


Abstract
Post-translational modification of histones and other chromosomal proteins regulates chromatin conformation and gene activity. Methylation and acetylation of lysyl residues are among the most frequently described modifications in these proteins. Whereas these modifications have been studied in detail, very little is known about a recently discovered chemical modification, the Nε-lysine formylation, in histones and other nuclear proteins. Here we mapped, for the first time, the sites of lysine formylation in histones and several other nuclear proteins. We found that core and linker histones are formylated at multiple lysyl residues located both in the tails and globular domains of histones. In core histones, formylation was found at lysyl residues known to be involved in organization of nucleosomal particles that are frequently acetylated and methylated. In linker histones and high mobility group proteins, multiple formylation sites were mapped to residues with important role in DNA binding. Nε-lysine formylation in chromosomal proteins is relatively abundant, suggesting that it may interfere with epigenetic mechanisms governing chromatin function, which could lead to deregulation of the cell and disease.
Mapping of post-translational modifications of histones has been an object of many recent studies (1–5). In depth identification of the modifications, their dynamics and comparative analyses of organismal differences, are important for understanding mechanisms of chromatin regulation. Proteins involved in DNA binding, regulation of chromatin condensation and DNA repair, appear to be modulated by sequential and cooperative interplay of a variety post-translational modifications including acetylation, methylation and phosphorylation (6,7). In addition to core histones (H2A, H2B, H3 and H4), proteins such as linker histone H1, high mobility group (HMG) and other chromosomal proteins can also influence the regulation of chromatin function. We and others recently showed that linker histones carry post-translational modification features similar to core histones (8,9). Analyses of post-translational modifications of mammalian linker histones revealed multiple sites of acetylation, methylation, phosphorylation and formylation. The latter modification was observed at residues that are known to be primarily involved in DNA binding and that were also found to be acetylated or monomethylated. As the observed modifications were restricted to only a few sites in H1, occurred in different cell culture models and tissues and with different extraction procedures, we suggested that formylation reflects a specific post-translational modification and is not an artifact resulting from extraction and analysis procedures (9).
Supporting this argument, more recently, Jiang et al. (10) have demonstrated that lysine formylation can arise from oxidative damage of DNA. They showed that 3′formylphosphate generated by oxidation of deoxyribose reacts with the Nε-moiety of lysine. Due to the spatial proximity to DNA, histones are the proteins primarily affected by this modification. The frequency of formylation in cultured human lymphoblastoid cells TK-6 has been estimated to be 0.1% of all lysines in acid soluble chromatin proteins. Furthermore, in histones this value can increase several-fold upon oxidative stress (10). Thus, formylated lysines can reach the abundance levels of more prominent histone modifications. Due to the chemical similarity of Nε-acetylation and Nε-methylation to Nε-formylation (Figure 1A), this modification may have functional consequences for histones.
Figure 1.

(A) Acetylated, dimethylated and formylated lysyl residues show similar structures. (B) Complete mass separation of the otherwise identical dimethylated and formylated peptides. Boxplot of mass deviations from theoretical masses for formylation and dimethylation in histone H3 measured using the LTQ-Orbitrap instrument. The box represents the interquartile range of 25–75%, the center line represents median, and ‘whiskers’ (small horizontal lines) extend to the most extreme ratio up to 1.5-fold interquartile range. The mass differences of dimethylated or formylated peptides are 28.0313 and 27.9949 Da, respectively, versus unmodified peptide. Mass deviations for dimethylation are taken from Supplementary Table S2.
High-resolution mass spectrometry has become an indispensable tool in elucidation of known and novel modification sites (11). In particular, the mass accuracy achievable with modern instruments such as the linear ion trap orbitrap mass spectrometer is at the low p.p.m. or sub-p.p.m. level. This accuracy makes it possible to unambiguously distinguish modifications that lead to the same nominal mass shift. We have recently used these mass spectrometric capabilities to investigate modifications in human linker histone H1. Among several other modifications, H1 proved to be formylated and we mapped the sites of this modification (9). In this study, we describe sites of formylation in histones and other nuclear proteins isolated by different extraction methods from a variety of cultured cell types as well as from human and mouse tissues. Our data show that the lysyl residues affected by this modification are also frequently found to be acetylated and/or methylated.
MATERIALS AND METHODS
Tissue
Human MCF7, HeLa and A549 cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were washed with PBS, pH 7.5 and stored at −80°C.
Extraction of nuclear proteins from mouse liver
Livers were dissected from mice of the inbred strain C57BL/6 that were sacrificed by decapitation. Nuclei were isolated according to the method of Blobel and Potter (12) with some modifications. Briefly, one liver was homogenized in 10 ml of 0.25 M sucrose in buffer A: 10 mM Mops/NaOH, pH 7.0, 10 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 1 mM DTT containing Complete Protease Inhibitor Cocktail (Roche Diagnostics, Mannheim, Germany). The homogenate was loaded onto the top of a 1.8 M sucrose layer and centrifuged at 28 000 g in 4°C for 30 min. The pellet containing nuclei was resuspended in 1 ml of 0.25 M sucrose in Buffer A. Nuclei were extracted with 1.4 M ammonium sulfate and the extracts were dialyzed against water.
Histone H1 extraction and purification
Frozen tissues were extracted with HClO4 as described previously (9). Briefly, ∼200−400 mg of each tissue was blended in 0.6−1.2 ml of 5% (v/v) HClO4 using an Ultra Turbax blender (IKA, Staufen, Germany) at the maximum speed of approx 25 000 r.p.m. for 30 s. The suspension was subjected to three freezing (−20°C), thawing and vortexing (10 s) cycles. The cultured cells were extracted in the same way but without initial homogenization. The final suspension was centrifuged at 15 000 g for 10 min. Pellets were re-extracted with 0.2 M H2SO4. Both supernatants were precipitated with ice cold 100% (v/v) CCl3COOH was added to a final concentration of 33% (v/v). The proteins were precipitated for 30 min on ice and collected by centrifugation at 15 000 g for 10 min. The pellets were washed with 0.2% HCl in acetone, then twice with pure acetone and vacuum-dried. The protein pellets were solubilized in 0.1% (v/v) CF3COOH in water and were separated on C18 reverse phase column as described previously (9).
Digestion and LC-MS/MS analysis
Proteins were resolved on 4–12% gradient Bis-Tris gels (NuPAGE, Novex, Invitrogen, Carlsbad, CA, USA) and stained using Colloidal Blue Stain Kit (Novex). Gel bands containing ∼0.5–2 µg protein were trypsinized as described previously (13). The resulting peptide mixtures were desalted using in-house made C18 STAGE tips (14), vacuum-dried and reconstituted in 0.5% acetic acid prior to analysis. In all steps any use of formic acid or formaldehyde was avoided.
The LC-MS/MS setup was similar to that described before (15). The samples were injected onto an in-house made fused-silica capillary column (15 cm, inner diameter 75 µm, packed with 3 µm ReproSil-Pur C18-AQ media (Dr Maisch GmbH, Ammerbuch-Entringen, Germany), using the Agilent 1100 nanoflow system (Agilent Technologies, Palo Alto, CA, USA). The LC setup was connected to either an LTQ Orbitrap or LTQ-FT mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with a nanoelectrospray ion source (Proxeon Biosystems, Odense, Denmark). The peptides were separated with 100 or 200 min gradients from 5 to 40% acetonitrile in 0.5% acetic acid. The data-dependent acquisition mode was employed. Survey MS scans were acquired either in the orbitrap with the resolution set to a value of 60 000 or in the FT with the resolution set to 100 000. Up to 5 most intense ions per scan were fragmented and analyzed in the linear trap. For accurate mass measurements of the survey MS scans the lock-mass option was used (16). Additional accurate mass measurements of the fragmentation products were performed in the orbitrap with the resolution set to 15 000.
The data were searched for post-translational modifications against the International Protein Index (IPI) database with the aid of the MASCOT (Matrix Science, London, UK) search engine (17). All relevant spectra were verified manually.
RESULTS
With the exception of the thiol-moiety of cysteine, the ε-amine of lysine is the strongest nucleophile in proteins and can undergo a variety of chemical reactions. These include the reaction with formaldehyde that results in formylation, and therefore procedures in which formaldehyde is used such as silver staining of gels, for example, have to be avoided (18). In our study, three different protein extraction methods were used to rule out the possibility that any of the reagents used were contaminated with formic acid or formaldehyde or could, directly or indirectly, generate Nε-formylation of lysines. For this reason histones were extracted either using 0.2 M sulfuric acid or from density gradient purified nuclei with ammonium sulfate. Linker histones were also extracted together with high mobility group proteins with 5% perchloric acid. To further rule out that formylation could be caused by particular cell culture conditions, our analyses of core histones were performed using three different cultured cell types: MCF7 breast cancer cells, HeLa cervical cancer cells and A549 lung cancer cells. Mouse livers also were used as a source of histones. Linker histones were prepared from MCF7 cells and from human cancer tissues.
Identification of Nε formylated lysines
Mass spectrometry (MS) is a powerful tool for the mapping of post-translational modifications in proteins, but the quality of the obtained data is highly dependent on the resolving power and mass-accuracy of the type of mass spectrometer employed. High accuracy MS instrumentation makes it possible to differentiate between moieties of the same nominal mass—thus helping to discern modifications such as trimethylation and acetylation. The presence of formylation on endogenous proteins was first suggested during in-depth profiling of H1 modifications with the LTQ-Orbitrap instrument (19) when some of the identified ‘dimethylated’ peptides had unusually high deviations from their theoretical masses (9). Dimethylation and formylation are modifications of the same nominal mass of 28 Da, but they differ by 0.0364 Da (Figure 1A). High-resolution mass spectrometers capable of mass measurement within a low p.p.m. window can easily distinguish these mass differences. Figure 1B shows the measured distribution of mass deviations of formylated and dimethylated peptides of histone H3 observed in different runs. The graph clearly demonstrates that these two modifications can be discriminated with high confidence by the accurate mass measurement. Fragmentation signatures of a histone H3 peptide that was identified as carrying either formylation or dimethylation are shown in Figure 2. Lysine-80 in the peptide 74–84 is the modification site. The fragmentation spectra appear almost identical when acquired in the low accuracy linear ion trap detection mode, and, without the knowledge gained from the high accuracy peptide mass measurement would likely have been misidentified. A lysine to arginine mutation could in principle also be the cause of a nominal mass increase of 28 Da (28.00615 Da). However, the difference of 0.01123 Da between this modification and formylation is easily discriminated by high accuracy mass spectrometry. In Figure 3, examples of high accuracy fragmentation spectra of peptides dimethylated and formylated at the same residue are shown. We have observed that trypsin cuts only weakly at the N-site of formylated lysine, what is in contrast to its activity on dimethylated lysine. Therefore identification of both modifications at the same residue was often possible only on peptides with different lengths. For this reason, it is difficult to make conclusions on the ratio of the abundances of dimethylation and formylation comparing the intensities of the relevant peptides in the chromatogram. Only in seldom cases, when a dimethylated peptide was miscleaved at the C-site of the dimethylated residue, a comparison of the ratios of both modifications at the same site was possible (Figure 4). For the peptide LLLPGELA*K from histone H2B (HeLa cells) formylation appears to be less abundant than dimethylation (Figure 4). Interestingly, the formylated peptide shows increased retention in comparison to its dimethylated version (Figure 4).
Figure 2.

The MS/MS fragmentation spectra of (A) EIAQDFfKTDLR formylated and (B) EIAQDFdmKTDLR dimethylated peptides of Histone H3. The major groups of ions produced on cleavage of peptide bonds by low energy collision fragmentation are b ions extending from the N-terminus of a peptide, and y ions extending from its C-terminal part. The comprehensive introduction to the peptide sequencing by mass spectrometry is given by Steen and Mann (33).
Figure 3.

High accuracy MS/MS spectra of peptides dimethylated (A, C, E) and formylated (B, D, F) at the same residue, respectively. (A and B) Modifications at K-108 in histone H2B. (C and D) Modifications at K-43 in histone H2B. (E and F) Modifications at K-77 in histone H4. In contrast to dimethylation, formylation was observed mainly on peptides that were not cut at the modified lysyl residue. An example of the MS and MS/MS data interpretation: after the MS scan, the ESYSVYVYKVLK peptide (B) was identified as carrying an extra mass of 27.995 Da that corresponds to the delta mass of formylation. Peptide fragmentation analysis reveals that, starting from y4 ion (KVLK), the formyl group is added to y-ion series. The observations, additionally supported by the presence of b10 (ESYSVYVYKV) and b11 (ESYSVYVYKVL) ions with the mass shift indicative of formylation, unambiguously position the modification at the middle lysyl residue—ESYSVYVYfKVLK. Average absolute mass accuracy of all assigned peaks was better than 2 p.p.m.
Figure 4.

Dimethylated (dm) and formylated (f) forms of the peptide LLLPGELA*K (from histone H2B; K-108) resolved by chromatography. Inserts show the mass envelopes of dimethylated and formylated peptides.
In this study, we isolated histones and other nuclear proteins from different cell types and tissues. These protein mixtures were digested with trypsin and online analyzed on the LTQ-Orbitrap as described in Materials and Methods section. Only the formylation sites observed at least in two analyses are presented in Table S1. In total we identified 165 peptides containing Nε-formyl lysine using high accuracy mass spectrometry (Supplementary Table S1). They represent 47 unique formylation sites in core and linker histones, high mobility group proteins and two other proteins (Table 1).
Table 1.
Formylation sites identified in human and mouse tissues
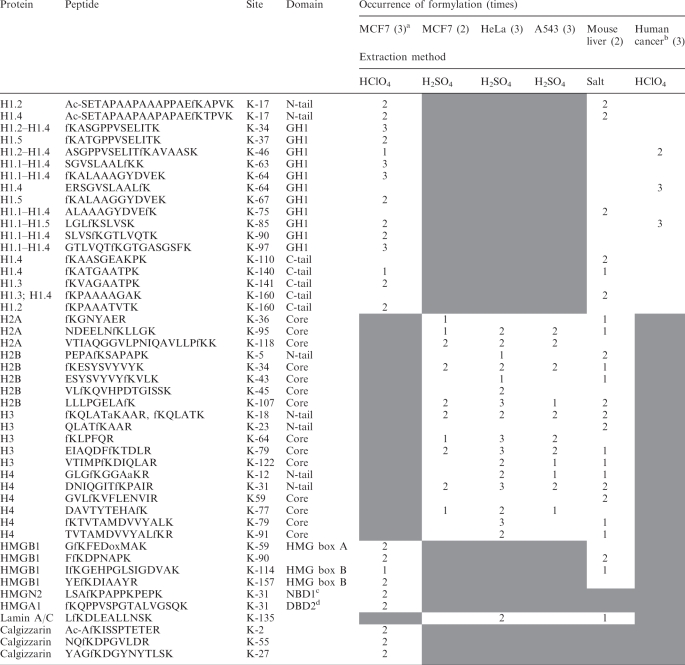 |
aNumber of independent experiments.
bSingle samples of breast ductal, colon, and ovarian cancer were analyzed.
cNBD, nucleosomes-binding domain.
dDBD, DNA-binding domain.
‘Shaded areas’ indicate that HClO4 extracts do not contain core histones and lamin A/C; in sulfuric acid extracts only protein bands containing core histones were analyzed; in salt extracts of mouse liver HMGN1 and HMGA1 were not identified; and in cancer samples only fractions containing linker histones were analyzed.
Core histones
Our analysis allowed identification of 19 Nε-lysine formylation sites in core histones that were located in their N-terminal tails and globular domains. Two sites of formylation were found in the tail of histone H3 and three in its globular domain (Table 1). The sites at K-18 and K-23 that are located in the tail of H3 are known to occur in both acetylated and methylated forms (3). In the core of H3 we identified formylation at K-64, K-79 and K-122. Methylation or dimethylation of these residues have been previously reported (20).
In histone H4, we identified one formylation site in the tail (K-12) and five sites in its globular domain. This site is functionally important because acetylated K-12 is selectively targeted by the BRD2 bromodomain and activates transcription from the cyclin E promoter (21). In histone H2A, three sites of formylation at K-36, K-95 and K-118 were mapped to the globular domain of the histone, whereas in histone H2B a site in the tail K-5 and four sites in the core of this histone were mapped at K-34, K-44, K-47 and K109. Lysine H2B-K-5 is known to be acetylated.
These data show that formylation often occurs at residues that can also be methylated and acetylated. Clearly, formylation of these sites prevents subsequent methylation and acetylation. Since many of those modifications are important in chromatin regulation, formylation could interfere with this process.
In the nucleosomal particles some of the histone lysyl moieties are involved in direct interactions with DNA (22). These include residues H3-K64, H4-K79, H2B-K34 that we identified in a formylated state and that create main chain interactions with DNA. Additionally, we measured formylation on H4-K77, which sits in the major groove of DNA (22). Thus, formylation of lysyl residues may also impair organization of the nucleosomes.
Linker histones
Analysis of linker histones revealed formylated lysines distributed over all three structural domains of these proteins: the N-terminal tail, the central domain and the C-terminal tail. For the most abundant H1 variant in human cells, the H1.4 protein, we identified 12 sites of formylation (Table 1). Seven of these sites are located in the central domain and three of them, K-64, K-85 and K-97 (H1.4 residue numbering), at residues involved in DNA binding (23,24). Since formylation, similar to acetylation, can interfere with DNA binding, it is possible that these modifications can significantly change the binding properties of H1. It is interesting in this context that nine of the formylated sites were previously identified as acetylated.
HMG proteins
Formylation sites were found in each on the dominant members of the high mobility group protein families HMGA, HMGB and HMGN. Formylation of K-31 in HMGA1 is located in one of the three DNA-binding domains of this protein. HMGN2 is the most abundant of the nucleosome-binding proteins and we found formylation in its nucleosome-binding domain. In the HMGB1 protein, four different formylation sites were mapped. Three sites are located in the HMG-box domains (K-59, K-114 and K-157 and in the linker between the domains (K-90). K-114 plays an important role in interaction with DNA, because mutation of this residue results in significant loss of DNA-binding affinity and supercoiling activity of the HMG-box B-domain (25).
Other proteins
Calgizzarin, also known as S100A11, is a calcium-binding protein implicated in a variety of biologic functions such as proliferation and differentiation, as well as in cancer. For a long time cellular localization of this protein was obscure, but recently calgizzarin was identified in nuclear fractions from cultured cells (26). We observed three formylation sites in this protein. Lamins are components of the nuclear lamina, a fibrous layer on the nucleoplasmic side of the inner nuclear membrane, which is thought to provide a framework for the nuclear envelope and may also interact with chromatin. Identification of formylation of K-135 in lamin A/C suggests that this residue may be located in the vicinity of DNA.
DISCUSSION
Recently, formylation has been recognized as a novel post-translational modification of lysine that occurs in histones (9,10). The two previous studies had mapped none (10) or three sites in a single protein (9). In this study, we have considerably extended our knowledge of the occurrence of this post-translational modification in histones and other nuclear proteins by proteomic mapping of 48 formylated sites in 10 different proteins. We took advantage of the high-mass accuracy of modern MS instrumentation that allowed unambiguous discrimination between dimethylation (28.0313 Da) and formylation (27.9949 Da). In addition, both modifications can be distinguished by different LC-retention times. We observed that formylation can affect lysyl residues important for interaction with DNA and also those modified by acetylation and methylation. Thus, formylation can impair the function of histones by interfering with the histone code-mediated formation and stabilization of binding sites for regulatory proteins such as transcription factors and regulators of chromatin activity.
Histone H1 was found to be the most frequently formylated among histones. Whereas in four core histones 19 formylation sites were identified altogether, in the most abundant linker histone variant H1.4 thirteen formyl-lysines were mapped. The higher number of H1 formylation sites correlates with the high degree of H1 formylation observed previously (10). The abundance of formylation in all three structural domains of H1 may reflect the fact that, similarly to the DNA-binding domain, both N-terminal and C-terminal tails are in close contact with DNA.
Since histones and some other chromosomal proteins have slow turnover rates (27–29), lysine formylation could accumulate with age and contribute to deregulation of chromatin function. Furthermore, lysine formylation is stimulated by oxidative stress (10) and, therefore, this modification may be involved in development of diseases linked to this stress, including cancer. In fact, lysine formylation may be involved in loss of protein function in the organism in an analogous way to other chemical modifications, such as glycation in diabetes (30) or oxidative damage in aging (31,32).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR online.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Piotr Ziółkowski (The Wroclaw Medical University, Poland) for the kind gift of human tissue biopsies, and Sonja Krüger for technical assistance. This work was supported by the Max-Planck Society for the Advancement of Science and by HEROIC, an Integrated Project funded by the European Union under the 6th Framework Programme (LSHG-CT-2005-018883). Funding to pay the Open Access publication charges for this article was provided by HEROIC.
Conflict of interest statement. None declared.
REFERENCES
- 1.Chu F, Nusinow DA, Chalkley RJ, Plath K, Panning B, Burlingame AL. Mapping post-translational modifications of the histone variant MacroH2A1 using tandem mass spectrometry. Mol. Cell. Proteomics. 2006;5:194–203. doi: 10.1074/mcp.M500285-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Medzihradszky KF, Zhang X, Chalkley RJ, Guan S, McFarland MA, Chalmers MJ, Marshall AG, Diaz RL, Allis CD, et al. Characterization of Tetrahymena histone H2B variants and posttranslational populations by electron capture dissociation (ECD) Fourier transform ion cyclotron mass spectrometry (FT-ICR MS) Mol. Cell. Proteomics. 2004;3:872–886. doi: 10.1074/mcp.M400041-MCP200. [DOI] [PubMed] [Google Scholar]
- 3.Hake SB, Garcia BA, Duncan EM, Kauer M, Dellaire G, Shabanowitz J, Bazett-Jones DP, Allis CD, Hunt DF. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J. Biol. Chem. 2006;281:559–568. doi: 10.1074/jbc.M509266200. [DOI] [PubMed] [Google Scholar]
- 4.Garcia BA, Barber CM, Hake SB, Ptak C, Turner FB, Busby SA, Shabanowitz J, Moran RG, Allis CD, et al. Modifications of human histone H3 variants during mitosis. Biochemistry. 2005;44:13202–13213. doi: 10.1021/bi050906n. [DOI] [PubMed] [Google Scholar]
- 5.Bonenfant D, Coulot M, Towbin H, Schindler P, van Oostrum J. Characterization of histone H2A and H2B variants and their post-translational modifications by mass spectrometry. Mol. Cell. Proteomics. 2006;5:541–552. doi: 10.1074/mcp.M500288-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Santos-Rosa H, Caldas C. Chromatin modifier enzymes, the histone code and cancer. Eur. J. Cancer. 2005;41:2381–2402. doi: 10.1016/j.ejca.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Garcia BA, Joshi S, Thomas CE, Chitta RK, Diaz RL, Busby SA, Andrews PC, Ogorzalek-Loo RR, Shabanowitz J, et al. Comprehensive phosphoprotein analysis of linker histone H1 from Tetrahymena thermophila. Mol. Cell. Proteomics. 2006;5:1593–1609. doi: 10.1074/mcp.M600086-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Wiśniewski JR, Zougman A, Krüger S, Mann M. Mass spectrometric mapping of linker histone H1 variants reveals multiple acetylations, methylations, and phosphorylation as well as differences between cell culture and tissue. Mol. Cell. Proteomics. 2007;6:72–87. doi: 10.1074/mcp.M600255-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Jiang T, Zhou X, Taghizadeh K, Dong M, Dedon PC. N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl Acad. Sci. USA. 2007;104:60–65. doi: 10.1073/pnas.0606775103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 12.Blobel G, Potter VR. Nuclei from rat liver: isolation method that combines purity with high yield. Science. 1966;154:1662–1665. doi: 10.1126/science.154.3757.1662. [DOI] [PubMed] [Google Scholar]
- 13.Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 14.Rappsilber J, Ishihama Y, Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 15.Olsen JV, Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc. Natl Acad. Sci. USA. 2004;101:13417–13422. doi: 10.1073/pnas.0405549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, et al. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics. 2005;12:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 18.Oses-Prieto JA, Zhang X, Burlingame AL. Formation of epsilon-formyllysine on silver-stained proteins: implications for assignment of isobaric dimethylation sites by tandem mass spectrometry. Mol. Cell. Proteomics. 2007;6:181–192. doi: 10.1074/mcp.M600279-MCP200. [DOI] [PubMed] [Google Scholar]
- 19.Makarov A, Denisov E, Kholomeev A, Balschun W, Lange O, Strupat K, Horning S. Performance evaluation of a hybrid linear ion trap/orbitrap mass spectrometer. Anal. Chem. 2006;78:2113–2120. doi: 10.1021/ac0518811. [DOI] [PubMed] [Google Scholar]
- 20.Garcia BA, Hake SB, Diaz RL, Kauer M, Morris SA, Recht J, Shabanowitz J, Mishra N, Strahl BD, et al. Organismal differences in post-translational modifications in histones H3 and H4. J. Biol. Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 21.Kanno T, Kanno Y, Siegel RM, Jang MK, Lenardo MJ, Ozato K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell. 2004;13:33–43. doi: 10.1016/s1097-2765(03)00482-9. [DOI] [PubMed] [Google Scholar]
- 22.Luger K, Richmond TJ. The histone tails of the nucleosome. Curr. Opin. Struct. Biol. 1998;8:33–40. doi: 10.1016/s0959-440x(98)80007-9. [DOI] [PubMed] [Google Scholar]
- 23.Goytisolo FA, Gerchman SE, Yu X, Rees C, Graziano V, Ramakrishnan V, Thomas JO. Identification of two DNA-binding sites on the globular domain of histone H5. EMBO J. 1996;15:3421–3429. [PMC free article] [PubMed] [Google Scholar]
- 24.Brown DT, Izard T, Misteli T. Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo. Nat. Struct. Mol. Biol. 2006;13:250–255. doi: 10.1038/nsmb1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Štros M, Muselikova E. A role of basic residues and the putative intercalating phenylalanine of the HMG-1 box B in DNA supercoiling and binding to four-way DNA junctions. J. Biol. Chem. 2000;275:35699–35707. doi: 10.1074/jbc.M007167200. [DOI] [PubMed] [Google Scholar]
- 26.Sakaguchi M, Miyazaki M, Kondo T, Namba M. Up-regulation of S100C in normal human fibroblasts in the process of aging in vitro. Exp. Gerontol. 2001;36:1317–1325. doi: 10.1016/s0531-5565(01)00097-3. [DOI] [PubMed] [Google Scholar]
- 27.Gurley LR, Hardin JM. The metabolism of histone fractions. II. Conservation and turnover of histone fractions in mammalian cells. Arch. Biochem. Biophys. 1969;130:1–6. doi: 10.1016/0003-9861(69)90002-2. [DOI] [PubMed] [Google Scholar]
- 28.Hancock R. Conservation of histones in chromatin during growth and mitosis in vitro. J. Mol. Biol. 1969;40:457–466. doi: 10.1016/0022-2836(69)90165-x. [DOI] [PubMed] [Google Scholar]
- 29.Ghidelli S, Claus P, Thies G, Wiśniewski JR. High mobility group proteins cHMG1a, cHMG1b, and cHMGI are distinctly distributed in chromosomes and differentially expressed during ecdysone dependent cell differentiation. Chromosoma. 1997;105:369–379. doi: 10.1007/BF02529752. [DOI] [PubMed] [Google Scholar]
- 30.Vlassara H, Palace MR. Diabetes and advanced glycation endproducts. J. Int. Med. 2002;251:87–101. doi: 10.1046/j.1365-2796.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 31.Johnson FB, Sinclair DA, Guarente L. Molecular biology of aging. Cell. 1999;96:291–302. doi: 10.1016/s0092-8674(00)80567-x. [DOI] [PubMed] [Google Scholar]
- 32.Huang H, Manton KG. The role of oxidative damage in mitochondria during aging: a review. Front Biosci. 2004;9:1100–1117. doi: 10.2741/1298. [DOI] [PubMed] [Google Scholar]
- 33.Steen H, Mann M. The ABC's (and XYZ's) of peptide sequencing. Nat. Rev. Mol. Cell. Biol. 2004;5:699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.