulian and Karin, 2002). While KSHV activates the positive regulators of MAPK pathways, including c-Jun, c-Fos and FosB during primary infection (Xie et al., 2005), it remains to be determined what components of AP-1 are activated during KSHV latency and reactivation. 3) LANA also has repressive functions for KSHV lytic replication. Binding of LANA to the episome results in epigenetic modifications of the viral genomes and repression of gene expression (Sakakibara et al., 2004; Lan et al., 2005; Lu et al., 2006). In addition, LANA binds to RTA and its promoter to repress its expression and transactivation function (Lan et al., 2004; Lan et al., 2005). 4) Other viral, host and environmental factors could play synergetic or opposing effects on KSHV lytic replication. Therefore, further defining the interactions of MAPK pathways with other viral, host and environmental factors during KSHV lytic replication could shed light on the molecular basis of KSHV life cycle. In this case, the application of reverse genetics approach to define the viral cis-element(s) responsive to MAPK pathways or the AP-1, or viral factors that could mediate these pathways would be particularly insightful (Zhou et al., 2002).
The identification of MAPK pathways and the downstream target AP-1 as important regulators of KSHV lytic replication is of particular interest. A number of inflammatory and angiogenic cytokines including: IL-6, bFGF, TNF-α, interferon-γ, VEGF, oncostatin M, and IL-1β are expressed in KS tumors (Ensoli et al., 2001a; Ensoli et al., 2001b), and regulate MAPK pathways (Chaturvedi and Sarkar, 2005; Kang et al., 2005; Kaplin et al., 2005; Weiss et al., 2005; Aoki et al., 2006; Boing et al., 2006; Lee et al., 2006). Thus, some of these cytokines could modulate KSHV reactivation. In fact, several inflammatory cytokines are capable of reactivating KSHV. Interferon-γ reactivates KSHV lytic replication and the expression of lytic genes in PEL cells and peripheral blood mononuclear cells isolated from KS patients (Monini et al., 1999; Blackbourn et al., 2000; Chang et al., 2000; Mercader et al., 2000). Oncostatin M also induces KSHV lytic replication in BCBL-1 cells (Mercader et al., 2000). We have previously shown that MAPK pathways modulate KSHV infectivity and subsequent productive lytic replication during KSHV primary infection (Pan et al., 2006). Thus, besides viral lytic replication, inflammatory and angiogenic cytokines can also modulate KSHV infectivity. Indeed, it was recently demonstrated that VEGF induced by Raf enhances KSHV infectivity (Hamden et al., 2004). It could be postulated that such VEGF-mediated enhancement of KSHV infectivity could be via the activation of MAPK pathways. Importantly, the expression of many inflammatory and angiogenic cytokines is also modulated by MAPK pathways and AP-1, which could have a positive feedback on KSHV lytic replication and infection (Xie et al., 2005; Ye et al., 2007; Qian el al., 2007). Therefore, as we have postulated (Xie et al., 2005), a sustaining reactivation and productive primary infection cycle in the host could contribute to the rapid progression of KS by not only producing large quantities of virions for infecting new cells, but also contributing to other aspects of KS pathogenesis, including the production of inflammatory and angiogenic cytokines, and promotion of cellular transformation, cell invasion and angiogenesis.
Materials and Methods
Plasmids
The full-length RTA promoter reporter construct, R-914 and its consecutive deletion constructs R-836, R-587, R-348, R-259 and R-15 were previously described (Pan et al., 2006). A mutant construct of the R-259 reporter, R-259mut, with the putative AP-1 site mutated from (-87)-CGACTCA-(-81) to (-87)-CGGATCC-(-81) was also used in the study. The following plasmids were previously described: pCEP4L-ERK1 and pCEP4L-HA-ERK1K71R are expression vectors for ERK1 and its DN, respectively (Frost et al., 1994); pcDNA3-wt-p38 is an expression vector for wild-type p38 (Thomas et al., 2002); pcDNA3-p38/AF is a DN of p38 (Han et al., 1995); HA-JNK/pCAGGGS is a JNK1 expression construct with JNK1 cloned into the pCAGGS vector (Fujii et al., 2004). HA-JNK[APF] is a DN of JNK (Derijard et al., 1994); A-Fos is a DN of c-Fos (Olive et al., 1997); pcDNA3Flag-c-Fos expresses an active form of c-Fos (Murphy et al., 2002); and RSV-c-Jun expresses an active form of c-Jun (Kim et al., 2003). DN-c-Jun is a DN construct of c-Jun construct (Pan et al., 2006).
Cell culture
BCBL-1 cells harboring a recombinant KSHV, BAC36, have been previously described (Zhou et al., 2002). BCBL-1 cells were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS, Sigma, St. Louis, MO), 50 μg/ml of gentamicin, and 2 mM L-glutamine. For virus titration, 293 cells were used as target cells, and were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, 50 μg/ml of gentamicin, and 2 mM L-glutamine.
Virus induction and titration
Virus induction and titration were carried out as described previously (Zhou et al., 2002; Gao et al., 2003). To induce KSHV lytic replication, BCBL-1 cells were treated with 20 ng/ml of TPA (Sigma) for different lengths of time depending on the experiments. In some experiments, specific inhibitors were used to inhibit the MAPK pathways. The inhibitors were U0126 for MEK, JNK Inhibitor II for JNK, and SB 203580 for p38, all purchased from Calbiochem (EMD Biosciences, Inc., San Diego, CA). Inhibitors were freshly reconstituted at 1000X of their recommended concentrations (10 μM for U0126, and 50 μM for both JNK Inhibitor II and SB203580) with dimethyl sulfoxide before use. The inhibitors were used at 1X of their recommended concentrations unless specified.
For virus titration, 293 cells at 60-70% confluency seeded in a 96-well plate one day before were infected with culture supernatants or virus inocula at 50 μl/well in the presence of 4 μg/ml of polybrene, and incubated at 37°C for 4 h. The viral inoculum was then removed and replaced with normal culture medium. The plates were examined with an inverted fluorescence microscope at 48 h post-infection for the percentage of cells expressing GFP, which was then converted to GFU to reflect the virus titer in the inoculum. For each treatment, virus titration was carried out in 4 wells. The number of GFP-positive cells for each well was obtained by averaging the results of 9 microscopic fields randomly selected with a 20X objective. In all the experiments involving the use of inhibitors to determine the relevance of the MAPK pathways in KSHV reactivation, the inocula were titrated at day 5 after the addition of the inhibitors to ensure that they no longer interfered with virus infectivity (see Fig. 2C).
Western-blot analysis
Protein preparations from BCBL-1 cells were separated by SDS-polyacrylamide gel electrophoresis (PAGE), and transferred to nitrocellulose membranes as previously described (Gao et al., 1996a). The membranes were incubated first with antibodies specific for phosphorylated forms of ERK, JNK, and p38, respectively (Santa Cruz Biotechnology, Santa Cruz, CA), and then with a goat anti-rabbit horseradish peroxidase (HRP) conjugate (Sigma). Antibodies specific to total ERK, JNK and p38 were used to detect the expression of the respective proteins (Santa Cruz Biotechnology). A mouse antibody to β-tubulin (Sigma) was used to monitor sample loading. Specific signals were revealed with chemiluminescence substrates, and recorded on films or with a Kodak Image Station 2000 MM Multi-Modal Imager (Eastman Kodak Company, Rochester, NY).
RNA extraction and reverse transcription quantitative real-time PCR (RT-qPCR)
RT-qPCR was performed as previously described (Yoo et al., 2005). Briefly, total RNA from BCBL-1 was prepared with TRI reagent as recommended by the manufacturer (Sigma). The RNA was treated with RQ1 RNase-free DNase (Promega, Madison, WI) and reverse transcribed to obtain the first-strand cDNA using a Superscript III first-stand synthesis system (Invitrogen, Carlsbad, CA). qPCR was carried out in a DNA engine Opiticon 2 continuous fluorescence detector (Bio-Rad, Hercules, CA). PCR was also carried out on the RNA samples without reverse transcription to exclude any possible genomic DNA contamination. The vFLIP primers were 5′GGATGCCCTAATGTCAATGC3′ (forward) and 5′GGCGATAGTGTTGGGAGTGT3′ (reverse), which amplified a product of 113 bp. The RTA primers were 5′CACAAAAATGGCGCAAGATGA3′ (forward) and 5′TGGTAGAGTTGGGCCTTCAGTT3′ (reverse), which amplified a product of 98 bp. The ORF59 primers were 5′CGAGTCTTCGCAAAAGGTTC3′ (forward) and 5′AAGGGACCAACTGGTGTGAG3′ (reverse), which amplified a product of 99 bp. The vIL-6 primers were 5′ACCCTTGCAGATGCCGG3′ (forward) and 5′GGATGCTATGGGTGATCGATG3′ (reverse), which amplified a product of 65 bp. The ORF-K8.1 primers were 5′AAAGCGTCCAGGCCACCACAGA3′ (forward) and 5′GGCAGAAAATGGCACACGGTTAC3′ (reverse), which amplified a product of 160 bp. Primers for glyceraldehydes-3-phosphate dehydrogenase (GAPDH) GAPDH-F (5′ACAGTCAGCCGCATCTTCTT3′) and GAPDH-R (5′ACGACCAAATCCGTTGACTC3′) that amplify a product of 94 bp were used to normalize the samples. Each sample was assayed with three repeats.
Immunofluorescence antibody assay (IFA)
IFA was carried out as previously described to determine the protein expression of ORF59 (Gao et al., 1996b). Briefly, BCBL-1 cells were washed 3 times with PBS by centrifugation and evenly spread onto 10-well slides. After fixation at 56 °C for 1 h, the slides were blocked with 3% bovine serum albumin (BSA) in PBS. The slides were incubated with a mouse anti-ORF59 monoclonal antibody kindly provided by Dr. Bala Chandran (Chan et al., 1998). Specific signals were revealed with a goat anti-mouse IgG-ALEXA568 conjugate (Molecular Probe, Eugene, OR). To detect LANA protein, a rat-anti-LANA monoclonal antibody (ABI, New York, NY) was used. A goat anti-rat IgG-ALEXA568 conjugate was used to reveal the specific signal. The cells were stained with DAPI at 1 μg/ml for 1 min at room temperature, and analyzed with a digitalized Zeiss fluorescent microscope.
Transient transfection and reporter assay
Transient transfection and reporter assays were carried out in Bjab cells. Cells (1 × 107) were harvested and used for Nucleofection electroporation using Nucleofator® Solution V according to the instructions of the manufacturer (Amaxa Inc., Gaithersburg, MD). To control the transfection efficiency, samples were normalized by cotransfection with a reporter plasmid, pSV-β-galactosidase (Promega). The cells were collected in 300 μl of 1X lysis buffer (Promega) at 48 h after transfection. An aliquot of the supernatant (60 μl) and a β-galactosidase kit (Promega) were used to measure the β-galactosidase activity. All the reporter assays were independently carried out three times with three repeats each time.
Electrophoretic mobility shift assay (EMSA)
EMSA was carried out with nuclear extracts from uninduced or TPA-induced BCBL-1 cells as previously described (Wang et al., 2002). Annealed double-stranded oligonucleotides (5′GGGAGCTACCGGCGACTCATTAAGC3′) from the putative AP-1 site in the RTA promoter and its mutant (5′GGGAGCTACCGGCGGATCCTTAAGC3′) with the mutated positions underlined were labeled with [γ-32P]ATP. For gel shift assays, 4 μg of nuclear extract was incubated for 20 min at room temperature with 5 × 105 cpm of the labeled probe in 20 μl of binding buffer, containing 10 mM Tris-HCl at pH 7.6, 50 mM NaCl, 1 mM EDTA, 1 mM DTT, 5% glycerol, 1 μg/μl of BSA, and 2 μg of poly-d(I-C). Competition assays were carried out in the same manner except that the above reaction mixture was pre-incubated with excess cold wild-type or mutant probe for 10 min at 4 °C before the addition of the labeled probe. Samples were separated on 6% non-denaturing PAGE.
Acknowledgments
This work was supported in part by grants from the National Institute of Health (CA096512, DE017333 and CA124332), an American Cancer Society Research Scholar Grant (#RSG-04-195), and a Type B Outstanding Abroad Young Scientist Award from the National Science Foundation of China (30328001) to S-J Gao. We thank Drs. Bala Chandran (Rosalind Franklin University of Medicine), Jiahua Han (Scripps Institute), Lin Mantell (New York University School of Medicine), Roger Davis (University of Massachusetts Medical School), Melanie Cobb (University of Texas Southwest Medical Center), Charles Vinson (National Institute of Health), John Blenis (Harvard Medical School), Laurice J. Goodyear (Harvard Medical School), and Francis Bernenbaum (Universite Pierre et Marie Curie) for kindly providing us reagents. We thank members of Dr. Gao's laboratory for technical assistance and helpful comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akula SM, Pramod NP, Wang FZ, Chandran B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell. 2002;108(3):407–419. doi: 10.1016/s0092-8674(02)00628-1. [DOI] [PubMed] [Google Scholar]
- An J, Lichtenstein A, G B, Rettig M. The Kaposi's sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element. Blood. 2002;99(2):649–654. doi: 10.1182/blood.v99.2.649. [DOI] [PubMed] [Google Scholar]
- An J, Sun Y, Rettig MB. Transcriptional coactivation of c-Jun by the KSHV-encoded LANA. Blood. 2004;103(1):222–228. doi: 10.1182/blood-2003-05-1538. [DOI] [PubMed] [Google Scholar]
- An J, Sun Y, Sun R, Rettig MB. Kaposi's sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-kappaB and JNK/AP1 pathways. Oncogene. 2003;22(22):3371–3385. doi: 10.1038/sj.onc.1206407. [DOI] [PubMed] [Google Scholar]
- Aoki H, Ohnishi H, Hama K, Ishijima T, Satoh Y, Hanatsuka K, Ohashi A, Wada S, Miyata T, Kita H, Yamamoto H, Osawa H, Sato K, Tamada K, Yasuda H, Mashima H, Sugano K. Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2006;290(4):C1100–1108. doi: 10.1152/ajpcell.00465.2005. [DOI] [PubMed] [Google Scholar]
- AuCoin DP, Volletti KS, Cei SA, M P, Tarrant M, Pari GS. Amplification of the Kaposi's sarcoma-associated herpsvirus/human herpesvirus 8 lytic origin of DNA replication in dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors (K-Rta) and K8 (b-zip) Virology. 2004;318(2):542–555. doi: 10.1016/j.virol.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Mesri EA. G-protein-coupled receptor of Kaposi's sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature. 1998;391(6662):86–89. doi: 10.1038/34193. [DOI] [PubMed] [Google Scholar]
- Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science. 1999;284(5414):641–644. doi: 10.1126/science.284.5414.641. [DOI] [PubMed] [Google Scholar]
- Blackbourn DJ, Fujimura S, Kutzkey T, Levy JA. Induction of human herpesvirus-8 gene expression by recombinant interferon gamma. Aids. 2000;14(1):98–99. doi: 10.1097/00002030-200001070-00017. [DOI] [PubMed] [Google Scholar]
- Boing I, Stross C, Radtke S, Lippok BE, Heinrich PC, Hermanns HM. Oncostatin M-induced activation of stress-activated MAP kinases depends on tyrosine 861 in the OSM receptor and requires Jak1 but not Src kinases. Cell Signal. 2006;18(1):50–61. doi: 10.1016/j.cellsig.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Brinkmann M, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, Schulz T. Activation of Mitogen-Activated protein kinase and NF-κB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J Virol. 2003;77(17):9246–9385. doi: 10.1128/JVI.77.17.9346-9358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. NF-kappaB inhibits gammaherpesvirus lytic replication. J Virol. 2003;77(15):8532–8540. doi: 10.1128/JVI.77.15.8532-8540.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun H, Gwack Y, Hwang S, Choe J. Kaposi's sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF 57 promoters via heterogeneous response elements. Mol Cells. 2002;14(2):185–191. [PubMed] [Google Scholar]
- Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. In vitro establishment and characterization of two AIDS-related lymphoma cell lines containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood. 1995;86(7):2708–2714. [PubMed] [Google Scholar]
- Chan SR, Bloomer C, Chandran B. Identification and characterization of human herpesvirus-8 lytic cycle-associated ORF59 protein and the encoding cDNA by monoclonal antibody. Virology. 1998;240(1):118–126. doi: 10.1006/viro.1997.8911. [DOI] [PubMed] [Google Scholar]
- Chang J, Renne R, Dittmer D, Ganem D. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology. 2000;266(1):17–25. doi: 10.1006/viro.1999.0077. [DOI] [PubMed] [Google Scholar]
- Chang M, Brown HJ, Collado-Hidalgo A, Arevalo JM, Galic Z, Symensma TL, Tanaka L, Deng H, Zack JA, Sun R, Cole SW. beta-Adrenoreceptors reactivate Kaposi's sarcoma-associated herpesvirus lytic replication via PKA-dependent control of viral RTA. J Virol. 2005;79(21):13538–13547. doi: 10.1128/JVI.79.21.13538-13547.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi K, Sarkar DK. Mediation of basic fibroblast growth factor-induced lactotropic cell proliferation by Src-Ras-mitogen-activated protein kinase p44/42 signaling. Endocrinology. 2005;146(4):1948–1955. doi: 10.1210/en.2004-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc Natl Acad Sci U S A. 2001;98(7):4119–4124. doi: 10.1073/pnas.051004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A, Brodie C, Sarid R. An essential role of ERK signalling in TPA-induced reactivation of Kaposi's sarcoma-associated herpesvirus. J Gen Virol. 2006;87(Pt 4):795–802. doi: 10.1099/vir.0.81619-0. [DOI] [PubMed] [Google Scholar]
- Davis DA, Rinderknecht AS, Zoeteweij JP, Aoki Y, Read-Connole EL, Tosato G, Blauvelt A, Yarchoan R. Hypoxia induces lytic replication of Kaposi's sarcoma-associated herpesvirus. Blood. 2001;97(10):3244–3250. doi: 10.1182/blood.v97.10.3244. [DOI] [PubMed] [Google Scholar]
- Deng H, Liang Y, Sun R. Regulation of KSHV lytic gene expression. Curr Top Microbiol Immunol. 2007;312:157–183. doi: 10.1007/978-3-540-34344-8_6. [DOI] [PubMed] [Google Scholar]
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76(6):1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Deutsch E, Cohen A, Kazimirsky G, Dovrat S, Rubinfeld H, Brodie C, Sarid R. Role of protein kinase C delta in reactivation of Kaposi's sarcoma-associated herpesvirus. J Virol. 2004;78(18):10187–10192. doi: 10.1128/JVI.78.18.10187-10192.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol Biol Rev. 2003;67(2):175–212. doi: 10.1128/MMBR.67.2.175-212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc Natl Acad Sci U S A. 1999;96(8):4546–4551. doi: 10.1073/pnas.96.8.4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensoli B, Sgadari C, Barillari G, Sirianni MC, Sturzl M, Monini P. Biology of Kaposi's sarcoma. Eur J Cancer. 2001a;37(10):1251–1269. doi: 10.1016/s0959-8049(01)00121-6. [DOI] [PubMed] [Google Scholar]
- Ensoli B, Sturzl M, Monini P. Reactivation and role of HHV-8 in Kaposi's sarcoma initiation. Adv Cancer Res. 2001b;81:161–200. doi: 10.1016/s0065-230x(01)81005-8. [DOI] [PubMed] [Google Scholar]
- Ford PW, Bryan BA, Dyson OF, Weidner DA, Chintalgattu V, Akula SM. Raf/MEK/ERK signalling triggers reactivation of Kaposi's sarcoma-associated herpesvirus latency. J Gen Virol. 2006;87(Pt 5):1139–1144. doi: 10.1099/vir.0.81628-0. [DOI] [PubMed] [Google Scholar]
- Frost JA, Geppert TD, Cobb MH, Feramisco JR. A requirement for extracellular signal-regulated kinase (ERK) function in the activation of AP-1 by Ha-Ras, phorbol 12-myristate 13-acetate, and serum. Proc Natl Acad Sci U S A. 1994;91(9):3844–3848. doi: 10.1073/pnas.91.9.3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii N, Boppart MD, Dufresne SD, Crowley PF, Jozsi AC, Sakamoto K, Yu H, Aschenbach WG, Kim S, Miyazaki H, Rui L, White MF, Hirshman MF, Goodyear LJ. Overexpression or ablation of JNK in skeletal muscle has no effect on glycogen synthase activity. Am J Physiol Cell Physiol. 2004;287(1):C200–208. doi: 10.1152/ajpcell.00415.2003. [DOI] [PubMed] [Google Scholar]
- Gao SJ, Deng JH, Zhou FC. Productive lytic replication of a recombinant Kaposi's sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J Virol. 2003;77(18):9738–9749. doi: 10.1128/JVI.77.18.9738-9749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao SJ, Kingsley L, Hoover DR, Spira TJ, Rinaldo CR, Saah A, Phair J, Detels R, Parry P, Chang Y, Moore PS. Seroconversion of antibodies against Kaposi's sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi's sarcoma. N Engl J Med. 1996a;335(4):233–241. doi: 10.1056/NEJM199607253350403. [DOI] [PubMed] [Google Scholar]
- Gao SJ, Kingsley L, Li M, Zheng W, Parravicini C, Ziegler J, Newton R, Rinaldo CR, Saah A, Phair J, Detels R, Chang Y, Moore PS. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi's sarcoma. Nat Med. 1996b;2(8):925–928. doi: 10.1038/nm0896-925. [DOI] [PubMed] [Google Scholar]
- Gonzalez CM, Wong EL, Bowser BS, Hong GK, Kenney S, Damania B. Identification and characterization of the Orf49 protein of Kaposi's sarcoma-associated herpesvirus. J Virol. 2006;80(6):3062–3070. doi: 10.1128/JVI.80.6.3062-3070.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene W, Kuhne K, Ye FC, Chen JG, Zhou FC, Lei XF, Gao SJ. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. In: Meyers C, editor. AIDS-Associated Viral Oncogenesis. Springer Science+Business Media; 2007. pp. 69–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamden KE, Ford PW, Whitman AG, Dyson OF, Cheng SY, McCubrey JA, Akula SM. Raf-induced vascular endothelial growth factor augments Kaposi's sarcoma-associated herpesvirus infection. J Virol. 2004;78(23):13381–13390. doi: 10.1128/JVI.78.23.13381-13390.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza M, Reyes R, Izumiya Y, Wisdom R, Kung HJ, Luciw P. ORF36 protein kinase of Kaposi's sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem. 2004;279(37):38325–38330. doi: 10.1074/jbc.M400964200. [DOI] [PubMed] [Google Scholar]
- Han J, Richter B, Li Z, Kravchenko V, Ulevitch RJ. Molecular cloning of human p38 MAP kinase. Biochim Biophys Acta. 1995;1265(23):224–227. doi: 10.1016/0167-4889(95)00002-a. [DOI] [PubMed] [Google Scholar]
- Kang HB, Kim JS, Kwon HJ, Nam KH, Youn HS, Sok DE, Lee Y. Basic fibroblast growth factor activates ERK and induces c-fos in human embryonic stem cell line MizhES1. Stem Cells Dev. 2005;14(4):395–401. doi: 10.1089/scd.2005.14.395. [DOI] [PubMed] [Google Scholar]
- Kaplin AI, Deshpande DM, Scott E, Krishnan C, Carmen JS, Shats I, Martinez T, Drummond J, Dike S, Pletnikov M, Keswani SC, Moran TH, Pardo CA, Calabresi PA, Kerr DA. IL-6 induces regionally selective spinal cord injury in patients with the neuroinflammatory disorder transverse myelitis. J Clin Invest. 2005;115(10):2731–2741. doi: 10.1172/JCI25141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Woolridge S, Biffi R, Borghi E, Lassak A, Ferrante P, Amini S, Khalili K, Safak M. Members of the AP-1 family, c-Jun and c-Fos, functionally interact with JC virus early regulatory protein large T antigen. J Virol. 2003;77(9):5241–5252. doi: 10.1128/JVI.77.9.5241-5252.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Robertson ES. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jkappa, the major downstream effector of the Notch signaling pathway. J Virol. 2005;79(6):3468–3478. doi: 10.1128/JVI.79.6.3468-3478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Kuppers DA, Verma SC, Robertson ES. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J Virol. 2004;78(12):6585–6594. doi: 10.1128/JVI.78.12.6585-6594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan K, Murakami M, Choudhuri T, Kuppers DA, Robertson ES. Intracellular-activated Notch1 can reactivate Kaposi's sarcoma-associated herpesvirus from latency. Virology. 2006;351(2):393–403. doi: 10.1016/j.virol.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Lee KY, Ito K, Hayashi R, Jazrawi EP, Barnes PJ, Adcock IM. NF-{kappa}B and Activator Protein 1 Response Elements and the Role of Histone Modifications in IL-1{beta}-Induced TGF-{beta}1 Gene Transcription. J Immunol. 2006;176(1):603–615. doi: 10.4049/jimmunol.176.1.603. [DOI] [PubMed] [Google Scholar]
- Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 2002;16(15):1977–1989. doi: 10.1101/gad.996502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Day L, Gao SJ, Lieberman PM. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J Virol. 2006;80(11):5273–5282. doi: 10.1128/JVI.02541-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J Virol. 2003;77(21):11425–11435. doi: 10.1128/JVI.77.21.11425-11435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF50 transactivator, a homolog of the EBV R protein. Virology. 1998;252(2):304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307(5710):739–741. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- Mercader M, Taddeo B, Panella JR, Chandran B, Nickoloff BJ, Foreman KE. Induction of HHV-8 lytic cycle replication by inflammatory cytokines produced by HIV-1-infected T cells. Am J Pathol. 2000;156(6):1961–1971. doi: 10.1016/S0002-9440(10)65069-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monini P, Colombini S, Sturzl M, Goletti D, Cafaro A, Sgadari C, Butto S, Franco M, Leone P, Fais S, Melucci-Vigo G, Chiozzini C, Carlini F, Ascherl G, Cornali E, Zietz C, Ramazzotti E, Ensoli F, Andreoni M, Pezzotti P, Rezza G, Yarchoan R, Gallo RC, Ensoli B. Reactivation and persistence of human herpesvirus-8 infection in B cells and monocytes by Th-1 cytokines increased in Kaposi's sarcoma. Blood. 1999;93(12):4044–4058. [PubMed] [Google Scholar]
- Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4(8):556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, Vinson C. A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem. 1997;272(30):18586–18594. doi: 10.1074/jbc.272.30.18586. [DOI] [PubMed] [Google Scholar]
- Orem J, Otieno MW, Remick SC. AIDS-associated cancer in developing nations. Curr Opin Oncol. 2004;16(5):468–476. doi: 10.1097/00001622-200409000-00010. [DOI] [PubMed] [Google Scholar]
- Pan H, Xie J, Ye F, Gao SJ. Modulation of Kaposi's sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J Virol. 2006;80(11):5371–5382. doi: 10.1128/JVI.02299-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Xie J, Ye F, Gao SJ. Kaposi's sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J Virol. 2007;81(13):7001–7010. doi: 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat Med. 1996;2(3):342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- Sakakibara S, Ueda K, Nishimura K, Do E, Ohsaki E, Okuno T, Yamanishi K. Accumulation of heterochromatin components on the terminal repeat sequence of Kaposi's sarcoma-associated herpesvirus mediated by the latency-associated nuclear antigen. J Virol. 2004;78(14):7299–7310. doi: 10.1128/JVI.78.14.7299-7310.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgarbanti M, Arguello M, tenOever BR, Battistini A, Lin R, Hiscott J. A requirement for NF-kappaB induction in the production of replication-competent HHV-8 virions. Oncogene. 2004;23(34):5770–5780. doi: 10.1038/sj.onc.1207707. [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4(5):E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71(1):715–719. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95(18):10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. Kinetics of Kaposi's sarcoma-associated herpesvirus gene expression. J Virol. 1999;73(3):2232–2242. doi: 10.1128/jvi.73.3.2232-2242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas B, Thirion S, Humbert L, Tan L, Goldring MB, Bereziat G, Berenbaum F. Differentiation regulates interleukin-1beta-induced cyclo-oxygenase-2 in human articular chondrocytes: role of p38 mitogen-activated protein kinase. Biochem J. 2002;362(Pt 2):367–373. doi: 10.1042/0264-6021:3620367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wu F, Chen H, Shamay M, Zheng Q, Hayward G. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J Virol. 2004;78(8):4248–4267. doi: 10.1128/JVI.78.8.4248-4267.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SE, Wu FY, Yu Y, Hayward GS. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J Virol. 2003;77(17):9590–9612. doi: 10.1128/JVI.77.17.9590-9612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XP, Zhang YJ, Deng JH, Pan HY, Zhou FC, Gao SJ. Transcriptional regulation of Kaposi's sarcoma-associated herpesvirus-encoded oncogene viral interferon regulatory factor by a novel transcriptional silencer. Tis J Biol Chem. 2002;277(4):12023–12031. doi: 10.1074/jbc.M108026200. [DOI] [PubMed] [Google Scholar]
- Weiss TW, Kvakan H, Kaun C, Zorn G, Speidl WS, Pfaffenberger S, Maurer G, Huber K, Wojta J. The gp130 ligand oncostatin M regulates tissue inhibitor of metalloproteinases-1 through ERK1/2 and p38 in human adult cardiac myocytes and in human adult cardiac fibroblasts: a possible role for the gp130/gp130 ligand system in the modulation of extracellular matrix degradation in the human heart. J Mol Cell Cardiol. 2005;39(3):545–551. doi: 10.1016/j.yjmcc.2005.03.015. [DOI] [PubMed] [Google Scholar]
- Xie JP, Pan HY, Yoo SM, Gao SJ. Kaposi's sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J Virol. 2005;79(24):15027–15037. doi: 10.1128/JVI.79.24.15027-15037.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, AuCoin DP, Huete AR, Cei SA, Hanson LJ, Pari GS. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J Virol. 2005;79(6):3479–3487. doi: 10.1128/JVI.79.6.3479-3487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye FC, Blackbourn DJ, Mengel M, Xie JP, Qian LW, Greene W, Yeh IT, Graham D, Gao SJ. Kaposi's sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J Virol. 2007;81(8):3980–3991. doi: 10.1128/JVI.02089-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye FC, Zhou FC, Yoo SM, Xie JP, Browning PJ, Gao SJ. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J Virol. 2004;78(20):11121–11129. doi: 10.1128/JVI.78.20.11121-11129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SM, Z FC, Ye FC, Pan HY, G SJ. Early and sustained expression of latent and host modulation genes in coordinated transcriptional program of KSHV productive primary infection of human primary endothelial cells. Virology. 2005;343(1):47–64. doi: 10.1016/j.virol.2005.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Wang H, Herndier B, Ganem D. Restricted expression of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi's sarcoma. Proc Natl Acad Sci U S A. 1996;93(13):6641–6646. doi: 10.1073/pnas.93.13.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J Virol. 2002;76(12):6185–6196. doi: 10.1128/JVI.76.12.6185-6196.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoeteweij JP, Moses AV, Rinderknecht AS, Davis DA, Overwijk WW, Yarchoan R, Orenstein JM, Blauvelt A. Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of Kaposi's sarcoma-associated herpesvirus. Blood. 2001;97(8):2374–2380. doi: 10.1182/blood.v97.8.2374. [DOI] [PubMed] [Google Scholar]